
Nanomaterial-based biosensors using dual transducing elements for solution phase detection
Ning
Li
a,
Xiaodi
Su
*a and
Yi
Lu
ab
aInstitute of Materials Research and Engineering, Agency for Science, Technology and Research (A*STAR), 3 Research Link, 117602 Singapore. E-mail: xd-su@imre.a-star.edu.sg
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA
First published on 25th February 2015
Abstract
Biosensors incorporating nanomaterials have demonstrated superior performance compared to their conventional counterparts. Most reported sensors use nanomaterials as a single transducer of signals, while biosensor designs using dual transducing elements have emerged as new approaches to further improve overall sensing performance. This review focuses on recent developments in nanomaterial-based biosensors using dual transducing elements for solution phase detection. The review begins with a brief introduction of the commonly used nanomaterial transducers suitable for designing dual element sensors, including quantum dots, metal nanoparticles, upconversion nanoparticles, graphene, graphene oxide, carbon nanotubes, and carbon nanodots. This is followed by the presentation of the four basic design principles, namely Förster Resonance Energy Transfer (FRET), Amplified Fluorescence Polarization (AFP), Bio-barcode Assay (BCA) and Chemiluminescence (CL), involving either two kinds of nanomaterials, or one nanomaterial and an organic luminescent agent (e.g. organic dyes, luminescent polymers) as dual transducers. Biomolecular and chemical analytes or biological interactions are detected by their control of the assembly and disassembly of the two transducing elements that change the distance between them, the size of the fluorophore-containing composite, or the catalytic properties of the nanomaterial transducers, among other property changes. Comparative discussions on their respective design rules and overall performances are presented afterwards. Compared with the single transducer biosensor design, such a dual-transducer configuration exhibits much enhanced flexibility and design versatility, allowing biosensors to be more specifically devised for various purposes. The review ends by highlighting some of the further development opportunities in this field.
 Ning Li | Ning Li obtained his B. Eng from National University of Singapore in 2013. He is an A*STAR scholar and performed his one-year research attachment in Institute of Materials Research and Engineering (IMRE), A*STAR, supervised by Dr Xiaodi Su. His current research interests focus on the design of nanomaterial-based biosensors and disease diagnosis techniques. |
 Xiaodi Su | Dr Xiaodi Su received her PhD in 1995 from Nankai University, China, and majored in Analytical Chemistry. Currently she is a senior scientist in the Institute of Materials Research & Engineering (IMRE), A*STAR, Singapore, heading the Materials Analysis and Characterization capability group. Her research interests include developing advanced analytical technologies for biomedical research and developing biosensors for disease diagnosis, environmental monitoring, and food analysis. |
 Yi Lu | Yi Lu received his BS degree from Peking University in 1986, and a PhD degree from University of California at Los Angeles in 1992. After 2 years of postdoctoral research with Professor Harry B. Gray's group at Caltech, he started an independent career in the Department of Chemistry at the University of Illinois at Urbana Champaign in 1994. He is now a Jay and Ann Schenck Professor of Chemistry in the Departments of Chemistry, Biochemistry, Bioengineering and Materials Science and Engineering. His research interests lie at the interface between chemistry and biology, including bioinorganic chemistry, biomaterials chemistry and bioanalytical chemistry. |
Introduction
Detection of biological agents and sensing of particular bio-reactions are of great importance for biochemical and biomedical applications,1 especially for early diagnosis of certain diseases, monitoring therapeutic prognosis and critical biomarker identification. While various types of bioassay methods are available, many are costly, labour intensive and involve tedious assaying steps. Thus, significant demands have still not been met, particularly on developing biosensors with high selectivity and sensitivity, as well as reasonable robustness, cost-effectiveness, versatility and portability.2 A typical biosensor features two basic components: a biological sensing probe that provides selective target binding and a transducing element that transforms the binding into detectable signals. Therefore, the performance of a biosensor, in terms of limit of detection (LOD), selectivity, response time and signal-to-noise ratio, depends heavily on the quality of biological sensing probes and transducing elements, as well as the interfaces between them. This requirement imposes major effort on the development of related materials.In 1959, Richard Feynman pointed out that “There's plenty of room at the bottom”. Decades later, nanoscale science and engineering emerges and exerts extraordinary impact on many technological fields. Owing to the quantum confinement effect induced by its 10−9 m scale, nanomaterials demonstrate various unprecedented and unique properties that can be applied broadly in improving the target-recognizing and signal-transducing mechanisms in biosensor design.3 In addition, they have comparable size with the common biomolecules such as DNA and proteins, affording integration of the nanomaterials with biomolecules for advanced biosensor development.4–6 To date, a large number of nanomaterial-based biosensors have been reported and reviewed from different perspectives, such as targets to be detected, types of transducing elements, detection principles, and sensing performance.5,7–29 Among all the reported works, however, many use a single nanomaterial sensing element to generate signals, such as the colour change of metal nanoparticles due to analyte-induced aggregation and dispersion,8,10,15,20,23 or the conductance change in 1-D nanowire (e.g., carbon nanotube or conducting polymer chain) induced by analyte adsorption.29
In addition to the biosensors involving a single nanomaterial transducer, biosensors using dual transducing elements of inorganic nanomaterials or a combination of an inorganic nanomaterial with an organic luminescent agent offers greater promise of advancing the biosensor research by enhancing both the sensitivity and selectivity of the sensors.9–11,15,19,20,23 However, few specific review articles have focused on and systematically covered this topic. We hereby provide a review focusing on solution-phase biosensors with dual transducing elements, particular on selection of transducing materials, design principle, application and performance. The dual transducing elements here are exclusive of the sensing probes that are often biomolecules. Benefiting from the dual transducers and their interplay with the sensing probe, we can therefore expect improved assay sensitivity and selectivity over single transducer design. Herein, the term “biosensor” refers to any assay method that can detect certain biological/biochemical substances or monitor biological reactions/mechanisms, with the use of one or more physicochemical transducer(s), although in some other studies stricter criteria apply (e.g., capable of continuously sensing a biological process or a target30). The “solution-phase” bioassay/biosensor sometimes is also called the “Mix-and-Measure” or “Separation-Free” detection method, in which both target binding and signal transducing occur in homogeneous solution, and the signal acquisition can be accomplished using common spectrophotometers and/or microplate readers, but not sophisticated equipment.
Within this review, we first introduce several important classes of nanomaterials, especially their unique chemical and optical properties appealing to biosensor construction. Then, the majority of the discussion will focus on the various dual-transducer-based mechanisms including Förster Resonance Energy Transfer (FRET), Amplified Fluorescence Polarization (AFP), Bio-barcode Assay (BCA) and Chemiluminescence (CL), as well as their representative examples that have been demonstrated by worldwide research communities recently. Due to the availability of the large variety of fluorescent transducers and labels, FRET, which describes the coupling energy transfer between two fluorescent agents, is highly versatile, and thus accounts for a considerably larger quantity of literature than the other three. We therefore divide its discussion based on the target analytes (e.g., metal ions and small molecules, DNA, proteins and enzymes). Schematic drawings are created in the FRET and AFP sections, to provide visualization of the design rules for different analytes under the respective principles. With the wide coverage of inorganic nanomaterials, ranging from quantum dots, metal nanoparticles, upconversion nanoparticles, and carbon based nanomaterials (e.g. graphene, graphene oxide, carbon nanotube, and carbon dots), this review provides a perspective of the emergence of different generations of nanomaterials for analytical usage in the fields of chemical sensing, biosensing and diagnosis. At the end of this review, we also try to answer the following reflective questions: what are the additional opportunities that nanomaterial-based dual-transducer biosensors can provide in comparison with the single transducer-based counterpart? And what are the advantages and disadvantages of each of the four dual-transducer sensing principles discussed in this review? (Supported by a Holland Vocational Interest Test illustration.)
Nanomaterials used for constructing biosensors
The unique chemical and physical properties of utilized nanomaterials, as well as their interaction with sensing probes and the other transducing elements, lay the foundation for biosensor construction and excellent performances. In this section, we aim to provide a brief introduction to several nanomaterials that have been intensively investigated for constructing dual-transducer based biosensors. The covered materials include quantum dots (QDs), metal nanoparticles (mNPs), upconversion nanoparticles (UCNPs), graphene oxide (GO), carbon nanotubes (CNTs), as well as carbon nanodots (CDs). Table 1 illustrates the favourable properties, major roles of each nanomaterial in dual-transducer based biosensors, as well as the challenges that need to be taken care of for sensor design.| Nanomaterial | Favorable properties | Major roles in design | Challenges | Other remarks |
|---|---|---|---|---|
| QDs | • Tunable fluorescence (Fig. 1) | • FRET donor (more preferential) | • Cytotoxicity43 | • Surface functionalization is required to increase aqueous solubility and biocompatibility |
| • Photostability | • FRET acceptor | • Fluorescence blinking | ||
| • Emission brightness | • No standard synthetic protocol | |||
| Metal NP | • LSPR absorption (Fig. 2) | • Fluorescence quencher/enhancer | • Non-specific aggregation (e.g. during particle surface modification or assaying procedures) | • Size-dependent optical behavior (e.g., absorption dominates for AuNPs <80 nm; Scattering for any larger ones)71 |
| • Light scattering | • Fluorescence polarization amplifier | • Various other particle shapes (e.g. rods, plates, triangles, stars.) to provide tunable absorption spectra | ||
| • Facile surface modification (e.g. Au–thiol interaction) | • Carrier for DNA barcode or CL reaction catalyzers | |||
| • Intrinsic affinity to ssDNA and proteins through coordination chemistry | ||||
| UCNPs | • NIR excitation (Fig. 3) | • FRET donor | • Low quantum yield77 | • Reduced background noises relative to organic dyes and QDs90 |
| • Photostability | • NIR heating effect80 | • Surface modification is required to increase aqueous solubility and biocompatibility | ||
| Graphene, GO, CNT | • Optical quenching (Fig. 4) | • Fluorescence quencher | • Non-specific adsorption | • GO is more commonly used than intact graphene. |
| • Different binding affinity with ds- and ssDNA | • Fluorescence polarization amplifier | • CNT structure heterogeneity | • Intrinsically suitable for multiplexed sensing because of large surface area | |
| • Enzyme mimicking | • CL reaction catalyzer | |||
| Carbon nanodot | • Tunable fluorescence | • FRET donor | • Lack of thorough understanding on physical and chemical properties | • Usually need further surface modification (e.g. ligand attachment or solid capping layers) for efficient emission115,117 |
| • Facile synthesis | ||||
| • Low fabrication cost | ||||
Quantum dots
Quantum dots (QDs), first reported by Brus and colleagues in 1983,31 is a family of semiconducting nanocrystallized fluorophore that is small enough to exhibit the quantum confinement effect (Fig. 1a and b).13 They have been widely explored in diverse biological fields primarily as fluorescent labelling agents, by virtue of their excellent brightness and photostability. Many reviews have been published regarding their physical properties, synthesis, surface chemistry, and various applications such as biosensors, clinical therapy and in vivo bioimaging.13,21,22,32–35 Of particular interest are their broad absorbance spectrum, narrow emission band and tunable emission wavelength, which afford high flexibility in choosing the excitation wavelength and modulating the emission spectra among different QDs (Fig. 1c). These optical properties are intrinsically favourable for designing biosensors with dual transducing elements. For example, coupling QDs into the FRET system is an effective approach to improve design versatility and performances, by virtue of the QDs compatibility with various other fluorophores and quenchers.36 In addition, QDs have multitude functionalization sites on their large surface area. This becomes advantageous when conjugation of several sensing probes is desired for multiplexed detection37 or for improving the detection sensitivity with multiple donors attached to a single acceptor and vice versa.13 | ||
| Fig. 1 (a) Schematic drawing and (b) TEM image of CdSe–ZnS core–shell QD. (c) Illustrative size, photograph and photoluminescence spectrum showing progressive colour change of CdSe–ZnS QD with increasing diameter. Reprinted with permission from ref. 13, © 2011 American Chemical Society. | ||
For many as-synthesized QDs, surface modification is necessary to improve their biocompatibility. There are two general strategies for modifying QDs’ surface, namely ligand exchange and surface coverage.38–42 In the former, bifunctional linker molecules are utilized to replace the hydrophobic capping agents on the as-synthesized QDs, affording both introduced aqueous stability and extra functional groups for further functionalization.42 In the latter, QDs’ hydrophobic surface is simply shielded by an additional layer of amphiphilic molecules, possibly also with a protective semiconducting shell in between.34,40 Some compositional elements of QDs, like Cd, are widely considered cytotoxic, hindering much of their practical applications, especially in vivo.43 However, no consensus on QDs’ cytotoxicity has been reached so far,44 as there have been reports of cytotoxicity being detected,45 but also others of it being absent.46,47 Bottrill and Green recently reviewed various aspects of QDs cytotoxicity, and concluded with an optimistic outlook about the future of QDs in biotechnology.43 One effective way to tackle the cytotoxicity problem is perhaps to encapsulate QDs with nontoxic layers (e.g., silica and ZnS).48 On the other hand, QDs made of cytotoxicity-free or less toxic materials, such as Si, InP and InGaP, could be another antidote to the cytotoxicity problem.49–52
Metal nanoparticles
Metal nanoparticles (mNP), particularly gold nanoparticles (AuNP), have long been attractive transducing elements for biosensor design, by virtue of their facile synthesis, non-linear optical property, fluorescence quenching and enhancement capability, as well as their versatile surface chemistry.17–20,53 A detailed discussion on AuNP synthesis, surface functionalization, and physical and chemical properties can be found in ref. 20. In favour of colloidal stability, the AuNP surface has to be covered with capping agents like citrate ions (Fig. 2a).54 These agents can be further replaced by other desired substances like oligonucleotides, peptides, polymers and proteins, which establish the foundation of AuNP versatility for biological applications. Optical absorption of colloidal AuNP solutions is governed by the well-known Localized Surface Plasmon Resonance (LSPR) (Fig. 2b). Originating from LSPR, two types of colorimetric biosensors have been well developed, relying on particle aggregation and change of ambient dielectric constant principles, respectively. Under the aggregation principle, it can be further divided into crosslinking55–58 and non-crosslinking59–64 schemes. Mirkin and co-workers55 pioneered the crosslinking research and were able to detect a femtomolar (fM = 10−15 M) level oligonucleotide target in solution. Lu and co-workers further advanced this field with reversed colour change using DNAzyme or aptamer as the sensing probe, which is able to cleave the substrate when cofactors (e.g. Pb2+, Uranyl) are present, or able to interact with the target to dehybridize the crosslinker that initially connects neighbouring AuNPs.65–70 Alternatively in the non-crosslinking scheme, the analytical target possesses the capability of agitating the colloidal stability and allows van der Waals attraction to irreversibly aggregate AuNPs. In the second design principle that relies on the ambient dielectric constant change, adsorption of complexes onto the AuNP surface is able to red-shift the LSPR band.71 Such band shifting depends on the adsorbent layer thickness but produces very limited colour change. Therefore, microplate readers might be needed to show the differences in the light absorption spectra.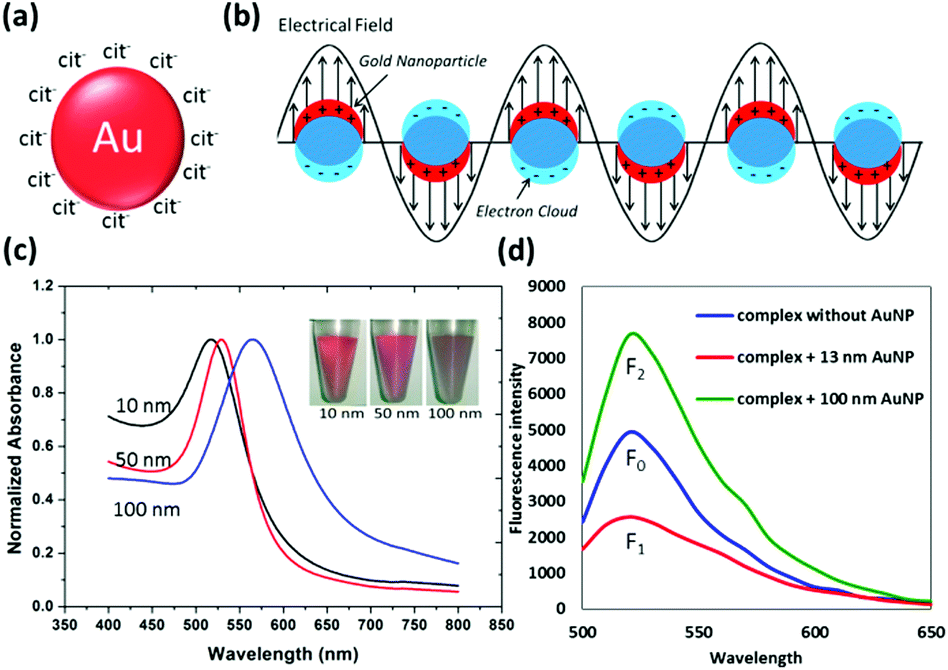 | ||
| Fig. 2 (a) Schematic drawing of citrate-capped AuNP; (b) simplified illustration of localized surface plasma resonance, where the free electrons in the conduction band are driven into oscillation due to the coupling with an electrical field of incident light; (c) aqueous absorbance spectra of citrate-capped AuNPs with diameters of 10 nm, 50 nm and 100 nm (inset is the digital photograph of AuNP solution); (d) effect of the fluorescence quenching (curve F1, for 13 nm AuNP) and enhancement (curve F2, for 100 nm AuNP) for the FAM-contained complex. Panel (d) is reprinted with permission from ref. 71, © 2014 Springer. | ||
It is worth highlighting that the optical behaviour of mNPs actually has two components: absorption and scattering that are responsible for fluorescence quenching and enhancement, respectively (Fig. 2d). Both of them are favourable for the biosensor construction with dual transducing elements and benefit from their capabilities of modulating proximal fluorophore emission. For example, the AuNP of appropriate size is able to silence the neighbouring fluorophore over a much longer distance than that for organic quenchers, rendering itself great competence to be a superquencher in those fluorescence based dual-transducer designs. In addition, the versatile surface chemistry of AuNP is another contributing factor for its prominent excellence. Thiol-containing molecules have demonstrated great suitability as an anchor onto AuNP, by virtue of a strong thiol–Au interaction and availability of thiol groups in many biomolecules and organic compounds. For instance, cysteine-tagged peptides are being widely utilized to functionalize AuNPs for different applications, including aqueous stability enhancement, matrilysin (MMP-7) detection, botulinum neurotoxin sensing, as well as gene regulation.72–75
Upconversion nanoparticles
Upconversion describes a phenomenon in which a fluorophore is excited at longer wavelength and then gives off shorter wavelength emission.76–80 Since most inorganic crystals do not demonstrate such a property, attention is mainly focused on a particular system that comprises a crystalline host and lanthanide dopants (Fig. 3a and b).77 Detailed discussion on the upconversion nanoparticle (UCNP) synthesis, surface modification, fluorescence tuning and various applications in biological research can be found in ref. 77–80. For a given host matrix like NaYF4, by carefully changing lanthanide dopant combination and concentration, the emission can be finely tuned from visible to near infrared (NIR) regions upon single wavelength excitation (Fig. 3c and d).81,82 However, it is also of note that the host matrix does play an important role in the overall optical behaviour. For example, it has been found that the hexagonal phase (β) NaYF4 exhibits orders of magnitude higher quantum efficiency than the cubic phase (α).83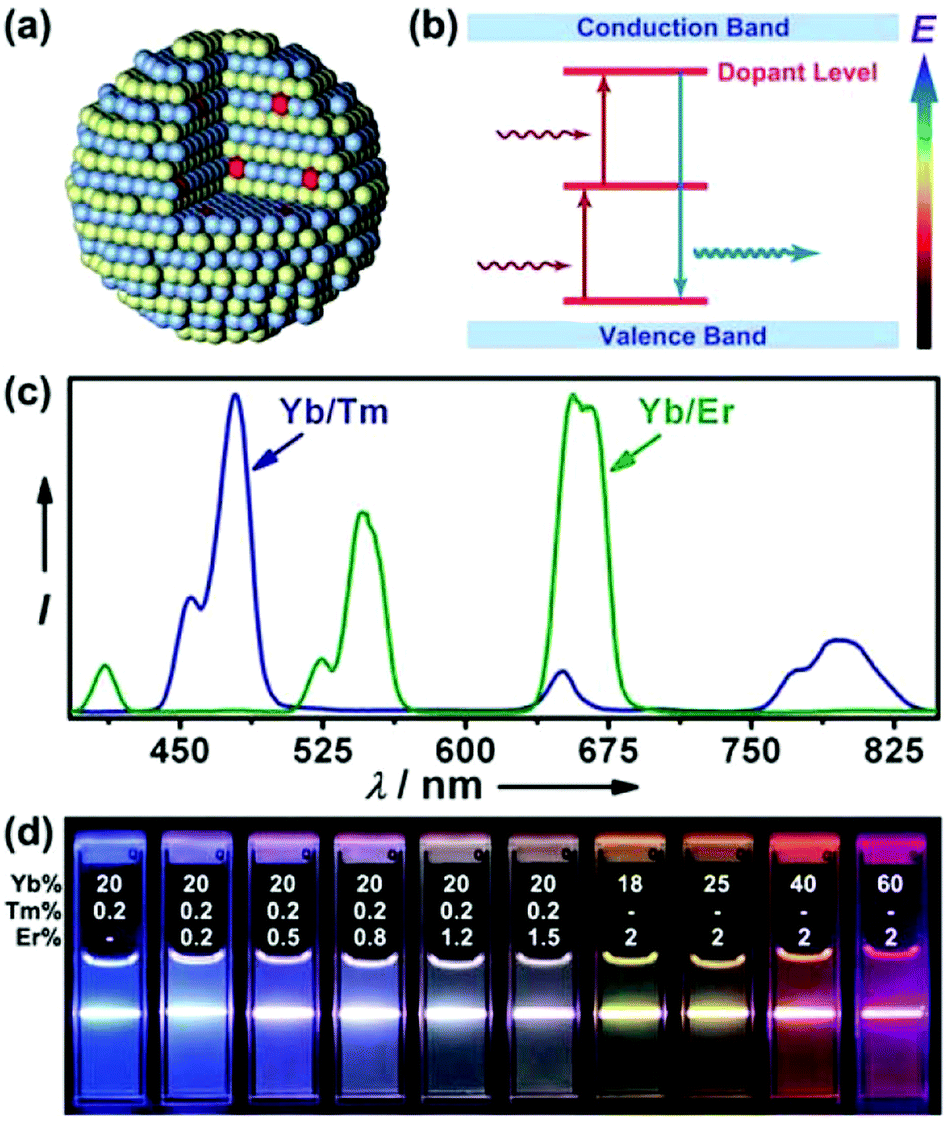 | ||
| Fig. 3 (a) Schematic drawing of UCNPs with lanthanide dopant ions embedded in the nanocrystal host, (b) simplified energy level diagram for the energy transfer mechanism between different lanthanide dopants, (c) emission spectra of NaYF4: Yb/Tm (20/0.2 mol%) and NaYF4: Yb/Er (18/2 mol%), (d) UCNP emission colour tuning by varying the dopant combination and concentration. Panel (a) and (b) are reproduced with permission from ref. 90, © 2010 Royal Society of Chemistry. Panel (c) and (d) are reprinted with permission from ref. 82, © 2008 American Chemical Society. | ||
Despite various synthetic approaches for diverse UCNP morphologies,76–78,84–86 many of them only produce the hydrophobic surface that cannot be directly used for biological applications. Currently, in the literature, two strategies are available for UCNP surface modification, namely encapsulation with SiO2 or amphiphilic copolymers and replacement of the original organic layer with hydrophilic ligands.87–89 Such a concept is similar to the QD surface modification that has been discussed in the corresponding section.
