
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
En route to traceable reference standards for surface group quantifications by XPS, NMR and fluorescence spectroscopy†
Andreas
Hennig
*ab,
Paul M.
Dietrich
*a,
Felix
Hemmann
a,
Thomas
Thiele
c,
Heike
Borcherding
c,
Angelika
Hoffmann
a,
Uwe
Schedler
c,
Christian
Jäger
a,
Ute
Resch-Genger
*a and
Wolfgang E. S.
Unger
a
aBAM Federal Institute for Materials Research and Testing, Richard-Willstaetter-Strasse 11, 12203 Berlin, Germany. E-mail: paul.dietrich@bam.de; ute.resch@bam.de
bJacobs University Bremen, School of Engineering and Science, Campus Ring 1, 28759 Bremen, Germany. E-mail: a.hennig@jacobs-university.de
cPolyAn GmbH, Rudolf-Baschant-Strasse 2, 13086 Berlin, Germany
First published on 28th January 2015
Abstract
The fluorine content of polymer particles labelled with 2,2,2-trifluoroethylamine was reliably quantified with overlapping sensitivity ranges by XPS and solid-state NMR. This provides a first step towards reference materials for the metrological traceability of surface group quantifications. The extension of this concept to fluorescence spectroscopy is illustrated.
The controlled functionalization of surfaces has moved into the focus of many material and life scientists as this paves the way for applications in biosensing, drug delivery, implantation medicine, separation sciences, optoelectronics, and solar energy conversion.1–3 The precise knowledge of the chemical nature, areic group density, and spatial distribution of surface functional groups is thus key for the broad application of existing and rational design of improved functional materials as well as for the public acceptance of new nanotechnology-based materials. It is similarly relevant for a reliable quality control during material fabrication and processing and thus, for their reproducible large-scale production. Consequently, numerous analytical methods have been used for the characterization of functionalized surfaces.2,4–7
Despite the overall importance of a reliable and quantitative surface analysis, we recently noted that surface group quantification methods are usually not mutually validated.5–7 In fact, even the application of more than one surface group quantification method to the same material is not always common practice. This is particularly severe for surface quantification methods relying on the use of chemical derivatization agents. Therein, a quantitative coupling yield is often presumed, which may not always hold true.5–9 In addition, certain surface functional group quantification methods such as X-ray photoelectron spectroscopy (XPS) have a limited information depth and require elaborated data analysis based on theoretical models to address layered structures, especially on curved surfaces.7 However, also these quantification models have so far only rarely been experimentally verified.10
A standard reference material applicable to various surface functional group quantification methods is thus highly desirable. This would allow mutual method validation as well as experimental testing of theoretical quantification models. A particular challenge in this endeavour is to provide overlapping sensitivity ranges for the various methods, which all have different limits regarding the highest and lowest concentrations that can be reliably detected. Moreover, certain surface analysis methods (e.g. XPS) are mainly applied to planar functionalized surfaces rather than to particles, while others (e.g. NMR) are commonly applied to bulk materials rather than to surface analysis.4
Herein, we now present the first step towards the development of such a reference material. We have previously extensively characterized polymer particles composed of a poly(methyl methacrylate) (PMMA) core with a grafted shell of poly(acrylic acid) (PAA),5,6 including a detailed characterization by XPS before and after labelling with 2,2,2-trifluoroethylamine (TFEA).7 The fluorine content of these particles was now additionally determined by 19F solid-state NMR, which provides an unprecedented link in the traceability chain between XPS and NMR. Furthermore, we illustrate that the detection sensitivity by 19F NMR is also sufficient to provide overlapping sensitivity ranges with fluorescence spectroscopy. We thus disclose herein a strategy towards reference materials for the surface functional group quantification by NMR, XPS, and fluorescence spectroscopy.
