
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirect arsenic(III) sensing by a renewable gold plated Ir-based microelectrode†
Romain
Touilloux
,
Mary-Lou
Tercier-Waeber
* and
Eric
Bakker
*
Department of Inorganic and Analytical Chemistry, University of Geneva, Quai Ernest-Ansermet 30, 1211 Geneva 4, Switzerland. E-mail: marie-louise.tercier@unige.ch; eric.bakker@unige.ch
First published on 23rd March 2015
Abstract
We aim to determine arsenic(III) in natural aquatic systems in the nanomolar range and at natural pH. In view of a future application of a gel integrated electrochemical detection approach to reduce fouling and to control mass transport, we introduce here a microelectrode capable of quantifying As(III) that consists of a gold plated Ir-based microelectrode (Au-IrM). The key advantage of this approach is the ability to renew the Au layer by electrochemical control for better robustness in the field. The microsensor was electrochemically characterized by Square Wave Anodic Stripping Voltammetry. The obtained results demonstrate that the stripping peaks exhibit reproducible linear calibration curves at pH 8 for As(III) concentrations from 10 to 50 nM and from 1 to 10 nM, using 3 and 36 min preconcentration times, respectively. The interference by copper and chloride is negligible for an As![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Cu concentration ratio of 1:20 and a chloride concentration of 0.6 M typically found in seawater. The gold layer exhibits a lifetime of 7 days. The measurements are reproducible over time for a given gold layer (RSD < 9%) and between renewed layers (RSD ≤ 12.5%). While this work forms the basis for further progress on gel coated microelectrode arrays, As(III) detection in freshwater samples was successfully demonstrated here.
:Cu concentration ratio of 1:20 and a chloride concentration of 0.6 M typically found in seawater. The gold layer exhibits a lifetime of 7 days. The measurements are reproducible over time for a given gold layer (RSD < 9%) and between renewed layers (RSD ≤ 12.5%). While this work forms the basis for further progress on gel coated microelectrode arrays, As(III) detection in freshwater samples was successfully demonstrated here.
Introduction
Arsenic originates from natural and anthropogenic sources and is ubiquitous in natural waters. The natural arsenic concentration is typically in a range of 1 to 130 nM (1 to 10 ppb) in freshwater and 1 to 30 nM (0.5 to 2 ppb) in seawater and can be up to ∼13 μM (1 ppm) in contaminated areas. Natural arsenic contamination of surface and ground waters occurs by liberation and dispersion processes via weathering of arsenic-bearing rocks and release of deep arsenic-rich thermal waters into aquifers. The major anthropogenic sources are related to mining activities and its use in the production of ceramics, pesticides and fungicides (vineyards).Arsenic is one of the most toxic elements. It is classified as a group 1 human carcinogen by the International Agency for Research on Cancer.1 The major intake of arsenic by humans is due to the pollution of water, either by drinking contaminated water or through food treated with contaminated water. For these reasons, arsenic has been classed as a hazardous substance under European legislation (List II of the Water Framework Directive) and recently most countries have reduced the threshold value of arsenic in drinking water from ∼660 nM (50 μg L−1) to ∼130 nM (10 μg L−1) as recommended by the World Health Organisation (WHO).2 However, the true extent of the health hazards from arsenic depends on its physicochemical speciation. Arsenic exists in four oxidation states, −III, 0, +III and +V, and under inorganic and organic forms. In aquatic ecosystems, inorganic As(III) (trivalent arsenite species: H3AsO3, H2AsO3−) and As(V) (pentavalent arsenate oxyanions: H2AsO4−, HAsO42−) are predominant in water, while organic (e.g. arsenobetaine, arsenosugars, and methylated arsenic acids) forms are the main arsenic species in aquatic organisms.3,4 As(III) species are 60 times as toxic as the pentavalent salts and several hundred times as toxic as methylated arsenicals.5 The concentration and the proportion of the inorganic arsenite and arsenate species in waters are a function of the physicochemical properties and may vary continuously in time and space. The development of robust, selective and sensitive analytical methods for on-site mapping of inorganic arsenic species, and in particular As(III), at appropriate time scales is therefore of prime interest.
Electrochemical techniques are in principle well suited for this purpose6 as they are inexpensive and can be miniaturized for in situ monitoring.7,8 In the last decade, the development of electroanalytical procedures for the measurement of arsenic speciation in natural water samples has been reported by several groups, using a variety of electrode materials.6,9 Gold electrodes appeared to be the most suitable choice owing to a high hydrogen overpotential, which reduces the problem of simultaneous evolution of hydrogen during the preconcentration step.10 Moreover, gold exhibits a better reversibility of the electrode reaction in both the plating and the stripping step than other electrode materials.10 Jena et al.11 obtained good reproducibility (0.17%) with macroelectrodes between two sets of 20 measurements performed in two consecutive days. Still, with one measurement every 6 h, the signal decreased by 7% after 7 days of use. Li et al.12 developed on macroelectrode a system where 70 consecutive measurements give a standard deviation of just 5%, and furthermore, the electrode can be stored for 14 days while remaining functional. In order to improve the analytical performance, different kinds of gold electrodes were developed in addition to the solid gold electrode, especially electrodes based on gold coatings. These electrodes involve multistep fabrication processes,13,14 polymeric,15 alkyl terminated reagents15 or a deposition of gold nanoparticles on a substrate.16–19
The studies discussed above were performed on macroelectrodes. Modern electrochemical sensors are based on microelectrodes,20 where micro-sized disks and spherical or hemispherical electrodes (r ≤ 10 μm) have a few unique characteristics for voltammetric environmental monitoring.7,20 Their low iR drop enables direct measurements in low ionic strength freshwater, without addition of an electrolyte; this avoids the sample perturbations required for speciation analysis. A non-zero steady-state mass transport, resulting from (hemi-) spherical diffusion, is quickly established at constant potential, even in quiescent solution. Stirring the solution is thus unnecessary during the pre-concentration step of stripping techniques, which greatly improves the reliability of the analysis. Finally, thanks to their increased mass-transport and lower capacitance, a significantly larger signal-to-noise (S/N) ratio is obtained, relative to macroelectrode configurations, resulting in better sensitivity and, therefore, detection limit.20 These enhancements on gold microelectrode arrays were put forward by the group of Kounaves13 and Mardegan et al.21 (interconnected micro- and nano-arrays, respectively). These studies show an improvement of sensitivity with a nanomolar to picomolar range detection limit. Unfortunately, these low detection limits were obtained in an acidic media (0 ≤ pH ≤ 1).