As a new class of luminescent material, UCNPs have drawn much scientific attention, and have been widely considered as a promising alternative to organic dyes and QDs for diverse applications. They offer tunable luminescence wavelength, high quantum yield, as well as excellent photostability. Therefore, those well-established biosensor design principles for QDs and organic dyes can be extended to UCNPs with ease. More importantly, the anti-Stokes shift allows infrared (IR) or near infrared (NIR) excitation, which can significantly lower the interfering noises from the background and also increase the penetration depth.90 All these characteristics make UCNPs highly suitable for bioanalytical studies in complex medium or even in vivo. Nevertheless, coupling UCNP with other transducing elements (e.g., luminescent reporters and fluorescence quenchers) facilitates more design versatilities possibly with further enhanced sensing performances.
Graphene, graphene oxide and carbon nanotube
Graphitic material is not new, considering its wide usage as pencils, lubricants and electrical conductors. However, after the Nobel Prize winning study on single- or bi-layer graphene,91 enormous attention has been re-focused on this star material as well as its various derivatives like graphene oxide (GO) and carbon nanotube (CNT). Thanks to their diverse favourable properties,92–94 graphene-derived nanomaterials have drawn enormous attention for biological studies including biosensors, disease diagnosis, drug delivery and cell imaging.95,96Despite the intact graphene that has been successfully synthesized via many approaches,91,97–103 it is graphene oxide that attracts more attention in biosensor studies (Fig. 4a). The epoxy, hydroxyl and carboxyl functional groups not only allow more versatile surface functionalization, but also contribute to excellent aqueous solubility and bio-compatibility. The carbon nanotube, as inferred by the name, is in a cylindrical configuration.104,105 Further classification divides CNT into single-walled carbon nanotube (SWNT) and multi-walled carbon nanotube (MWNT) based on their specific structure (Fig. 4b).106 It is of particular note that the functional groups on CNT mainly locate at the end tips, leaving the side walls highly aromatic.
 | ||
| Fig. 4 (a) Schematic structure of graphene and graphene oxide. (b) Schematic structure of single-walled carbon nanotube and multi-walled carbon nanotube. (c) Illustration of GO quenching: fluorescence spectra of 50 nM FAM in the absence (black curve) and presence of GO with various concentrations (from top to bottom: 5, 10, 15, 20, 25, 30, 35 μg mL−1). (d) Illustration of CNT quenching: fluorescence spectra of 50 nM ssDNA-tethered FAM in the presence of a CNT quencher and various concentration of target DNA (target DNA can hybridize with an ssDNA probe, and recovers the FAM fluorescence from CNT quenching). (e) SWNT fluorescence spectra in the absence (Curve 1) and presence (Curve 2) of a quencher. Panel (a) is reproduced with permission from ref. 103, © 2008 WILEY. Panel (b) is reproduced with permission from ref. 107, © 2007 Elsevier. Panel (c) is reprinted with permission from ref. 108, © 2010 WILEY. Panel (d) is reprinted with permission from ref. 109, © 2008 American Chemical Society. Panel (e) is adapted with permission from ref. 110, © 2007 Nature Publishing Group. | ||
In dual-transducer based biosensors, GO and CNT mainly perform as fluorescence quenchers, working cooperatively with various fluorophores and sensing probes for both “light-on” and “light-off” sensor construction (Fig. 4c and d). Among many of the demonstrated characteristics, different binding affinities towards ssDNA and dsDNA are under intensive consideration. By virtue of the accessibility of nucleobases in flexible ssDNA sequences, they can easily adsorb onto the aromatic surface; whereas the rather rigid phosphate backbone in dsDNA hinders its effective binding with the aromatic basal plane. Stemming from such affinity differences, many novel biosensor designs with dual transducing elements have been reported. Among some special cases, biomolecules in vertical orientations can also be established onto the aromatic surface, with the assistance of linkers or specially designed functional groups.111 The crosslinking-based sensing principle can therefore be employed.
Despite the similarities, GO and CNT also differ in many aspects. A thorough and detailed comparative study was reported by Braet and co-workers recently, with particular focus on material structures, properties and applications in biosensor design.105 Although most GO or CNT-based optical assays rely on the fluorescence quenching phenomenon, there are also examples in which GO/CNT enzyme-mimicking catalytic capability and CNT NIR photoluminescence (Fig. 4e) are employed for biosensor construction.105,110,112
Carbon nanodots
Carbon nanodots (CDs), as a new class of carbon allotrope material, have attracted much scientific attention worldwide.113 Most CDs are quasi-spherical nanoparticles (<10 nm in size) with numerous carboxylic acid moieties on the surface (Fig. 5a and b). They inherit the favourable characteristics of QDs including emission tunability and photobleaching resistance, but are exempt from the shortcomings that conventional QDs suffer from. Following its serendipitous discovery in 2004,114 many synthetic approaches have been developed for effectively fabricating CDs of different sizes and surface functional groups.115,116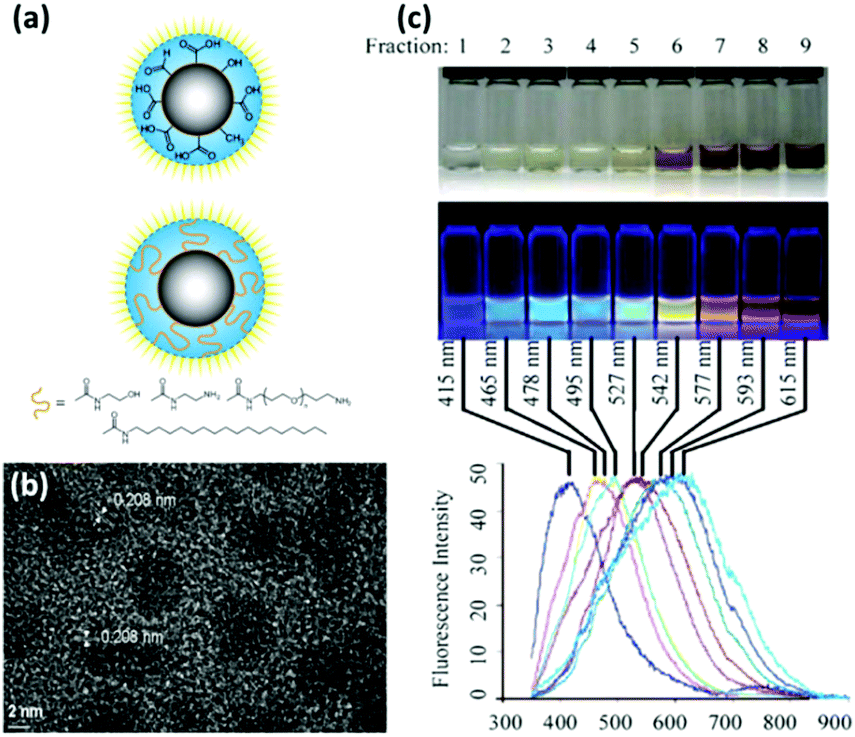 | ||
| Fig. 5 (a) Schematic drawing of a carbon nanodot after surface oxidative treatment and further functionalization with surface-passivation agents. (b) High resolution TEM image of carbon nanodot. (c) Photograph of a polyacrylamide gel electrophoresis (PAGE)-separated carbon nanodot produced from candle soot under white and UV excitation, as well as the corresponding fluorescence emission spectra. Panel (a) is reproduced with permission from ref. 115, © 2010 WILEY. Panel (b) is reprinted with permission from ref. 122, © 2009 American Chemical Society. Panel (c) is adapted with permission from ref. 124, © 2007 WILEY. | ||
The carbon atoms within CDs have a great sp2 character that indicates its nanocrystalline graphite signature. Typically, CDs demonstrate strong absorption in the UV region, with a tail well extended to the visible part.117–121 Although the origin of CDs’ photoluminescence has not yet been fully understood, accumulating evidence points to the fact that it arises from the radiative recombination of excitons located at surface energy traps.117–119 Similarly, mechanisms behind CDs’ surface passivation and quantum yield are also not clear at this moment and more effort should be devoted to them.121–124
Recently, various studies on CDs have demonstrated many of their biologically favourable characteristics, such as photostability and tunable photoluminescence.125–133 As a promising alternative to QDs, CDs contain no cytotoxic element, rendering more application possibilities in biotechnology, especially in the cases involving live cells or in vivo studies. In addition to the well-established role as bioimaging labels, the dual-transducer biosensor design has witnessed CDs’ involvement as FRET donors and acceptors,134 as well as enzyme mimics for chemiluminescence reactions.135 The use of CDs in biosensor research is just starting to emerge. By virtue of all the favourable characteristics discussed above, intensive explorations and many more appealing achievements can be expected in the near future.
Mechanisms and applications of dual-transducer principles
As aforementioned, a typical biosensor features two fundamental components: a sensing probe for target recognition, and a transducing element for transforming biological binding events into detectable signals. The target recognition step generally relies on the bioaffinity interactions, including but not limited to the reactions between complementary nucleic acid strands, antibody and antigen, aptamer and target, biotin and avidin, enzyme and substrate, as well as DNA molecules and their binders (e.g. drugs, metal ions, and proteins). These specific interactions are then transduced into the change of fluorescence intensity and/or polarization, solution colour, or some other optical signals. Such signals are readily detectable using ordinary lab equipment and even the naked eye in some cases. In this section, dual-transducer based biosensor designs are discussed. The content is organized according to the transducing mechanisms, namely Förster Resonance Energy Transfer (FRET), Amplified Fluorescence Polarization (AFP), Bio-barcode Assays (BCA) and Chemiluminescence (CL) principles. With the assistance of schematic drawings of various assay principles and representative examples, we attempt to provide an overall picture of the dual-transducer based biosensor research, with particular focus on the general design rules and respective performances for the desired target analytes.Förster resonance energy transfer (FRET)
Förster Resonance Energy Transfer (FRET) was first reported by Theodor Förster in 1948.136 It describes an energy transfer mechanism between proximal fluorophores via transition dipole–dipole coupling. Given aligned dipole orientation and appropriate overlap between donor emission and acceptor absorption, energy transfer efficiency follows the correlation of 1/[1 + (R/R0)6]. R is the distance between a donor and an acceptor. R0 is the Förster radius defined as the distance at which 50% energy is transferred. This radius is a function of ambient refractive index, degree of spectra overlap, quantum yield of donor–acceptor pair, as well as the dipole-orientation factor. Due to the inverse 6th power relationship, small variation of the inter-fluorophore distance can lead to a tremendous change in the energy transfer efficiency. This makes FRET a sensitive and convenient technique for sensing the distance variation and conformational change of biomolecules. However, the effective FRET distance is less than 10 nm, which is usually too short for large proteins, protein complexes or DNAs of moderate length.137 Therefore, it is greatly desired to increase the distance over which effective FRET can occur. Theoretical work done in the 1980s suggested that noble metal particles are able to improve the FRET efficiency of its proximal donor–acceptor pairs.138 Experimentally, Lakowicz and co-workers demonstrated that by attaching a silver nanoparticle (20 nm diameter) onto the donor, the Förster radius can be increased from 8.3 to 13.0 nm and the FRET rate constant near the particle is found to be 21 times higher than that without the silver nanoparticle.139,140Though FRET was originally defined as the energy transfer phenomenon between two fluorophores,136,141 nowadays researchers tend to name a system FRET as long as the fluorescence energy is transferred to a proximal unit, regardless of whether it is a fluorophore or fluorescence quencher.142 To avoid any ambiguity in this review, we follow the latter definition of FRET, in which energy transfer could happen in either fluorophore/fluorophore or fluorophore/quencher combination. Of note, the FRET efficiency between a fluorophore and a metallic quencher is inversely related to the 4th power of the distance, instead of 6th in the conventional design.143–145 This is possibly due to the surface isotropic distribution of the dipole vectors on metal nanoparticles. Such energy transfer is sometimes referred as nanosurface energy transfer (NSET).146 Herein, we merely treat it as a sub-category of FRET. To quantify the quenching effect, plots of donor fluorescence intensity change I/I0 and quenching efficiency (1 − I/I0) are commonly utilized, in which I0 and I are the donor fluorescence intensities in the absence and presence of the quencher, respectively. In some cases, the fluorescence data can also be fitted into the Stern–Volmer equation, (I0/I) − 1 = KSVCA, where CA is the quencher molar concentration and KSV is the corresponding Stern–Volmer quenching constant.147 However, it is worth mentioning that such a Stern–Volmer relation is more commonly used to describe diffusion-driven collision between the donors and acceptors.
Given the flourishing research on nanomaterial,148–150 FRET, as a flexible fluorescence-based design principle, has been customized into various configurations for much wider applications than ever before. This results in the exceedingly larger quantity of FRET related literature than that on the other three dual-transducer mechanisms in later sections. To make these versatile FRET designs easy to follow, discussions in this section are divided into three parts according to their sensing target, including metal ions and small molecules, DNA, proteins and enzymes. In each part, schematic drawings are provided to illustrate how the presence of analyte can be transduced into detectable signals. As a general trend, the quenching-based principle is more preferentially applied, mainly because of its higher energy transfer efficiency than the conventional designs with energy transferred from one fluorophore to the other. In addition, the light-on assay is usually more sensitive than the light-off, due to its smaller standard deviation (δ) of the blank sample signal. Such a δ value is often used to calculate the limit of detection according to the 3δ rule.