We selected four different PMMA particle batches with varying amounts of surface PAA (0, 35, 99, and 946 μmol g−1, referred to as P0, P35, P99, and P946). The number of surface COOH groups was previously determined by conductometry,5–7 and the amount of surface-grafted PAA on P946 was now confirmed with solid-state 13C NMR by comparing the integrated peak areas of the carboxy region and the methyl group. This gave PAA surface densities of (1600 ± 1000) μmol g−1 for P946, which agree well with the results from conductometry within the stated measurement uncertainties.‡
The different PAA-functionalized particles were analysed by XPS, which gave the elemental composition within the first 10 nm of the surface (XPS information depth).7 The survey spectra as well as the high-resolution C 1s and O 1s XP spectra of the different PMMA/PAA microparticles were all perfectly consistent with the varying amounts of surface-grafted PAA of the unlabelled particles obtained by our previous studies.5–7 Successful covalent labelling with TFEA according to a previously established protocol (Scheme S1a in ESI†),5 was confirmed by significantly altered photoelectron spectra, in particular by the appearance of a new peak in the survey and core-level spectra corresponding to the CF3 group (cf. Fig. 1a and b).7
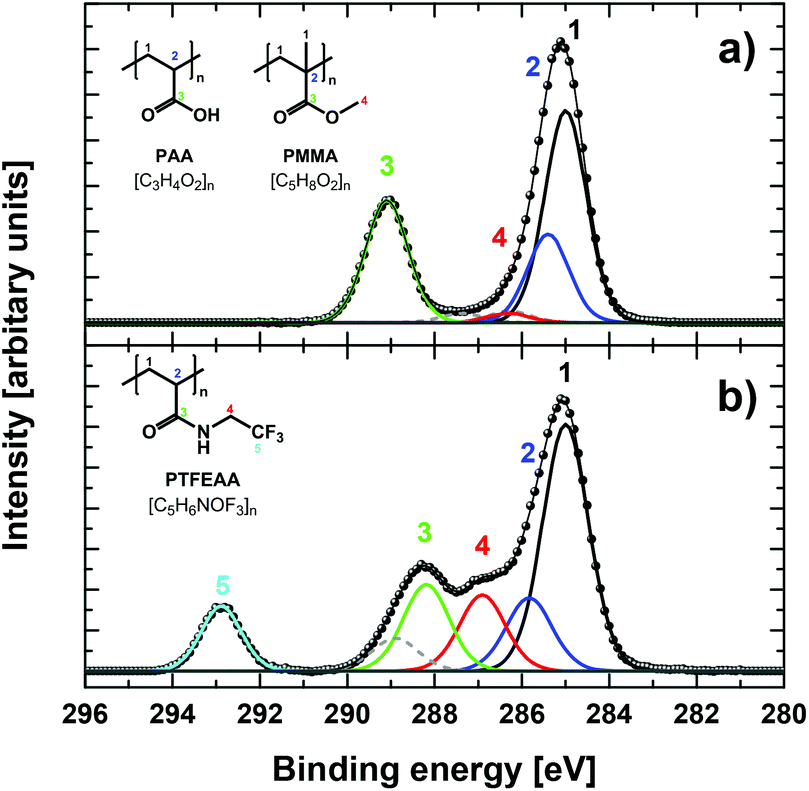 | ||
| Fig. 1 High-resolution C 1s core-level spectra of PMMA/PAA particles P946 (a) before and (b) after chemical derivatization with TFEA. Additional components originating from poly(N-vinylpyrrolidone) encapsulated in the PMMA core are shown as dashed lines in grey. | ||
Successful covalent surface labelling with TFEA was also independently confirmed by solid-state 19F NMR. All NMR spectra showed a peak at δ = (−70.7 ± 0.3) ppm with MAS spinning side bands, which is the typical peak position of the CF3 group of trifluoroethylamide (Fig. 2). This is a striking result, since it demonstrates that even the particles with the lowest amount of surface-grafted PAA could be successfully analysed by 19F NMR within reasonable time. This is due to the high gyromagnetic ratio and natural abundance of the 19F nucleus, which is only outperformed by 1H NMR in terms of its relative sensitivity. The latter is, however, inapplicable to routine surface group quantifications because of the omnipresence of hydrogen atoms and a comparably small frequency range of typical 1H NMR resonances in combination with line broadening in solid-state NMR.
