
Real-time microfluidic recombinase polymerase amplification for the toxin B gene of Clostridium difficile on a SlipChip platform†
M.-N.
Tsaloglou
*a,
R. J.
Watson
b,
C. M.
Rushworth
a,
Y.
Zhao
a,
X.
Niu
c,
J. M.
Sutton
b and
H.
Morgan
c
aFaculty of Physical Sciences and Engineering, Institute for Life Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: M.Tsaloglou@soton.ac.uk
bHealthcare Biotechnology, Public Health England, Porton Down, Salisbury, SP4 0JG, UK
cFaculty of Engineering and the Environment, and Institute for Life Sciences, University of Southampton, Highfield, Southampton, SO17 1BJ, UK
First published on 23rd October 2014
Abstract
Clostridium difficile is one of the key bacterial pathogens that cause infectious diarrhoea both in the developed and developing world. Isothermal nucleic acid amplification methods are increasingly used for identification of toxinogenic infection by clinical labs. For this purpose, we developed a low-cost microfluidic platform based on the SlipChip concept and implemented real-time isothermal recombinase polymerase amplification (RPA). The on-chip RPA assay targets the Clostridium difficile toxin B gene (tcdB) coding for toxin B, one of the proteins responsible for bacterial toxicity. The device was fabricated in clear acrylic using rapid prototyping methods. It has six replicate 500 nL reaction wells as well as two sets of 500 nL control wells. The reaction can be monitored in real-time using exonuclease fluorescent probes with an initial sample volume of as little as 6.4 μL. We demonstrated a limit of detection of 1000 DNA copies, corresponding to 1 fg, at a time-to-result of <20 minutes. This miniaturised platform for pathogen detection has potential for use in resource-limited environments or at the point-of-care because of its ease of use and low cost, particularly if combined with preserved reagents.
Introduction
Infectious diarrhoea following nosocomial and antibiotic treatment is mainly caused by Gram-positive bacteria, like Clostridium difficile.1 In the US alone this accounted for 337![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 infections and 14000 deaths in 2012.2 Infection from ingested spores of the bacterium is an emerging issue in in-patient healthcare facilities such as hospitals and nursing homes in the developed world. Overpopulation of a patient's colon by C. difficile typically occurs after treatment with a broad-spectrum antibiotic, which depletes the natural gut flora. The hostile bacteria can then produce the potent toxins A and B, as well as a binary toxin, which are highly pathogenic to humans, causing infectious diarrhoea.3 Toxins A and B are structurally similar and their crystal structures have been fully characterised.4 Toxin B is the most potent and binds to disaccharides found on the I, X and Y blood antigens.5 Diagnosis of infection is therefore a priority and clinical labs in many parts of the world are increasingly using nucleic acid amplification tests because of improved diagnostic sensitivity for mild and severe cases. Toxin B is one of the virulence factors of C. difficile and is the product of the tcdB gene. This is the principle target of molecular diagnostics in hospitals, and for this reason we have chosen tcdB as the molecular target of our assay.
000 infections and 14000 deaths in 2012.2 Infection from ingested spores of the bacterium is an emerging issue in in-patient healthcare facilities such as hospitals and nursing homes in the developed world. Overpopulation of a patient's colon by C. difficile typically occurs after treatment with a broad-spectrum antibiotic, which depletes the natural gut flora. The hostile bacteria can then produce the potent toxins A and B, as well as a binary toxin, which are highly pathogenic to humans, causing infectious diarrhoea.3 Toxins A and B are structurally similar and their crystal structures have been fully characterised.4 Toxin B is the most potent and binds to disaccharides found on the I, X and Y blood antigens.5 Diagnosis of infection is therefore a priority and clinical labs in many parts of the world are increasingly using nucleic acid amplification tests because of improved diagnostic sensitivity for mild and severe cases. Toxin B is one of the virulence factors of C. difficile and is the product of the tcdB gene. This is the principle target of molecular diagnostics in hospitals, and for this reason we have chosen tcdB as the molecular target of our assay.
Microfluidic micro-systems can provide simple and low-cost alternatives to hospital instrumentation for pathogen detection, without the need for trained personnel and expensive facilities.6 In the developed world, microfluidic devices are being used for point-of-care applications. In the developing world, low-cost diagnostics are being developed to comply with the guidelines of the World Health Organization for diagnostics in resource-poor settings, defined by the ASSURED acronym: affordable, sensitive, specific, user friendly, rapid and robust, equipment-free, delivered to those who need it.7
An example of a very low-cost microfluidic microsystem that has been used for molecular assays is the SlipChip, developed by the Ismagilov lab.8 First introduced in 2009, it is very simple and does not require pumps or valves to move fluids. It can be used for multiplex reactions, similar to microplate-based systems. It consists of two plates into which a series of nanolitre volume wells and channels are machined. The top and bottom plates are brought together and slipped relative to each other so that when wells overlap fluids and samples can exchange. Liquid is confined in the wells by a thin layer of lubricating fluorinated oil.
Among the molecular tools available for genetic analysis, isothermal nucleic acid amplification technologies (INAAT) are ideal candidates for miniaturised point-of-care applications.9 They operate at lower temperatures than polymerase chain reaction (PCR),10 need less precise temperature control and importantly for micro-devices they do not require thermo-cycling.
INAAT methods amplify either DNA or RNA targets. One example is loop-mediated amplification (LAMP) which amplifies DNA with high specificity at 65 °C.11 Auto-cycling of DNA synthesis is achieved using three sets of ∼20–45 nucleotide-long primers, operating at inner and outer sites, as well as the loop regions. An amplification efficiency of 109 occurs in 60 minutes and the amplicons are long looped DNA catenates. Recombinase polymerase amplification (RPA) can reach a similar amplification efficiency to LAMP within 30 minutes at lower incubation temperatures, in the range of 37 to 41 °C.12 The RPA reaction is summarised in Fig. 1. An E. coli recombinase ATPase, RecA, forms complexes with two primers specific to the DNA target which scan the DNA target for complementary sequences.13 A T4 bacteriophage polymerase, Bsu,14 extends the strand in a characteristic D-loop structure. Real-time fluorescence detection is used to monitor the assay quantitatively by means of fluorescently labelled probes with a tetrahydrofuran (THF) moiety. An exonuclease III, present in the RPA reaction master-mix, digests the THF spacer when the probes are hybridised to the target.