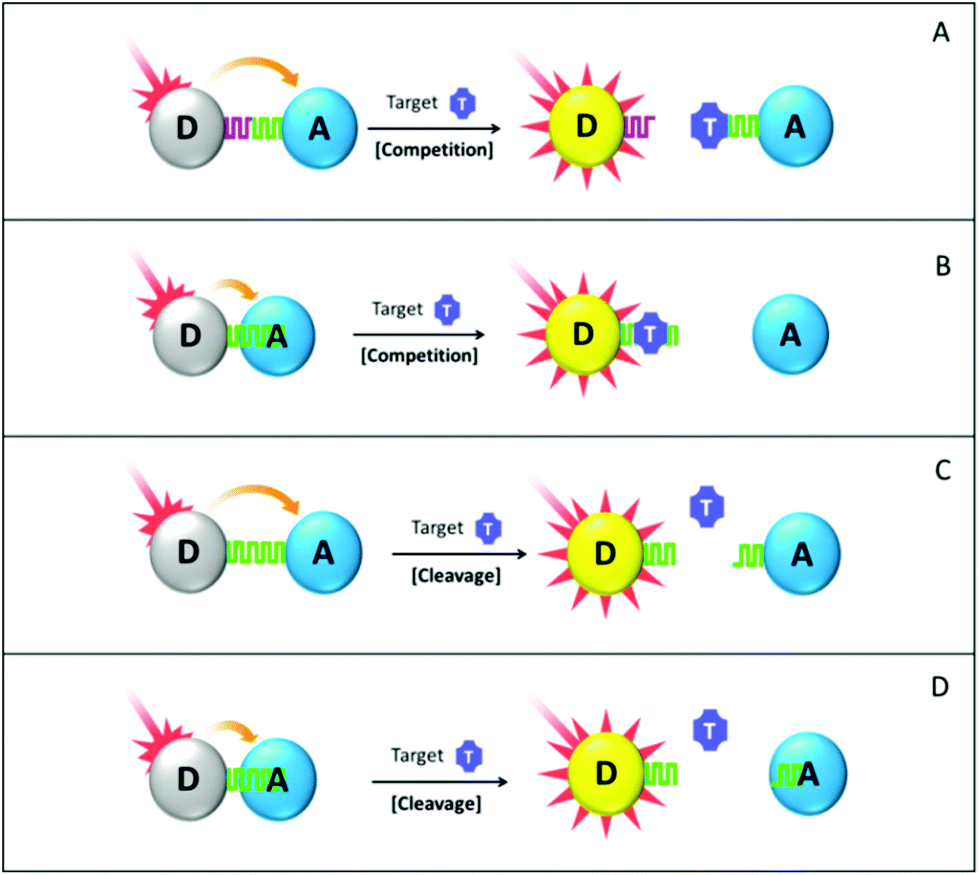 | ||
| Fig. 6 Schematic drawings of FRET biosensors for detecting metal ions and small molecules: (A & B) competition-based assays with crosslinking and nonspecific adsorption-based FRET configurations respectively; (C & D) cleavage-based assays with crosslinking and nonspecific adsorption-based FRET configurations, respectively. In principle, the positions of donor “D” and acceptor “A” are interchangeable in all cases. The drawings are not representing actual dimension of the respective biosensor components. | ||
| Assay type | Target | Donor | Acceptor | Sensing probe | LOD | Ref. |
|---|---|---|---|---|---|---|
| Fig. 6 scheme A [Competition] | Cholesterol | Fluorescein | AuNP | β-Cyclodextrin | 9 nM | 151 |
| Glucose | CdTe QDs | AuNP | Concanavalin A | 50 nM | 152 | |
| Glucose | UCNPs | GO | ConA and chitosan | 25 nM | 153 | |
| K+ | CDs | rGO | 18-Crown-6 ether | 10 μM | 134 | |
| Pb2+ | CdTe QDs | AuNP | 11-Mercaptoundecanoic acid | 30 ppb (∼146 nM) | 154 | |
| Adenosine; cocaine | QDs | AuNP | Adenosine aptamer; cocaine aptamer | 50 μM; 120 μM | 155 | |
| Hg2+ | Rhodamine B | AuNP | Nil | 10 pM | 156 and 157 | |
| Hg2+ | CDs | Hg2+ | Nil | 4.2 nM | 158 | |
| Biothiols | CDs | Hg2+ | Nil | 4.9–8.5 nM | 158 | |
| Glucose | FITC | AuNP | Dextran | 5 nM | 159 | |
| F− | CdTe QDs | AuNP | Boronate ester | 50 nM | 160 | |
| Fig. 6 scheme B [Competition] | Ag+ | FAM | GO | C-rich ssDNA | 5 nM | 161 |
| Hg2+ | FAM | GO | T-rich ssDNA | 30 nM | 108 and 162 | |
| Hg2+ | UCNP | GO | T-rich ssDNA | 0.5 nM | 163 | |
| ATP | Cy5 | AuNP | Aptamer | mM level | 164 | |
| ATP | FAM | GO | Aptamer | μM level | 165 | |
| Adenosine | FAM | GO | Aptamer | 10 μM | 108 | |
| ATP | UCNPs | GO | Aptamer | 80 nM | 163 | |
| Hemin | Acridine orange | GO | Aptamer | 50 nM | 166 | |
| Mycotoxins | UCNPs | GO | Aptamer | fM level | 167 | |
| Fig. 6 scheme C [Cleavage] | ˙OH | FAM | AuNP | ssDNA | 2.4 nM | 168 |
| Uranyl | Cy3 | AuNP and BHQ-2 | DNAzyme substrate | μM level | 69 | |
| Fig. 6 scheme D [Cleavage] | ˙OH | FAM | GO | ssDNA | N.A. | 169 |
| BLM | FAM | GO | ssDNA | 0.2 nM | 170 | |
| Pb2+ | FAM | GO | DNAzyme substrate | 300 pM | 171 |
In the bioaffinity competition assay, the FRET system has to be carefully designed to fulfill the following two requirements. First, in the absence of a target analyte, energy transfer between the donor and the acceptor is well facilitated and donor fluorescence is readily quenched. Second, analyte–probe interaction has a higher binding constant than the donor/probe or acceptor/probe combination, so that an added target analyte is able to displace the donor or the acceptor out of the FRET system and recover the donor fluorescence subsequently. Based on the interaction between the sensing probe and the donor/acceptor, such a design principle can be further categorized into two cases. In the first (Fig. 6 scheme A), the donor and the acceptor are functionalized with their respective sensing probe (e.g. Concanavalin A and thiolated β-cyclodextrins,152 cysteamine and 11-mercaptoundecanoic acid154) or a single molecule with two distinctive epitopes (e.g. 18-crown-6 ether,134 β-cyclodextrin151) that are able to bring the donor and the acceptor into the FRET-sensitive distance in the initial state. Then by displacing the donor or the acceptor out of the established FRET system, the added target analyte helps us to recover the quenched donor fluorescence. Of note, competition herein occurs between the analyte target and the sensing probe, which does not destroy the pre-assembled donor–probe or acceptor–probe conjugate.
In the second case, the association between the sensing probe and the acceptor (or donor) is achieved via nonspecific interaction (Fig. 6 scheme B). The target analyte then competes with the sensing probe to non-specifically bind with the acceptor. Such a design is mainly employed in the cases where the sensing probe is a linear biomolecule like ssDNA, and the acceptor contains aromatic surfaces. Interaction between ssDNA and the aromatic surface is mainly via π–π stacking and hydrophobic reaction between nucleobases and aromatic rings. The mechanically flexible structure of ssDNA makes its nucleobases readily accessible for the nonspecific stacking, whereas those in dsDNA are shielded by the rigid phosphate backbone, resulting in poor structure accessibility. Therefore, in the presence of the aromatic surface, ssDNA can be adsorbed but not for dsDNA (see Fig. 7). Provided such affinity difference, target detection can be achieved if it is able to induce DNA structural change from single-stranded to double-stranded or hairpin shape. An example of this design could be the Hg2+ and Ag+ detection using an ssDNA sensing probe.108,161,163 Hg2+ and Ag+ can hybridize T-rich and C-rich mismatched ssDNA sequences, respectively. The presence of the target ion can therefore be translated into reduced binding affinity due to the ssDNA-to-dsDNA transformation, and further, to the recovered fluorescence intensity of the ssDNA-tagged fluorophore. Moreover, utilization of aptamer as a sensing probe further emphasises the significance of such affinity change, making it capable of detecting various target analytes, including ATP, adenosine, small molecular drugs and peptides.108,165,166 Aptamer, with the name coming from the Latin aptus (meaning “fit”) and meros (meaning “part”), has emerged in recent years as a powerful and versatile sensing probe material. It exhibits antibody-comparable selectivity but with a more robust structure. Many of the commonly used aptamers are ssDNA and they behave just like ordinary oligonucleotides if the target is absent. While upon interacting with the target analyte, it will undergo significant conformational change, wrapping around the target or forming hairpin structures. These restricted structures have much reduced aromatic binding affinity due to poor accessibility of the nucleobases, as discussed before. Of particular interest is that in some other special cases the sensing probe can be omitted and donor–acceptor association is realized solely via their surface ligand interaction. For example, direct mixing of unmodified AuNP with Rhodamine B (RB) leads to significant RB fluorescence quenching due to its binding with AuNP. The Hg2+ target is able to displace the RB molecule out of the AuNP-RB composite and recover its fluorescence intensity in a Hg2+ concentration-related manner.157
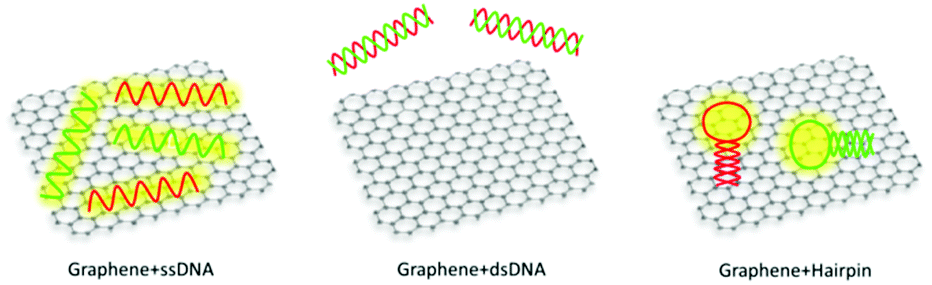 | ||
| Fig. 7 Interaction of graphene with ssDNA, dsDNA and hairpin structured DNA. ssDNA is able to adsorb onto the aromatic graphene surface (left); dsDNA shows little affinity (middle); hairpin structured DNA demonstrates moderate binding affinity due to the simultaneous presence of single-stranded and double-stranded portions. | ||
In the probe cleavage-based assay design (Fig. 6 schemes C and D), the added target analyte possesses the capability of digesting sensing probes that connect the donor and the acceptor in the pre-established FRET system.168,169 Following such digestion, the FRET system is largely disturbed and donor fluorescence is therefore recovered with an increment value directly proportional to target concentration. The aforementioned classification based on the ways of probe–acceptor association still applies here. In scheme C, the two epitopes of the sensing probe are anchored onto the donor and acceptor surface. Hence, probe cleavage does not involve any on-particle reaction as the cleavage site is on the sensing probe. Yet in scheme D, the sensing probe is non-specifically adsorbed onto one of the FRET components (e.g. acceptor), which leads to the probe-cleavage happening on the acceptor surface although the cleavage site still locates on the sensing probe itself. As the strength of nonspecific binding is proportional to the length of the sensing probe (e.g. ssDNA or polypeptide), the shortened probe fragment after cleavage is no longer able to retain the donor in close proximity with the acceptor.171 Therefore, it departs and leaves the donor fluorescence greatly recovered. However, one has to pay special attention to ensure that such probe cleavage assays are intrinsically limited to the target analyte which possesses probe-cleaving capability. To further broaden the application spectrum, enzymes or equivalent substances that are able to digest certain substrate can be introduced. The enzymes have to be well selected to make sure enzymatic digestion only occurs in the presence of specific cofactors (e.g. metal ions, small molecules). With the help of such enzymes, the presence of the target analyte is surrogated by the donor fluorescence recovery although the donor–acceptor linkage is not directly cleaved by the target metal ions or small molecules. As an illustrative example, Zhang and colleagues reported a Pb2+ detection assay with 300 pM LOD, using a FAM donor, a GO quencher and a DNAzyme substrate as a sensing probe.171 The used GR-5 DNAzyme is not activated until Pb2+ is added in, which then assists the substrate cleavage and FAM fluorescence recovery.
Bearing in mind the above-discussed principles, some useful guidelines can be drawn to guide the design of FRET biosensors for metal ions and small molecules in general. First, most of the reported LODs fall in the range of micromolar (μM = 10−6 M) to nanomolar (nM = 10−9 M), with some down to picomolar (pM = 10−12 M). Beyond this range, FRET might not be a suitable method and some other options have to be considered. This limitation may arise from the intrinsic nature of FRET, either light-on or light-off without further signal amplification. Second, although no overall advantages can be claimed for any of the four design principles shown in Fig. 6, some points are still worth highlighting. For example, with adenosine as the modal target, scheme B possesses much lower LOD than scheme A (10 μM vs. 50 μM),108,155 mainly because aptamer–GO interaction is much weaker than aptamer–ssDNA hybridization, which makes it easier for target adenosine to interact with the aptamer in the scheme A design. In contrast, lower sensitivity of scheme A than scheme B (10 pM vs. 0.5 nM) is observed when looking at Hg2+ as a target analyte.157,163 This might be due to the fact that the oligonucleotide-modified UCNPs in scheme B are adsorbed onto the GO by multitude contacting points, so that multiple Hg2+ are required to free one UCNP from the GO quenching. However, in scheme A the Hg2+-RB displacement may follow a lower ratio. Third, GO and AuNP are commonly used as FRET acceptors by virtue of their high quenching efficiency and well understood chemical and physical properties. As for the donors, nanometer-sized fluorophores (e.g. UCNPs,163 QDs152,155 and CDs134,158) start to play active roles despite the still dominant usage of organic dyes (e.g., FAM,108,161,162,165,168–171 cyanine dyes,68,164 Rhodamine B156,157). Fourth, given similar conditions, biosensors with nanomaterial donors (e.g., UCNPs) demonstrate superior sensitivity over those with organic dyes.108,163,165 UCNP has a much lower fluorescence background because of its IR or NIR excitation. Therefore, detection sensitivity can be significantly improved because the LOD is often calculated using a 3δ rule where δ is the standard deviation of the background signal. Fifth, use of the aptamer as a sensing probe is always worth thorough consideration for detecting metal ions and small molecules, especially for a target analyte that does not possess any usable physical or biochemical properties. Structure-wise, many of the aptamers are merely ssDNA which can effectively interact with GO or CNT in a sequence-independent manner. Function-wise, it is able to specifically bind with the desired target and undergo dramatic structural transformation. Such functional and structural characteristics make the aptamer much suitable and attractive for the design of bioaffinity competition based FRET assays. Sixth, the FRET system utilizing nonspecific association between the sensing probe and the acceptor (Fig. 6 schemes B and D) oversteps their counterpart assays (schemes A and C) in terms of facile material synthesis and lower assaying cost, since only single labelling is required for probe modification. However, such merits can be balanced against the poor detection selectivity, particularly when target or sensing probe analogues are present, which is able to non-specifically disassociate the sensing probe out of the FRET system.
Relative to single-target detection, the multiplexed assay also draws enormous attention due to its advantages of reduced analyzing time/cost and less amount of required sample volume. More importantly, it facilitates feasible, reproducible and reliable comparative analysis. As a proof of concept, Fan, Song and co-workers proposed an aptamer–AuNP based multicolour design with adenosine, potassium ion and cocaine as modal targets.172 The AuNP surface was functionalized with three types of DNA sequences that were complementary to the three utilized aptamers. Each type of aptamer was labelled by a specific dye molecule. Initially, DNA hybridization between the aptamer and the AuNP-tethered oligonucleotide brought the dye molecules into close proximity with AuNP and therefore had the dye fluorescence readily quenched. However, in the presence of the target analyte, DNA dehybridization took place due to the higher binding affinity between the aptamer and its corresponding target analyte. The respective dye fluorescence was therefore recovered in a target concentration-correlated manner. In view of the fact that the disease diagnosis and biomedical study sometimes require the information from multiple targets for pattern recognition, multiplexed assays are thus becoming increasingly crucial to complement the advances in healthcare application to monitor several physiological parameters or to detect various target analytes simultaneously.
 | ||
| Fig. 8 Schematic drawings of a FRET biosensor for detecting DNA molecules: (A) DNA crosslinking light-off assay; (B–F) analyte induced light-on assays. (B, C & D) strands competition based assays with different acceptor attachment configurations; (E & F) DNA probe conformational alternation-based designs with hairpin and ordinary ssDNA sensing probes, respectively. In principle, the positions of donor “D” and acceptor “A” are interchangeable for all drawings. The drawings are not representing actual dimensions of the biosensor components. | ||
The target crosslinking mechanism generally leads to the light-off FRET assay, in which target addition results in donor fluorescence quenching. In a typical crosslinking design (scheme A), a FRET donor and an acceptor are modified with ssDNA sensing probes that have nucleobase sequences complementary with the target. Initially, appropriate excitation can only trigger signatory emission of the donor, because it is well separated from the acceptor. Following the addition of the target DNA, hybridization between sensing probes and the target analyte takes place, crosslinking the donor and the acceptor to form the sandwich-like structure. Such crosslinking brings the donor and the acceptor into a FRET-sensitive distance, so that the donor is able to transfer its fluorescence energy to the proximal acceptor and has its own fluorescence intensity significantly reduced. Fig. 8 scheme A is a drawing of the crosslinking principle using a single donor–acceptor pair model. In practice, the system could have huge network structures due to multiple DNA probes on the donor and acceptor surfaces. The LOD of the picomolar (pM = 10−12 M) to femtomolar level (fM = 10−15 M) has been reported by using either fluorophore/fluorophore or fluorophore/quencher crosslinking designs.111,173 It is also worth mentioning that such crosslinking could be easily applied for multiplexed target detection using different sensing probes and respective fluorophores.111 Due to the high specificity of DNA hybridization, the presence of certain target oligonucleotides can only lead to the fluorescence quenching of its corresponding donor while leaving the others unaffected. Hence, it intrinsically imposes no negative influences on the detection limit for each individual target. Experimental results from Cui and co-workers have validated this point.111
DNA strand competition assays (see schemes B, C and D) rely on the principle that the added DNA target competes with transducing elements or sensing probes for binding with the others. In the first configuration (scheme B), both the donor and the acceptor are modified with an ssDNA probe and the two probes are complementary to each other. Before target DNA addition, the donor and the acceptor are in close proximity via probe hybridization. Following the addition of a target analyte that has an identical nucleobase sequence with one of the probes, bioaffinity competition takes place and the pre-established FRET system is disturbed.175 To further improve detection sensitivity, mismatched base pairs could be introduced into the hybridized probes. As a perfectly complementary ssDNA target has a higher binding constant, it is able to displace more of the donors or acceptors out of the FRET system. The second configuration (scheme C) is based on the binding affinity difference between ssDNA and dsDNA towards aromatic acceptors (e.g. GO and CNT). ssDNA can be readily adsorbed onto the aromatic surface and have its tethered donor quenched. Following the target addition, dsDNA is formed and departs from the aromatic surface, and the donor fluorescence subsequently recovers. In addition, multiplexed target detection using the competition principle has also been reported by Fan and co-workers using dye-labelled ssDNA as the sensing probe and GO as the common quencher by virtue of its large surface area for multitude probe adsorption.108 The third configuration (scheme D) is much similar to the second, except for the utilized hairpin sensing probe. As a special member of the DNA family, the hairpin DNA structure has been intensively investigated regarding their working mechanism and diverse applications.184,185 Structure-wise, the hairpin DNA is a oligonucleotide loop flanked by two self-complementary ssDNA tails. In the stem-close state, the self-complementary tails hybridize with each other, forming a “stem-loop” configuration. The target analyte then hybridizes with the loop portion, forcing the stem part to open up and inducing the formation of linear dsDNA. From stem-loop to dsDNA, its binding affinity towards aromatic surface decreases. Exploiting such a decrease, DNA biosensors with LOD in the nanomolar (nM = 10−9 M) to picomolar (pM = 10−12 M) range have been constructed by several groups.108,109,176 However, one should be aware that the stem part in the hairpin structure becomes single-stranded after the target-loop hybridization. Therefore, the ratio of nucleobase numbers in the stem and loop portions have to be carefully designed to ensure the overall dsDNA percentage increase after hybridization.177 In some particular cases, the ssDNA target also contains a stem-complementary sequences, which leaves no dangling ssDNA tails after hybridization. Such a design is kinetically favourable due to the elimination of ssDNA tails, but its practical application is much restricted simply because the desired target oligonucleotide does not necessarily contain the stem-complementary sequences.
DNA conformational alternation based design (see schemes E and F) generally involves on-particle interaction without any sensing probe cleavage or strand displacement processes. In scheme E, hairpin DNA is dually labelled with the donor and the acceptor at two far ends of the self-complementary tails. Hence, donor fluorescence is quenched in the stem-close state because of its proximity with the acceptor. Such a configuration is usually termed ‘molecule beacon’ (MB) in the literature.184 Following the addition of target DNA, sequence hybridization opens up the stem-loop structure and pushes the donor far away from the acceptor. With donor fluorescence recovery, the presence of the target analyte is therefore translated into the detectable optical signals. Compared with assays using an ordinary ssDNA probe, one of the most significant advantages that MB-based design possesses is probably the higher detection sensitivity towards a base-pair mismatch. However, several shortcomings are also identified, including low quenching efficiency of organic quenchers, a complicated synthetic process and vulnerability to endogenous nuclease degradation or corresponding DNA binders.186,187 To address these challenges, especially the first two, various nanomaterials have been thoroughly examined for their potential roles in MB assay design. In 2001, it was the Dubertret group that first used AuNP (1.4 nm diameter) to replace the conventional organic quencher and successfully constructed a novel MB-based sensor for ssDNA detection.181 Benefiting from the high quenching efficiency of AuNP, eight times higher detection sensitivity towards a single-base mismatch was achieved. Fan and co-workers further improved such a design by using larger AuNP (15 nm diameter).182 More importantly, they demonstrated the feasibility of detecting a multiplexed target in a single assay. In their hairpin-AuNP design, it is critical to introduce the short “helper” oligonucleotides in between the hairpin sensing probes on the AuNP surface because of the poor aqueous stability of the hairpin-AuNP composite. Alternatively, such nanometre-sized quenchers can also work cooperatively with conventional MB, involving both the original organic donor and acceptor.179,188 In addition to the improved LOD, such a nanomaterial-MB hybrid DNA sensor exhibits higher thermal stability than the conventional MB counterpart, making itself much suitable for biodetection tasks under stringent experimental conditions.179
Scheme F illustrates another DNA conformational alternation based sensor design, utilizing ordinary ssDNA as a sensing probe. Some organic dyes (e.g. fluorescein) are able to reversibly adsorb onto the AuNP surface via ligand coordination or electrostatic interaction.189 Therefore, the ssDNA probe connecting them is constrained into an arch shape at the initial state. Upon target DNA addition, probe-target hybridization forms the much more rigid dsDNA and the dye molecule is separated far from the AuNP surface. Donor fluorescence is therefore recovered in a target concentration-dependent manner with LOD around 40 nM under favourable conditions.183 Compared with the hairpin-based assay shown in scheme E, this design possesses the advantages of facile material synthesis and low assaying cost, whereas it might suffer from sacrificed sensitivity for detecting a single base-pair mismatch, as well as the false positive signals that arise from possible nonspecific interactions between the ssDNA probe and its various binders.