 | ||
| Fig. 2 Solid-state 19F MAS NMR spectrum of TFEA-labelled P946. | ||
After characterization of the TFEA-labelled particles, the amount of surface-bound TFEA was quantified by XPS and solid-state 19F NMR spectroscopy (Table 1). By XPS, the fraction of TFEA-labelled COOH groups was determined by comparing the integrated peak areas of carbon and fluorine atoms in the survey spectra.7,11 This gave coupling yields of 32% for P35, 54% for P99, and 47% for P946, respectively. Subsequently, the quantification of surface-bound TFEA by solid-state 19F NMR was performed. Typically, known amounts of sample and standard are mixed and the integrated peak areas (including the areas of the MAS side bands) of the sample are referenced to the signal derived from a known amount of the 19F intensity standard.12 This is, however, in the present case not feasible, because only a very low fraction of the sample is TFEA-labelled, namely just the surface groups. The required amount of standard, i.e., 4-(trifluoromethyl) benzoic acid, is thus too small to reliably afford a homogeneous mixture of sample and reference. As an alternative, reference and sample were measured consecutively in different rotors under the same experimental conditions, particularly using the same receiver gain. This procedure was repeated several times to confirm the spectrometer stability. The results indicated a maximum deviation between individual measurements of about 1%. This analysis gave TFEA coupling yields of 30%, 26%, and 39% for P35, P99, and P946, respectively.
| Sample | [COOH]a (μmol g−1) | XPSb | 19F-NMR | |||
|---|---|---|---|---|---|---|
| F (at%) | Yield (%) | [CF3] (μmol g−1) | Yieldc (%) | [CF3] (μmol g−1) | ||
| a Total number of surface COOH groups as determined by conductometry (uncertainty ca. 9%, see ref. 5). b Taken from ref. 7 (uncertainty 10%). c Coupling yield calculated using 4-(trifluoromethyl) benzoic acid as reference. | ||||||
| P35 | 35 | 15.8 ± 0.2 | 32 ± 1 | 11 ± 1 | 30 ± 1 | 10.4 ± 0.5 |
| P99 | 99 | 20.8 ± 1.3 | 54 ± 3 | 53 ± 3 | 26 ± 1 | 26 ± 1 |
| P946 | 946 | 19.9 ± 0.4 | 47 ± 2 | 443 ± 24 | 39 ± 2 | 367 ± 18 |
The coupling yields determined by XPS and solid-state 19F NMR are in good agreement for P35 and P946, and they are significantly higher than those previously reported for the fluorescein derivative FL-A (Chart S1 in ESI†) and adamantylmethylamine (both around 5%).5,6 This is probably due to the smaller size of TFEA and to a much lower pKa of the amino group of TFEA (pKa = 5.7) compared to aliphatic amines (pKa ∼ 10) leading to a significantly increased reactant concentration, yet they do not reach the maximum coupling yield of 50% claimed for PAA.13 The results for P99 obtained by XPS, however, exceed this value and are also significantly higher than those determined by NMR and should consequently be treated with caution. We believe that several factors contribute to this discrepancy, which are all related to the intermediary thickness of the TFEA-labelled PAA layer on randomly distributed, spherical P99 particles. For example, in case that the thickness of the probed layer is smaller than the XPS information depth, the PMMA core additionally contributes to the amount of carbon used for quantification of TFEA-labelled COOH groups. In addition, a recent XPS depth profiling study has revealed an increased concentration of the labelling reagent within the top few nanometers of grafted polymers, which is indiscernible in bulk measurements like NMR but clearly influences the XPS results.5,14 For this combination of factors, common quantification models should usually not be applied, and we are currently developing alternative quantification models to address this issue. Nonetheless, even when including P99, the relative error of the average of all coupling yields (38 ± 10%) is still in accordance with our previous surface quantification method comparisons with much larger sample sets.5,6
Most important, our results clearly demonstrate the potential of fluorine as an attractive heteroatom for the development of reference materials for traceable surface functional group quantifications. The utilization of our TFEA-functionalized polymer particles now allows the combined use of XPS and solid-state 19F NMR spectroscopy for surface group quantifications on the same material. Although a combination of XPS and NMR has been used complementary to characterize particle-based sample materials such as hybrid organic–inorganic materials and nanodiamonds,15 it has, to the best of our knowledge, neither been used to mutually validate both methods nor for establishing a metrological traceability chain. With the identification of a very pure and suitable reference standard containing both 19F and 1H, XPS could even be traced back to a certified NMR reference standard and thus to the SI unit mole via the quantitative method solid-state 19F NMR as shown in Fig. 3a.