Not only does an acidification step complicate the required instrumental protocol, but a perturbation of the sample pH will invariably also change the speciation equilibria of the sample, making such tools difficult to use in speciation analysis. This limitation has been recognized, and arsenite detection at neutral pH in synthetic electrolytes and seawater using a gold microwire electrode was recently demonstrated by Salaun et al.,22,23 and in freshwater by Gibbon-Walsh et al.24 These studies show a subnanomolar detection limit (0.2 nM and 0.5 nM, respectively) using a vibrating gold microwire, demonstrating enhanced mass transfer properties owing to the single micron-sized dimension of the electrode. A remaining drawback is the requirement of stirring during the pre-concentration step as a true steady state current is not observed at microwire shaped electrodes due to the associated hemicylindrical diffusion.25,26 Other remaining challenges in view of future in situ environmental studies include a limited reproducibility of measurements over time, due to an irreversible process between arsenic and gold.27 Alves et al.28 and others23,29 solved this problem by cleaning the electrode in acidic media at regular intervals. A system with a longer lifetime would demand less maintenance.
This work aims to develop the detection basis for the electrochemical quantification of As(III) in unperturbed water samples at pH 8 (in the range of natural pH) in view of an eventual gel integrated in situ environmental monitoring application with a long lifetime. As gold microelectrodes would too easily corrode in the SWASV detection of arsenite, the goal of this work is to develop a fully electrochemical protocol to deposit, remove and renew a gold coating on an iridium microelectrode which exhibits good affinity to gold. Once successful, this protocol will be used to control the deposition and re-oxidation of the gold layer across the gel coating to allow one to achieve reliable arsenite detection in natural waters, presented here as preliminary results.
Experimental
Chemicals and instrumentation
Suprapur water (Milli-Q; 18.2 MΩ.cm) was used for preparing all solutions. Tetrachloroauric acid (HAuCl4, 99.99%) used for the gold film deposition was purchased from Aldrich (Saint Louis, MO, USA). KSCN and As2O3 were products of Fluka (Deisendorf, Germany). Mercury acetate (Hg(CH3COO)2) was produced by Acros Organics (Geel, Belgium). Phosphate buffer (di-Potassium hydrogen phosphate form), HNO3 suprapur, NaNO3 suprapur (99.99%), NaCl suprapur (99.99%) and NaOH·H2O suprapur (99.99%) were purchased from Merck (Darmstadt, Germany). LGL agarose was purchased from Biofinex (Switzerland).Two stock solutions of As(III) of 13.3 mM and 0.1 mM were prepared in the presence of 8.6 mM NaOH and refrigerated at 4 °C before use. These stock solutions were used to prepare all arsenic solutions on the day of use. The 10 mM phosphate buffer was adjusted to pH 8 with 0.1 M HNO3 solution in the presence of 0.01 M NaNO3.
Electrochemical experiments were conducted with a μAutolab type II and a PGSTAT101 workstation (Metrohm AG, Switzerland) using the software NOVA v1.10 and a conventional three-electrode system. Potentials were referenced to a Ag/AgCl (3 M KCl) reference electrode (Metrohm AG, Switzerland) protected by an additional bridge of 0.1 M NaNO3 to avoid contamination. All experiments were conducted at room temperature in a home-made Faraday cage. All synthetic solutions were deoxygenated using a nitrogen stream for 10 min before measurement. A nitrogen atmosphere was maintained over the solution during the experiments. Scanning Electron Microscopy (SEM) images were achieved with a JEOL JSM7001F.