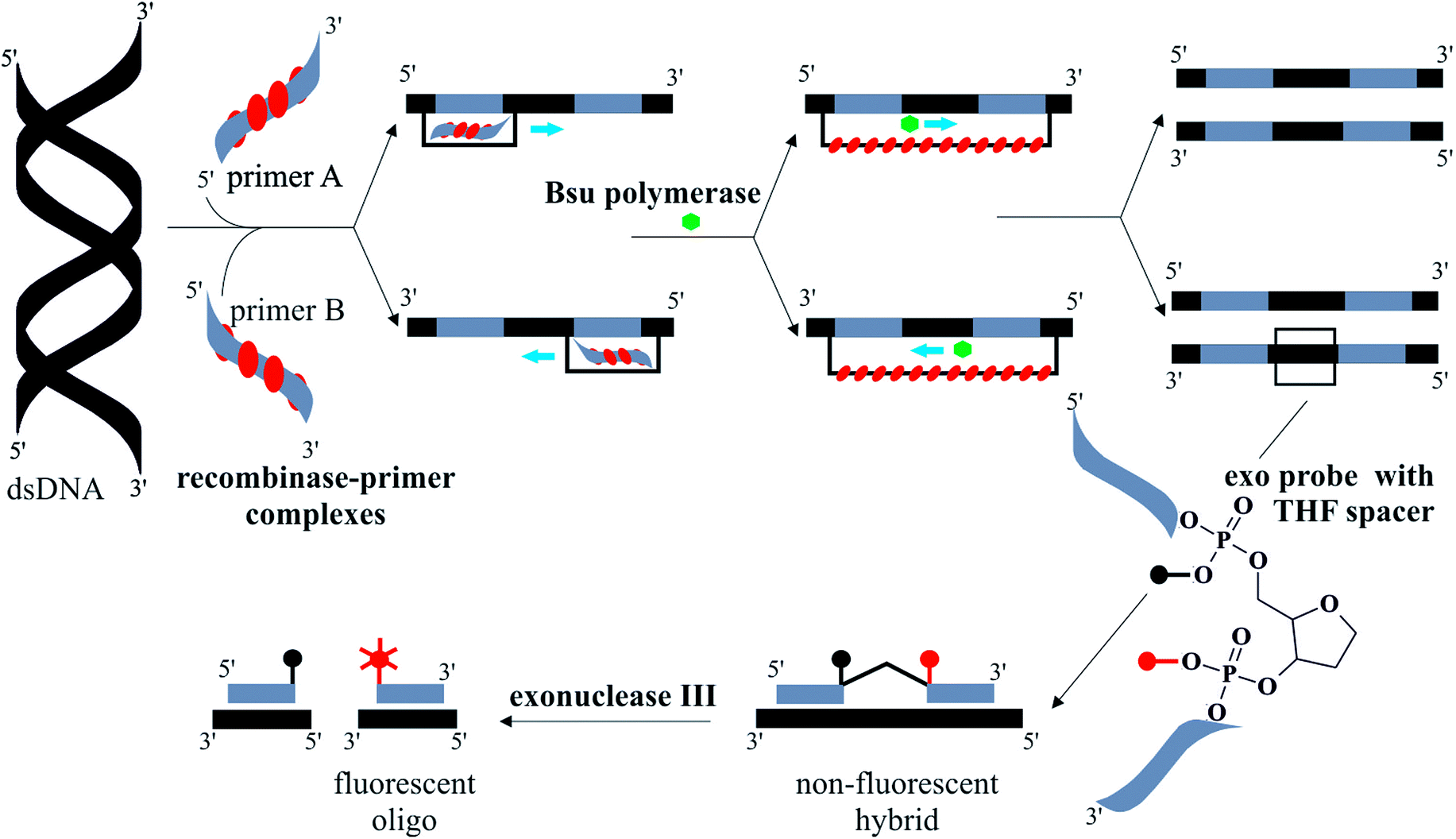 | ||
| Fig. 1 Illustration summarising the steps of the RPA reaction, with real-time exonuclease probes. Double-stranded (ds) template DNA hybridises with recombinase–primer complexes. Primers are extended by Bsu polymerase in D-loop structure from both 5′ and 3′ directions. This results in two copies of the original ds DNA. The exonuclease probe anneals to a complementary sequence on the template DNA. When bound, an exonuclease III digests the tetrahydrofuran (THF) spacer on the probe causing real-time fluorescence generation. Part of diagram adapted from ref. 12. | ||
The SlipChip has been used for protein and nucleic acid amplification assays. A multiplexed bead-based heterogeneous immunoassay for insulin using 48 individual 9 nL droplets was reported with a limit of detection (LOD) of 13 pM.15 This was an improvement over existing nL-volume on-chip immunoassays16 and all steps from sample addition to fluorescence detection occurred on the device. A high-throughput nL multiplex PCR platform was demonstrated for up to 16 pathogens, including E. coli and MRSA, in 40 and 384 wells.17 In the latter, 384 primer pairs were dispensed in the wells and individual PCR reactions were run in parallel from a single 10 μL sample of genomic DNA. Total reaction time was ∼75 minutes and end-point fluorescence was measured with an epi-fluorescence microscope. A similar publication reported 1280 parallel digital reactions in 2.6 nL wells for detection of Staphylococcus aureus.18 Shen et al. demonstrated digital reverse-transcriptase (RT) PCR, targeting HIV and hepatitis C RNA in as little as 1 nL wells in 160 parallel reactions with no cross-contamination.19 Digital RT-LAMP has been shown for HIV RNA detection in spiked plasma on a SlipChip.20 A two-step modification of the bench-top LAMP protocol was used for 42 parallel end-point reactions in ∼3 nL wells. During the first step RNA molecules were compartmentalised and DNA was produced by RT; LAMP was performed on each individual RT product. This approach overcame the underestimation of target copies typically encountered by digital nucleic acid amplification methods. Digital (end-point) RPA has also been demonstrated on a SlipChip.21
All these assays were performed on SlipChip devices, made from etched glass and fabricated using standard methods of photolithography and wet chemical etching in a clean room environment.22 This type of fabrication requires trained personnel, often toxic processes and access to expensive facilities. Recently we described a polymer-chip-based SlipChip platform for protein separation using isoelectric focusing.23 This device was made from poly-methyl methacrylate (PMMA) material and features were micro-milled without the need for clean room facilities using computer numeric control (CNC) micromachining. In this work, we have employed the same low-cost fabrication technique to produce PMMA SlipChip devices for RPA on-chip. Access vials were easily fabricated for sample loading and collection, and multiple layer chips were feasible.