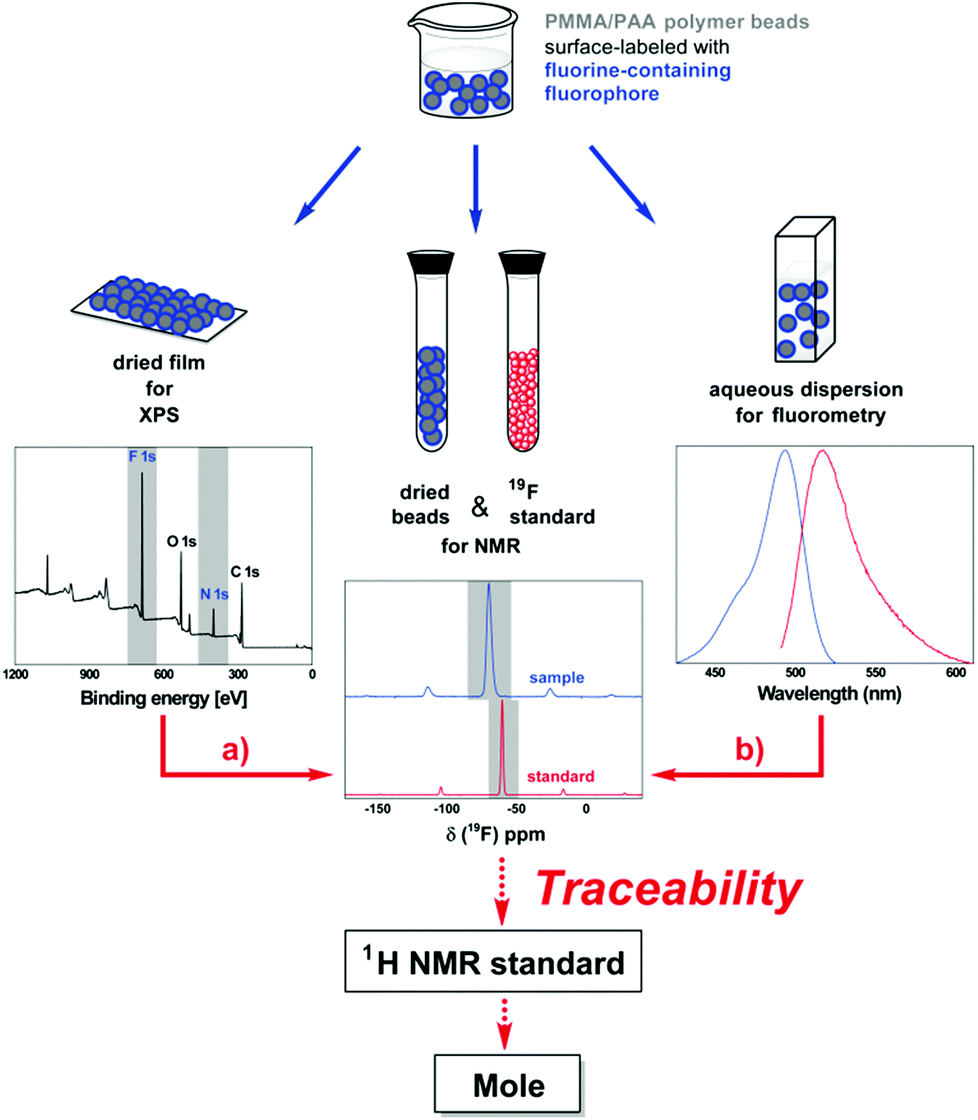 | ||
| Fig. 3 Traceability chain for surface group quantification, linking measurements (blue arrows) of (a) XPS and (b) fluorometry to quantitative solid-state 19F NMR (solid red arrows). The use of a certified NMR reference standard containing both 19F and 1H provides the link to the SI unit mole (dotted red arrows). | ||
We have previously prepared and comprehensively characterized the same selection of polymer particles labelled with a mixture of the fluorescein derivative FL-A and varying amounts of H2N–CH2(OCH2)3–CO2H (added to prevent particle aggregation).5 The P946 particles contain the highest practically relevant amounts of surface-bound fluorophores, which have been successfully quantified by absolute fluorometry and the surface concentrations of fluorophores were in the range of 0.42 to 6.3 μmol g−1. However, a tentative recording of XPS spectra did not show any peaks assignable to the thiourea group of FL-A. Even evaporating a drop of a solution containing suspended unlabelled particles and unbound FL-A, that gave a spot containing the polymer particles and a FL-A concentration of ca. 13 μmol g−1, showed the sulfur peak for the thiourea group only with an inacceptable signal-to-noise ratio. This clearly indicates that the combination of maximum achievable surface concentration of fluorophores and sensitivity of XPS for sulfur is insufficient, while detection of fluorine would be unproblematic in this concentration range by XPS and even by NMR. This is not only ascribed to the higher content of heteroatoms in TFEA (three fluorine atoms) compared to FL-A (one sulfur atom), but also to the enhanced sensitivity of XPS for fluorine.16 This is consistent with previous results on plasma-chemically modified polypropylene films, in which the nitrogen atom (which has a XPS sensitivity comparable to fluorine) of the fluorescent label dansyl was qualitatively detected by fluorescence and XPS.17
Overall, we believe that fluorine-containing fluorophores will present ideal candidates to additionally link fluorescence spectroscopy to the traceability chain now established for XPS and NMR (Fig. 3b). Fortunately, fluorine is included in several fluorescent dyes.18 In fact, we recently reported the determination of particle-encapsulated fluorophores by absolute fluorometry and 19F NMR.12 In the case of surface-bound fluorophores, the sensitivity ranges could additionally be fine-tuned by adjusting the number of fluorine atoms per fluorophore through the use of fluoroalkyl linkers with varying lengths and numbers of fluorine atoms. The resulting increase in number of fluorine atoms per molecule would even enable decreasing the amount of surface fluorophores to concentrations, at which fluorescence self-quenching does not apply. Thereby, we could not only link XPS and NMR to absolute fluorometry,12 which accounts for varying fluorescence quantum yields, but also to the much more widely used relative fluorometry.5