Working microelectrode
The working electrode consisted of a gold plated Iridium-based Microelectrode (Au-IrM). The IrM was a homemade single microelectrode composed of an electroetched iridium wire sealed in a glass capillary as shown by Tercier et al.30 The Ir microdisk radius was determined as 2.1 μm by cyclic voltammetry in a deoxygenated 6 mM K3Fe(CN)6 in 1 M NaNO3 solution.30 The electroplated Au layers were characterized using SEM images. As the geometry of the glass microelectrode used does not allow its incorporation into the scanning electron microscope, gold layers were deposited, under similar conditions, on microelectrode arrays made of 5 × 20 interconnected Ir microdisks of 5 μm diameter and a center-to-center spacing of 150 μm.31A 1.5% LGL agarose gel layer of 375 (±25) μm was added on top of the Au-IrM only for the preliminary results for environmental samples.32
Gold deposition and renewal
A solution of 1 mM Au(III), 0.1 M NaNO3 at pH 2 (HNO3), was used for gold chronoamperometric deposition. Gold film removal was achieved in a solution containing 5 mM Hg(CH3COO)2 and 1 M KSCN by Linear Sweep Voltammetry (LSV) between +300 mV and +800 mV with a scan rate of 5 mV s−1 and a step potential of 2.5 mV.Stripping voltammetric detection of As(III)
The electrochemical protocol used for the quantification of As(III) was Square Wave Anodic Stripping Voltammetry (SWASV). The SWASV parameters used were as follows: preconcentration potential (Eprec) of −1.0 V; preconcentration times (tprec) of 3 min or 36 min, for As(III) concentrations ≥10 nM or ≤10 nM respectively; 10 s equilibration time; 200 Hz frequency (f); 25 mV potential pulse amplitude (Esw); 8 mV potential step height (Es); and a potential stripping ramp from −1.0 V to 0.3/0.5 V. This step was preceded by a conditioning step at +500 mV for 30 s. Each measurement was followed by a second scan (background scan) using the same conditions but without the preconcentration and conditioning steps. This background scan was subtracted from the analytical scan to obtain a corrected scan.Sample collection
River freshwater was collected from the Arve river in Geneva; some samples were filtered on 0.45 μm pore size nitrocellulose membrane (Whatman, Dassel, Germany). All river samples were stored in polyethylene containers. Analyses were performed within 30 min. A 100 ml aliquot was bubbled with a mixture of N2 and CO2 gas for 10 min before measurement to remove oxygen while adjusting the pH value at 8.0. A N2/CO2 atmosphere was maintained over the solution during the experiments. The sampling polyethylene bottles were pre-cleaned using the following procedures: 24 h in 0.1 M HNO3 suprapur, 2 times 24 h in 10−2 M HNO3 suprapur; followed by dipping in Milli-Q water for 12 h after each acid washing step. They were then dried in a laminar flow hood and stored in double polyethylene bags.Results and discussion
Electro-deposition and renewal of gold film
We explore here the deposition of a gold layer, its use in arsenite detection, and the possibility of its subsequent removal and recoating as needed. All steps are performed by electrochemical control and without any mechanical treatment of the surface. If successful, this should provide the chemical basis for the development of gel integrated sensors for the in situ long-term detection of arsenite in aquatic systems. Different parameters were explored to optimize the electro-deposition and renewal of the gold film. The gold electrodeposition procedure requires an acceptable reproducibility for each new gold layer. The deposition potential was chosen from the cyclic voltammogram to observe only the current associated with gold reduction from Au(III) to Au(0). In the background electrolyte used for deposition (HNO3, pH 2), the potential window extends to −350 mV (Fig. S1†) and is limited by hydrogen reduction. The selected potential was therefore chosen as −300 mV to avoid parasitic reduction currents. Nevertheless, consecutive chronoamperograms performed at a potential of −300 mV showed poor reproducibility (Fig. 1a). | ||
| Fig. 1 Consecutive chronoamperograms (1–4) of Au deposition on an IrM obtained before (a) and after (b) the application of ten consecutive pulses (+800 mV, 50 ms) to oxidize/desorb impurities at the Iridium microdisk substrate. Edep = −300 mV; solution: 1 mM Au(III), 0.1 M NaNO3, HNO3 pH 2. | ||
The growth of gold films is known not to be homogeneous but depends on the numbers of nuclei formed in the first few seconds of the deposition process.16 As the presence of impurities is thought to promote or hinder nucleus formation, ten consecutive pulses of 50 ms at +800 mV were applied before deposition of the gold layer to help oxidize and desorb such impurities and prepare the iridium substrate. The success of this electrochemical treatment is demonstrated in Fig. 1, which shows consecutive chronoamperograms before and after the application of this pulse protocol in the presence (Fig. 1b) and absence (Fig. S2b†) of Au(III) in the solution. No residual parasitic current is observed after this pulse protocol in the electrolyte solution. As shown in Fig. 1b, the obtained chronoamperograms are almost identical, suggesting a reproducible coating approach. It is consequently possible to use a fixed time (86 s) to obtain reproducible gold layers in consecutive runs, see Table 1. The associated reduction charge, Qred, is used to determine the average layer thickness based on Faraday's law as follows:
 | (1) |
| Au layer no. | Q red/μC | Thickness/μm |
|---|---|---|
| 1 | 0.464 | 1.086 |
| 2 | 0.435 | 1.017 |
| 3 | 0.453 | 1.061 |
| 4 | 0.458 | 1.071 |
| Average | 0.453 ± 0.013 | 1.059 ± 0.030 |