PMMA compares very well to glass in both optical properties and physical adsorption. First, PMMA has excellent optical properties24 and shows very little auto-fluorescence; as low as glass.25 Second, both glass and PMMA require surface passivation for efficient nucleic acid amplification on-chip.26 PMMA is a hydrophilic polymer with a water contact angle of ∼68° compared to ∼73° for glass. In previous work using glass SlipChip devices for RPA, a dichloromethyl silane treatment was used to render the surface hydrophobic. For PMMA chips, we employed a rapid vapour polishing method,27 developed in our lab, to remove any surface roughness caused by micromachining. We then treated the PMMA with a hydrophobic surface coating, followed by surface priming with an inert protein. Overall, PMMA is suitable for rapid prototyping, which makes it an ideal candidate for proof-of-concept studies. Once a suitable design for a system has been proven, low-cost and high throughput methods such as injection moulding could be used to produce single-use disposable chips.
Here we present a PMMA-chip-based SlipChip platform for quantitative real-time fluorescence detection of isothermal amplification of the tcdB gene of C. difficile. Bacterial DNA was detected to 1000 copies (or 1 fg) with a total reaction time of less than 20 minutes.
Experimental
1 Chemicals and materials
Nuclease-free water (DEPC-treated) was purchased from Invitrogen (Paisley, UK). Isopropyl alcohol (BioReagent, for molecular biology, ≥99.5%), bovine serum albumin (BSA, BioReagent, for molecular biology, 20 mg mL−1 in water) and Tween20 (molecular biology grade) were sourced from Sigma Aldrich (Dorset, UK). PMMA at different thicknesses, neodymium block magnets (25 mm × 10 mm × 5 mm), a silicone rubber patch thermocouple (50 to +150 °C, K Type), a 0.075 mm probe thermocouple (250 °C maximum, K Type) and M6 hexagonal nuts and bolts were supplied from RS Components (Northants, UK). The 0.01 mm micrometer (10 μm graduations) with a spherical tip was from Thor Labs (Cambridgeshire, UK).2 SlipChip design and fabrication
The device consists of two separate plates with wells, ducts and holes patterned in each half. The top plate (55 mm × 60 mm) and bottom plate (64 mm × 45 mm) were fabricated from 1.0 mm-thick PMMA. Fig. 2 shows the design of the two plates which together form three separate fluidic paths for loading the sample, RPA master-mix and magnesium acetate when assembled together. The assembled device has six replicate reaction wells for sample amplification and two additional duplicate control wells. All three wells had a surface area of 1 mm2.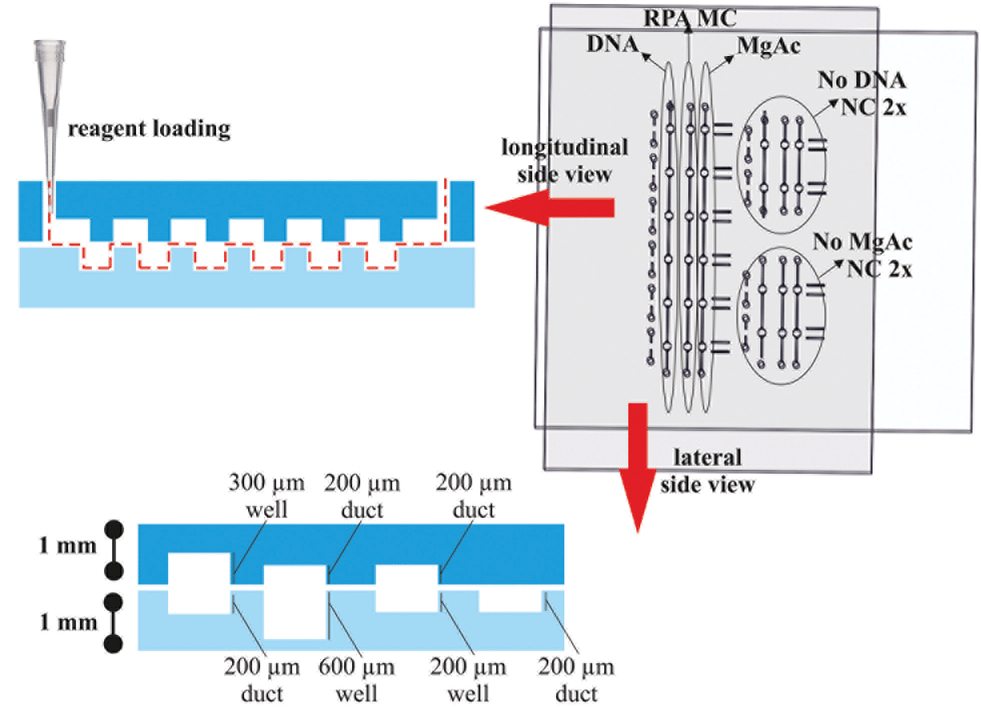 | ||
| Fig. 2 Device loading diagram. The top and bottom plates have six replicate reaction wells and two sets of negative control (NC) wells, one for no DNA and one for no magnesium acetate. In the longitudinal side view, the loading configuration is demonstrated. A pipette is used to load the wells with DNA sample (4.6 μL), RPA master-mix (MC, 6.4 μL) and magnesium acetate (6.8 μL) in the replicate sample wells. Equivalent volumes in the control wells were 2.9, 3.5 and 5 μL, respectively. In the lateral side view, the dimensions and features of the devices are shown with micro-milled wells and ducts ranging from 200 to 600 μm in depth fabricated in 1 mm thick clear acrylic. | ||
Features at 200 μm, 300 μm and 600 μm depth were formed by CNC micromachining using an automated LPKF Protomat S100 micro-mill. Prior to use, the plates were washed with a decontamination solution (RNaseZap, Invitrogen, UK), nuclease-free water and isopropyl alcohol. They were then dried under nitrogen, dehydrated at 60 °C for 5 minutes and exposed to chloroform vapour for 3 minutes at room temperature to polish the surface by reflowing.27 A hydrophobic coating (Duxcoat Nano solution, Duxback Ltd, Somerset, UK) was applied and the plates were dried at 60 °C for 5 minutes. To minimise non-specific binding, the plates were passivated in an aqueous solution of 0.1% (v/v) Tween20 and 0.1% (w/v) BSA overnight at 4 °C.