With the target DNA of comparable length, it can be seen from Table 3 that the crosslinking principle demonstrates higher sensitivity than the competition or probe alternation based designs, possibly because of the better accessibility of the sensing probe. In crosslinking (scheme A), the sensing probes on donor and acceptor surfaces all extend outwards, making them much easier for target DNA to approach and bind with. Yet in the other designs, sensing probes initially all bind with some elements involved in nonspecific interaction, complementary hybridization or structural restriction. This largely retards the efficiency of the desired target–probe interaction. Despite the advantageous detection sensitivity, crosslinking design suffers from tedious material synthesis (e.g., dual labelling) and high assaying cost. The same drawbacks also apply for the structure alternation-based assays (schemes E and F). As for the strand competition design (schemes C and D), only single labelling on the sensing probe is necessary, with the other labelling surrogated by nonspecific adsorption. However, DNA hybridization in the presence of GO or CNT takes much longer time than that in the typical hybridization buffer. Elongated assaying time is detrimental to convenience of application and deteriorates the dye photobleaching problem as well. As discussed before, such nonspecific adsorption is also vulnerable to the complex biological matrix and sensing probe analogues, which may lead to false positive signals. To add on, experiment stoichiometry involving DNA adsorption on the aromatic surface has to be carefully selected, as there is evidence showing the remaining dsDNA on the SWNT surface after hybridization.190
| Assay type | Target DNA length | Donor | Acceptor | LOD | Ref. |
|---|---|---|---|---|---|
| Fig. 8 scheme A [Crosslinking] | 30 bp | CdSe–ZnS QDs | Cy5 | 4.8 fM | 173 |
| 26 bp | UCNP | TAMRA | 1.3 nM | 174 | |
| 20 bp | CdSe QDs | MWNT | 0.2 pM | 111 | |
| Fig. 8 scheme B [Competition] | 24 bp | CdTe QDs | AuNP | N.A. | 175 |
| Fig. 8 scheme C [Competition] | 23 bp | FAM | SWNT | ∼5 nM | 109 |
| 17 bp | FAM | GO | 100 pM | 108 | |
| 23 bp | FAM | GO | ∼10 nM | 176 | |
| Fig. 8 scheme D [Competition] | 15 bp | FAM or Cy5 | GO | 2.0 nM | 177 |
| 22 bp | CdTe QDs | GO | 12 nM | 178 | |
| 15 bp | FAM | GO (and TAMRA) | 0.1 nM | 179 | |
| 19 bp | FAM | SWNT | 4 nM | 180 | |
| Fig. 8 scheme E [Alternation] | 16 bp | Rhodamine 6G | AuNP | nM level | 181 |
| 15 bp | Organic dye | AuNP | nM level | 182 | |
| Fig. 8 scheme F [Alternation] | 24 bp | Fluorescein | AuNP | 40 nM | 183 |
 | ||
| Fig. 9 Schematic drawings of a FRET biosensor for detecting protein molecules: (A & B) bioaffinity competition based light-on assays with crosslinking and nonspecific adsorption FRET configurations, respectively; (C) target-induced crosslinking light-off assay; (D & E) cleavage enzyme light-on assay with crosslinking and nonspecific adsorption configuration, respectively. The positions of donors and acceptor are interchangeable in all cases. The drawings are not representing actual size of the biosensor components. | ||
| Assay type | Target | Donor | Acceptor | Sensing probe | LOD | Ref. |
|---|---|---|---|---|---|---|
| Fig. 9 scheme A [Competition] | Avidin | QDs | AuNP | Biotin | 10 nM | 191 |
| Avidin | SWNT | Organic dyes | Biotin | 1 nM | 110 | |
| Fig. 9 scheme B1 [Competition] | Thrombin | FAM | GO | Aptamer | 31.3 pM | 192 |
| Thrombin | FAM | GO | Aptamer | 2.0 nM | 176 | |
| HIV antibody | UCNP | GO | Polypeptide | 2.0 nM | 193 | |
| Cyclin A2 | FITC | GO | Polypeptide | 0.5 nM | 194 | |
| Fig. 9 scheme B2 [Competition] | ERα, ERβ | CPEs | AuNP | ER binding site (DNA) | N.A. | 195 |
| FoxA1, AP-2γ | CPEs | AuNP | Protein binding site (DNA) | N.A. | 196 | |
| Fig. 9 scheme C [Linking] | ERα, ERβ | CPEs | AuNP | ER binding site | N.A. | 195 |
| FoxA1, AP-2γ | CPEs | AuNP | Protein binding site (DNA) | N.A. | 196 | |
| Fig. 9 scheme D [Cleavage] | MMP | Cy5.5 | AuNP | Polypeptide | 1.0 nM | 197 |
| MMP-2 | CdSe–ZnS QDs | GO | Polypeptide | nM level | 198 | |
| S1 nucleases | Cy3 | AuNP | ssDNA | N.A. | 146 | |
| Caspase-3 | FAM | GO | Polypeptide | 0.4 nM | 199 | |
| Caspase-3 | QDs | mCherry fluorescent protein | Peptide | 20 pM | 200 | |
| MMP-2 | Bioluminescent protein | QD | Peptide | 19 ng mL−1 | 201 | |
| Protease (Caspase 1, collagenase, chymotrypsin, thrombin) | CdSe–ZnS QDs | Cy3 | Peptide | N.A. | 202 | |
| Kallikrein | CdSe/ZnS QDs | Cy3-maleimide | Peptide | N.A. | 203 | |
| Fig. 9 scheme E [Cleavage] | MMP-2 | FITC | GO | Polypeptide | 50 pM | 204 |
The bioaffinity competition assay for protein detection (Fig. 9 scheme A) is similar to that for detecting small molecules and DNA sequences described in previous sections. Both the FRET donor and acceptor are functionalized with the sensing probe that can specifically bind to each other, bringing them into a FRET sensitive distance. The target protein is able to compete with one probe for binding with the other, which breaks the pre-established FRET system and recovers the donor fluorescence accordingly. As a representative example, biotin–avidin interaction has been well understood and commonly used for designing competition-based FRET biosensors.191,205 The reverse mechanism has also been employed, in which the target analyte is able to crosslink the donor and the acceptor, and hence establish the FRET system.206 Another design under the competition principle (see scheme B1) involves the usage of an aptamer as the sensing probe. The aptamer is anchored onto the donor surface at one end, but associated with the acceptor via nonspecific adsorption. Upon binding with the protein target, the aptamer undergoes a dramatic structural change and loses its affinity towards the aromatic quencher. Donor fluorescence can therefore be recovered.176,192 A similar principle can be further adopted by using a polypeptide sensing probe. However, the polypeptide has to be specially designed so that it is able to interact with the target protein and induce the formation of the polypeptide–protein complex. Analogues with the aptamer, such polypeptides also contain aromatic groups and demonstrate high binding affinity towards the aromatic quencher via π–π stacking. Formation of the polypeptide–protein complex much reduces the affinity and induces the donor fluorescence recovery.193,194 Moreover, hairpin peptide beacon (HPB), as a sensing probe, has been proposed as well for protein target detection.207–209 HPB, similar to DNA molecular beacon (MB), comprises a protein-specific polypeptide loop flanked by two self-complementary peptide nucleic acid (PNA) sequences, as well as a donor and an acceptor tagged at the PNA far ends.210 In the absence of a target protein, PNA hybridization brought the donor and the acceptor into close proximity and facilitated the donor fluorescence quenching. The protein target interacts with the central polypeptide sequence, opening up the hybridized PNA and forcing the donor and the acceptor far apart. As a result, donor fluorescence intensity recovers. The superiority of HBP over MB is perhaps the possibility of removing the self-complementary PNA portion, leaving polypeptide conformational changes solely dependent on the amino acid sequence and its interaction with the target protein.207–209 Unfortunately, nanomaterial involvement in the HBP-based biosensor design is still lacking so far, and awaits intensive efforts to be devoted to it.
Our group has recently developed a suit of hybrid sensors for detecting the protein–DNA interaction, using double stranded DNA–gold nanoparticle (dsDNA–AuNP) composite as the acceptor and water soluble conjugated polyelectrolytes (CPEs) as the donor.195,196 Both “light-on” (scheme B2) and “light-off” (scheme C) schemes have been designed, in which CPE fluorescence is recovered and quenched respectively upon adding in the corresponding target protein. Water soluble CPEs, as a new type of fluorescent material, have a better competitive edge than the others by virtue of their high quantum yield, excellent light harvesting capability and more importantly, unique linear conformation. The involved CPEs have to be properly selected, so that their charge properties match well with the dsDNA–AuNP composite and their emission wavelength overlap with the AuNP LSPR absorption. In principle, the protein target should bind with the dsDNA at a specific DNA binding site, inducing a change of electrostatic property of the dsDNA–AuNP composite. Such a change modulates the composite electrostatic interaction with CPEs, leading to either stronger or weaker CPE fluorescence quenching by AuNP. A “two-way” model, either “light-on” or “light-off”, is constructed as well, which allows the detection of proteins with unknown charge properties. Experimentally, such principles have been exploited to study the binding behaviour of various proteins (e.g. ERα, ERβ, FoxA1, AP-2γ) with their corresponding DNA binding site.195,196 It is further able to differentiate the binding affinities of the two Estrogen Receptor (ER) subtypes (ERα and ERβ), which have about 96% similarity in their DNA binding domain but substantially differ in physiological distribution and bio-functionality.195 We also demonstrated that such hybrid sensors are highly sensitive to detect site- and nucleotide-specific single base variations on the ERα binding region. Screening of 15 single mutations on the ERα binding site (3 possible substitutions in each of the 5 nucleobases of the binding site) gives rise to an in vitro binding energy model. Such a model correlates well with the energy matrix obtained from in vivo genome-wide ERα binding data using Thermodynamic Modeling of ChIP-seq. This renders our assay design a highly reliable alternative for understanding in vivo protein binding mechanisms.
Enzymes, as a special group of protein molecules, are capable of catalysing biological reactions and/or inducing structural changes of their respective substrates. The malfunction of a certain enzyme is usually responsible for severe biological abnormalities and disorders, which makes the design of enzyme sensors exceedingly important and highly desirable for diverse purposes. In the following discussion of this section, we will focus on the FRET assay designs primarily for peptide and DNA cleavage enzymes. According to the cleavage site, two commonly used sensor design configurations are elaborated, as shown in schemes D and E of Fig. 9.
In scheme D, the donor and the acceptor are linked via the linear sensing probe, whose length falls in the range of an effective FRET distance. Donor fluorescence is therefore quenched at the initial state. The target enzyme then digests the sensing probe at the specific location, leaving two short fragments tethered on the donor and acceptor surfaces. Consequentially, the FRET mechanism collapses and donor fluorescence recovers. With an appropriate cleavage substrate, detection of various enzymes including matrix metalloproteinase (MMP),197,198 caspase-3,199 S1 nucleases,146 as well as the multiplexed detection of various targets in a single assay211 has been well demonstrated with remarkable sensitivity, selectivity as well as rapidity. Alternatively, in scheme E a sensing probe is associated with the acceptor via nonspecific interaction. Due to decreased binding affinity, the shortened probe fragment is released back into the solution and has its tethered donor fluorescence recovered. LOD of 50 pM for MMP-2 was reported by Ma and co-workers using a FITC donor, a GO quencher and a polypeptide sensing probe.204 This example confers a better optical sensing design and sensitivity competitiveness over electrochemical, liquid chromatography and mass spectrometry approaches.212,213 Due to limited space, it is impractical to cover all assay designs here, even merely for peptide and DNA cleavage enzymes. However, the two design principles discussed above are widely used and lay the foundation of designing many other FRET enzyme biosensors.24,214–218
However, detection sensitivity and assaying rapidity of the above examples can never exceed the reaction kinetics between target enzymes and their substrates due to the lack of signal amplification. To resolve such limitations, Chung and co-workers proposed a cascade method involving DNA–peptide and DNA–RNA cleavages by MMP-2 and RNase H, respectively.219 As shown in Fig. 10, digestion of the DNA–peptide composite by MMP-2 generates free DNA fragments. They are released from the AuNP surface and enter the cycling loop to form a heteroduplex with RNA molecules labelled by FITC. RNase H has no influence on single-stranded RNA but is able to degrade the RNA chain in the DNA/RNA duplex. Therefore, the FITC dye is disassociated from AuNP in the cycle and has its fluorescence intensity significantly recovered. Relying on such a novel cycling loop for signal amplification, this design permits assessment of the MMP-2 activity at a concentration of 10 pM within 4 hours, about 100-fold more sensitive and 2-time faster than the design without amplification. Such an improvement results from the fact that RNase has a higher turnover rate and reaction affinity towards its substrate compared with MMP-2.
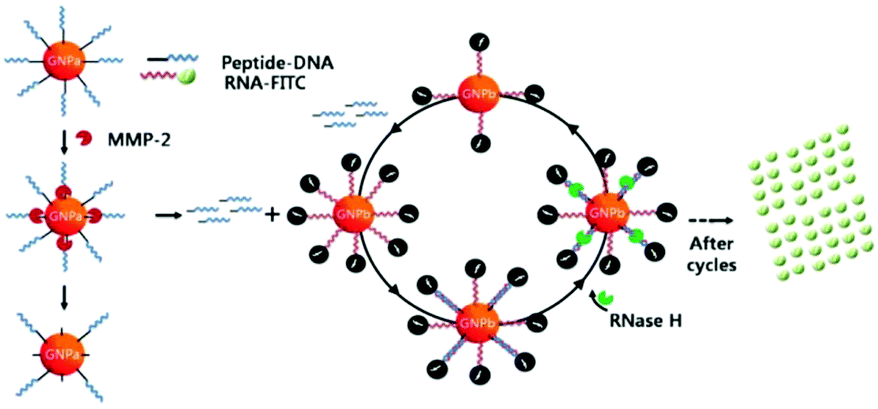 | ||
| Fig. 10 Schematic diagram showing the cascade procedure for fluorescence signal amplification. Reprinted with permission from ref. 219, © 2010 WILEY. | ||
Amplified fluorescence polarization (AFP)
Fluorescence polarization (FP) or fluorescence anisotropy is a phenomenon in which fluorophore emission has unequal intensities along different directions. These two terms describe the same physical process, using different quantitative measurements with correlation shown below, where P denotes the value of fluorescence polarization and r is the fluorescence anisotropy.220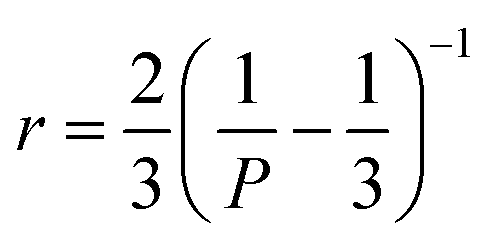
Under light irradiation, fluorophore absorbs photons and excites itself to a higher energy level. The absorbed energy is lost via photon emission and heat dissipation mechanisms after a certain time period called “fluorophore lifetime”. Since the excitation process involves redistribution of electrons about the fluorophore molecule, polarized excitation can only excite fluorophores along certain orientation. Subsequently, light is emitted in the same polarized plane, provided the fluorophores are kept stationary throughout the excitation/emission process. However, fluorophores in aqueous solution undergo constant Brownian motion. They keep moving and rotating due to the thermodynamic collisions. In such a case, light is emitted along different planes from polarized excitation and the difference is dependent on the relationship between fluorophore lifetime and rotational correlation time. The rotational correlation time of a fluorophore alone or as a small complex is usually much shorter than the fluorophore lifetime. Hence, the fluorophores become randomly oriented and give off depolarized fluorescence emission. In contrast, polarized emission can be observed for fluorophore-containing complexes of a larger size, because of the longer rotational correlation time than the fluorophore lifetime. With an amplifier, the fluorophore-containing complex becomes even larger and the rotation is further slowed down, giving rise to a more significant FP value (see Fig. 11). Via such a principle, the size of the fluorophore-containing complex at the micro-level is reflected by the FP value at the macro-level. This makes the FP principle highly suitable for biosensor design, because of the molecular binding and/or complex size change in many biological processes.221,222
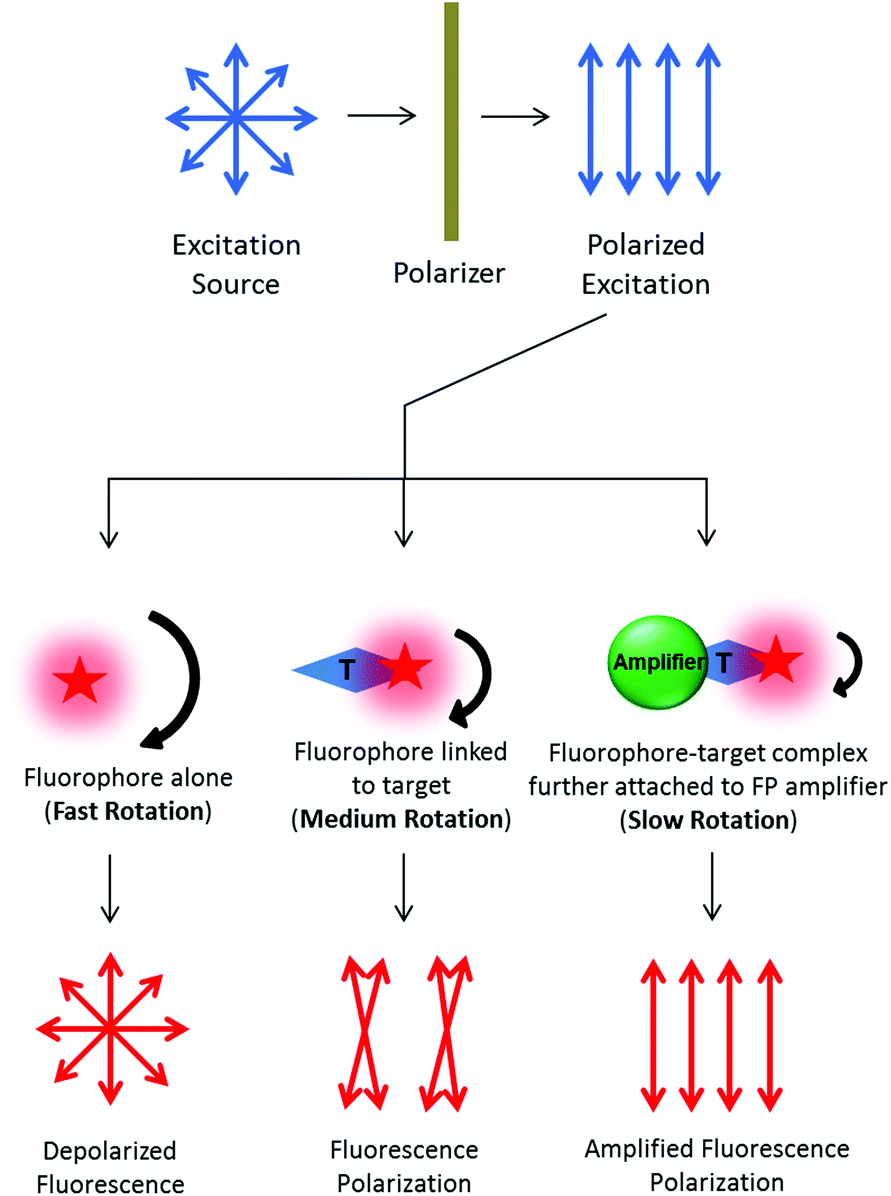 | ||
| Fig. 11 Schematic illustration of the fluorescence polarization principle and FP amplification mechanism. The curly arrow size indicates rotational speed of the corresponding complex. | ||
For instance, dye-labelled aptamers have been readily utilized for quantitative analysis of biomolecules.223–225 Binding of a target macromolecule with a dye-labelled aptamer leads to much slower complex rotation and a higher FP value. However, it is generally not applicable for detecting small molecules due to the limited size increase after target-probe binding. FP signal amplification is therefore required to make such design suitable for various targets detection, especially those of small size. One effective approach is to magnify the size difference before and after target binding. Thrombin, ssDNA binding protein and antibody have been widely reported in the literature.226–228 In recent years, nanomaterials have also started to reveal their excellence for FP signal amplification. Such excellence stems from their tunable size/shape and superb interaction affinity with various biomolecules.