The quality and characteristics of the gold layer were evaluated visually with a Scanning Electron Microscope (Fig. 2) and electrochemically by detecting As(III) (Fig. S3†). The desired properties are a gold film that does not extend over the edge of the Ir microdisk substrate and that gives a well-defined As(III) stripping peak. From the observations, the best result was obtained with a  ratio of 97959 C m−2 corresponding to a thickness of ∼1.1 μm (Qred = 0.432 μC for a rIr = 2.1 μm). Under these conditions, the SEM images revealed the presence of gold nanoparticles, with a size range of 60 to 100 nm, dispersed homogeneously on the interconnected Ir microdisk arrays (Fig. 2). A larger deposition charge gives wider and likely thicker films, and the associated stripping peaks become ill-defined and are not suitable for quantification of As(III) as shown in the example of Fig. S3c.†
ratio of 97959 C m−2 corresponding to a thickness of ∼1.1 μm (Qred = 0.432 μC for a rIr = 2.1 μm). Under these conditions, the SEM images revealed the presence of gold nanoparticles, with a size range of 60 to 100 nm, dispersed homogeneously on the interconnected Ir microdisk arrays (Fig. 2). A larger deposition charge gives wider and likely thicker films, and the associated stripping peaks become ill-defined and are not suitable for quantification of As(III) as shown in the example of Fig. S3c.†
 | ||
| Fig. 2 Scanning Electron Microscopy (SEM) image of an Au layer electroplated on a IrM-arrays using the following conditions: Edep = −300 mV; solution: 1 mM Au(III), 0.1 M NaNO3, HNO3 pH 2. | ||
For field-based instrumentation, any renewal of the Au-IrM needs to be as fast and as simple as possible. With solid gold microelectrodes, the electrode can be renewed chemically by cleaning with H2SO4,22 or mechanically by polishing.12 Since in the future we aim to apply these electrodes under a gel coating to control mass transport and reduce fouling, the removal of the gold film is performed only by electrochemical control under appropriate solution conditions (5 mM Hg(CH3COO)2 and 1 M KSCN). Mercury forms an amalgam with gold and helps to remove the gold film.33Fig. 3 shows the LSV response for the reoxidation of the film. When the current is integrated over time, the gold reoxidation charge (Qreox) is determined to be 0.645 μC, which is slightly higher than the original charge used to deposit the gold film (Qred = 0.432 μC for rIr = 2.1 μm) and may therefore suggest some co-oxidation of mercury. The gold removal takes just a few minutes to complete and allows one to renew the gold layer in a short period of time. The number of scans necessary to completely remove the Au layer is found to be always the same (2 ± 1) and confirms the reproducibility of the deposited gold film.
 | ||
| Fig. 3 Linear sweep voltammograms on Au-IrM resulting in complete reoxidation of the Au. Parameters: Ei = +300 mV, Ef = +800 mV, Eincrement = 2.5 mV, scan rate E = 5 mV s−1. solution: 5 mM Hg(CH3COO)2 in 1 M KSCN. | ||
Reproducibility and reliability of As(III) voltammetric measurement at pH 8
SWASV was used to detect As(III) on Au-IrM with an optimal gold layer thickness of ∼1.1 μm. The stripping peak potential corresponding to the reoxidation of the pre-concentrated As(0) into As(III) for the solution at pH 8, was −0.10 V. By using a short preconcentration time of just 3 min, the limit of quantification was found to be 10 nM (Fig. 4), and was decreased to 1 nM by prolonging the preconcentration time to 36 min (Fig. 5). Typical SWASV calibration curves, obtained by successive standard additions of As(III) in concentration ranges of 10 to 50 nM and 1 to 10 nM and using a pre-concentration time of 3 and 36 min, respectively, are reported in Fig. 4 and 5. The data shown are from five and three replicates, respectively, for each concentration. Good reproducibility and linearity (slopes: 0.0239 ± 0.0001 nA nM−1 and 0.1975 ± 0.0033 nA nM−1, respectively) were obtained for both concentration ranges. The average normalized slope obtained for the 10–50 nM (tprec = 3 min) range is 8.7 pA nM−1 min−1, whereas the one obtained for the 1–10 nM range is 5.5 pA nM−1 min−1. This suggests that the electrodes must be calibrated for a given preconcentration time. The limit of detection, determined as 3σ of the smallest quantifiable concentration, was found to be 2.7 nM and 0.5 nM for tprec of 3 min and 36 min, respectively. The calibration in the concentration range of 10 to 50 nM was repeated several times with different gold films and the observed slopes are shown in Table 2. Individually, the slopes exhibit a small error (RSD < 9%); while averaged, the standard deviation is somewhat higher (12.5%). | ||
| Fig. 4 Calibration plot (n = 5) of stripping peak currents as a function of increasing As(III) concentration (0, 10, 20, 30, 40 and 50 nM) in 0.01 M NaNO3 buffered at pH 8 (10 mM phosphate buffer). Inset: corresponding calibration curves. SWASV conditions: Eprecleaning = +500 mV (30 s); Eprec = −1000 mV; tprec = 3 min; Ei = −1000 mV; Ef = +300 mV; f = 200 Hz; ESW = 25 mV; Es = 8 mV. | ||
 | ||
| Fig. 5 Calibration plot (n = 3) of stripping peak currents as a function of increasing As(III) concentration (0, 1, 3, 5 and 10 nM), sample otherwise as in Fig. 4. Inset: corresponding calibration curves. SWASV conditions as in Fig. 4, except tprec = 36 min. | ||
| Slope/nA nM−1 | RSD/% | |
|---|---|---|
| Calibration 1 | 0.0239 ± 0.0001 | 0.4 |
| Calibration 2 | 0.0271 ± 0.0014 | 5.2 |
| Calibration 3 | 0.0230 ± 0.0020 | 8.7 |
| Calibration 4 | 0.0302 ± 0.0011 | 3.6 |
| All Data | 0.0261 ± 0.0032 | 12.5 |
The short-term stability of different Au layers was evaluated for three different films where the As(III) stripping peak current was determined in an identical solution containing 50 nM of As(III) for 20 measurements (Fig. 6). The first Au layer gave an As(III) peak current of 1.05 ± 0.01 nA (RSD 1.3%), the second Au layer, 0.99 ± 0.04 nA (RSD 4.1%) and the third layer, 0.99 ± 0.02 nA (RSD 2.4%), which demonstrates good interlayer reproducibility. If the results from these three films are taken together, the current is found to be 1.01 ± 0.03 nA, giving an uncertainty of 2.6%.