3 Assembly of SlipChip platform
The two plates were clamped together using two small neodymium magnets and the entire assembly was held in a custom-made holder as shown in Fig. 3a. Fluorinated aliphatic oil (50 μL, Fluorinert FC-40, 3M, Sigma Aldrich, UK) was added to the bottom plate before clamping the top plate. This layer of oil facilitated slipping of the plates and prevented leakage. The holder was made in PMMA using laser micromachining (Epilog Mini 24 Laser System, Epilog, USA). It measured 197 mm × 93 mm and was fabricated from four sheets of 3 mm PMMA were held together with nuts and bolts (Fig. 3b). The bottom plate of the device was slipped into different positions using a micrometer drive with a spherical tip to provide accurate alignment of the plates. A custom-made thermo-regulating plate fitted at the top plate controlled the RPA at 39 ± 0.1 °C. For detailed description, see ESI.† | ||
| Fig. 3 (a) Photograph of the SlipChip device and base holder. The base holder measures 197 × 93 mm and is fabricated from four sheets of 3 mm poly(methyl methacrylate). The sheets are held together with M6 hexagonal nuts and bolts. The bottom plate of the device is moved using a 0.01 mm micrometer drive. (b) Diagram showing all device and holder components. | ||
4 DNA template preparation
Cells of C. difficile (strain R20291) were inoculated from a Fastidious Anaerobe Agar (FAA) plate (Oxoid, Fisher Scientific, UK) into 10 mL pre-reduced Brain Heart Infusion (BHI) broth. The BHI broth was based on the original formulation28 but prepared in-house at Public Health England. Cells were grown for 24 hours at 37 °C in a 2.5 L anaerobe jar (Oxoid, Fisher Scientific, UK) with a 3.5 L AnaeroGen sachet (Oxoid, Fisher Scientific, UK) to generate anaerobic conditions. After 24 hours, a culture aliquot (1 mL) containing approximately 1 × 108 colony forming units (CFU) was removed and genomic DNA was extracted (Protocol G, Promega Wizard, Promega, UK). The purified 1168 bp amplicon of tcdB was then amplified further by PCR. A forward primer, 5′-TCT TTT TAT GGT TCT GGA GGA ACT TAT-3′, and a reverse primer, 5′-GTT TAT TTC ATC TGT ATA TAT ATT TGG C-3 were used. The PCR reaction was performed using a commercial master-mix (GoTaq Hot Start Green Master Mix, Promega, UK) as per the manufacturer's instructions on a thermal cycler (BIOER LifePro, China) with 5 minutes hot start at 95 °C, 30 s denaturation at 95 °C, 30 s annealing at 55 °C, 5 minutes extension at 72 °C, repeated for 30 cycles. A final extension was performed at 72 °C for 5 minutes. The PCR product was then purified using a commercial PCR purification kit (QIAquick Qiagen, UK) as per the manufacturer's instructions. The pure DNA eluent was then quantified using the Qubit double stranded DNA quantification kit and the Qubit 2.0 fluorimeter (Life Technologies, UK) as per the manufacturer's instructions.5 Benchtop RPA
RPA was performed in a final volume of 50 μL using an exonuclease kit (Exo Kit, TwistDx, Cambridge, United Kingdom). The final solution of the rehydrated mastermix included 0.42 μM forward primer, 0.42 μM reverse primer, 0.12 μM Cy5-labelled RPA exo probe, 14 mM magnesium acetate and 1× rehydration buffer. The RPA primers were 35 base pair oligonucleotides that were HPLC purified (IDT, Belgium). The real-time probe was a 47 base pair sequence (ADT Bio, Southampton, UK). All the reagents except the template DNA and magnesium acetate were added to the purchased lyophilised pellet to give a final volume of 46.5 μL. The DNA template (1 μL) was added to each reaction at a variable concentration. The magnesium acetate (2.5 μL, 280 mM) was added last to initiate the reaction. The reaction was incubated at 39 °C in a thermal cycler (ABI7500, Life Technologies, United Kingdom) and fluorescence was monitored real-time at 670 nm.6 Optical and mechanical system
A custom fluorescence detection system was made as shown in Fig. 4. | ||
| Fig. 4 Schematic of optical geometry, where EO 624/40 is a band-pass filter and PMT is photomultiplier tube. | ||
This system measures the fluorescence from each well an excitation wavelength of 635 nm and an emission wavelength of 692 nm. Each well is imaged sequentially using a linear translation stage. Fluorescence was recorded during 15 ms exposure to reduce photo-bleaching. A total of 100 data points were recorded for each well over approximately 45 minutes, using a custom LABVIEW™ program. A detailed description of the system is included in ESI.†
Results and discussion
An 1186 base-pair sequence was identified as the target of the RPA reaction. This was designed to be homologous across multiple strains of C. difficile. The genome for C. difficile R20291 has been annotated.29 The presence of either tcdA or tcdB is reportedly critical for virulence,30 but the majority of clinical isolates always harbour the tcdB gene. The first ever report of a tcdA positive and tcdB negative clinical strain was only shown in 2012 from rabbit samples,31 thus making tcdB the most stable of the two toxins to target.The RPA reactions were performed both on the 500 nL well SlipChip platform and in 50 μL bench-top assays. The bench-top protocol was adapted to the SlipChip format as follows. First, the DNA sample was mixed with the RPA master-mix, which consisted of all the necessary reagents for amplification except the magnesium acetate chemical initiator. The concentration of primers and exo probes was increased in the final 500 nL SlipChip reaction to match the bench-top concentration. The reaction components and DNA sample were brought together and allowed to mix at room temperature for 5 minutes. Second, the magnesium acetate was added to initiate the reaction. The on-chip protocol is summarised in the experimental section and Fig. 5.