Six distinctive design principles of nanomaterial-amplified FP (AFP) assays are shown in Fig. 12 schemes A–F. In scheme A, the target analyte has the capability to assemble sensing probes. These sensing probes are initially tethered onto the fluorophore and nano-amplifier, respectively. Without the target analyte, the FP value of the probe-tethered fluorophore is small. Following the target addition, the formed complex contains both the fluorophore and the sensing probe, as well as the nano-amplifier. The overall size increases significantly and molecule rotational speed decreases, resulting in a higher FP value relative to the initial state. As an example, Hg2+ detection has been demonstrated using an AuNP amplifier. By virtue of the capability of hybridizing T-rich mismatched ssDNA, Hg2+ helps us to crosslink the FAM and the AuNP amplifier, inducing a large increase in the FAM FP value. LOD of 1.0 nM (0.2 ppb) was reported. As a control group, the LOD value was merely in the micromolar (μM) range in the design without an AuNP amplifier.229
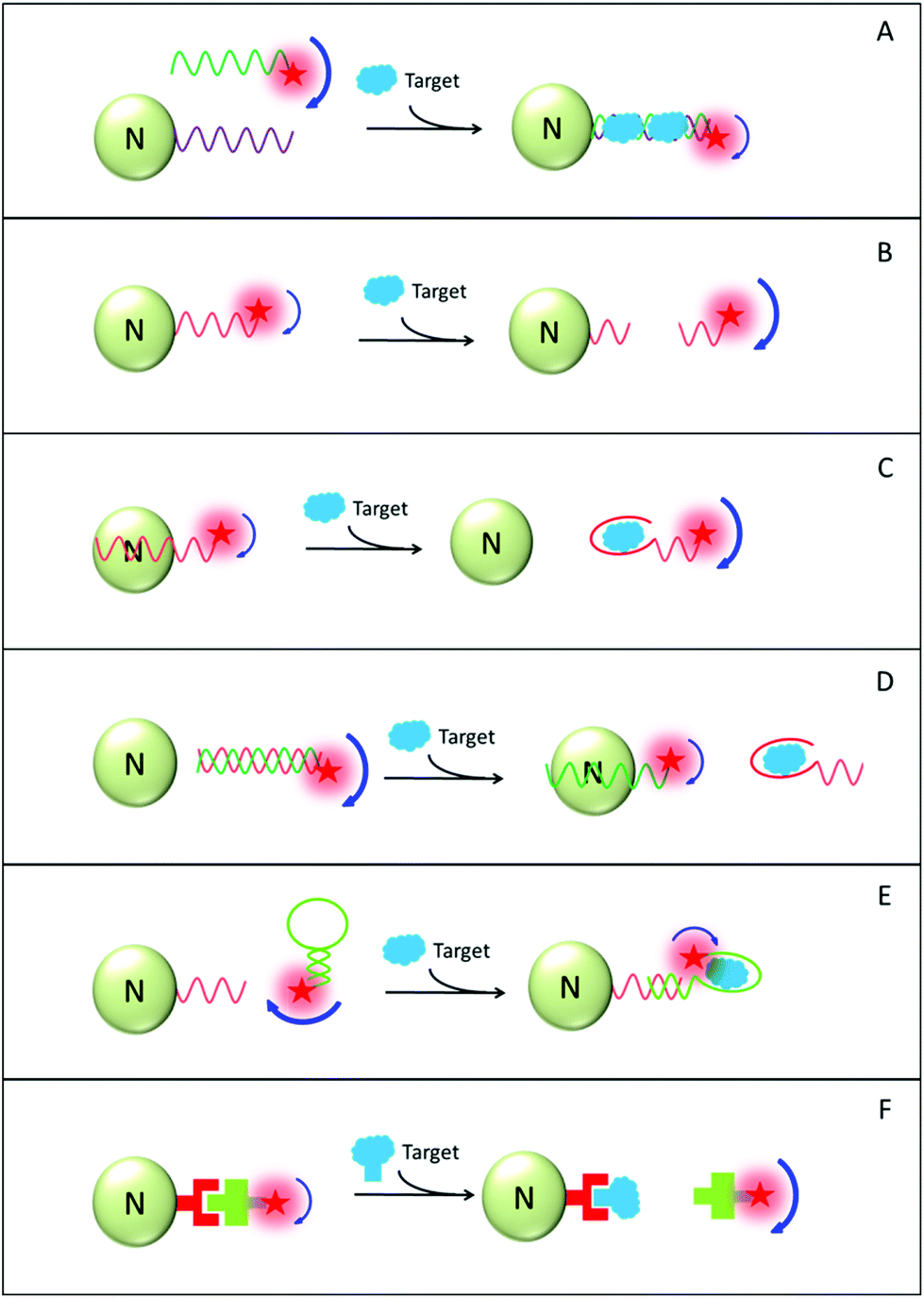 | ||
| Fig. 12 Schematic drawing of nanomaterial-amplified FP (AFP) assays, using principles of: (A) target-induced hybridization; (B) target-induced cleavage; (C) aptamer-based bioaffinity competition; (D) target-induced dehybridization; (E) hairpin-based bioaffinity competition; (F) target-based displacement. “N” stands for the utilized nanomaterial. The drawings do not reflect actual dimension of the biosensor components. | ||
In scheme B, initially the fluorophore is associated with a nano-amplifier and the system displays a high FP value. Upon adding a target analyte with probe digestion capability (e.g., enzymes or oxygen radicals), the fluorophore and the nano-amplifier are separated and the overall FP value decreases subsequently.230 Moreover, other analytes such as enzymatic cofactors, can also be detected with the assistance of corresponding enzymes. Very recently, Huang and co-workers designed a FP-based Cu2+ detection assay using GO as the amplifier and DNAzyme as the sensing probe.231 The DNAzyme cleaves substrate DNA only in the presence of Cu2+ at a nanomolar level. Other carbon-based nanomaterials (e.g. carbon nanoparticles and MWNT) were also tested as nano-amplifiers. Yet their performance was inferior, possibly due to their low efficiency of releasing the cleaved DNA fragments.
In schemes C and D, aptamers are exploited as sensing probes for detecting small molecules. In the following discussion, we will use adenosine triphosphate (ATP) as a model target to elaborate the detailed design principles.232 As previously discussed, an aptamer exhibits strong binding affinity towards aromatic surfaces, but such affinity largely decreases after aptamer–target interaction. Based on this principle, both “signal-on” and “signal-off” AFP-based designs are made available. The “signal-off” design (decreasing of FP value) is relatively simple as it only involves a GO amplifier and a fluorophore-tethered aptamer (see scheme C). Initially, an aptamer sequence is adsorbed onto GO and the overall FP value is high. Upon target addition, the formed aptamer–target complex departs from GO, leading to a reduced FP value. As for the “signal-on” design, additional ssDNA with dye labelling needs to be introduced (see scheme D). This ssDNA sequence is complementary to the aptamer. Therefore, the target-free condition gives rise to a low FP value due to the low affinity of a hybridized aptamer–ssDNA duplex towards GO. Following target addition, stronger aptamer–target interaction dehybridizes the duplex. Free ssDNA molecules then get adsorbed onto the aromatic surface and the overall FP value increases. Benefiting from the highly selective aptamer, such AFP-based biosensors demonstrate excellent detection selectivity, even in the presence of ATP analogues like UTP, CTP and GTP with much higher concentrations.232
Scheme E also involves the aptamer as the sensing probe, but with a slightly different design. Unlike the conventional linear conformation, the aptamer used in this case includes a hairpin structure at one terminus.233 Only upon binding with the target analyte, the stem part of the hairpin opens up and hybridizes with its complementary nucleotide sequence that has been pre-attached onto nano-amplifier surface. Via such a mechanism, the aptamer-tethered fluorophore is incorporated in a much larger complex, so that the overall FP value increases significantly. The LOD of this assay design for detecting ATP and thrombin can be as low as 20 pM and 0.3 pM respectively. They are more than six orders of magnitude lower than the control group without FP amplification.233
The assay design shown in scheme F is based on the competition between the target analyte and its fluorophore-modified analogue for binding with the sensing probe. In the initial state, the composite structure comprising a fluorophore-modified target analogue, a sensing probe and a nano-amplifier demonstrates a high FP value. Then following target addition, the fluorophore-modified target analogue is displaced out of the composite. The degree of displacement depends on the similarity between the target and its analogue, as well as its stoichiometry ratio. Along with the displacement, the overall FP value of the system decreases. Though a large sensing probe such as an antibody or another macromolecule might be adequate to induce considerable FP change upon target binding, inclusion of a nano-amplifier can always further improve detection sensitivity and afford this FP-based assay competitiveness in practical application.228
Besides the six configurations discussed above, there are many other AFP-based assays in the literature that exploit much delicate designs.234 However, the majority of them rely on one or a combination of the fundamental principles we have covered here (schemes A–F). Moreover, one may realize that the FP-based assay shares numerous similarities with the FRET-based designs discussed in the previous section, since both (1) involve nanomaterials as either a FP amplifier or a fluorescence quencher, (2) are homogeneous assays that require no separation or washing steps, (3) are based on non-adulterated measurement. However, compared with FRET, the FP-based assay design possesses the additional advantage of higher reliability due to its ratio-based nature. This makes it much more resistant to the inner filter effect, dye photobleaching, nonspecific quenching, and non-uniform illumination problems. However, its downsides, such as the requirement of expensive FP spectrometers and vulnerability to non-specific interaction with macromolecules, may balance against the advantage. Therefore, it is perhaps impossible to provide a universal guideline for the selection of assay principles and transducing elements for both AFP- and FRET-based biosensor designs, since it highly depends on the actual assay requirements including the type of target analyte, sensitivity expectation, material availability, and potential application.
Bio-barcode assay (BCA)
Bio-barcode assay (BCA) was first demonstrated by Mirkin and co-workers in 2003.235 Strictly speaking, it is not a solution phase detection since washing and substrate-based target analysis may be involved.236 However, we prefer to include it in the present review because its novel barcode amplification step occurs in solution and the signal transducing mechanism can vary from case to case, with some of them based in solution as well. More importantly, its detection sensitivity is extremely low, ranging from attomolar (aM = 10−18 M) to zeptomolar (zM = 10−21 M) levels, which may provide additional opportunities for clinical diagnosis applications.In a typical BCA design, one copy of the target analyte can be surrogated into multiple copies of DNA molecules called “barcode”.237 It is these barcode DNAs that undergo quantitative analysis and give off detectable signals. Via such target-based amplification, much improved detection sensitivity can be achieved. And the improvement primarily depends on the quantity ratio between the target surrogate and the target itself. In general, BCA is in the hybrid form, involving two basic components. First is the magnetic microparticle (MMP) modified with capturing probes. Second is the gold nanoparticle (AuNP) co-functionalized with another type of capturing probe and dsDNA molecule. This dsDNA is the hybridization product of barcode ssDNA and its complementary sequences that have been pre-tethered onto AuNP (see Fig. 13). The two capture probes can recognize the same target, but at different epitopes. Therefore, a sandwich-like structure can be formed and extracted out of the assaying solution by applying an external magnetic field. The barcode DNA sequences are then discharged from the AuNP surface via water or buffer washing at elevated temperature, and subject to analysis using various DNA analysis techniques. Following such a protocol, the target analyte is successfully surrogated by multiple copies of ssDNA barcode sequences that can be detected with ultrahigh sensitivity.238
 | ||
| Fig. 13 The bio-barcode assay method. (A) Illustration of the two key components: AuNP modified with sensing probe I and barcode DNA; MMP functionalized with sensing probe II; (B) typical bio-barcode assay procedures for PSA detection. Reprinted with permission from ref. 235, © 2003 American Association for the Advancement of Science. | ||
The first demonstrated example of BCA was for detecting protein molecules (e.g. prostate-specific antigen, PSA) using antibodies (anti-PSA-1 and anti-PSA-2) as capturing probes.235 The PSA target was first recognized by the anti-PSA-1 coated MMP, and then anti-PSA-2 coated AuNP probes were added in to form the sandwich-like complex (Fig. 13). With scanometric barcode (DNA) analysis, LOD of 30 aM was achieved. And alternative DNA analysis using PCR allowed even lower LOD of 3 aM, six orders of magnitude lower than that of clinically accepted conventional assays. Some succeeding exploration further extends the BCA application in serum-based detection,239 as well as for some other target analytes including amyloid-β-derived diffusible ligands, HIV p24 Gag protein and bluetongue virus.240–242 Furthermore, Liu and co-workers managed to transfer this bio-barcode design onto a single disposable chip device.243 An LOD of 500 aM was successfully demonstrated using PSA as the model target.
With slight modification, BCA can also be utilized for DNA detection while attaining its ultrahigh sensitivity. In the modified design, two types of ssDNA sequences replaced the two antibody capturing probes on MMP and AuNP, respectively.244 Each sequence was perfectly complementary with half of the desired target analyte. Therefore, DNA hybridization catalysed the sandwich structure formation. The achieved LOD of 500 zeptomolar (zM = 10−21 M) using the scanometric technique was equivalent to about 10 copies of DNA molecules in the entire 30 μL sample volume.244 Such detection sensitivity was comparable with that of PCR. Similar to PCR, this BCA design is implemented in homogeneous solution as well. It therefore allows people to utilize high concentration of AuNP and MMP probes to push the reaction equilibrium toward the formation of sandwich complexes and to enhance the target binding kinetics.
In spite of all the merits and corresponding examples above, BCA assays suffer from numerous drawbacks as well. For instance, it requires many experimental and analytical steps, especially the synthetically demanding scanometric or PCR techniques for barcode analysis, which make such an assay very tedious and rather costly. Also, in the scanometric analysis, it is difficult to achieve consistent and complete barcode loading on the surface-support strands via hybridization. Such inefficiency, to a large extent, increases the experimental variability. Moreover, the dose response, defined as the net amount of generated signals for unit increase of target concentration, is relatively low. In other words, a large increase in target concentration can only lead to limited signal increment. In order to circumvent these downsides, intensive effort has been devoted in recent years to simplifying the assay process and also to exploring other convenient readout possibilities. To date, several new versions of BCA assays have been devised as shown in later discussion, inheriting the ultrahigh sensitivity from the very original design but averting some of the critical shortcomings.
Following the bio-barcode assay invention, Mirkin and co-workers reported another new AuNP probe for BCA that only required modification with single type of DNA, instead of three in the original design (including target-specific probe strand, thiolated barcode-capturing strand and barcode itself).245,246 The new probing DNA was single stranded with thiol-labels for AuNP tethering. It performs as target-specific sensing probe, as well as the barcode oligonucleotide. After target crosslinking and magnetic extraction, the thiolated ssDNA can be discharged from AuNP using dithiothreitol (DTT), followed by scanometric analysis as usual. Experiment showed successful detection of the mock mRNA target with 7 aM LOD under favourable conditions. Compared with the original design, the present example exhibits a simplified synthetic process and enhanced quantitative capability. However, its dose response is rather shallow, with a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold increase in target concentration leading to only <10 times of signal increase. Müller and co-workers demonstrated the possibility of a thousand-fold increase in dose response without sacrificing the detection sensitivity.247 Such an effort makes the BCA principle more analytically useful, as a more accurate calibration curve can be obtained with this improved dose response.
000-fold increase in target concentration leading to only <10 times of signal increase. Müller and co-workers demonstrated the possibility of a thousand-fold increase in dose response without sacrificing the detection sensitivity.247 Such an effort makes the BCA principle more analytically useful, as a more accurate calibration curve can be obtained with this improved dose response.
Instead of scanometric and PCR techniques, many alternative methods for barcode DNA analysis have been made available and have demonstrated several competitive edges (see Fig. 14). For example, Groves and co-workers reported a colorimetric-based BCA design for ultrasensitive detection of cytokine (e.g. interleukin-2).248,249 AuNP in conventional design was replaced with porous silica particle of 3 μm diameter. Its large size and high porosity permitted millions of barcode DNAs to be attached on the surface. After magnetic extraction and barcode discharge, the single-stranded barcode sequence was analysed using ssDNA functionalized AuNP. Barcode DNA herein can crosslink the AuNP and induce colour change from red to blue (or purple). Quantitative analysis of AuNP aggregation was accomplished using a Thin-Layer Chromatography (TLC) plate, with 30 aM LOD achieved both in buffer and in human serum samples. Such a colorimetric assay design might be of great clinical use in the diagnosis of Alzheimer's disease, as cytokines have been widely considered as the Alzheimer biomarker with maximum allowable concentration around 100 aM.240 In addition, Oh and others developed fluorescence-based BCA by attaching luminescent tags onto barcode DNA.250–254 Therefore, the barcode could be directly quantified by measuring the fluorescence emission intensity. Such a fluorescence-based design had been successfully demonstrated for detecting various bio-targets, including PSA,250 DNA,251 avian influenza virus,252 and Salmonella enterica serovar Enteritidis.253
 | ||
| Fig. 14 Various techniques for analysing the barcode DNA after discharged from the AuNP probe. | ||
The electrochemical read-out is another alternative of particular attention.255–260 However, the detailed assay design differs from case to case. In the first, all the procedures were similar to the conventional assay, except for the barcode analysis step. Barcode sequences in this case were specially designed, either poly-A or poly-G. They underwent hydrolysis in a heated H2SO4 environment. The obtained nucleobases were then electrochemically detected by virtue of their respective redox properties.255,256 In the second case, barcode DNA was labelled by some tracers like PbS or CdS.259 These tracers would be released by appropriate treatment and detected electrochemically using screen-printed carbon electrodes. In the third case, the functionalized AuNP composite was immobilized onto a bulky substrate. The AuNP-tethered DNA was hybridized by PbSNP-labelled barcode DNA. Released Pb2+ was detected through differential pulse anodic stripping voltammetry.257 The fourth case resembled the third. The only difference was, instead of PbSNP, the barcode sequences were bound with [Ru(NH3)6]3+. Such a Ru-based complex could produce detectable chronocoulometric signals under appropriate conditions.258 However, as mentioned at the beginning of this section, BCA is not purely a solution phase, especially in the third and fourth cases above, in which a flat substrate is involved. They are included here, only for the purpose of providing a complete overview of this ultrasensitive principle.
In addition, Hill et al. reported another novel BCA design for detecting double-stranded genomic DNA from Bacillus subtilis.261 The functionalized MMP- and AuNP-based composites were prepared as usual. A critical step in this new design was the dehybridization of target dsDNA and the utilization of blocker oligonucleotides. These blockers were bound to specific regions of the dehybridized target DNA upon cooling and prevented it from re-hybridization. Therefore, the produced ssDNA sequences were available as pseudo-targets, transforming the dsDNA detection task into ssDNA based one. Multiplexed target detection, such as various protein cancer markers,262 different oligonucleotides256,263 and gene sequences,259 has been realized as well, using BCA principles with the same scheme but different sensing probes.