Conclusions
We demonstrated that fluorine on the surface of polymer microparticles labelled with TFEA could be detected by XPS and solid-state 19F NMR with overlapping detection ranges. Consistent coupling yields of the small reporter TFEA were determined by XPS and quantitative solid-state 19F NMR spectroscopy establishing a very important step towards the metrological traceability of surface group quantifications. Furthermore, we disclosed a strategy to include fluorometry as an additional surface quantification method with comparable detection ranges and currently pursue the identification of fluorine-containing fluorophores with suitable spectroscopic, photophysical and chemical properties.5,6c,18 Such fluorine-containing fluorescent dyes would be key for a prospective reference material and could additionally be utilized as a chemical derivatization reagent for a reliable and traceable quantification of chemically addressable surface functional groups.5,8,9Notes and references
- (a) K. Chatterjee, S. Sarkar, K. Jagajjanani Rao and S. Paria, Adv. Colloid Interface Sci., 2014, 209, 8–39 CrossRef CAS PubMed; (b) S. Rödiger, C. Liebsch, C. Schmidt, W. Lehmann, U. Resch-Genger, U. Schedler and P. Schierack, Microchim. Acta, 2014, 181, 1151–1168 CrossRef; (c) R. A. Sperling and W. J. Parak, Ther. Innovation Regul. Sci., 2013, 47, 1333–1383 Search PubMed; (d) C. Xu and S. Sun, Adv. Drug Delivery Rev., 2013, 65, 732–743 CrossRef CAS PubMed; (e) C. T. Adkins, J. N. Dobish, S. Brown and E. Harth, ACS Macro Lett., 2013, 2, 710–714 CrossRef CAS PubMed; (f) J. M. Montenegro, V. Grazu, A. Sukhanova, S. Agarwal, J. M. de la Fuente, I. Nabiev, A. Greiner and W. J. Parak, Adv. Drug Delivery Rev., 2013, 65, 677–688 CrossRef CAS PubMed; (g) K. Hoffmann, T. Behnke, D. Drescher, J. Kneipp and U. Resch-Genger, ACS Nano, 2013, 7, 6674–6684 CrossRef CAS PubMed; (h) K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed; (i) J. M. Baumes, J. J. Gassensmith, J. Giblin, J. J. Lee, A. G. White, W. J. Culligan, W. M. Leevy, M. Kuno and B. D. Smith, Nat. Chem., 2010, 2, 1025–1030 CrossRef CAS PubMed; (j) A. I. Abdelrahman, S. Dai, S. C. Thickett, O. Ornatsky, D. Bandura, V. Baranov and M. A. Winnik, J. Am. Chem. Soc., 2009, 131, 15276–15283 CrossRef CAS PubMed; (k) L. Barner, Adv. Mater., 2009, 21, 2547–2553 CrossRef CAS; (l) U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- (a) K. E. Sapsford, W. R. Algar, L. Berti, K. B. Gemmill, B. J. Casey, E. Oh, M. H. Stewart and I. L. Medintz, Chem. Rev., 2013, 113, 1904–2074 CrossRef CAS PubMed.
- (a) J. E. Gagner, S. Shrivastava, X. Qian, J. S. Dordick and R. W. Siegel, J. Phys. Chem. Lett., 2012, 3, 3149–3158 CrossRef CAS; (b) N. B. Shah, G. M. Vercellotti, J. G. White, A. Fegan, C. R. Wagner and J. C. Bischof, Mol. Pharm., 2012, 9, 2146–2155 CAS; (c) K. E. Sapsford, K. M. Tyner, B. J. Dair, J. R. Deschamps and I. L. Medintz, Anal. Chem., 2011, 83, 4453–4488 CrossRef CAS PubMed; (d) D. F. Moyano and V. M. Rotello, Langmuir, 2011, 27, 10376–10385 CrossRef CAS PubMed; (e) S. J. Tan, N. R. Jana, S. J. Gao, P. K. Patra and J. Y. Ying, Chem. Mater., 2010, 22, 2239–2247 CrossRef CAS.
- D. R. Baer, M. H. Engelhard, G. E. Johnson, J. Laskin, J. Lai, K. Mueller, P. Munusamy, S. Thevuthasan, H. Wang, N. Washton, A. Elder, B. L. Baisch, A. Karakoti, S. V. N. T. Kuchibhatla and D. Moon, J. Vac. Sci. Technol., A, 2013, 31, 050820 Search PubMed.
- A. Hennig, H. Borcherding, C. Jaeger, S. Hatami, C. Würth, A. Hoffmann, K. Hoffmann, T. Thiele, U. Schedler and U. Resch-Genger, J. Am. Chem. Soc., 2012, 134, 8268–8276 CrossRef CAS PubMed.
- (a) A. Hennig, A. Hoffmann, H. Borcherding, T. Thiele, U. Schedler and U. Resch-Genger, Chem. Commun., 2011, 47, 7842–7844 RSC; (b) A. Hennig, A. Hoffmann, H. Borcherding, T. Thiele, U. Schedler and U. Resch-Genger, Anal. Chem., 2011, 83, 4970–4974 CrossRef CAS PubMed; (c) A. Hennig, S. Hatami, M. Spieles and U. Resch-Genger, Photochem. Photobiol. Sci., 2013, 12, 729–737 RSC.
- P. M. Dietrich, A. Hennig, M. Holzweber, T. Thiele, H. Borcherding, A. Lippitz, U. Schedler, U. Resch-Genger and W. E. S. Unger, J. Phys. Chem. C, 2014, 118, 20393–20404 CAS.
- (a) B. Panella, A. Vargas, D. Ferri and A. Baiker, Chem. Mater., 2009, 21, 4316–4322 CrossRef CAS; (b) S. Huang, R. Joso, A. Fuchs, L. Barner and S. V. Smith, Chem. Mater., 2008, 20, 5375–5380 CrossRef CAS; (c) Y. Xing, N. Dementev and E. Borguet, Curr. Opin. Solid State Mater. Sci., 2007, 11, 86–91 CrossRef CAS PubMed; (d) K. Hoffmann, M. Mix, U. Resch-Genger and J. F. Friedrich, Langmuir, 2007, 23, 8411–8416 CrossRef CAS PubMed; (e) K. Qi, Q. Ma, E. E. Remsen, C. G. Clark Jr. and K. L. Wooley, J. Am. Chem. Soc., 2004, 126, 6599–6607 CrossRef CAS PubMed; (f) V. B. Ivanov, J. Behnisch, A. Holländer, F. Mehdorn and H. Zimmermann, Surf. Interface Anal., 1996, 24, 257–262 CrossRef CAS.
- (a) A. Chilkoti and B. D. Ratner, in Surface Characterization of Advanced Polymers, ed. L. Sabbatini and P. G. Zambonin, Wiley VCH, 1993, p. 221 Search PubMed; (b) A. Chilkoti and B. D. Ratner, Surf. Interface Anal., 1991, 17, 567–574 CrossRef CAS; (c) A. Chilkoti, B. D. Ratner and D. Briggs, Chem. Mater., 1991, 3, 51–61 CrossRef CAS; (d) C. D. Batich, Appl. Surf. Sci., 1988, 32, 57–73 CrossRef CAS; (e) H. Kondo and Y. Nishida, Bull. Chem. Soc. Jpn., 2007, 80, 1405–1412 CrossRef CAS; (f) V. I. Povstugar, S. S. Mikhailova and A. A. Shakov, J. Anal. Chem., 2000, 55, 405–416 CrossRef CAS; (g) J. Vickerman and I. S. Gilmore, Surface Analysis - The Principal Techniques, Wiley, 2nd edn, 2009 Search PubMed.
- (a) P.-L. Girard-Lauriault, T. Gross, A. Lippitz and W. E. S. Unger, Anal. Chem., 2012, 84, 5984–5991 CrossRef CAS PubMed; (b) A. G. Shard, J. Phys. Chem. C, 2012, 116, 16806–16813 CrossRef CAS; (c) G. Zorn, S. R. Dave, X. Gao and D. G. Castner, Anal. Chem., 2011, 83, 866–873 CrossRef CAS PubMed; (d) S. Techane, D. R. Baer and D. G. Castner, Anal. Chem., 2011, 83, 6704–6712 CrossRef CAS PubMed; (e) M. Mohai and I. Bertóti, Surf. Interface Anal., 2012, 44, 1130–1134 CrossRef CAS; (f) M. Mohai and I. Bertóti, Surf. Interface Anal., 2004, 36, 805–808 CrossRef CAS.
- T. Gross, F. Pippig, B. Merz, R. Merz, U. Vohrer, R. Mix, H. Steffen, W. Bremser and W. E. S. Unger, Plasma Processes Polym., 2010, 7, 494–503 CrossRef CAS.
- A. Huber, T. Behnke, C. Wurth, C. Jaeger and U. Resch-Genger, Anal. Chem., 2012, 84, 3654–3661 CrossRef CAS PubMed.
- (a) E. R. Bissell and M. J. Finger, J. Org. Chem., 1959, 24, 1256–1259 CrossRef CAS; (b) G. T. Hermanson, Bioconjugate Techniques, Academic Press (Elsevier), London, 2nd revised edn, 2008, p. 1202 Search PubMed; (c) N. Nakajima and Y. Ikada, Bioconjugate Chem., 1995, 6, 123–130 CrossRef CAS.
- R. Barbey, V. Laporte, S. Alnabulsi and H.-A. Klok, Macromolecules, 2013, 46, 6151–6158 CrossRef CAS.
- (a) H. Virieux, M. Le Troedec, A. Cros-Gagneux, W.-S. Ojo, F. Delpech, C. Nayral, H. Martinez and B. Chaudret, J. Am. Chem. Soc., 2012, 134, 19701–19708 CrossRef CAS PubMed; (b) O. Shenderova, A. M. Panich, S. Moseenkov, S. C. Hens, V. Kuznetsov and H.-M. Vieth, J. Phys. Chem. C, 2011, 115, 19005–19011 CrossRef CAS.
- J. H. Scofield, J. Electron Spectrosc. Relat. Phenom., 1976, 8, 129–137 CrossRef CAS.
- K. Hoffmann, U. Resch-Genger, R. Mix and J. F. Friedrich, J. Fluoresc., 2006, 16, 441–448 CrossRef CAS PubMed.
- (a) B. R. Renikuntla, H. C. Rose, J. Eldo, A. S. Waggoner and B. A. Armitage, Org. Lett., 2004, 6, 909–912 CrossRef CAS PubMed; (b) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed; (c) M. Hecht, T. Fischer, P. Dietrich, W. Kraus, A. B. Descalzo, W. E. S. Unger and K. Rurack, ChemistryOpen, 2013, 2, 25–38 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, details on quantification, and supplementary figures. See DOI: 10.1039/c4an02248c |
| ‡ The large uncertainty for P946 is attributed to the signal-to-noise ratio of the 13C MAS NMR spectra, i.e. the (COOH + COOMe)/C-CH3 ratio was 1.16 ± 0.10 for P946 and subtracting the value of 1.00 for the unmodified PMMA core gives 0.16 ± 0.10 moles of surface COOH per mole of PMMA monomer in the core. |
| This journal is © The Royal Society of Chemistry 2015 |