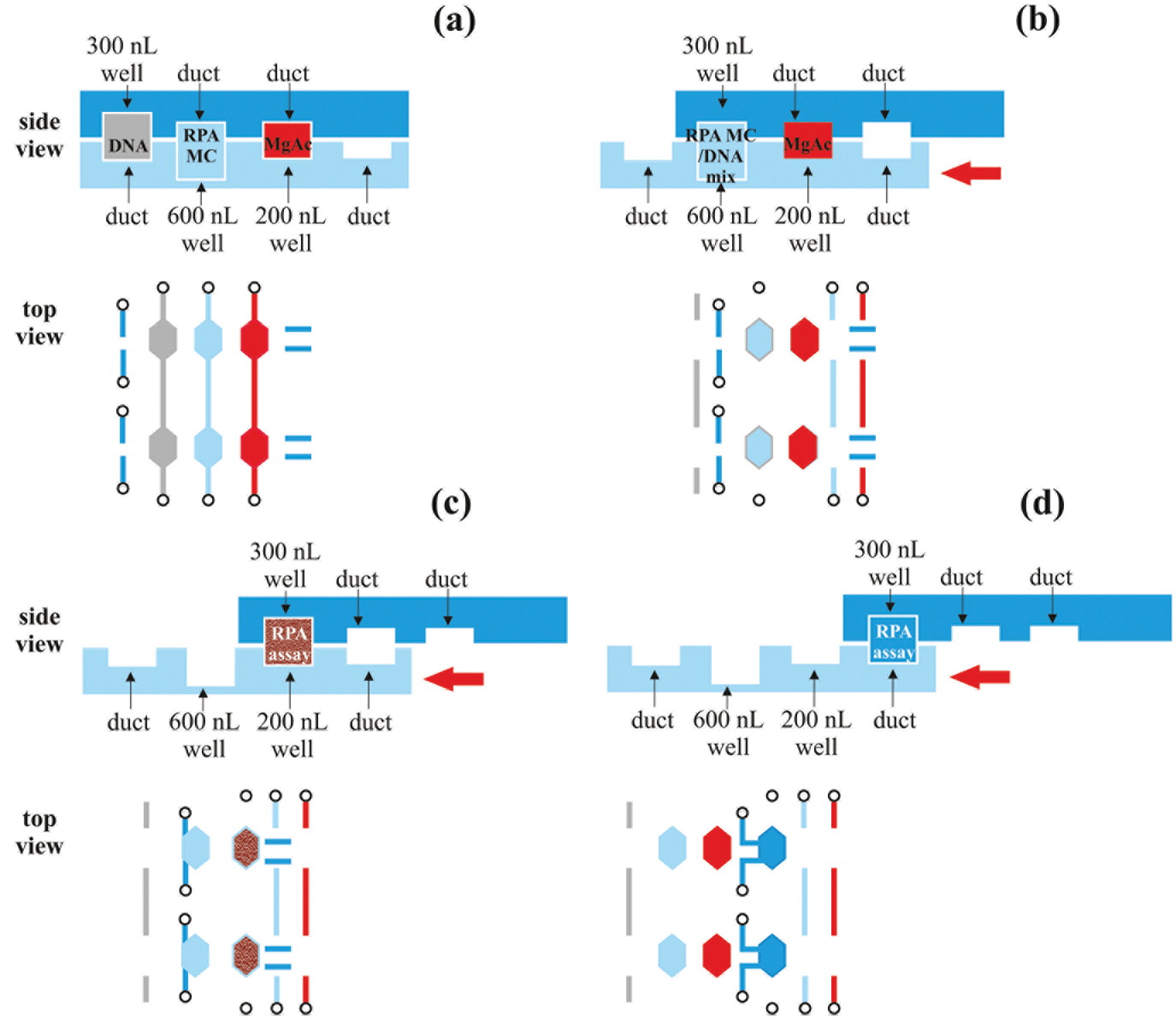 | ||
| Fig. 5 Top and bottom plates of the SlipChip during the RPA reaction. (a) Loading configuration: DNA, RPA master-mix (MC) and magnesium acetate (MgAc) are loaded into the channels and the separate chambers are filled with 300 nL, 600 nL and 200 nL, respectively. (b) Mixing configuration: The bottom plate is slipped, aligning the DNA and RPA MC chambers. The reagents are incubated for 5 minutes in room temperature for diffusive mixing. (c) Incubation configuration: the bottom plate is slipped to align the reaction chamber with the MgAc chamber. The RPA reaction is chemically activated with magnesium acetate and is monitored real-time for up to 1 h under a fluorescence microscope. (d) Collection configuration: the bottom plate is slipped to align the reaction chamber with two output ports. This allows 300 nL sample collection for further analysis which is feasible in the case of non-real-time RPA. | ||
Titration results comparing the on-chip with a bench-top assay are shown in Fig. 6. After an initial lag phase of ∼5 minutes, the fluorescence intensity increases exponentially. The final level of fluorescence varies with copy number since kinetics after 15 minutes into the RPA reaction are ill-defined. The LOD was at 1000 copies of the tcdB gene achieved at 15.7 minutes on-chip and 13.4 minutes on bench-top. The fastest reaction was observed for 107 copies at 5.0 minutes on-chip and 4.2 minutes on bench-top, respectively.