Compared with the FRET or AFP-based assays, it is the ultrahigh detection sensitivity that makes the bio-barcode principle highly outstanding among the various assay designs. It offers great potential for clinical application of biological maker identification and therapeutic purposes. More importantly, when it becomes possible to detect specific target(s) at extremely low levels, detection sensitivity is no longer the driving force of the research. Instead, it will be the time to answer questions of how this technology can be applied and benefit the human community. In modern clinical practices, a cut-off concentration is usually set for specific biomarker(s) to differentiate the health and disease situations. Such a concentration is limited by current technology. With the application of an ultrasensitive bio-barcode assay, that cut-off concentration can be significantly lowered, which not only contributes to the even earlier diagnostics of associated diseases, but also provides a more informative target monitoring approach that can greatly assist clinicians in their examination of patients.
Chemiluminescence (CL)
Chemiluminescence (CL), as inferred by its name, is the luminescent emission from chemical reactions. Perhaps the most well-known example of biosensor application is the substrate oxidation catalysed by horseradish peroxidase (HRP),264–266 which has been intensively explored in the conventional microplate ELISA. Intrinsically, CL can only take charge of the signal transducing process, leaving target recognition open to any of the aforementioned mechanisms, including DNA hybridization, antibody–antigen binding, and aptamer–target interaction. For example, by exploiting DNA hybridization, Fan and co-workers264 reported a magnetic separation-based CL sensor with pM sensitivity (Fig. 15). The design comprised two main components: ssDNA functionalized magnetic particle (MP) and the AuNP-based composite. The AuNP herein is triply functionalized with ssDNA, HRP and BSA as a sensing probe, a signal transducer and a surface blocker, respectively. The added ssDNA target is able to crosslink the two components by hybridizing with the two probes. The formed sandwich-like structure is then magnetically extracted out of solution and undergoes optical analysis in a medium that contains a HRP-catalysing substrate. In other words, only in the presence of target ssDNA, HRP could be isolated from the assaying solution and catalyse the substrate oxidation for optical signal generation. A target of 100 pM concentration showed naked eye detectable colour contrast. Instrument-assisted reading further lowered the LOD to 1.0 pM. Moreover, tethering secondary AuNP-composite onto the MP-AuNP complex could further increase the HRP loading density, which led to another 10-fold improvement in detection sensitivity.267 | ||
| Fig. 15 The magnetic bead-AuNP based chemiluminescence biosensor for ssDNA detection. Added target ssDNA is able to crosslink the MMP and AuNP composites, facilitating the subsequent magnetic extraction of the sandwich-like complex out of assaying solution and further optical signal generation by reacting with the oxidation substrate. Reproduced with permission from ref. 264, © 2008 WILEY. | ||
Recently, many nanomaterials (e.g. GO, CNT and their various derivatives) have been found to exhibit peroxidase-like activity, which provides additional opportunities to design a CL-based biosensor. Yang and co-workers reported a solution phase sensor for PSA detection using a magnetic particle (MP) and GO.268 MP was functionalized with one type of PSA-specific antibody as a sensing probe and GO was functionalized with another type. In the presence of PSA, the sandwich-like structure was formed and magnetically isolated from the assay solution. Therefore, the target PSA concentration was translated into the amount of extracted GO. Such GO was further employed to catalyse hydroquinone oxidation reaction by H2O2. The colour change could be clearly observed for PSA concentrations of above and below 4 ng mL−1, which was the cut-off value to distinguish those possibly with prostate cancer from the healthy. As another example, Qu and colleagues developed a highly sensitive (1 μM LOD) solution phase copper ion (Cu2+) sensor, using the enhanced peroxidase-like activity of MWNT.269 In principle, azide-functionalized magnetic silica nanoparticles can conjugate with acetylene-functionalized MWNT in the presence of Cu2+ and sodium ascorbate. The obtained hybrid composite exhibited much higher peroxidase-like activity than MWNT alone, which was evident from the faster colour change upon addition of H2O2 and reaction substrate (TMB). As control, no detectable signals could be seen in the absence of the Cu2+ target.
Moreover, nanomaterials’ peroxidase-like activity can work collaboratively with its other characteristics, thereby providing much more versatile design possibilities for CL-based biosensor construction. By exploiting the peroxidase-like activity of graphene–AuNP interface and the different binding affinities of ssDNA/dsDNA towards the aromatic surface, Quan and co-workers proposed a novel sensing scheme that could be used for diverse applications, including detection of DNA, sensing of protein–aptamer interaction and monitoring of enzymatic DNA cleavage.270 The peroxidase-like activity was found at the graphene–AuNP interface, although graphene or AuNP alone showed little catalysing capability. Due to the strong affinity, added ssDNA or aptamer would adsorb onto graphene and prevent the peroxidase substrate from diffusing to, and binding with, the active interface. Catalytic reaction was therefore largely retarded. Introducing target analytes (e.g. complementary DNA sequence, aptamer-specific substrate or DNA enzymes) could liberate ssDNA from the graphene surface in respective manners and make the graphene–AuNP interface accessible to the peroxidase substrate. Catalytic reaction was switched on and started producing luminescent signals.
In some other special cases, the nanomaterial did not contribute to the peroxidase-like activity at all, but solely performed as a sensing probe or a signal amplifier. Dong and co-workers reported a novel colorimetric assay for detecting the ssDNA target and single-nucleotide polymorphism, using hemin–graphene hybrid assembly.271 In the assembly, it was the hemin molecule that exhibited peroxidase-like activity. Graphene only performed as a precipitation inducer in the design. In the presence of dsDNA, the hemin–graphene complex was easily precipitated by adding an electrolyte, whereas a mixture of the complex and ssDNA showed no precipitation under identical ionic strength. As follows, the un-precipitated solution was utilized to catalyse CL reactions and produce detectable signals. Experimentally, the solution absorbance value decreased linearly with the concentration of target ssDNA up to 100 nM. LOD was reported as 2 nM based on the 3δ rule.
The last example presented in this section is rather complicated, involving mechanisms of CL, fluorescence energy transfer, aptamer–target interaction, as well as enzymatic digestion (see Fig. 16).266 The system comprised FAM-labelled DNA functionalized-AuNP, hairpin-structured aptamer, exonuclease III (Exo III) and luminol–H2O2–HRP complex. In the absence of the target analyte (e.g. thrombin), the produced CL energy from CL was transferred to FAM via chemiluminescence resonance energy transfer (CRET). Such an energy was then quenched by AuNP via FRET due to FAM-AuNP proximity. However, in the presence of the target, the hairpin-structured aptamer would open up and form an aptamer–target complex with an oligonucleotide tail at the 5′-end. The tail could hybridize with the DNA sequence which was pre-attached onto AuNP at one end and pre-modified with FAM at the other. The formed dsDNA then activated selective cleavage of the FAM-labelled DNA sequences by Exo III, liberating FAM and the aptamer–target complex back into solution. The free FAM now demonstrated fluorescence via the aforementioned CRET mechanism. The released aptamer–target complex could hybridize with another FAM-labelled DNA, and initiated the hybridization–digestion-release cycle all over again. Ultrahigh sensitivity (2 fM LOD) of the present design was reported, benefiting from the cyclic signal amplification mechanism (see Fig. 16).
 | ||
| Fig. 16 (a) Schematic illustration of the amplified CRET aptasensor based on bi-resonance energy transfer, (b) the CL spectra of the CRET aptasensor for analysing target thrombin at different concentrations, (c) illustration of detection selectivity of the fabricated CRET aptasensor. Reproduced with permission from ref. 266, © 2012 Royal Society of Chemistry. | ||
Comparison of design principles
After going through all the design principles of the dual-transducer biosensors covered in this review, it is time to look back at the two questions we posed in the introduction, and set about answering them.What are the additional opportunities that nanomaterial based dual-transducer biosensors can provide relative to the single nanomaterial based counterpart?
First and foremost, the combined use of dual transducing elements contributes to the well-enhanced design versatility. In general, each type of nanomaterial possesses several fascinating and unique characteristics. With the single nanomaterial transducer, only the individual feature of that particular nanomaterial or interaction among neighbouring nanomaterials of the same identity can be employed, which largely restricts the design flexibility and versatility. On the other hand, in the dual-transducer design, utilization of different types of nanomaterials or combining the nanomaterial with other transducing elements can significantly broaden the spectrum of possible design configurations. Taking the metal nanoparticles as examples, when the AuNP or AgNP alone is used as the transducing element, the majority of the reported assays are based on the aggregation-induced colour change that arises from the LSPR band shift. In comparison, in the dual-transducer designs illustrated above, the AuNP or AgNP exhibits various roles, including a fluorescence quencher in FRET,154,164,168,175,181–183 a fluorescence enhancer,71 a polarization amplifier in AFP,228–230 and a probe carrier in both BCA and CL.235,239,240,244,245,264,267 Nowadays, biosensors in different fields have different requirements and restrictions. For example, in the initial stage of drug screening, assay designs with low cost and easy operation are preferred, in order to screen the drug candidates in a high throughput and rapid manner. However, in cancer diagnosis, ultrahigh sensitivity and selectivity are always the most primary concerns, overstepping all the other factors like cost-effectiveness and portability. Hence, with the additional design possibilities, we can always select the most suitable one that best accommodates the practical requirements.Moreover, the idea of enhancing design versatility can be further extended to the signal transducing step, such as the dual-signal based (both fluorescent and colorimetric) design for Hg2+ detection.162 With dual transducing elements, biosensors can be constructed in such a way that multiple signals are generated by a single target via different mechanisms. These signals can verify, support and reinforce each other, resulting in improved and more reliable sensing capability. The multiplexed detection discussed in the FRET and BCA sections of this review is another example of sensor design with versatility, in which various targets are mixed with a single assay kit and generate discrete signals for respective targets.108,111,172,182,211,256,259,262,263 Such a design not only reduces the assaying time and cost, but also makes the sample comparison feasible, reproducible and much reliable as well.
Second, in terms of performance, it is impractical to claim superiority for either the single nanomaterial based or the dual-transducer based schemes because similar LODs in the pM to nM range are observed for both. However, it is worth highlighting that the dual-transducer design can always employ the single-transducer sensing principle, but possibly equipped with an additional signal amplification mechanism to further enhance detection sensitivity. For instance, one of the modified BCA designs was based on ssDNA-induced AuNP crosslinking and subsequent solution colour change. What makes it outstanding is that those ssDNA sequences are not the real target analytes. Instead, they are merely barcode surrogates released from the target-induced sandwich-like structure. With such an amplification mechanism, the LOD of aM (10−18 M) level was demonstrated, which was significantly lower than those from direct target-induced aggregation designs. Nevertheless, it should also be noted that the dual-transducer based biosensors are in general more synthetically demanding, because multitude bio-functionalization procedures may be required for different transducing elements. This will jeopardize the cost-effectiveness and convenience of the dual-transducer biosensors in practical application and further possible commercialization.
What are the advantages and disadvantages of each of the four discussed dual-transducer biosensing principles?
Comparison of the four design principles is summarized in Fig. 17 in terms of detection sensitivity, selectivity, versatility, cost-effectiveness, design robustness, and portability. According to Giljohann et al.,2 these six factors are the drivers for biodiagnostic development, which we believe can also be used for assessing the respective biosensor designs. For each factor, four levels are set (from 1 to 4), with 1 being the worst and 4 being the best. These numbers are arbitrarily assigned based on the comparative merits of each principle. We herein borrow the concept of the “Holland Vocational Interest Test” to illustrate the results of the comparison. The plot shown in Fig. 17 clearly indicates the merits and demerits for each principle, with overall performance well reflected from the hexagon shape, area and orientation.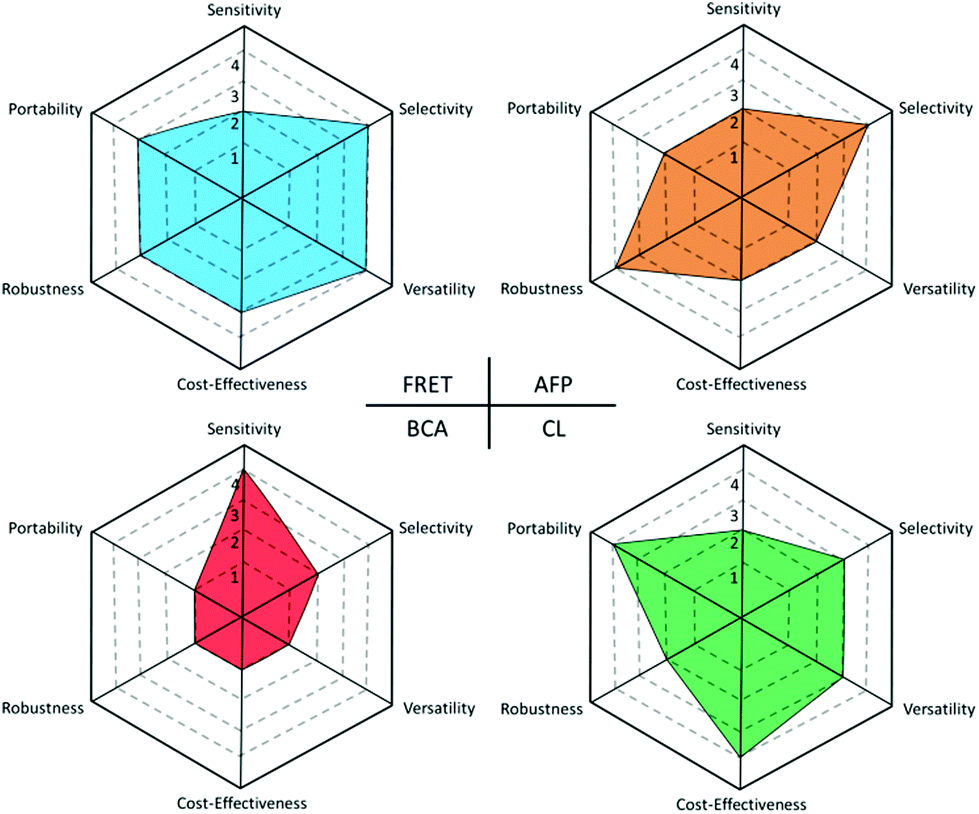 | ||
| Fig. 17 Comparison of the four dual-transducer design principles: Förster Resonance Energy Transfer (FRET), Amplified Fluorescence Polarization (AFP), Bio-barcode Assays (BCA) and Chemiluminescence (CL). | ||
It is not difficult to see that detection selectivity of direct transduction is superior to the indirect scheme, because the latter scenario is more vulnerable to environmental interferences. Concreting such a concept to the four principles, AFP and FRET assays are direct transduction, hence exhibiting better selectivity than the CL-based assays, the majority of which are indirect transduction. BCA is perhaps the least selective among the four, mainly because its target recognition and signal generation are well separated by multiple procedures in between. Therefore, the four principles should follow the selectivity merit order of FRET ≈ AFP (level 4) > CL (level 3) > BCA (level 2). In addition, it is worth highlighting that most of the reported assays involving non-specific adsorption on acceptor “A” (e.g., Fig. 6B, D, 8C, D, 9B and 12C, D) under FRET and AFP principles are only carried out in biological buffers (e.g., PBS,109,169,170,180,193 Tris-HCl,162,166,177 HEPES,171 and MOPS161) where no or limited interfering molecules are present as in a real biological matrix (e.g., serum or urine). Therefore, the selectivity of such designs in complex environments remains unclear. However, the aptamer, as a highly specific sensing probe, has demonstrated its advantageous edge of binding to the target analyte in complex samples like serum233 or living cells.164,165 This renders the aptasensor family with high potential for real life clinical applications.
Summary and outlook
The incorporation of a nanomaterial transducer with collaborative transducing elements opens up a new era for further development of novel biosensors for detecting a broad range of target analytes. With the assistance of representative examples of Förster Resonance Energy Transfer (FRET), Amplified Fluorescence Polarization (AFP), Bio-barcode Assay (BCA) and Chemiluminescence (CL) principles, we have demonstrated the basic design rules and performance of dual-transducer based biosensors. Within this review, we did not intend to cover the entire literature in this research field. Even within the four principles, we only include representative papers with significant breakthroughs either in sensing performance or design concept. We hope this review can serve the role of summarizing the recent advances in the research field, as well as stimulating more interest in dual-transducer based biosensor development.In spite of the remarkable progress achieved, there still exist several challenges (we would call them opportunities) that could perform as guidelines for further development in this field. First and foremost, incorporation of inorganic nanomaterials introduces heterogeneous interfaces in the homogeneous bio-system. This may lead to slow binding kinetics, low recognition efficiency, and some more severe problems associated with nanotoxicity.272,273 Additional effort is therefore required to resolve this concern. Second, most efforts on bioassay development are devoted to the improvement of target recognition and signal amplification steps, but very limited attention is focused on the post-data processing. Sometimes suitable data acquisition (e.g. kinetic monitoring) and in-depth data analysis (e.g. pattern recognition) are able to enhance the sensor performance and provide adequate information for target identification and quantification.274,275 Third, near infrared (NIR) detection has been underexplored, but deserves particular attention, because the NIR signal can penetrate through complex biological samples, such as whole blood, serum and urine. For demonstration of concept, the majority of the reported assays are based in biological buffer, or even DI water; but ultimately target detection should be conducted in body fluid or in vivo, which renders NIR-based assays of particular importance and significance. Last but not least, translation of various assay designs from laboratory based proof-of-concept to commercial products in the marketplace requires long-term endeavour from both academic and industry communities, in order to promote the landscape of disease diagnosis, healthcare, and environment monitoring to the next level.
Acknowledgements
SuX would like to acknowledge the Agency for Science, Technology and Research (A*STAR), Singapore, for the financial support of JCO 14302FG096 and ETPL/13-R15GAP-0011.References
- P. Alivisatos, Nat. Biotechnol., 2003, 22, 47 CrossRef PubMed.
- D. A. Giljohann and C. A. Mirkin, Nature, 2009, 462, 461 CrossRef CAS PubMed.
- D. F. Moyano and V. M. Rotello, Langmuir, 2011, 27, 10376 CrossRef CAS PubMed.
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042 CrossRef CAS PubMed.
- D. Alili and M. M. Stevens, Chem. Soc. Rev., 2010, 39, 3358 RSC.
- D. F. Moyano and V. M. Rotello, Langmuir, 2011, 27, 10376 CrossRef CAS PubMed.
- R. Wilson, Chem. Soc. Rev., 2008, 37, 2028 RSC.
- W. Zhao, M. A. Brook and Y. Li, ChemBioChem, 2008, 9, 2363 CrossRef CAS PubMed.
- Z. Wang and Y. Lu, J. Mater. Chem., 2009, 19, 1788 RSC.
- Y.-W. Lin, C.-W. Liu and H.-T. Chang, Anal. Methods, 2009, 1, 14 RSC.
- W. Zhong, Anal. Bioanal. Chem., 2009, 394, 47 CrossRef CAS PubMed.
- S. Song, Y. Qin, Y. He, Q. Huang, C. Fan and H.-Y. Chen, Chem. Soc. Rev., 2010, 39, 4234 RSC.
- W. R. Algar, K. Susumu, J. B. Delehanty and I. L. Medintz, Anal. Chem., 2011, 83, 8826 CrossRef CAS PubMed.
- X. Cao, Y. Ye and S. Liu, Anal. Biochem., 2011, 417, 1 CrossRef CAS PubMed.
- D. Liu, Z. Wang and X. Jiang, Nanoscale, 2011, 3, 1421 RSC.
- Y.-W. Lin, C.-C. Huang and H.-T. Chang, Analyst, 2011, 136, 863 RSC.
- V. K. K. Upadhyayula, Anal. Chim. Acta, 2012, 715, 1 CrossRef CAS PubMed.
- L. M. Zanoli, R. D'Agata and G. Spato, Anal. Bioanal. Chem., 2012, 402, 1759 CrossRef CAS PubMed.
- H. Jans and Q. Huo, Chem. Soc. Rev., 2012, 41, 2849 RSC.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739 CrossRef CAS PubMed.