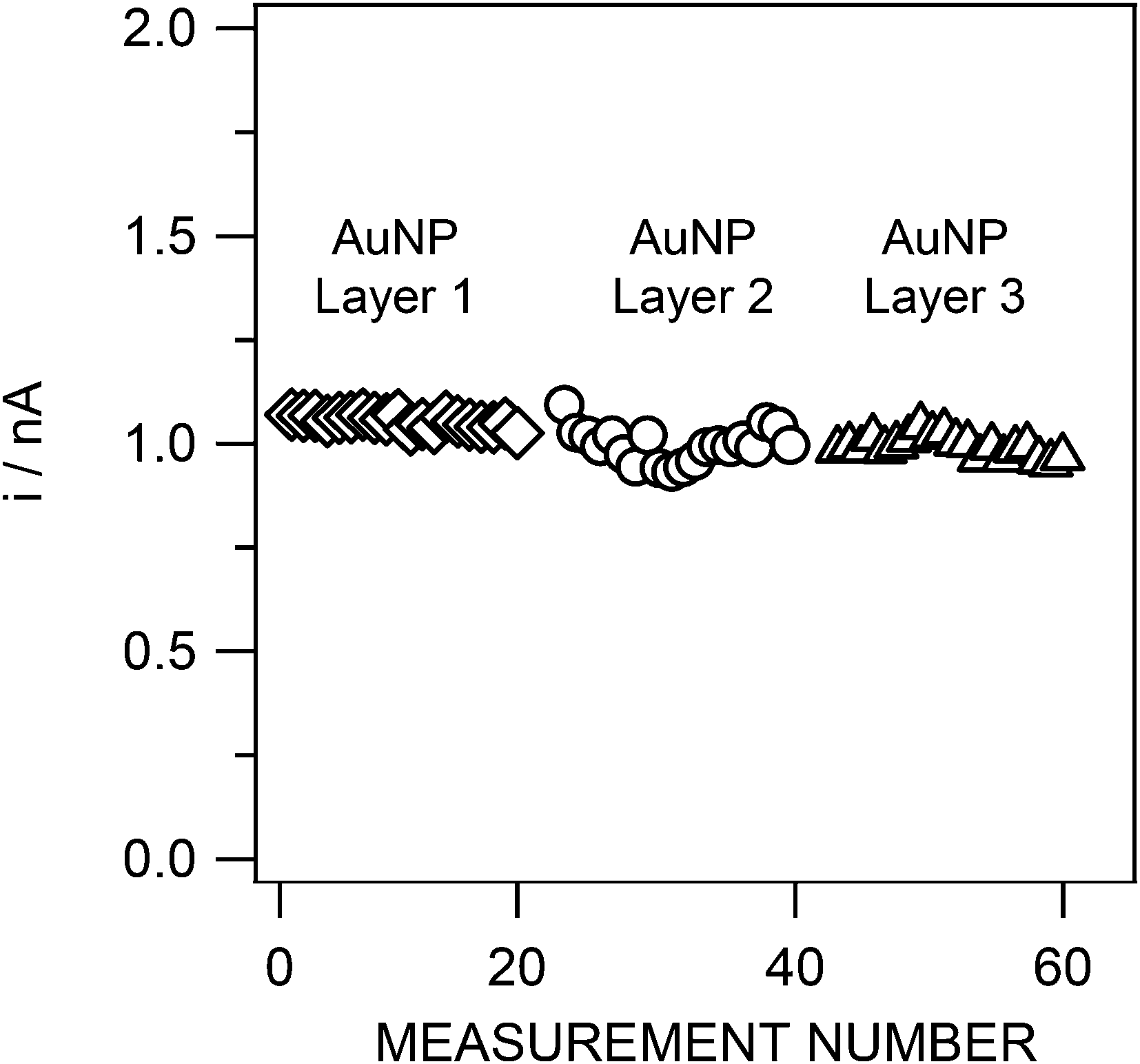 | ||
| Fig. 6 Reproducibility of the As(III) stripping peak current for three different gold layers (20 measurements for each layer). Sample: 50 nM As(III), otherwise as in Fig. 4. SWASV conditions as in Fig. 4. | ||
Long-term stability was studied by performing consecutive replicate SWASV measurements, in a pH 8 buffered (10 mM phosphate) 0.01 M NaNO3 solution spiked with 50 nM of As(III), until a significant decrease in the stripping As(III) peak current intensity was observed. On each day the solution was renewed to overcome any limited stability of As(III) over time. The results revealed an excellent reliability of the Au-IrM over a period of 7 days (Fig. 7). 3063 measurements, with a standard deviation of 2.4% (average of the associated As(III) peak current intensities: 1.04 ± 0.02 nA), were achieved during this period. This reproducibility is even more striking when the voltammogram of the first measurement is superposed to a voltammogram obtained seven days later (Fig. 7b); the two As(III) peaks are almost similar.
 | ||
| Fig. 7 (a) Reproducibility of the As(III) stripping peak currents of 3063 consecutive measurements performed over a period of 7 days; (b) Superposition of first (solid line) and 7 days later (dashed line) voltammograms. Sample and SWASV conditions as in Fig. 6. | ||
For comparison with the behaviour of a traditional gold electrode, a similar experiment was conducted with a solid gold disk microelectrode of 5 μm radius. For this purpose, the microelectrode was first polished and cleaned electrochemically over 15 s at −1.7 V in H2SO4.24 Consecutive SWASV measurements were then performed under the same conditions as reported above for the Au-IrM with the exception of the conditioning step, which was increased to 2 min (instead of 30 s). This was required as a memory effect between measurements was observed for conditioning time <2 min. Good reproducibility was observed for the first 50 replicates (RSD = 3.4%), whereas the peak current started to decrease for further measurements (Fig. S4†), where the Au-IrM continued to work reproducibly (Fig. 7). Moreover, two calibrations were performed using this solid gold microelectrode for two consecutive days under the same conditions. Two different slopes were obtained (0.0935 nA nM−1 and 0.0566 nA nM−1) that differed by ± 40% (Fig. S5†). This behaviour may be explained by incomplete reoxidation of As(III) between measurements. The longevity of this solid gold microelectrode can be related to the studies of Gibbon-Walsh et al.24 In that work, 20 replicates over 10 h for a solution of 10 ppb (130 nM) at pH 9 using a vibrating gold microwire were described, but reproducibility data were not given. The improved repeatability of 3000 consecutive measurements at pH 8 for 7 days in this study is welcome, but is not easily explained and might be due to the nanostructured gold coating. Based on these results, it is conceivable that this system could be applied to the real time data acquisition in environmental samples during at least few days with a single gold layer, and for a much longer period just by repeatedly renewing the gold layer, as demonstrated here.
Interferences
Interferences may manifest themselves by an overlap of the As(III) stripping peak with an interferent peak or by a decrease and/or broadening of the As(III) peak owing to adsorption of the interferent on the Au layer. Various species are known to interfere with electrochemical As(III) detection as anions (Cl−, Br−, I−, SO42−)11,24 and copper23 as well as colloidal/particulate matters and (bio)polymeric substances (humic and fulvic acids; extracellular polymeric substances). The latter are expected to be efficiently excluded by the protective gel layer31 which is shown here as a preliminary work. The focus was therefore to identify small dissolved molecule interferences, especially by copper and chloride. Copper interference in the determination of As(III)12,22 may manifest itself owing to the overlapping of the two metal stripping peaks. Chloride ions can interfere with gold34 or copper electrochemistry, resulting in potential decrease and/or broadening of the As(III) and Cu(II) peaks and shift in their peak potentials.35–37Stripping voltammograms obtained for 5 nM As(III) measurements in the presence of Cu(II) up to a molar ratio of As:Cu of 1:20 at pH 8 show good resolution of the two metal stripping peaks (Fig. 8). The As(III) stripping peak current is constant while the copper peak height varies. Fig. S6† shows linearity between the copper peak current and its concentration from 0 to 30 nM before reaching the saturation evidenced by a plateau. This suggests that copper interference is well controlled under the measurement conditions used here.
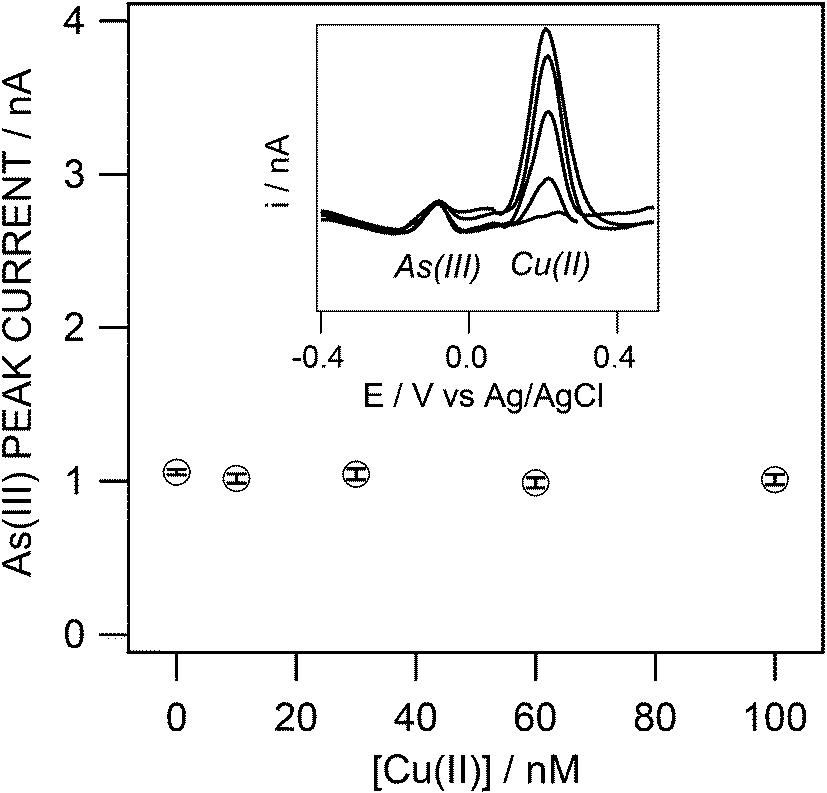 | ||
| Fig. 8 SWASV peak currents obtained for 5 nM As(III) in the presence of increasing concentrations of Cu(II) (0, 10, 30, 60 and 100 nM), average of 3 replicate measurements. Inset: corresponding curves. Sample and SWASV conditions as in Fig. 5. | ||
For chloride, experiments were conducted in the presence of 5 nM of As(III) and 30 nM of Cu(II), followed by the addition of 0.6 M of Cl− to study any variation of the As/Cu peak separation also. Chloride has almost no effect on the As stripping current and potential, shifting the peak by just +19.6 mV (Fig. 9a). Instead, the copper peak broadened and shifted to more positive potentials (Fig. 9a). This broadening suggests a stepwise Cu oxidation process (Cu(0)/Cu(I) and Cu(I)/Cu(II)).21 The positive shift of the copper peak may be due to a change in the adsorption affinity of the copper on the gold substrate in the presence of chloride.38 It was demonstrated that the adsorption effect was minimized by the application of a preconcentration potential below −0.8 V.38 For the solid gold microelectrode the copper shifted to positive potentials while diminishing in amplitude, but there was no evidence of peak broadening. For As(III) a somewhat larger positive potential shift of +51.6 mV along with a broadening of the peak was observed (Fig. 9b). For both types of electrodes, this resulted in an improved separation of the peaks between As and Cu in the presence of Cl− (ΔAu-IrMAs/Cu = +30.1 mV and +33.0 mV, ΔAu-solidAs/Cu = +34.9 mV and +42.9 mV, before and after addition of 0.6 M Cl−, respectively) on a gold surface as equally observed by Alves et al.28 and Bonfil et al.37
 | ||
| Fig. 9 SWASV peak currents obtained for 5 nM As(III) in the presence of 30 nM Cu(II) with (dashed line) and without (solid line) 0.6 M Cl− on the Au-IrM (a) and a solid gold microelectrode (b). Sample and SWASV conditions as in Fig. 5. | ||
Application to As(III) determination in natural waters
The SWASV method was tested to determine As(III) in unfiltered and filtered Arve river water samples. These experiments, as before, were performed without agitation. The samples were previously degassed by a mixture of N2/CO2 to remove O2 and to adjust the pH to 8.7 Owing to the nature of the sampled oxic surface water, the natural As(III) concentration was below the limit of quantification of the Au-IrM and explained why no As(III) was measured before spiking. A well-defined peak appears after the addition of 5 nM (tprec = 36 min). Nevertheless, the peak intensity was found to be lower than the one obtained in pH 8 phosphate buffered 0.01 M NaNO3 electrolyte with a recovery of only 71% (Table 3; Fig. S7a†). When the sample was filtered (Fig. S7a†), or when a LGL agarose gel layer was coated over the Au-IrM surface (Fig. S7b†), the recovery increased to 97% and 99.3%, respectively, (Table 3). These findings demonstrated that the method developed allows direct As(III) detection in natural waters, provided that natural fouling materials are excluded from the sensor surface. This can be successfully achieved by covering the sensor with an LGL agarose gel layer as previously reported7,8,31,32,39 and confirmed here.| Sample | 1.5% LGL agarose coating | As(III) added/nM | As(III) meas./nM | % Recovery |
|---|---|---|---|---|
| NF Arve river | No | 0 | 0 | 0 |
| NF Arve river | No | 5 | 3.55 ± 0.03 | 71 ± 0.8 |
| F Arve river | No | 5 | 4.85 ± 0.03 | 97.0 ± 0.6 |
| NF Arve river | Yes | 5 | 4.93 ± 0.03 | 99.3 ± 0.6 |
Conclusions
This work shows the development of a reproducible and electrochemically renewable Au-IrM with the ability to quantify As(III) using SWASV. The As(III) stripping peak currents observed at pH 8 show good reproducibility and reliability up to 7 days. The gold layer can be renewed by electrochemical control. The calibration curves show linearity until a limit of quantification of 1 nM (LOD = 0.5 nM) using a 36 min pre-concentration time. Moreover, the interference of copper and chloride is negligible for the As:Cu concentration ratio of 1:20 and the chloride concentration of 0.6 M typically found in seawater. The presence of chloride actually improves peak separation. This system was successfully applied in a filtered fresh water matrix with a low concentration of As(III). In analogy to our previous work on voltammetric gel integrated systems7,8,31,32 the long term goal is to use interconnected microelectrode arrays covered by a hydrogel film to control mass transport by pure diffusion, and to minimize interferences by natural inorganic colloids/particles and (bio-)polymeric substances (fouling problem). We are confident that this strategy is the first step of an attractive basis for further development and eventual deployment of an electrochemical sensor probe for direct arsenite analysis in aquatic systems.
Acknowledgements
This work is supported by the European Union Seventh Framework Programme (FP7-OCEAN 2013.2 SCHeMA project – Grant Agreement 614002), the Swiss National Science Foundation and the University of Geneva. The authors would also like to thank Prof. Rossana Martini and Agathe Martignier, University of Geneva, Department of Earth Sciences, for the SEM images, as well as Stéphane Noël and Adrien Matter, University of Geneva master students, for preliminary work.References
- Some Metals and Metallic Compounds, IARC Monogr. Eval. Carcinog. Risk Chem. Hum., 1980, 23.
- World Health Organisation, Guidelines for Drinking Water Quality, 1993, p. 41 Search PubMed.
- B. K. Mandal and K. T. Suzuki, Talanta, 2002, 58, 201–235 CrossRef CAS.
- P. L. Smedley and D. G. Kinniburgh, Appl. Geochem., 2002, 17, 517–568 CrossRef CAS.
- G. M. P. Morrison, G. E. Batley and T. M. Florence, Chem. Br., 1989, 25, 91–96 Search PubMed.
- J. H. T. Luong, E. Lam and K. B. Male, Anal. Methods, 2014, 6, 6157–6169 RSC.
- J. Buffle and M. L. Tercier-Waeber, TrAC, Trends Anal. Chem., 2005, 24, 172–191 CrossRef CAS PubMed.
- M. L. Tercier-Waeber and M. Taillefert, J. Environ. Monit., 2008, 10, 30–54 RSC.
- J. Ma, M. K. Sengupta, D. X. Yuan and P. K. Dasgupta, Anal. Chim. Acta, 2014, 831, 1–23 CrossRef CAS PubMed.
- G. Forsberg, J. W. Olaughlin, R. G. Megargle and S. R. Koirtyohann, Anal. Chem., 1975, 47, 1586–1592 CrossRef CAS.
- B. K. Jena and C. R. Raj, Anal. Chem., 2008, 80, 4836–4844 CrossRef CAS PubMed.
- D. Y. Li, J. Li, X. F. Jia, Y. C. Han and E. K. Wang, Anal. Chim. Acta, 2012, 733, 23–27 CrossRef CAS PubMed.
- R. Feeney and S. P. Kounaves, Anal. Chem., 2000, 72, 2222–2228 CrossRef CAS.
- R. Baron, B. Sljukic, C. Salter, A. Crossley and R. G. Compton, Russ. J. Phys. Chem. A, 2007, 81, 1443–1447 CrossRef CAS.
- Y. S. Song, G. Muthuraman, Y. Z. Chen, C. C. Lin and J. M. Zen, Electroanalysis, 2006, 18, 1763–1770 CrossRef CAS.
- X. Dai, O. Nekrassova, M. E. Hyde and R. G. Compton, Anal. Chem., 2004, 76, 5924–5929 CrossRef CAS PubMed.
- L. Rassaei, M. Sillanpaeae, R. W. French, R. G. Compton and F. Marken, Electroanalysis, 2008, 20, 1286–1292 CrossRef CAS.
- L. Xiao, G. G. Wildgoose and R. G. Compton, Anal. Chim. Acta, 2008, 620, 44–49 CrossRef CAS PubMed.
- A. O. Simm, C. E. Banks, S. J. Wilkins, N. G. Karousos, J. Davis and R. G. Compton, Anal. Bioanal. Chem., 2005, 381, 979–985 CrossRef CAS PubMed.
- Microelectrodes: Theory and Application, ed. M. I. Montenegro, M. A. Queiros and J. L. Daschbach, 1991 Search PubMed.
- A. Mardegan, P. Scopece, F. Lamberti, M. Meneghetti, L. M. Moretto and P. Ugo, Electroanalysis, 2012, 24, 798–806 CrossRef CAS.
- P. Salaun, K. B. Gibbon-Walsh, G. M. S. Alves, H. M. V. M. Soares and C. M. G. van den Berg, Anal. Chim. Acta, 2012, 746, 53–62 CrossRef CAS PubMed.
- P. Salaun, B. Planer-Friedrich and C. M. G. van den Berg, Anal. Chim. Acta, 2007, 585, 312–322 CrossRef PubMed.
- K. Gibbon-Walsh, P. Salaun and C. M. G. van den Berg, Anal. Chim. Acta, 2010, 662, 1–8 CrossRef CAS PubMed.
- P. M. Kovach, M. R. Deakin and R. M. Wightman, J. Phys. Chem., 1986, 90, 4612–4617 CrossRef CAS.
- P. J. Gellings and H. J. Bouwmeester, Handbook of Solid State Electrochemistry, Taylor & Francis, 2010 Search PubMed.
- Z. Jia, A. O. Simm, X. Dai and R. G. Compton, J. Electroanal. Chem., 2006, 587, 247–253 CrossRef CAS PubMed.
- G. M. S. Alves, J. M. C. S. Magalhaes, P. Salaun, C. M. G. van den Berg and H. M. V. M. Soares, Anal. Chim. Acta, 2011, 703, 1–7 CrossRef CAS PubMed.
- R. Prakash, R. C. Srivastava and P. K. Seth, Electroanalysis, 2003, 15, 1410–1414 CrossRef CAS.
- M. L. Tercier, N. Parthasarathy and J. Buffle, Electroanalysis, 1995, 7, 55–63 CrossRef CAS.
- C. Belmont-Hebert, M. L. Tercier, J. Buffle, G. C. Fiaccabrino, N. F. de Rooij and M. Koudelka-Hep, Anal. Chem., 1998, 70, 2949–2956 CrossRef CAS.
- M. L. Tercier and J. Buffle, Anal. Chem., 1996, 68, 3670–3678 CrossRef CAS.
- Standard Potentials in Aqueous Solution, ed. A. J. Bard, R. Parsons and J. Jordan, Marcel Dekker, Inc., 1985 Search PubMed.
- J. Wang, Stripping Analysis: Principles, Instrumentation, and Applications, VCH Verlagsgesellschaft, 1985 Search PubMed.
- A. Cavicchioli, M. A. La-Scalea and I. G. R. Gutz, Electroanalysis, 2004, 16, 697–711 CrossRef CAS.
- J. P. Arnold and R. M. Johnson, Talanta, 1969, 16, 1191–1207 CrossRef CAS.
- Y. Bonfil, M. Brand and E. Kirowa-Eisner, Anal. Chim. Acta, 2000, 424, 65–76 CrossRef CAS.
- Z. C. Shi and J. Lipkowski, J. Electroanal. Chem., 1996, 403, 225–239 CrossRef.
- M. L. Tercier-Waeber, F. Confalonieri, G. Riccardi, A. Sina, S. Noel, J. Buffle and F. Graziottin, Mar. Chem., 2005, 97, 216–235 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an00151j |
| This journal is © The Royal Society of Chemistry 2015 |