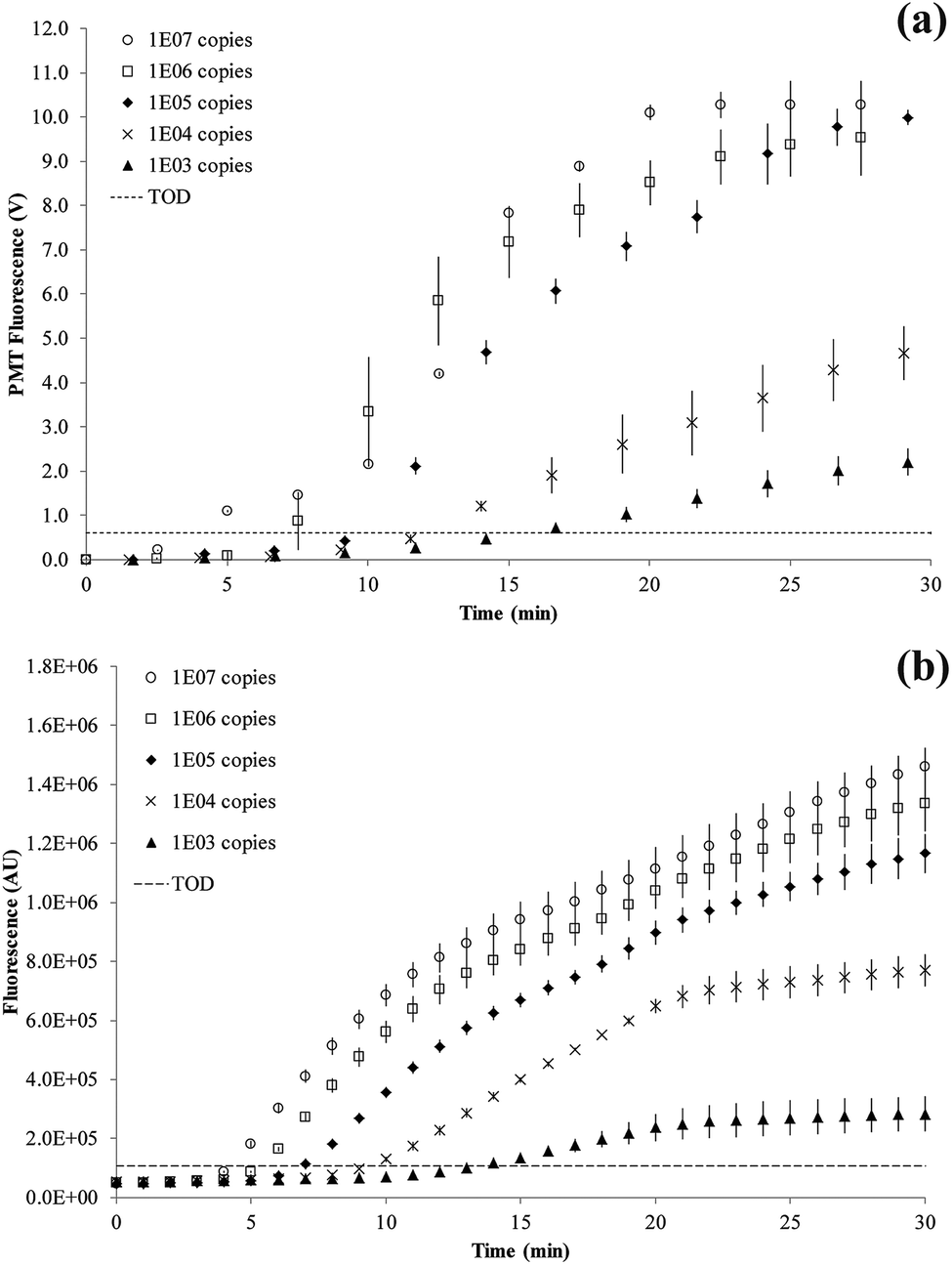 | ||
| Fig. 6 RPA amplification curves. The dashed line defines the threshold of detection (TOD) estimated as 3σ above the mean of two negative control reactions. The error bars denote standard deviation of replication experiments. (a) SlipChip results (n = 6) showing curves for 1 × 107 to 1 × 103 copies of C. difficile DNA. (b) Bench-top results (n = 3) showing curves for 1 × 107 to 1 × 103 copies of C. difficile DNA. | ||
The RPA reaction can be quantified from the time for onset of amplification12 or threshold time.32 This is the time point at which the amplification curve rises above a threshold of detection (TOD) and is equivalent to CT (or CQ) in qPCR.33 There is no set method for defining TOD as long as it is consistently applied for all data. In our experiments, the TOD has been set at 3σ above the mean of two negative control reactions, with no DNA template present. RPA reactions were performed at variable copy numbers of C. difficile DNA.
A plot of the onset of amplification against the logarithm of DNA concentration is shown in Fig. 7. A linear relationship was demonstrated for the RPA assay, and the SlipChip results are comparable with the bench-top method. The R2 values for on-chip and bench-top were at 0.99 and 0.93, respectively. Bench-top RPA was also performed for C. difficile R20291 genomic DNA using the same primers and conditions as for the PCR product. The LOD was 1000 copies at 12.6 minutes (data not shown).
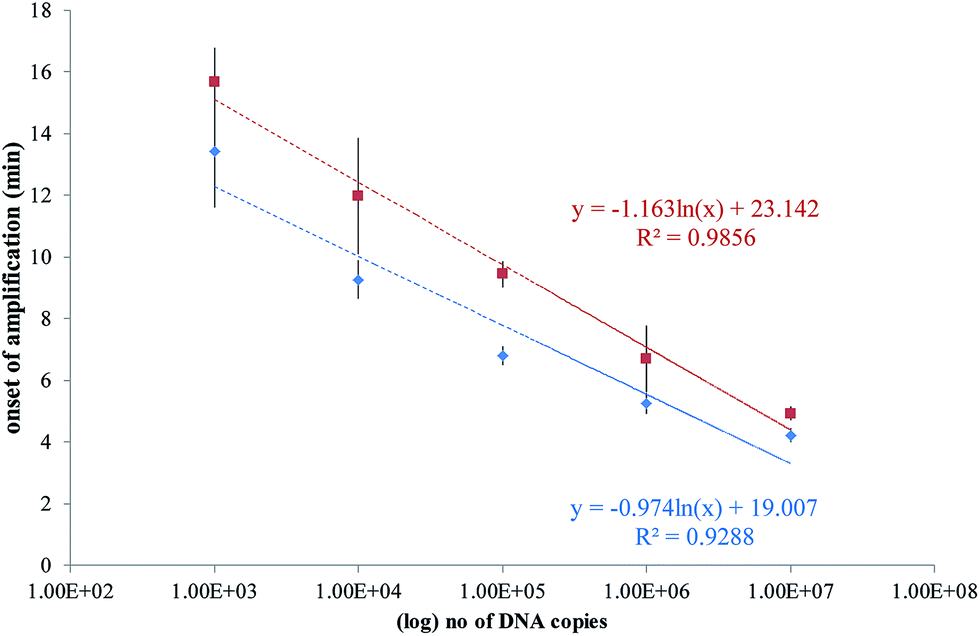 | ||
| Fig. 7 Plots of the onset of amplification of reactions shown in Fig. 6 against the logarithm of the template copy. The onset of amplification is defined as passing the threshold of detection (TOD). TOD is estimated as 3σ above the mean of two negative control reactions. (a) SlipChip results shown as red squares, with error bars denoting standard deviation of n = 6. (b) Bench-top results shown as blue diamonds, with error bars denoting standard deviation of n = 3. A linear relationship is demonstrated in both sets of results. | ||
It is informative to relate the number of DNA copies to fg of DNA or CFU. The C. difficile R20291 genome is 4191339 bp, so that one copy of this genome in theory equates to one CFU, and will correspond to ∼5 fg. However, since we are using a PCR amplicon for a sequence within the tcdB gene, a direct conversion from copies to CFU is not possible, as the LOD would be <1 CFU.
The RPA reaction is particularly attractive for microfluidic devices. It has been used for real-time detection of Staphylococcus aureus on a CD format, employing a cyclic olefin copolymer (COC) device.34 A total of 32 10 μL parallel reactions were run together with a time-to-result of ∼20 minutes and an LOD of 100 copies (or 0.1 fg). In our work, the reaction volume was reduced 20-fold to 500 nL with similar time-to-results.
Previous work by the Ismagilov group has shown RPA on a SlipChip format, employing a glass device fabricated by soft lithography.21 Reaction volumes were 9 nL in 1550 replicates. A 1 hour reaction was demonstrated for yes/no endpoint digital detection of Staphylococcus aureus. Our work demonstrates real-time quantitative analysis in nL volumes. This is advantageous because during the first exponential part of the amplification curve, accuracy is higher as reagents are in excess. Absolute quantification is based on an estimate of the abundance of the target relative to that of a standard curve. End-point detection used in nucleic acid amplification assays can be much more variable as just a few molecules difference at the reaction start can affect the final fluorescence greatly. The variation of endpoint fluorescence in RPA is even more pronounced since reaction kinetics after 30 minutes are still poorly understood.12
Future work will include first the integration of an amplified chemical readout, alleviating the need for a fluorescent detector as previously reported for a paper-based microfluidic device using RPA.35 This could involve using a biotin-labelled reverse primer, forming RPA amplicons that can be detected with commercial test strips lateral flow strips containing anti-biotin antibodies conjugated to gold nanoparticles. Second, very low cost devices will be fabricated using injection moulding of PMMA. The devices will be single-use and disposable.
Conclusions
A SlipChip platform with six parallel 500 nL reaction chambers was fabricated using rapid prototyping (laser cutting and micromilling) in PMMA. We demonstrated real-time RPA of the tcdB gene for C. difficile with 1 fg (1000 copies) of DNA. Our simple and low cost microfluidic isothermal nucleic acid amplification method could be developed into a point-of-care tool for intestinal infection without the need for lengthy laboratory tests, providing both rapid lab diagnostics and clinical decision-making at a single location.Contribution of authors
MNT designed and fabricated the device and thermoregulation mechanics, optimised on-chip RPA conditions, performed on-chip experiments and analysed the data. RW designed the RPA assay and performed bench-top experiments. CR assembled the optical setup and wrote the LABVIEW™ program. YZ designed and fabricated the base holder. XN, JMS and HM secured funding. MNT and HM wrote the paper.Acknowledgements
This report represents independent research by the National Institute for Health Research (Invention for Innovation (i4i), Rapid detection of infectious agents at point of triage (PoT), II-ES-0511-21002). The views expressed in this publication are those of the author(s) and not necessarily those of the NHS, the National Institute for Health Research or the Department of Health. The authors would like to thank Mark Long, Shilong Lu, Stephen Bettles, Daniel Spencer and Greg Elliott for technical support.Notes and references
- A. K. Deisingh and M. Thompson, Analyst, 2002, 127, 567–581 RSC.
- L. Clifford McDonald, F. Lessa, D. Sievert, M. Wise, R. Herrera, C. Gould, P. Malpiedi, M. Dudeck, A. Srinivasan, S. Fridkin and D. Cardo, Vital Signs: Preventing Clostridium difficile Infections, National Center for Emerging and Zoonotic Infectious Diseases, CDC, Morbidity and Mortality Weekly Report, 2012 Search PubMed.
- N. M. Thielman and R. L. Guerrant, N. Engl. J. Med., 2004, 350, 38–47 CrossRef CAS PubMed.
- D. J. Reinert, T. Jank, K. Aktories and G. E. Schulz, J. Mol. Biol., 2005, 351, 973–981 CrossRef CAS PubMed.
- D. E. Voth and J. D. Ballard, Clin. Microbiol. Rev., 2005, 18, 247–263 CrossRef CAS PubMed.
- P. Yager, T. Edwards, E. Fu, K. Helton, K. Nelson, M. R. Tam and B. H. Weigl, Nature, 2006, 442, 412–418 CrossRef CAS PubMed.
- M. Urdea, L. A. Penny, S. S. Olmsted, M. Y. Giovanni, P. Kaspar, A. Shepherd, P. Wilson, C. A. Dahl, S. Buchsbaum, G. Moeller and D. C. Hay Burgess, Nature, 2006, 444(suppl. 1), 73–79 CrossRef PubMed.
- W. Du, L. Li, K. P. Nichols and R. F. Ismagilov, Lab Chip, 2009, 9, 2286–2292 RSC.
- C.-M. Chang, W.-H. Chang, C.-H. Wang, J.-H. Wang, J. D. Mai and G.-B. Lee, Lab Chip, 2013, 13, 1225–1242 RSC.
- K. B. Mullis, F. A. Faloona and W. Ray, in Methods in Enzymology, Academic Press, 1987, vol. 155, pp. 335–350 Search PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, e204 Search PubMed.
- T. Shibata, R. P. Cunningham, C. Dasgupta and C. M. Radding, Proc. Natl. Acad. Sci. U. S. A., 1979, 76, 5100–5104 CrossRef CAS.
- T. Okazaki and A. Kornberg, J. Biol. Chem., 1964, 239, 259–268 CAS.
- W. Liu, D. Chen, W. Du, K. P. Nichols and R. F. Ismagilov, Anal. Chem., 2010, 82, 3276–3282 CrossRef CAS PubMed.
- R. S. Sista, A. E. Eckhardt, V. Srinivasan, M. G. Pollack, S. Palanki and V. K. Pamula, Lab Chip, 2008, 8, 2188–2196 RSC.
- F. Shen, W. Du, E. K. Davydova, M. A. Karymov, J. Pandey and R. F. Ismagilov, Anal. Chem., 2010, 82, 4606–4612 CrossRef CAS PubMed.
- F. Shen, W. Du, J. E. Kreutz, A. Fok and R. F. Ismagilov, Lab Chip, 2010, 10, 2666–2672 RSC.
- F. Shen, B. Sun, J. E. Kreutz, E. K. Davydova, W. Du, P. L. Reddy, L. J. Joseph and R. F. Ismagilov, J. Am. Chem. Soc., 2011, 133, 17705–17712 CrossRef CAS PubMed.
- B. Sun, F. Shen, S. E. McCalla, J. E. Kreutz, M. A. Karymov and R. F. Ismagilov, Anal. Chem., 2013, 85, 1540–1546 CrossRef CAS PubMed.
- F. Shen, E. K. Davydova, W. Du, J. E. Kreutz, O. Piepenburg and R. F. Ismagilov, Anal. Chem., 2011, 83, 3533–3540 CrossRef CAS PubMed.
- R. Zaouk, B. Y. Park and M. J. Madou, in T Microfluidic Techniques, 2005, vol. 321, pp. 5–15 Search PubMed.
- Y. Zhao, F. Pereira, A. J. deMello, H. Morgan and X. Niu, Lab Chip, 2014, 14, 555–561 RSC.
- P. C. H. Li, Microfluidic lab-on-a-chip for chemical and biological analysis and discovery, Taylor & Francis, New York, 2006 Search PubMed.
- A. Piruska, I. Nikcevic, S. H. Lee, C. Ahn, W. R. Heineman, P. A. Limbach and C. J. Seliskar, Lab Chip, 2005, 5, 1348–1354 RSC.
- M. C. Carles and N. J. Sucher, in Microfluidic Techniques, 2005, vol. 321, pp. 131–140 Search PubMed.
- I. R. G. Ogilvie, V. J. Sieben, C. F. A. Floquet, R. Zmijan, M. C. Mowlem and H. Morgan, J. Micromech. Microeng., 2010, 20, 065016 CrossRef.
- E. C. Rosenow, J. Dent. Res., 1919, 1, 205–249 CrossRef PubMed.
- R. A. Stabler, M. He, L. Dawson, M. Martin, E. Valiente, C. Corton, T. D. Lawley, M. Sebaihia, M. A. Quail, G. Rose, D. N. Gerding, M. Gibert, M. R. Popoff, J. Parkhill, G. Dougan and B. W. Wren, Genome Biol., 2009, 10, R102 CrossRef PubMed.
- S. A. Kuehne, S. T. Cartman, J. T. Heap, M. L. Kelly, A. Cockayne and N. P. Minton, Nature, 2010, 467, 711–U797 CrossRef CAS PubMed.
- I. Drigo, L. Bano, C. Bacchin, E. Tonon, T. Ferro, F. Barbanti, P. Spigaglia and F. Agnoletti, 4th International Clostridium difficile Symposium, Bled, Slovenia, 2012 Search PubMed.
- M. Euler, Y. Wang, P. Otto, H. Tomaso, R. Escudero, P. Anda, F. T. Hufert and M. Weidmann, J. Clin. Microbiol., 2012, 50, 2234–2238 CrossRef CAS PubMed.
- S. A. Bustin, V. Benes, J. A. Garson, J. Hellemans, J. Huggett, M. Kubista, R. Mueller, T. Nolan, M. W. Pfaffl, G. L. Shipley, J. Vandesompele and C. T. Wittwer, Clin. Chem., 2009, 55, 611–622 CAS.
- S. Lutz, P. Weber, M. Focke, B. Faltin, J. Hoffmann, C. Muller, D. Mark, G. Roth, P. Munday, N. Armes, O. Piepenburg, R. Zengerle and F. von Stetten, Lab Chip, 2010, 10, 887–893 RSC.
- B. A. Rohrman and R. R. Richards-Kortum, Lab Chip, 2012, 12, 3082–3088 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4an01683a |
| This journal is © The Royal Society of Chemistry 2015 |