- J. Li and J.-J. Zhu, Analyst, 2013, 138, 2506 RSC.
- Y. Wang, R. Hu, G. Lin, I. Roy and K.-T. Yong, ACS Appl. Mater. Interfaces, 2013, 5, 2786 CAS.
- J. Wang and X. Qu, Nanoscale, 2013, 5, 3589 RSC.
- H. Jang, J. Lee and D.-H. Min, J. Mater. Chem. B, 2014, 2, 2452 RSC.
- J.-J. Xu, W.-W. Zhao, S. Song, C. Fan and H.-Y. Chen, Chem. Soc. Rev., 2014, 43, 1601 RSC.
- L.-C. Ho, C.-W. Wang, P. Roy and H.-T. Chang, J. Chin. Chem. Soc., 2014, 61, 163 CrossRef CAS.
- P.-C. Chen, P. Roy, L.-Y. Chen, R. Ravindranath and H.-T. Chang, Part. Part. Syst. Charact., 2014, 31, 917 CrossRef CAS.
- W. Deng and E. M. Goldys, Analyst, 2014, 139, 6321 Search PubMed.
- J. Wang, Analyst, 2005, 130, 421 RSC.
- Y. Yonemori, E. Takahashi, H. Ren, T. hayashi and H. Endo, Anal. Chim. Acta, 2009, 633, 90 CrossRef CAS PubMed.
- R. Rossetti, S. Nakahara and L. E. Brus, J. Chem. Phys., 1983, 79, 1086 CrossRef CAS PubMed.
- L. Shao, Y. Gao and F. Yan, Sensors, 2011, 11, 11736 CrossRef PubMed.
- F. A. Esteve-Turrillas and A. Abad-Fuentes, Biosens. Bioelectron., 2013, 41, 12 CrossRef CAS PubMed.
- P. Reiss, M. Protière and L. Li, Small, 2009, 5, 154 CrossRef CAS PubMed.
- D. A. Hines and P. V. Kamat, ACS Appl. Mater. Interfaces, 2014, 6, 3041 CAS.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17 RSC.
- Z. Jin and N. Hildebrandt, Trends Biotechnol., 2012, 30, 394 CrossRef CAS PubMed.
- W. C. W. Chan and S. Nie, Science, 1998, 281, 2016 CrossRef CAS.
- M. Bruchez Jr., M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS.
- C. Luccardini, C. Tribet, F. Vial, V. Marchi-Artzner and M. Dahan, Langmuir, 2006, 22, 2304 CrossRef CAS PubMed.
- K. Susumu, H. T. Uyeda, I. L. Medintz, T. Pons, J. B. Delehanty and H. Mattoussi, J. Am. Chem. Soc., 2007, 129, 13987 CrossRef CAS PubMed.
- E. Gravel, C. Tanguy, E. Cassette, T. Pons, F. Knittel, N. Bernards, A. Garofalakis, F. Ducongé, B. Dubertret and E. Doris, Chem. Sci., 2013, 4, 411 RSC.
- N. Chen, Y. He, Y. Su, X. Li, Q. Huang, H. Wang, X. Zhang, R. Tai and C. Fan, Biomaterials, 2012, 33, 1238 CrossRef CAS PubMed.
- M. Bottrill and M. Green, Chem. Commun., 2011, 47, 7039 RSC.
- C. Kirchner, T. Liedl, S. Kudera, T. Pellegrino, A. M. Javier, H. E. Gaub, S. Stolzle, N. Fertig and W. J. Parak, Nano Lett., 2005, 5, 331 CrossRef CAS PubMed.
- F. Chen and D. Gerion, Nano Lett., 2004, 4, 1827 CrossRef CAS.
- S. T. Selvan, T. T. Tan and J. Y. Ying, Adv. Mater., 2005, 17, 1620 CrossRef CAS.
- F. M. Winnik and D. Maysinger, Acc. Chem. Res., 2013, 46, 672 CrossRef CAS PubMed.
- K.-T. Yong, H. Ding, I. Roy, W.-C. Law, E. J. Bergey, A. Maitra and P. N. Prasad, ACS Nano, 2009, 3, 502 CrossRef CAS PubMed.
- S. Kim, H. Mohseni, M. Erdtmann, E. Michel, C. Jelen and M. Razeghi, Appl. Phys. Lett., 1998, 73, 963 CrossRef CAS PubMed.
- J.-H. Park, L. Gu, G. v. Maltzahn, E. Ruoslahti, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2009, 8, 331 CrossRef CAS PubMed.
- F. Erogbogbo, K.-T. Yong, I. Roy, G. Xu, P. N. Prasad and M. T. Swihart, ACS Nano, 2008, 2, 873 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740 RSC.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20 CrossRef CAS.
- C. A. Mirkin, R. L. Letsinger, R. C. Mucic and J. J. Storhoff, Nature, 1996, 382, 607 CrossRef CAS PubMed.
- Y. N. Tan, K. H. Lee and X. Su, RSC Adv., 2013, 3, 21604 RSC.
- Y. N. Tan, X. Su, Y. Zhu and J. Y. Lee, ACS Nano, 2010, 4, 5101 CrossRef CAS PubMed.
- X. Xie, R. Deng, F. Liu, W. Xu, S. F. Y. Li and X. Liu, Anal. Methods, 2013, 5, 1116 RSC.
- X. Su and R. Kanjanawarut, ACS Nano, 2009, 3, 2751 CrossRef CAS PubMed.
- R. Kanjanawarut and X. Su, Anal. Chem., 2009, 81, 6122 CrossRef CAS PubMed.
- Y. N. Tan, X. Su, E. T. Liu and J. S. Thomsen, Anal. Chem., 2010, 82, 2759 CrossRef CAS PubMed.
- Y. N. Tan, K. H. Lee and X. Su, Anal. Chem., 2011, 83, 4251 CrossRef CAS PubMed.
- X. Xie, W. Xu, T. Li and X. Liu, Small, 2011, 7, 1393 CrossRef CAS PubMed.
- Z. Wang, J. H. Lee and Y. Lu, Adv. Mater., 2008, 20, 3263 CrossRef CAS.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2004, 126, 12298 CrossRef CAS PubMed.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2005, 127, 12677 CrossRef CAS PubMed.
- J. H. Lee, Z. Wang, J. Liu and Y. Lu, J. Am. Chem. Soc., 2008, 130, 14217 CrossRef CAS PubMed.
- P. Wu, K. Hwang, T. Lan and Y. Lu, J. Am. Chem. Soc., 2013, 135, 5254 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2006, 45, 90 CrossRef CAS PubMed.
- L. Sutarlie, K. M. Moh, M. G. L. Lim, S. Lukman, E. Cheung and X. Su, Plasmonics, 2014, 9, 753 CrossRef CAS.
- R. Levy, N. T. K. Thanh, R. C. Doty, I. Hussain, R. J. Nichols, D. J. Schiffrin, M. Brust and D. G. Fernig, J. Am. Chem. Soc., 2004, 126, 10076 CrossRef CAS PubMed.
- P. Chen, R. Selegard, D. Aili and B. Liedberg, Nanoscale, 2013, 5, 8973 RSC.
- X. Liu, Y. Wang, P. Chen, Y. Wang, J. Zhang, D. Aili and B. Liedberg, Anal. Chem., 2014, 86, 2345 CrossRef CAS PubMed.
- P. C. Patel, D. A. Giljohann, D. S. Seferos and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 17222 CrossRef CAS PubMed.
- H. S. Mader, P. Kele, S. M. Saleh and O. S. Wolfbeis, Curr. Opin. Chem. Biol., 2010, 14, 582 CrossRef CAS PubMed.
- F. Wang and X. Liu, Chem. Soc. Rev., 2009, 38, 976 RSC.
- J. Zhou, Z. Liu and F. Li, Chem. Soc. Rev., 2012, 41, 1323 RSC.
- L. Sun, Y. Wang and C. Yan, Acc. Chem. Res., 2014, 47, 1001–1009 CrossRef CAS PubMed.
- C. T. Xu, Q. Zhan, H. Liu, G. Somesfalean, J. Qian, S. He and S. Andersson-Engels, Laser Photonics Rev., 2013, 7, 663 CrossRef CAS.
- L. Wang and Y. Li, Chem. Mater., 2007, 19, 727 CrossRef CAS.
- F. Wang and X. Liu, J. Am. Chem. Soc., 2008, 130, 5642 CrossRef CAS PubMed.
- K. W. Kramer, D. Biner, G. Frei, H. U. Gudel, M. P. Hehlen and S. R. Muthi, Chem. Mater., 2004, 16, 1244 CrossRef.
- G. S. Yi and G. M. Chow, Adv. Funct. Mater., 2006, 16, 2324 CrossRef CAS.
- H.-X. Mai, Y.-W. Zhang, R. Si, Z.-G. Yan, L.-D. Sun, L.-P. You and C.-H. Yan, J. Am. Chem. Soc., 2006, 128, 6426 CrossRef CAS PubMed.
- X. Wang, J. Zhuang, Q. Peng and Y. Li, Inorg. Chem., 2006, 45, 6661 CrossRef CAS PubMed.
- Z. Li and Y. Zhang, Angew. Chem., Int. Ed., 2006, 45, 7732 CrossRef CAS PubMed.
- Z. Chen, H. Chen, H. Hu, M. Yu, F. Li, Q. Zhang, Z. Zhou, T. Yi and C. Huang, J. Am. Chem. Soc., 2008, 130, 3023 CrossRef CAS PubMed.
- L.-L. Li, P. Wu, K. Hwang and Y. Lu, J. Am. Chem. Soc., 2013, 135, 2411 CrossRef CAS PubMed.
- F. Wang, D. Banerjee, Y. Liu, X. Chen and X. Liu, Analyst, 2010, 135, 1839 RSC.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666 CrossRef CAS PubMed.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- H.-H. Lu, C.-L. Zhu, J. Li, J.-J. Liu, X. Chen and H.-H. Yang, Chem. Commun., 2010, 46, 3116 RSC.
- Y. Wang, Z. Li, J. Wang, J. Li and Y. Lin, Trends Biotechnol., 2011, 29, 205 CrossRef CAS PubMed.
- L. Feng, L. Wu and X. Qu, Adv. Mater., 2013, 25, 168 CrossRef CAS PubMed.
- D. Li and R. B. Kaner, Science, 2008, 320, 1170 CrossRef CAS PubMed.
- S. Park and R. S. Ruoff, Nat. Nanotechnol., 2009, 4, 127 Search PubMed.
- Q. H. Wang and M. C. Hersam, Nat. Chem., 2009, 1, 40 Search PubMed.
- X. Li, W. Cai, J. An, S. Kim, J. Nah, D. Yang, R. Piner, A. Velamakanni, I. Jung, E. Tutuc, S. K. Banerjee, L. Colombo and R. S. Ruoff, Science, 2009, 324, 1312 CrossRef CAS PubMed.
- M. J. Allen, V. C. Tung and R. B. Kaner, Chem. Rev., 2010, 110, 132 CrossRef CAS PubMed.
- M. Choucair, P. Thordarson and J. A. Stride, Nat. Nanotechnol., 2009, 4, 30 CrossRef CAS PubMed.
- X. Fan, W. Peng, Y. Li, X. Li, S. Wang, G. Zhang and F. Zhang, Adv. Mater., 2008, 20, 4490 CrossRef CAS.
- D. Tasis, N. Tagmatarchis, A. Bianco and M. Prato, Chem. Rev., 2006, 106, 1105 CrossRef CAS PubMed.
- W. Yang, K. R. Ratinac, S. P. Ringer, P. Thordarson, J. J. Gooding and F. Braet, Angew. Chem., Int. Ed., 2010, 49, 2114 CrossRef CAS PubMed.
- S. Lijima, Nature, 1991, 354, 56 CrossRef.
- L. Lacerda, S. Raffa, M. Prato, A. Bianco and K. Kostarelos, Nano Today, 2007, 2, 38 CrossRef.
- S. He, B. Song, D. Li, C. Zhu, W. Qi, Y. Wen, L. Wang, S. Song, H. Fang and C. Fan, Adv. Funct. Mater., 2010, 20, 453 CrossRef CAS.
- R. Yang, Z. Tang, J. Yan, H. Kang, Y. Kim, Z. Zhu and W. Tan, Anal. Chem., 2008, 80, 7408 CrossRef CAS PubMed.
- B. C. Satishkumar, L. O. Brown, Y. Gao, C.-C. Wang, H.-L. Wang and S. K. Doorn, Nat. Nanotechnol., 2007, 2, 560 CrossRef CAS PubMed.
- D. Cui, B. Pan, H. Zhang, F. Gao, R. Wu, J. Wang, R. He and T. Asahi, Anal. Chem., 2008, 80, 7996 CrossRef CAS PubMed.
- D. A. Heller, H. Jin, B. M. Martinez, D. Patel, B. M. Miller, T.-K. Yeung, P. V. Jena, C. Hobartner, T. Ha, S. K. Silverman and M. S. Strano, Nat. Nanotechnol., 2009, 4, 114 CrossRef CAS PubMed.
- H. Li, Z. Kang, Y. Liu and S.-T. Lee, J. Mater. Chem., 2012, 22, 24230 RSC.
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736 CrossRef CAS PubMed.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726 CrossRef CAS PubMed.
- X. Hu, L. Cheng, N. Wang, L. Sun, W. Wang and W. Liu, RSC Adv., 2014, 4, 18818 RSC.
- S.-L. Hu, K.-Y. Niu, J. Sun, J. Yang, N.-Q. Zhao and X.-W. Du, J. Mater. Chem., 2009, 19, 484 RSC.
- J. Zhou, C. Booker, R. Li, X. Zhou, T.-K. Sham, X. Sun and Z. Ding, J. Am. Chem. Soc., 2007, 129, 744 CrossRef CAS PubMed.
- Y.-P. Sun, X. Wang, F. Lu, L. Cao, M. J. Meziani, P. G. Luo, L. Gu and L. M. Veca, J. Phys. Chem. C, 2008, 112, 18295 CAS.
- H. Zhu, X. Wang, Y. Li, Z. Wang, F. Yang and X. Yang, Chem. Commun., 2009, 5118 RSC.
- H. Peng and J. Travas-Sejdic, Chem. Mater., 2009, 21, 5563 CrossRef CAS.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803 CrossRef CAS.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455 CrossRef CAS PubMed.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539 CrossRef CAS.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473 CrossRef CAS PubMed.
- Q.-L. Zhao, Z.-L. Zhang, B.-H. Huang, J. Peng, M. Zhang and D.-W. Pang, Chem. Commun., 2008, 5116 RSC.
- Y.-P. Sun, B. ZHou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. Wang, P. G. Luo, H. Yang, M. E. Kose, B. Chen, L. M. Veca and S.-Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756 CrossRef CAS PubMed.
- X. Wang, L. Cao, F. Lu, M. J. Meziani, H. Li, G. Qi, B. Zhou, B. A. Harruff, F. Kermarrec and Y.-P. Sun, Chem. Commun., 2009, 3774 RSC.
- S. C. Ray, A. Saha, N. R. Jana and R. Sarkar, J. Phys. Chem. C, 2009, 113, 18546 CAS.
- S.-T. Yang, L. Cao, P. G. Luo, F. Lu, X. Wang, H. Wang, M. J. Meziani, Y. Liu, G. Qi and Y.-P. Sun, J. Am. Chem. Soc., 2009, 131, 11308 CrossRef CAS PubMed.
- S.-T. Yang, X. Wang, H. Wang, F. Lu, P. G. Luo, L. Cao, M. J. Meziani, J.-H. Liu, M. Chen, Y. Huang and Y.-P. Sun, J. Phys. Chem. C, 2009, 113, 18110 CAS.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S.-Y. Xie and Y.-P. Sun, J. Am. Chem. Soc., 2007, 129, 11318 CrossRef CAS PubMed.
- H. X. Zhao, L. Q. Liu, Z. D. Liu, Y. Wang, X. J. Zhao and C. Z. Huang, Chem. Commun., 2011, 47, 2604 RSC.
- W. Wei, C. Xu, J. Ren, B. Xu and X. Qu, Chem. Commun., 2012, 48, 1284 RSC.
- W. Shi, Q. Wang, Y. Long, Z. Cheng, S. Chen, H. Zheng and Y. Huang, Chem. Commun., 2011, 47, 6695 RSC.
- T. Förster, Ann. Phys., 1948, 6, 55 CrossRef.
- T. Rantanen, M.-L. Jarvenpaa, J. Vuojola, K. Kuningas and T. Soukka, Angew. Chem., Int. Ed., 2008, 47, 3811 CrossRef CAS PubMed.
- X. M. Hua, J. I. Gersten and A. Nitzan, J. Chem. Phys., 1985, 83, 3650 CrossRef CAS PubMed.
- J. Zhang, Y. Fu and J. R. Lakowicz, J. Phys. Chem. C, 2007, 111, 50 CAS.
- J. Zhang, Y. Fu, M. H. Chowdhury and J. R. Lakowicz, J. Phys. Chem. C, 2007, 111, 11784 CAS.
- T. Förster, Discuss. Faraday Soc., 1959, 27, 7 RSC.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562 CrossRef CAS PubMed.
- T. Sen, K. K. Haldar and A. Patra, J. Phys. Chem. C, 2008, 112, 17945 CAS.
- C. S. Yun, A. Javier, T. Jennings, M. Fisher, S. Hira, S. Peterson, B. Hopkins, N. O. Reich and G. F. Strouse, J. Am. Chem. Soc., 2005, 127, 3115 CrossRef CAS PubMed.
- T. L. Jennings, M. P. Singh and G. F. Strouse, J. Am. Chem. Soc., 2006, 128, 5462 CrossRef CAS PubMed.
- P. C. Ray, A. Fortner and G. K. Darbha, J. Phys. Chem. B, 2006, 110, 20745 CrossRef CAS PubMed.
- L. Feng, H. Li, Y. Qu and C. Lv, Chem. Commun., 2012, 48, 4633 RSC.
- W. R. A. U. J. Krull, Anal. Bioanal. Chem., 2008, 391, 1609 CrossRef PubMed.
- S. Mayilo, M. A. Kloster, M. Wunderlich, A. Lutich, T. A. Klar, A. Nichtl, K. Kurzinger, F. D. Stefani and J. Feldmann, Nano Lett., 2009, 9, 4558 CrossRef CAS PubMed.
- S. Y. Lim, J. H. Kim, J. S. Lee and C. B. Park, Langmuir, 2009, 25, 13302 CrossRef CAS PubMed.
- N. Zhang, Y. Liu, L. Tong, K. Xu, L. Zhuo and B. Tang, Analyst, 2008, 133, 1176 RSC.
- B. Tang, L. Cao, K. Xu, L. Zhuo, J. Ge, Q. Li and L. Yu, Chem. – Eur. J., 2008, 14, 3637 CrossRef CAS PubMed.
- C. Zhang, Y. Yuan, S. Zhang, Y. Wang and Z. Liu, Angew. Chem., Int. Ed., 2011, 50, 6851 CrossRef CAS PubMed.
- X. Wang and X. Guo, Analyst, 2009, 134, 1348 RSC.
- J. Liu, J. H. Lee and Y. Lu, Anal. Chem., 2007, 79, 4120 CrossRef CAS PubMed.
- C.-C. Huang and H.-T. Chang, Anal. Chem., 2006, 78, 8332 CrossRef CAS PubMed.
- G. K. Darbha, A. Ray and P. C. Ray, ACS Nano, 2007, 1, 208 CrossRef CAS PubMed.
- L. Zhou, Y. Lin, Z. Huang, J. Ren and X. Qu, Chem. Commun., 2012, 48, 1147 RSC.
- L. Li, F. Gao, J. Ye, Z. Chen, Q. Li, W. Gao, L. Ji, R. Zhang and B. Tang, Anal. Chem., 2013, 85, 9721 CrossRef CAS PubMed.
- M. Xue, X. Wang, L. Duan, W. Gao, L. Ji and B. Tang, Biosens. Bioelectron., 2012, 36, 168 CrossRef CAS PubMed.
- Y. Wen, F. Xing, S. He, S. Song, L. Wang, Y. Long, D. Li and C. Fan, Chem. Commun., 2010, 46, 2596 RSC.
- H. Wang, Y. Wang, J. Jin and R. Yang, Anal. Chem., 2008, 80, 9021 CrossRef CAS PubMed.
- C. Liu, Z. Wang, H. Jia and Z. Li, Chem. Commun., 2011, 47, 4661 RSC.
- D. Zheng, D. S. Seferos, D. A. Giljohann, P. C. Patel and C. A. Mirkin, Nano Lett., 2009, 9, 3258 CrossRef CAS PubMed.
- Y. Wang, Z. Li, D. Hu, C.-T. Lin, J. Li and Y. Lin, J. Am. Chem. Soc., 2010, 132, 9274 CrossRef CAS PubMed.
- Y. Shi, W. T. Huang, H. Q. Luo and N. B. Li, Chem. Commun., 2011, 47, 4676 RSC.
- S. Wu, N. Duan, X. Ma, Y. Xia, H. Wang, Z. Wang and Q. Zhang, Anal. Chem., 2012, 84, 6263 CrossRef CAS PubMed.
- B. Tang, N. Zhang, Z. Chen, K. Xu, L. Zhuo, L. An and G. Yang, Chem. – Eur. J., 2008, 14, 522 CrossRef CAS PubMed.
- M. Liu, Q. Zhang, H. Zhao, S. Chen, H. Yu, Y. Zhang and X. Quan, Chem. Commun., 2011, 47, 4084 RSC.
- F. Li, Y. Feng, C. Zhao, P. Li and B. Tang, Chem. Commun., 2012, 48, 127 RSC.
- X.-H. Zhao, R.-M. Kong, X.-B. Zhang, H.-M. Meng, W.-N. Liu, W. Tan, G.-L. Shen and R.-Q. Yu, Anal. Chem., 2011, 83, 5062 CrossRef CAS PubMed.
- J. Zhang, L. Wang, H. Zhang, F. Boey, S. Song and C. Fan, Small, 2010, 6, 201 CrossRef CAS PubMed.
- C.-Y. Zhang, H.-C. Yeh, M. T. Kuroki and T.-H. Wang, Nat. Mater., 2005, 4, 826 CrossRef CAS.
- P. Zhang, S. Rogelj, K. Nguyen and D. Wheeler, J. Am. Chem. Soc., 2006, 128, 12410 CrossRef CAS PubMed.
- Z. Dai, J. Zhang, Q. Dong, N. Guo, S. Xu, B. Sun and Y. Bu, Chin. J. Chem. Eng., 2007, 15, 791 CrossRef CAS.
- C.-H. Lu, H.-H. Yang, C.-L. Zhu, X. Chen and G.-N. Chen, Angew. Chem., Int. Ed., 2009, 48, 4785 CrossRef CAS PubMed.
- C.-H. Lu, J. Li, J.-J. Liu, H.-H. Yang, X. Chen and G.-N. Chen, Chem. – Eur. J., 2010, 16, 4889 CrossRef CAS PubMed.
- H. Dong, W. Gao, F. Yan, H. Ji and H. Ju, Anal. Chem., 2010, 82, 5511 CrossRef CAS PubMed.
- F. Li, Y. Huang, Q. Yang, Z. Zhong, D. Li, L. Wang, S. Song and C. Fan, Nanoscale, 2010, 2, 1021 RSC.
- R. Yang, J. Jin, Y. Chen, N. Shao, H. Kang, Z. Xiao, Z. Tang, Y. Wu, Z. Zhu and W. Tan, J. Am. Chem. Soc., 2008, 130, 8351 CrossRef CAS PubMed.
- B. Dubertret, M. Calame and A. J. Libchaber, Nat. Biotechnol., 2001, 19, 365 CrossRef CAS PubMed.
- S. Song, Z. Liang, J. Zhang, L. Wang, G. Li and C. Fan, Angew. Chem., Int. Ed., 2009, 48, 8670 CrossRef CAS PubMed.
- D. J. Maxwell, J. R. Taylor and S. Nie, J. Am. Chem. Soc., 2002, 124, 9606 CrossRef CAS PubMed.
- K. Wang, Z. Tang, C. J. Yang, Y. Kim, X. Fang, W. Li, Y. Wu, C. D. Medley, Z. Cao, J. Li, P. Colon, H. Lin and W. Tan, Angew. Chem., Int. Ed., 2008, 48, 856 CrossRef PubMed.
- N. E. Broude, Trends Biotechnol., 2002, 20, 249 CrossRef CAS.
- S. Tyagi, D. P. Bratu and F. R. Kramer, Nat. Biotechnol., 1998, 16, 49–53 CrossRef CAS PubMed.
- S. Tyagi and F. R. Kramer, Nat. Nanotechnol., 1996, 14, 303 CrossRef CAS PubMed.
- H. Li, M. Wang, C. Wang, W. Li, W. Qiang and D. Xu, Anal. Chem., 2013, 85, 4492 CrossRef CAS PubMed.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102 CrossRef CAS PubMed.
- E. S. Jeng, A. E. Moll, A. C. Roy, J. B. Gastala and M. S. Strano, Nano Lett., 2006, 6, 371 CrossRef CAS PubMed.
- E. Oh, M.-Y. Hong, D. Lee, S.-H. Nam, H. C. Yoon and H.-S. Kim, J. Am. Chem. Soc., 2005, 127, 3270 CrossRef CAS PubMed.
- H. Chang, L. Tang, Y. Wang, J. Jiang and J. Li, Anal. Chem., 2010, 82, 2341 CrossRef CAS PubMed.
- Y.-M. Wu, Y. Cen, L.-J. Huang, R.-Q. Yu and X. Chu, Chem. Commun., 2014, 50, 4759 RSC.
- X. Wang, C. Wang, K. Qu, Y. Song, J. Ren, D. Miyoshi, N. Sugimoto and X. Qu, Adv. Funct. Mater., 2010, 20, 3967 CrossRef CAS.
- S. Lukman, K. M. M. Aung, J. Liu, B. Liu and X. Su, ACS Appl. Mater. Interfaces, 2013, 5, 12725 CAS.
- S. Lukman, K. M. M. Aung, M. G. L. Lim, S. Hong, S. K. Tan, E. Cheung and X. Su, RSC Adv., 2014, 4, 8883 RSC.
- S. Lee, E.-J. Cha, K. Park, S.-Y. Lee, J.-K. Hong, I.-C. Sun, S. Y. Kim, K. Choi, I. C. Kwon, K. Kim and C.-H. Ahn, Angew. Chem., Int. Ed., 2008, 2804 CrossRef CAS PubMed.
- J. Li, C.-H. Lu, Q.-H. Yao, X.-L. Zhang, J.-J. Liu, H.-H. Yang and G.-N. Chen, Biosens. Bioelectron., 2011, 26, 3894 CrossRef CAS PubMed.
- H. Wang, Q. Zhang, X. Chu, T. Chen, J. Ge and R. Yu, Angew. Chem., Int. Ed., 2011, 50, 7065 CrossRef CAS PubMed.
- K. Boeneman, B. C. Mei, A. M. Dennis, G. Bao, J. R. Deschamps, H. mattoussi and I. L. Medintz, J. Am. Chem. Soc., 2009, 131, 3828 CrossRef CAS PubMed.
- H. Yao, Y. Zhang, F. Xiao, Z. Xia and J. Rao, Angew. Chem., Int. Ed., 2007, 46, 4346 CrossRef CAS PubMed.
- I. L. Medintz, A. R. Clapp, F. M. Brunel, T. Tiefenbrunn, H. T. Uyeda, E. L. Chang, J. R. Deschamps, P. E. Dawson and H. Mattoussi, Nat. Mater., 2006, 5, 581 CrossRef CAS PubMed.
- J. C. Breger, K. E. Sapsford, J. Ganek, K. Susumu, M. H. Stewart and I. L. Medintz, ACS Appl. Mater. Interfaces, 2014, 6, 11529 CAS.
- D. Feng, Y. Zhang, T. Feng, W. Shi, X. Li and H. Ma, Chem. Commun., 2011, 47, 10680 RSC.
- L. Wang, R. Yan, Z. Huo, L. Wang, J. Zeng, J. Bao, X. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2005, 44, 6054 CrossRef CAS PubMed.
- M. Wang, W. Hou, C. Mi, W. Wang, Z. Xu, H. Teng, C. Mao and S. Xu, Anal. Chem., 2009, 81, 8783 CrossRef CAS PubMed.
- K. J. Oh, K. J. Cash, V. Hugenberg and K. W. Plaxco, Bioconjugate Chem., 2007, 18, 607 CrossRef CAS PubMed.
- K. J. Oh, K. J. Cash and K. W. Plaxco, Chem. – Eur. J., 2009, 15, 2244 CrossRef CAS PubMed.
- K. J. Oh, K. J. Cash and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 14018 CrossRef CAS PubMed.
- S. Thurley, L. Roglin and O. Seitz, J. Am. Chem. Soc., 2007, 129, 12693 CrossRef CAS PubMed.
- M. Suzuki, Y. Husimi, H. Komatsu, K. Suzuki and K. T. Douglas, J. Am. Chem. Soc., 2008, 130, 5720 CrossRef CAS PubMed.
- G. Liu, J. Wang, D. S. Wunschel and Y. Lin, J. Am. Chem. Soc., 2006, 128, 12382 CrossRef CAS PubMed.
- Y. Wang, D. V. Zagorevski, M. R. Lennartz, D. J. Loegering and J. A. Steken, Anal. Chem., 2009, 81, 9961 CrossRef CAS PubMed.
- H. Jang, Y.-K. Kim, H.-M. Kwon, W.-S. Yeo, D.-E. Kim and D.-H. Min, Angew. Chem., Int. Ed., 2010, 49, 5703 CrossRef CAS PubMed.
- J. E. Ghadiali, S. B. Lowe and M. M. Stevens, Angew. Chem., Int. Ed., 2011, 123, 3479 CrossRef.
- W. Wu, H. Hu, F. Li, L. Wang, J. Gao, J. Lu and C. Fan, Chem. Commun., 2011, 47, 1201 RSC.
- W. R. Algar, M. G. Ancona, A. P. Malanoski, K. Susumu and I. L. Medintz, ACS Nano, 2012, 6, 11044 CAS.
- P. Biswas, L. N. Cella, S. H. Kang, A. Mulchandani, M. V. yates and W. Chen, Chem. Commun., 2011, 47, 5259 RSC.
- J. H. Kim and B. H. Chung, Small, 2010, 6, 126 CrossRef CAS PubMed.
- D. M. Jameson and J. A. Ross, Chem. Rev., 2010, 110, 2685 CrossRef CAS PubMed.
- X. Fang, Z. Cao, T. Beck and W. Tan, Anal. Chem., 2001, 73, 5752 CrossRef CAS.
- G. Gokulrangan, J. R. Unruh, D. F. Holub, B. Ingram, C. K. Johnson and G. S. Wilson, Anal. Chem., 2005, 77, 1963 CrossRef CAS PubMed.
- D. Zhang, Q. Zhao, B. Zhao and H. Wang, Anal. Chem., 2012, 84, 3070 CrossRef CAS PubMed.
- W. Li, K. Wang, W. Tan, C. Ma and X. Yang, Analyst, 2007, 132, 107 RSC.
- M. Zou, Y. Chen, X. Xu, H. Huang, F. Liu and N. Li, Biosens. Bioelectron., 2012, 32, 148 CrossRef CAS PubMed.
- L. Cui, Y. Zou, N. Lin, Z. Zhu, G. Jenkins and C. J. Yang, Anal. Chem., 2012, 84, 5535 CrossRef CAS PubMed.
- Z. Zhu, C. Ravelet, S. Perrier, V. Guieu, E. Fiore and E. Peyrin, Anal. Chem., 2012, 84, 7203 CrossRef CAS PubMed.
- Y. Qiao, H. Tang, G. R. Munske, P. Dutta, C. F. Ivory and W.-J. Dong, J. Fluoresc., 2011, 21, 2101 CrossRef CAS PubMed.
- B.-C. Ye and B.-C. Yin, Angew. Chem., Int. Ed., 2008, 47, 8386 CrossRef CAS PubMed.
- Y. Huang, S. Zhao, Z.-F. Chen, Y.-C. Liu and H. Liang, Chem. Commun., 2011, 47, 4763 RSC.
- Y. Yu, Y. Liu, S. J. Zhen and C. Z. Huang, Chem. Commun., 2013, 49, 1942 RSC.
- J. Liu, C. Wang, Y. Jiang, Y. Hu, J. Li, S. Yang, Y. Li, R. Yang, W. Tan and C. Z. Huang, Anal. Chem., 2013, 85, 1424 CrossRef CAS PubMed.
- Y. Huang, S. Zhao, Z.-F. Chen, M. Shi and H. Liang, Chem. Commun., 2012, 48, 7480 RSC.
- Y. Huang, J. Chen, M. Shi, S. Zhao, Z.-F. Chen and H. Liang, J. Mater. Chem. B, 2013, 1, 2018 RSC.
- J.-M. Nam, C. S. Thaxton and C. A. Mirkin, Science, 2003, 301, 1884 CrossRef CAS PubMed.
- C. S. Thaxton, D. G. Georganopoulou and C. A. Mirkin, Clin. Chim. Acta, 2006, 363, 120 CrossRef CAS PubMed.
- S. Brakmann, Angew. Chem., Int. Ed., 2004, 43, 5730 CrossRef CAS PubMed.
- T. A. Taton, C. A. Mirkin and R. L. Letsinger, Science, 2000, 289, 1757 CrossRef CAS.
- C. S. Thaxton, R. Elghanian, A. D. Thomas, S. I. Stoeva, J.-S. Lee, N. D. Smith, A. J. Schaeffer, H. Klocker, W. Horninger, G. Bartsch and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 18437 CrossRef CAS PubMed.
- D. G. Georganopoulou, L. Chang, J.-M. Nam, C. S. Thaxton, E. J. Mufson, W. L. Klein and C. A. Mirkin, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 2273 CrossRef CAS PubMed.
- E.-Y. Kim, J. Stanton, B. T. Korber, K. Krebs, D. Bogdan, K. Kunstman, S. Wu, J. P. Phair, C. A. Mirkin and S. M. Wolinsky, Nanomedicine, 2008, 3, 293 CrossRef CAS PubMed.
- H.-q. Yin, M.-x. Jia, L.-j. Shi, S. Yang, L.-y. Zhang, Q.-m. Zhang, S.-q. Wang, G. Li and J.-g. Zhang, J. Virol. Methods, 2011, 178, 225 CrossRef CAS PubMed.
- E. D. Goluch, J.-M. Nam, D. G. Georganopoulou, T. N. Chiesl, K. A. Shaikh, K. S. Ryu, A. E. Barron, C. A. Mirkin and C. Liu, Lab Chip, 2006, 6, 1293 RSC.
- J.-M. Nam, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2004, 126, 5932 CrossRef CAS PubMed.
- C. S. Thaxton, H. D. Hill, D. G. Georganopoulou, S. I. Stoeva and C. A. Mirkin, Anal. Chem., 2005, 77, 8174 CrossRef CAS PubMed.
- H. D. Hill and C. A. Mirkin, Nat. Protoc., 2006, 1, 324 CrossRef CAS PubMed.
- Y. P. Bao, T.-F. Wei, P. A. Lafebvre, H. An, L. He, G. T. Kunkel and U. R. Müller, Anal. Chem., 2006, 78, 2055 CrossRef CAS PubMed.
- J.-M. Nam, A. R. Wise and J. T. Groves, Anal. Chem., 2005, 77, 6985 CrossRef CAS PubMed.
- J.-M. Nam, K.-J. Jang and J. T. Groves, Nat. Protoc., 2007, 2, 1438 CrossRef CAS PubMed.
- B.-K. Oh, J.-M. Nam, S. W. Lee and C. A. Mirkin, Small, 2006, 2, 103 CrossRef CAS PubMed.
- D. Zhu, Y. Tang, D. Xing and W. R. Chen, Anal. Chem., 2008, 80, 3566 CrossRef CAS PubMed.
- C. Cao, R. Dhumpa, D. D. Bang, Z. Ghavifekr, J. Hogberg and A. Wolff, Analyst, 2010, 135, 337 RSC.
- D. Zhang, D. J. Carr and E. C. Alocilja, Biosens. Bioelectron., 2009, 24, 1377 CrossRef CAS PubMed.
- M. Trevisan, M. Schawaller, G. Quapil, E. Souteyrand, Y. Merieux and J.-P. Cloarec, Biosens. Bioelectron., 2010, 26, 1631 CrossRef CAS PubMed.
- P. He, L. Shen, Y. Cao and D. Li, Anal. Chem., 2007, 79, 8024 CrossRef CAS PubMed.
- X. Zhang, H. Su, S. Bi, S. Li and S. Zhang, Biosens. Bioelectron., 2009, 24, 2730 CrossRef CAS PubMed.
- X. Zhang, B. Qi, Y. Li and S. Zhang, Biosens. Bioelectron., 2009, 25, 259 CrossRef CAS PubMed.
- Y. Li, B. Liu, X. Li and Q. Wei, Biosens. Bioelectron., 2010, 25, 2543 CrossRef CAS PubMed.
- D. Zhang, M. C. Huarng and E. C. Alocilja, Biosens. Bioelectron., 2010, 26, 1736 CrossRef CAS PubMed.
- L. Shen, Z. Chen, Y. Li, S. He, S. Xie, X. Xu, Z. Liang, X. Meng, Q. Li, Z. Zhu, M. Li, M. Li, X. C. Le and Y. Shao, Anal. Chem., 2008, 80, 6323 CrossRef CAS PubMed.
- H. D. Hill, R. A. Vega and C. A. Mirkin, Anal. Chem., 2007, 79, 9218 CrossRef PubMed.
- S. I. Stoeva, J.-S. Lee, J. E. Smith, S. T. Rosen and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 8378 CrossRef CAS PubMed.
- M. He, K. Li, J. Xiao and Y. Zhou, J. Virol. Methods, 2008, 151, 126 CrossRef CAS PubMed.
- J. Li, S. Song, X. Liu, L. Wang, D. Pan, Q. Huang, Y. Zhao and C. Fan, Adv. Mater., 2008, 20, 497 CrossRef CAS.
- S. Bi, Y. Yan, X. Yang and S. Zhang, Chem. – Eur. J., 2009, 15, 4704 CrossRef CAS PubMed.
- Y. Huang, S. Zhao, Z.-F. Chen, M. Shi, J. Chen and H. Liang, Chem. Commun., 2012, 48, 11877 RSC.
- J. Li, S. Song, D. Li, Y. Su, Q. Huang, Y. Zhao and C. Fan, Biosens. Bioelectron., 2009, 24, 3311 CrossRef CAS PubMed.
- F. Qu, T. Li and M. Yang, Biosens. Bioelectron., 2011, 26, 3927 CrossRef CAS PubMed.
- Y. Song, K. Qu, C. Xu, J. Ren and X. Qu, Chem. Commun., 2010, 46, 6572 RSC.
- M. Liu, H. Zhao, S. Chen, H. Yu and X. Quan, ACS Nano, 2012, 6, 3142 CrossRef CAS PubMed.
- Y. Guo, L. Deng, J. Li, S. Guo, E. Wang and S. Dong, ACS Nano, 2011, 5, 1282 CrossRef CAS PubMed.
- H. F. Krug and P. Wick, Angew. Chem., Int. Ed., 2011, 50, 1260 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26 CrossRef CAS PubMed.
- S. A. Minaker, K. D. Daze, M. C. F. Ma and F. Hof, J. Am. Chem. Soc., 2012, 134, 11674 CrossRef CAS PubMed.
- S. S. Chou, M. De, J. Luo, V. M. Rotello, J. Huang and V. P. Dravid, J. Am. Chem. Soc., 2012, 134, 16725 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |