
Making pyrophosphate visible: the first precipitable and real-time fluorescent sensor for pyrophosphate in aqueous solution†
Shu-Yan
Jiao
,
Kun
Li
*,
Xin
Wang
,
Zeng
Huang
,
Lin
Pu
and
Xiao-Qi
Yu
*
Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, Chengdu, 610064, China. E-mail: kli@scu.edu.cn; xqyu@scu.edu.cn; Fax: +86-28-85415886; Tel: +86-28-85415886
First published on 21st October 2014
Abstract
An in situ generated BINOL–DPA–Zn(II) complex is presented as a chemo-sensing ensemble for the recognition of phosphate-based molecules. The ensemble showed high sensitivity and selectivity for pyrophosphates (PPi), and it could be successfully applied in imaging PPi in living cells. Notably, the R1–Zn(ii) ensemble exhibited a very low detection limit (95 nM) for PPi and could realize the real time detection of PPi by the naked eye through precipitate experiments. The ensemble also showed good selectivity towards ATP, and the selectivity coefficient for PPi and ATP was calculated to be 4.1/2.8.
Introduction
Anions such as phosphate-based inorganic as well as organic molecules play important roles in a wide range of chemical and biological processes of human life,1–3 hence, considerable attention has been given to the development of synthetic receptors for the recognition and sensing of such anions. Among the phosphates, adenosine triphosphate (ATP) and inorganic pyrophosphates (PPi) have been of particular interest because they are involved in energy transduction in organisms, and they control metabolic processes through participation in various enzymatic reactions. These processes play important roles in biological processes such as energy storage, signal transduction and DNA sequencing.4 Therefore, developing phosphate-based probes that could be applied in bioimaging with high selectivity and sensitivity are highly needed.In a classical chemical analysis, color change and precipitate formation are the two most convenient visual detection methods. In the past decades, significant progress has been made in the recognition of phosphate-based bioactive anions, and many examples of visuals that rely only on color change have been reported.5,6 Analyte treatment with suitable probes or sensors often involves the formation of an acid–base or a host–guest complex that may precipitate from solution. However, such precipitation has not been utilized to date for the development of PPi sensors. Meanwhile, compared to the traditional chemosensors, precipitable sensors could realize the real-time detection of the analytes, which would give a corresponding signal immediately and provide the information in a short time.
On the other hand, it is challenging to selectively recognize bioactive anions in an aqueous system because of the strong hydration effects of anions. In this regard, metal–ligand receptors that have a stronger affinity for anions in competitive water are widely used in designing anion sensors within the past decade. Among them, the di-(2-picolyl) amine (DPA) unit has been widely exploited as a motif within metal complex-based anion sensor designs, particularly for the detection of phosphate-based molecules.7,8 Most notably, the Zn(II) complex of DPA exhibits an affinity for phosphate-bearing moieties, which can lead to either an increase in the fluorescence or quenching of a tethered fluorophore and the binding mode adopted by the interacting anion.9 So far, the fluorescent sensors for phosphate-based anions based on the Zn(II) complex of DPA mainly employed anthracene,10 naphthalimide,11 acedan,12 perylene,13 naphthalene diimide,14,15 pyrene,16 naphthalene,17 NBD,18etc., as the fluorophores.
Based on the number of DPA units incorporated, the DPA–fluorophore conjugate could be divided into two classes: mono- and di-DPA sensors. Two mono-DPA–Zn(II) complexes could coordinate with one PPi by forming an excimer, which causes fluorescent changes. Compared with mono-DPA–Zn(II) sensors, the interaction modes between di-DPA–Zn(II) sensors and PPi are complicated. It might form excimers between the two sensors after coordinating with two PPi ions; it is also possible to form supermolecules via one PPi coordinating with two DPA–Zn(II) units at two different sensors; additionally, two DPA–Zn(II) units in one sensor could also collaboratively coordinate with one PPi, if the distance between the units is allowable. Obviously, the location and the distance of the binding site are important to the interaction mode. Two binding sites could supply a higher binding energy, therefore, di-DPA–Zn(II) sensors have attracted much attention. It's worth noting that most di-DPA based sensors used the same skeleton. Since the binding mode is closely related to the framework, developing a novel skeleton for di-DPA units are highly need. Lippard et al. reported the first fluorescein platform with two DPA–Zn(II) units, which was recently proven to be coordinated with one PPi.19 Yoon reported a 8-naphthalenetetracarboxylic dianhydride based di-DPA–Zn(II) complex, however, due to the planar rigidity, the sensor formed a 2 + 2 type excimer with PPi rather than a monomer coordination.20 These results indicated that the distance and the rigidity of the fluorophore are important to the binding mode with PPi (Scheme 1).
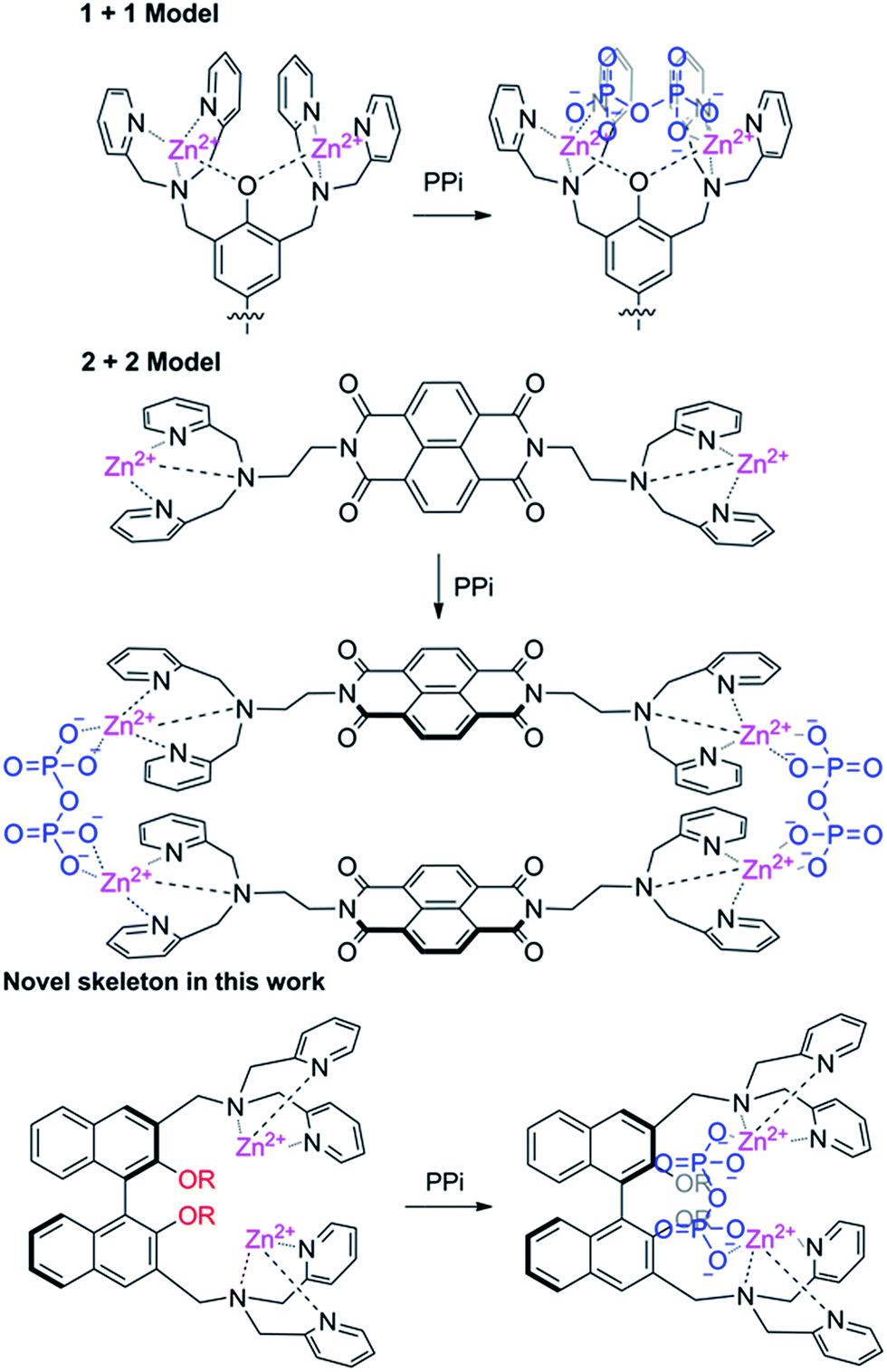 | ||
| Scheme 1 The structures of chemosensing probes and their proposed binding modes with PPi. | ||
1,1′-Bi-2-naphthol (BINOL) and its derivatives have attracted particular attention in fluorescence-based molecular recognition. Previously, we presented the first example of an enantioselective gel collapsing, as a new means of visual chiral sensing utilizing a BINOL–terpyridine-based Cu(II) complex.21 More recently, we reported a BINOL-based fluorescent sensor for Zn2+ and a ratiometric fluorescent sensor for Cu2+ in aqueous solution.23 The two 2-naphthol units in BINOL are not on the same plane. The introduction of a DPA unit into the BINOL skeleton could affect the axial chirality of the BINOL, which might provide better conditions for PPi after coordination with Zn(II). In fact, Ahn reported a naphthol based mono-DPA–Zn(II) complex which could selectively respond to PPi, however, it needed a large quantity of PPi (200 equiv.) and a longer time (6 h).22 We doubt whether the reactivity and selectivity could be improved by introducing two-DPA units at the 3,3′position of BINOL. In connection with our continuing research on fluorescent sensors for biologically and environmentally important ions, herein, we present a BINOL–DPA–Zn(II) complex as the first precipitable and real-time fluorescent sensor for PPi.
In this manuscript, we report R1, R2, R3 (Scheme 2) conjugates which employ the BINOL skeleton as the fluorophores and DPA–Zn(II) as the binding unit for the selective detection of phosphate-based anions in aqueous solution. Delightfully, the in situ generated R1–Zn(ii) ensemble could serve as the first precipitable and real-time fluorescent sensor for PPi, and it could further successfully be applied in imaging PPi in living cells. Moreover, the R1–Zn(ii) ensemble also exhibited a very low detection limit for PPi.
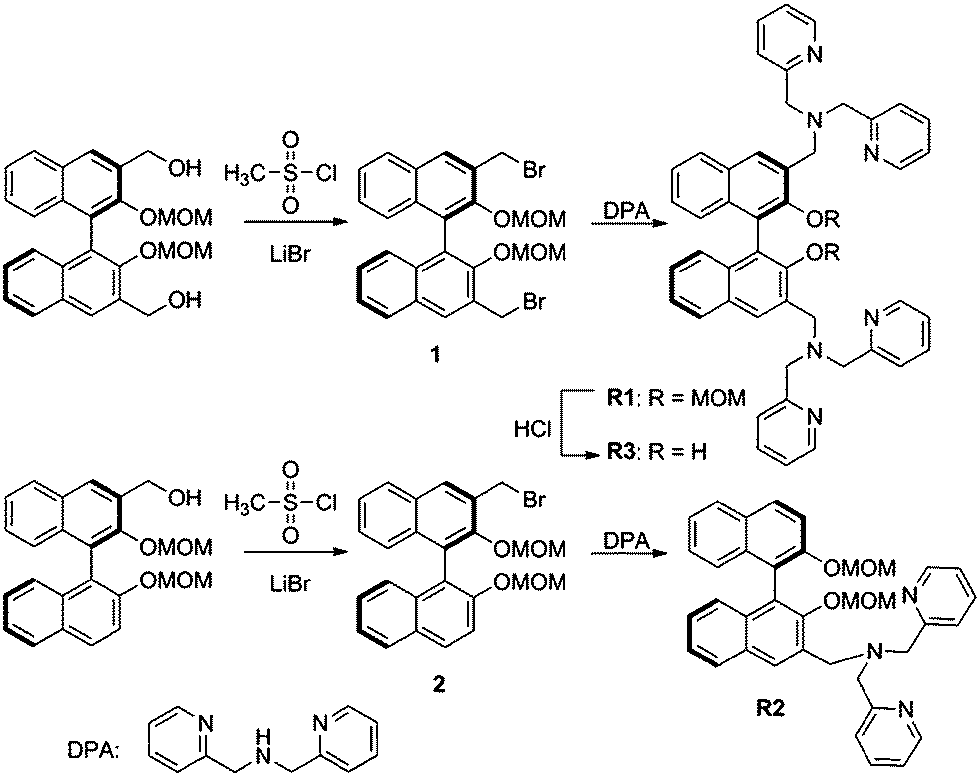 | ||
| Scheme 2 Synthesis route for the sensors R1/R2/R3. | ||
Results and discussion
The sensor R1 was synthesized from (R)-(2,2-bis(methoxymethoxy)-1,1′-binaphthyl-3,3′-diyl)dimethol (Scheme 2). The intermediate 1 was obtained by the reaction of (R)-(2,2-bis(methoxymethoxy)-1,1′-binaphthyl-3,3′-diyl)dimethol with methylsulfonyl chloride and lithium bromide, which was further transformed into the R1 reaction with di(2-picolyl) amine. R2 was synthesized by similar procedures to R1, while R3 was synthesized by removing the MOM group (–CH2OCH3) of R1 using concentrated HCl in a CH2Cl2/C2H5OH solution. The precursors and final products were characterized by analytical and spectral techniques (see the Experimental section and ESI†).The recognition of Zn2+ by sensors R1, R2, R3 were investigated by fluorescence titration. The sensor R1 was excited at 316 nm, and its fluorescence emission was collected in the range of 336–612 nm. The experiments were carried out in HEPES buffer by maintaining the sensor concentration of 10 μM throughout the experiment and varying the mole ratio of the added Zn2+; R1 is a weak emitter as its fluorescence is quenched due to the photoelectron transfer (PET) process from the lone pair of the nitrogen of the DPA group on BINOL. During the titration with Zn2+, the fluorescence intensity of sensor R1 was monitored as a function of the increase in the mole ratio of [Zn2+]/[R1] (Fig. 1). The reason for the resulting fluorescence enhancement upon the addition of Zn2+ is due to the suppression of the photoelectron transfer effect (PET) from the lone pair of electrons on the DPA group. Upon the titration of Zn2+, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 stoichiometry between R1 and Zn2+ was confirmed by the Job's plot analysis (Fig. 1, inset) and mass spectral analysis (Fig. S1, ESI†). The constant between R1 and Zn2+, 5.06 × 1010 M−1, was calculated by the Benesi–Hildebrand equation (Fig. S2, ESI†). The proposed binding mode of R1 and Zn2+ is shown in Fig. 2. Moreover, the responses for Zn2+ of the sensors R2 and R3 were also investigated by the fluorescence study. As shown in Fig. S3,† when Zn2+ was added into the solution of R2, a little red shift in the maximal emission was found, and the emission intensity increased gradually with the increase of Zn2+. These changes in the fluorescence spectrum stopped when the concentration of Zn2+ reached 1.2 equiv. of R2 (Fig. S3, ESI†). However, there was no change of R3 under the same conditions (Fig. S4, ESI†).
:2 stoichiometry between R1 and Zn2+ was confirmed by the Job's plot analysis (Fig. 1, inset) and mass spectral analysis (Fig. S1, ESI†). The constant between R1 and Zn2+, 5.06 × 1010 M−1, was calculated by the Benesi–Hildebrand equation (Fig. S2, ESI†). The proposed binding mode of R1 and Zn2+ is shown in Fig. 2. Moreover, the responses for Zn2+ of the sensors R2 and R3 were also investigated by the fluorescence study. As shown in Fig. S3,† when Zn2+ was added into the solution of R2, a little red shift in the maximal emission was found, and the emission intensity increased gradually with the increase of Zn2+. These changes in the fluorescence spectrum stopped when the concentration of Zn2+ reached 1.2 equiv. of R2 (Fig. S3, ESI†). However, there was no change of R3 under the same conditions (Fig. S4, ESI†).
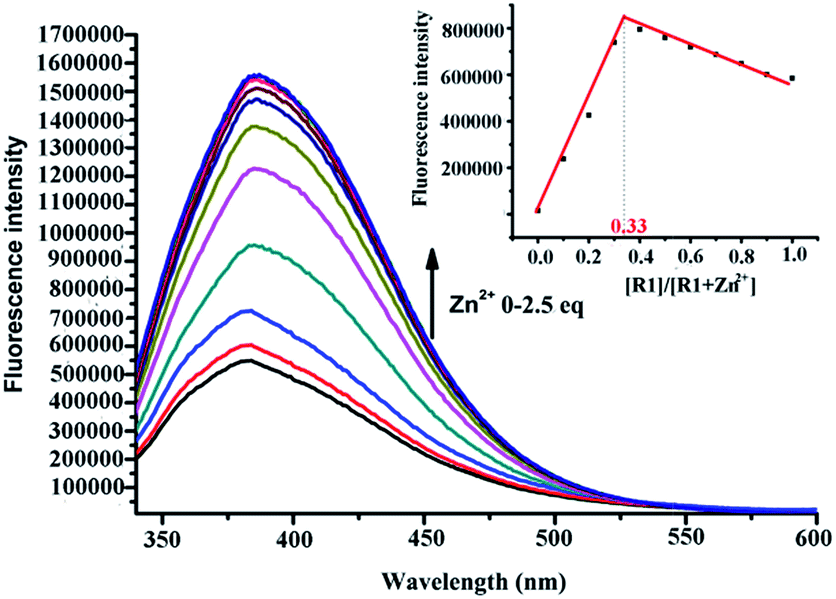 | ||
| Fig. 1 Fluorescence emission spectra of R1 upon addition of Zn2+ in HEPES (10 mM, pH = 7.4) [R1] = 10 μM, [Zn2+] = (0 μM, 1 μM, 2 μM, 3 μM, 4 μM, 6 μM, 8 μM, 10 μM, 15 μM, 20 μM, 25 μM). λex = 316 nm. Inset: Job's plot obtained from the fluorescent titration of R1 with Zn2+ in HEPES, the total concentration is 10 μM. | ||
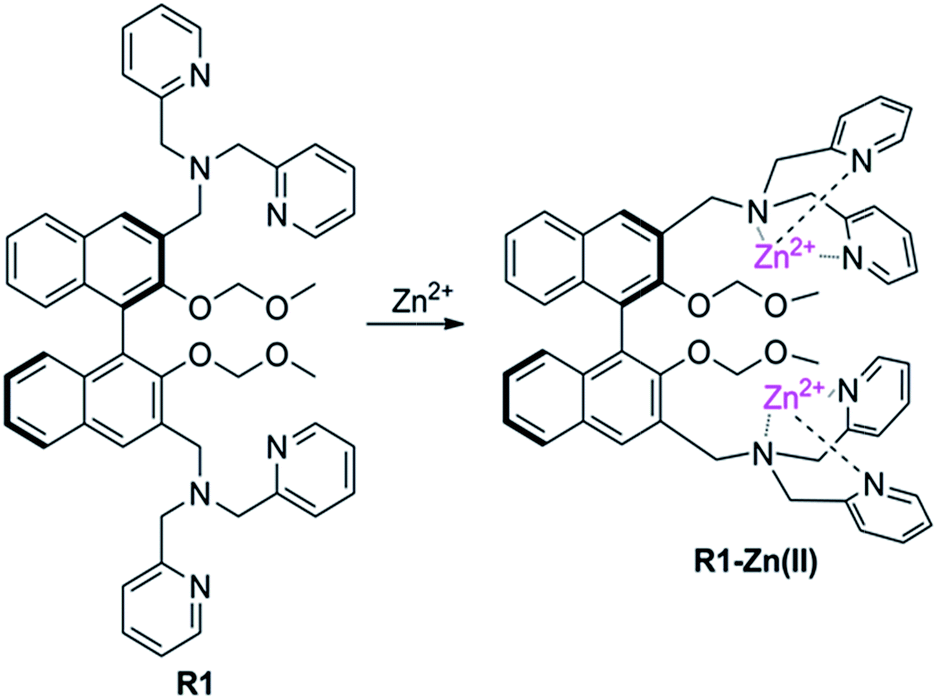 | ||
| Fig. 2 Proposed binding mode of R1 and Zn2+. | ||
To further understand the coordination properties of R1 towards Zn2+, DFT calculations with the B3LYP exchange functional were performed using the Gaussian 03 package. The geometries were optimized and energies were estimated at the B3LYP/6-31G(d)&LANL2DZ level. The optimized configuration is shown in Fig. 3, which indicates that the two Zn2+ were well chelated by the N1, N2, N3, and N3, N4, N5 and N6 of R1. The Zn1–N1 bond length is 2.008 Å, the Zn1–N2 bond length is 2.018 Å, the Zn1–N3 bond length is 2.022 Å, the Zn2–N4 bond length is 2.082 Å, the Zn2–N5 bond length is 2.054 Å, and the Zn2–N6 bond length is 2.118 Å. After the calculation, we found the binding energy of R1 and Zn2+ was −74.3 kcal mol−1, which was in agreement with the other experimental results.
 | ||
| Fig. 3 Optimized geometries of the R1–Zn(ii) complex. | ||
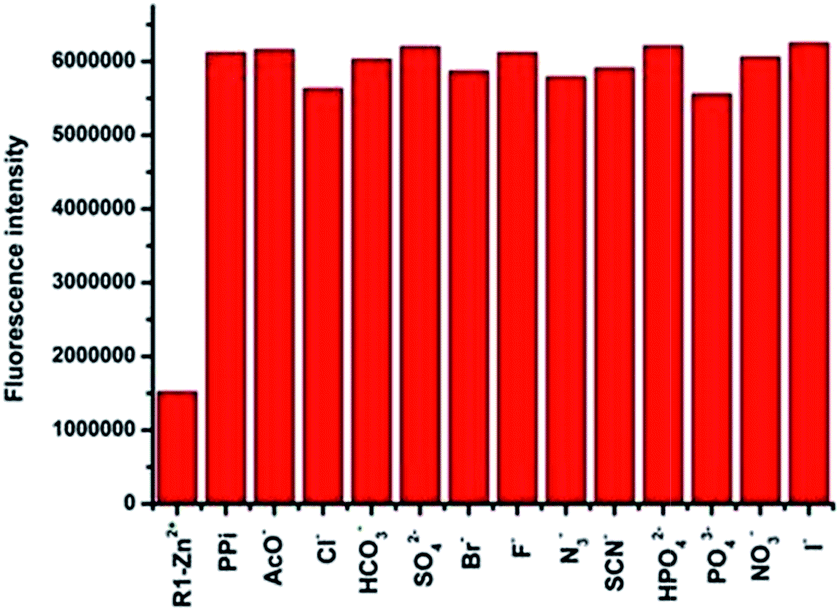
The anion recognition studies were carried out by titrating an in situ generated complex R1–Zn(ii), R2–Zn(ii), R3–Zn(ii) by different anions, viz., PPi, F−, SCN−, H2PO4−, HCO3−, Br−, PO43−, NO3−, AcO−, citrate, I−, N3−, SO42−. As shown in Fig. 4, for the sensor R1–Zn(ii), all the anion titrations exhibited no significant change in the fluorescence intensity except for PPi (Fig. 4, green and Fig. 5). The fluorescence of the in situ generated R1–Zn(ii) was increased gradually with the addition of PPi, wherein the intensity was saturated at 3 μM (0.3 equiv.) (Fig. 6). This clearly suggests that R1–Zn(ii) shows a stronger affinity with phosphates than other anions (F−, SCN−, H2PO4−, HCO3−, Br−, PO43−, NO3−, AcO−, citrate, I−, N3−, SO42−) and it is a sensitive chemo-sensing ensemble for PPi among the thirteen anions in aqueous buffer solution. Under the same conditions, almost no fluorescence intensity changes of the in situ generated complexes R2–Zn(ii) and R3–Zn(ii) were observed in the presence of various anions (Fig. 4 yellow and red). These results suggest that the number of DPA–Zn(II) units and the MOM group is important for the binding ability.
 | ||
| Fig. 4 Fluorescence responses of the in situ prepared R1–Zn(ii)/R2–Zn(ii)/R3–Zn(ii) towards 100 μM (10 equiv.) various anions at 383 nm in HEPES(10 mM, pH = 7.4, 1% CH3OH as the assistant solvent). Excitation at 316 nm: (1) R1–Zn(ii), R2–Zn(ii), R3–Zn(ii) (2) F− (3) SCN− (4) H2PO4− (5) HCO3− (6) Br−(7) PO43− (8) NO3− (9) AcO− (10) citrate (11) I− (12) Cl− (13) N3− (14) SO42− (15) PPi; λex = 316 nm. | ||
 | ||
| Fig. 5 Fluorescence responses of the in situ prepared R1–Zn(ii) complex towards 100 μM (10 equiv.) of various anions in HEPES (pH = 7.4, 10 mM, 1% CH3OH as the assistant solvent), λex = 316 nm. | ||
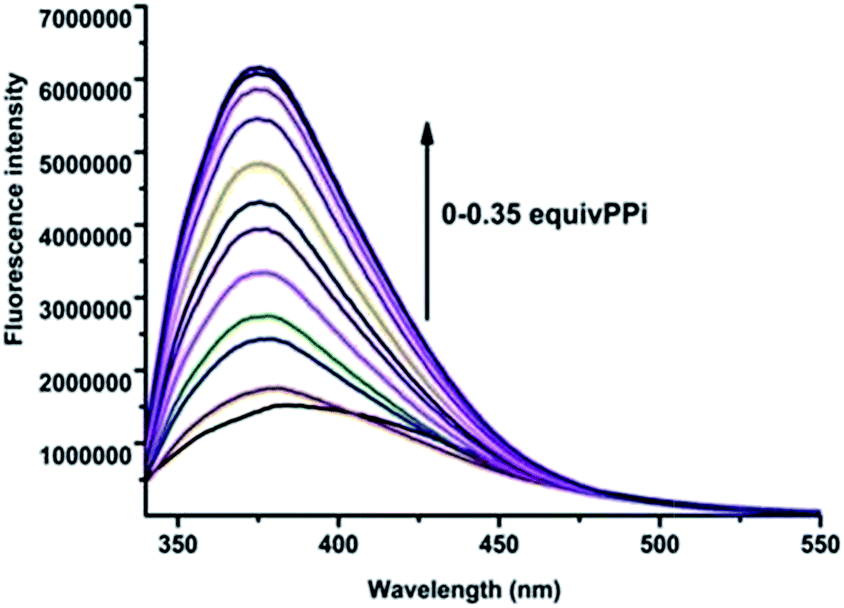 | ||
| Fig. 6 Fluorescence emission spectra of the in situ prepared R1–Zn(ii) complex upon addition of PPi in HEPES (10 mM, pH = 7.4) [R1–Zn(ii)] =10 μM, [PPi] = (0 μM, 0.1 μM, 0.2 μM, 0.3 μM, 0.4 μM, 0.6 μM, 0.8 μM, 1.0 μM, 1.5 μM, 2.0 μM, 3.0 μM, 4.0 μM). λex = 316 nm. | ||
To better investigate the practical applicability of the in situ prepared R1–Zn(ii) complex, competitive experiments were carried out in the presence of PPi (10 μM) mixed with F−, SCN−, H2PO4−, HCO3−, Br−, PO43−, NO3−, AcO−, citrate, I−, N3−, and SO42− (50 μM), as shown in Fig. 7. There was no significant change in the fluorescence intensity when R1–Zn(ii) was placed in the mixture of PPi (10 μM) and other test anions (Fig. S5, ESI†), therefore there was almost no interference of the R1–Zn(ii) complex recognition of PPi in the presence of other anions. The results of the competitive experiments suggest that the in situ prepared R1–Zn(ii) complex shows good selectivity for PPi among the other tested anions. The quantitative analytical behaviour of the in situ prepared R1–Zn(ii) complex for the analysis of PPi was examined by fluorescence titration. As displayed in Fig. 7, the in situ prepared R1–Zn(ii) complex displayed a weak fluorescence emission at 383 nm, and upon gradual addition of PPi to the solution of R1–Zn(ii), the emission intensity at 383 nm increased significantly. There was almost a 4-fold enhancement of fluorescence intensity compared to the intensity of the in situ prepared R1–Zn(ii) complex itself. The Job's plot of the data revealed a 1:1 complex formation of R1–Zn(ii) and PPi (Fig. S6, ESI†), which was also supported by the HRMS experiment (Fig. S7, ESI†). The association constant and detection limit of R1–Zn(ii) for PPi was calculated to be 6.7 × 105 M−1 and 95 nM, respectively (Fig. S8 and S9, ESI†).25 The UV-vis absorption spectra of R1–Zn(ii) in the presence of various concentrations of PPi also further proved the complexation between R1–Zn(ii) and PPi (Fig. S10, ESI†).
 | ||
| Fig. 7 Emissions of the in situ prepared R1–Zn(ii) complex with various anions (100 μM) in the absence and presence of PPi (10 μM) at 383 nm in HEPES(10 mM, pH = 7.4), the red bar: from left to right: R1–Zn2+ (10 μM); R1–Zn2+ (10 μM) + PPi (10 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + AcO− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + Cl− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + HCO3− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + SO42− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + Br− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + F−(50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + N3−(50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + SCN− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + HPO42− (50 μM); R1–Zn2+ (10 μM) + PPi (10 μM) + PO43− (50 μM); R1–Zn2+ (10 μM)+ PPi (10 μM) + NO3− (50 μM); R1–Zn2+(10 μM) + PPi (10 μM) + I− (50 μM). λex = 316 nm. | ||
As the in situ prepared R1–Zn(ii) exhibited good selectivity to PPi among general anions, the studies were further extended to organic biorelevant molecules bearing a phosphate moiety, viz., ATP, ADP, GTP, CTP, dUTP and dTTP. As displayed in Fig. 8, when 100 μM (10 equiv.) ATP and ADP was added to the R1–Zn(ii) (10 μM) solution, fluorescence enhancement was observed, and the enhancement in the presence of ATP is larger than that of ADP, while GTP, CTP, dUTP and dTTP didn't induce any change of the fluorescence intensity of R1–Zn(ii). We also found that the fluorescence enhancement caused by PPi is larger than that caused by ATP, and the selectivity coefficient for PPi and ATP is calculated to be 4.1/2.8, suggesting that the enhancement is dependent on both the bulkiness of the organic moiety and the number of phosphates. Therefore, we suspect that this selectivity for binding with anions is connected with the distance and the rigidity of the two DPA–Zn(II) units of the sensor. In order to assess whether the in situ prepared R1–Zn(ii) complex can be of any use in the recognition of anions by naked eye detection, R1–Zn(ii) (2.5 mM, when the concentrations of the sensor and analytes are μM, they cannot form precipitates, so we choose the concentration of the sensor as 2.5 mM) was titrated with 0.15 equiv. various anions, viz., PPi, ATP, ADP, GTP, CTP, dUTP and dTTP in HEPES. It was observed that in the presence of PPi, and ATP, the R1–Zn(ii) complex solution formed a precipitate. However, in the presence of GTP, CTP, dTTP, dUTP, ADP, the R1–Zn(ii) complex solution didn't form a precipitate; this result suggests that the in situ prepared R1–Zn(ii) complex can show selectivity for PPi and ATP over other anions, which is observable by the naked eye. In addition, we investigated how the different concentrations of PPi influence the precipitate formation. As shown in Fig. 9, we can see that with the increasing concentration of PPi, the quantity of the precipitate increased. This precipitate formation experiment further explained R1–Zn(ii) and PPi formed complex. Furthermore, this precipitation could be formed at the interface of the organic solvents and water (video S1†). This is the visual precipitate detection for PPi.
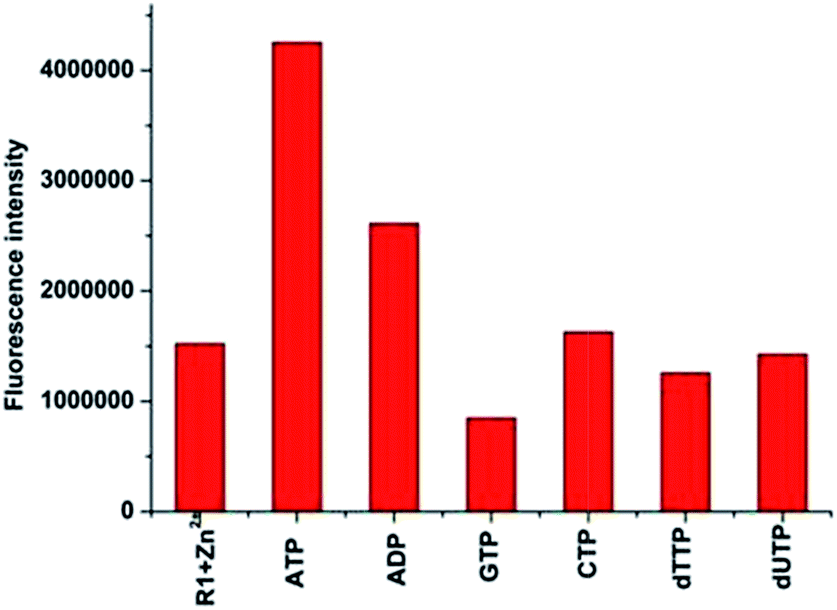 | ||
| Fig. 8 Fluorescence responses of the in situ prepared R1–Zn(ii) complex (10 μM) towards 100 μM (10 equiv.) of phosphate-based organic molecules at 383 nm in HEPES (pH = 7.4, 10 mM, 1% CH3OH as the assistant solvent), λex = 316 nm. | ||
 | ||
| Fig. 9 (a) The responses of the R1–Zn(ii) ensemble (2.5 mM) in the absence and presence of anions, from left to right: absence of anions, presence of PPi, ATP, ADP, CTP, GTP, dUTP, dTTP (0.275 mM); (b) the responses of the R1–Zn(ii) ensemble in the presence of different concentrations of PPi. | ||
To better understand this precipitation, scanning electron microscopy (SEM) was used to investigate the process (Fig. 10). The results indicated that R1 was uniformly dispersed in the solution by forming small particles (0.8–1.3 μm), due to its suitable solubility in the HEPES solution. We believed the MOM group and the two DPA units were responsible for the formation of these small particles, which could not be observed by the naked eye in the solution. However, upon the addition of Zn2+ to the system, the solution became clear and no small particles were found, which suggested that R1–Zn(ii) was totally soluble in the solution. Once PPi was added, rough and scaly solids with large enormous surfaces were observed. This result also suggests that the interaction occurred between R1–Zn(ii) and PPi.
 | ||
| Fig. 10 SEM micrographs, (a) for R1 (b) for R1–Zn(ii), (c) for R1-Zn(ii)–PPi. | ||
To further understand the in situR1–Zn(ii) complex, DFT calculations with the B3LYP exchange functional were performed by using the Gaussian 03 package. The geometries were optimized and the energies were estimated by the B3LYP/6-31G(d) & LANL2DZ level. The optimized configuration is shown in Fig. 11, which indicates that the O1, O2, O3 and O4 were well chelated by the Zn1 and Zn2 of the R1–Zn(ii) complex. The Zn1–O1 bond length is 2.066 Å, the Zn1–O2 bond length is 1.989 Å, the Zn1–N2 bond length is 2.126 Å, the Zn2–O3 bond length is 2.034 Å, the Zn2–O4 bond length is 1.997 Å, the Zn2–N4 bond length is 2.114 Å, the Zn2–N5 bond length is 2.159 Å, but the Zn1–N3 bond and the Zn2–N6 bond were broken, the results of the bonds breaking were supported by the Hong groups,24 after the calculation, we found the binding energy of R1-Zn(ii) and PPi was −173.8 kcal mol−1, which was also in agreement with the other experimental results.
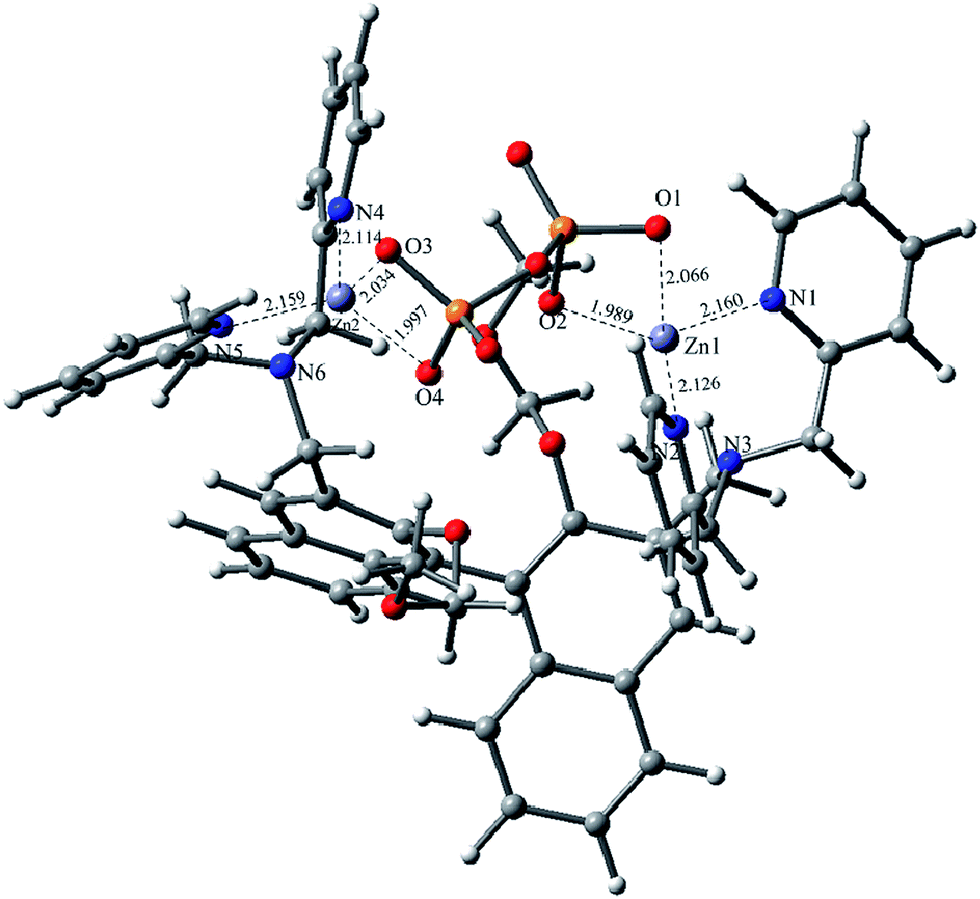 | ||
| Fig. 11 Optimized geometries of R1–Zn(ii) and PPi complex. | ||
In order to show the practical utility of R1–Zn(ii) for the detection of PPi in a biological medium, fluorescence microscopy studies were carried out using Hela cells. Hela cells were incubated in PBS buffer (pH = 7.4) containing 10 μM of R1 for 30 min at 37 °C, followed by washing the cells with the same buffer, at this stage, the Hela cells displayed almost no fluorescence (Fig. 12d), upon the addition of Zn2+/pyrithione to the cells exogenously via incubation for another 30 min at 37 °C, the cells exhibited a weak blue fluorescence. However, when R1 and Zn2+ were preincubated, and the Hela cells were further treated with PPi for 30 min at 37 °C, the cells exhibited highly intense blue fluorescence, therefore, the cellular studies clearly indicate that R1 exhibits good cell permeability and shows effective intracellular fluorescence emission by forming an in situ complex, which in turn increases its fluorescence intensity upon treatment with PPi.
 | ||
| Fig. 12 Fluorescence from images obtained from Hela cells upon treatment in PBS buffer at pH = 7.4: (a) bright field image when treated with R1 (10 μM); (b) bright field image when treated with R1 followed by 20 μM of Zn2+/pyrithione (1:1) solution; (c) bright field image of the Hela cells treated with R1–Zn(ii) followed by 20 μM of PPi; (d) fluorescence image of that given under (a); (e) fluorescence image of that given under (b); (f) fluorescence image of that given under (c). | ||
Experimental
General
Mass spectrometry (ESI-MS) and High Resolution Mass Spectrometry (HRMS) data were recorded on a Finnigan LCQDECA and a Bruker Daltonics Bio TOF mass spectrometer, respectively. The 1H NMR and 13C NMR spectra were measured on a Bruker AM400 NMR spectrometer and the δ scale in ppm was referenced to residual solvent peaks or internal tetramethylsilane (TMS). Absorption spectra were recorded on a Hitachi U1900 spectrophotometer at 298 K. Fluorescence emission spectra were obtained using a FluoroMax-4 Spectrofluorophotometer (HORIBA JobinYvon) at 298 K. The MDA-MB-231 cells were analyzed in an Array Scan VTI HCS Reader (Thermo Fisher Scientific, Inc.). Unless otherwise indicated, all syntheses and manipulations were carried out under N2 atmosphere. All of the solvents were dried according to the standard methods prior to use. All of the solvents were either HPLC or spectroscopic grade in the optical spectroscopic studies.Preparation of (R)-3,3′-bis (bromomethyl)-2,2′-bis(methoxy-methyl)-1,1′binaphol
To an ice-cooled solution of (R)-(2,2′-bis(methoxymethoxy)–1,1′-binaphthyl-3,3′-diyl)dimethanol (1.0 g, 2.3 mmol) in toluene (30 mL) were successively added Et3N (3.3 g, 23 mmol) and MsCl (2.62 g, 23 mmol). The mixture was stirred at 0 °C for 90 min and then LiBr (2.0 g, 23 mmol) in 30 mL DMF was added. The mixture was stirred at room temperature overnight. It was then diluted with ethyl acetate (120 mL) and washed with saturated brine and finally dried over MgSO4 and condensed in vacuo to give crude product; purification by a silica column afforded the product as a yellow solid. 1H NMR (400 Hz, CDCl3) δ (ppm): 8.12 (s, 2H), 7.91 (d, 2H, J = 8.0 Hz), 7.44 (t, 2H, J = 8.0 Hz), 7.17–7.27 (m, 5H), 7.30 (t, 1H, J = 8.0 Hz), 7.36 (d, 1H, J = 12.0 Hz), 7.89 (t, 2H, J = 8.0 Hz), 7.29 (t, 2H, J = 8.0 Hz), 7.18 (d, 2H, J = 8.0 Hz),4.98 (q, 4H, J =12.0 Hz, 16.0 Hz), 4.64 (d, 2H, J = 4.0 Hz), 4.64 (d, 2H, J = 4.0 Hz), 2.98 (s, 6H). 13C NMR (100 MHz, CDCl3), δ (ppm): 42.4, 56.9, 99.5, 125.5, 123.4, 125.6, 126.0, 127.2, 128.15, 130.6, 131.0, 131.2, 129.4, 134.2, 130.7, 152.2.Preparation of sensors R1, R2, R3
:CH3OH =100:1 as the eluent to give a yellow oil in 82% yield. HRMS (m/z): calcd for C50H48N6NaO4, [M + Na+], 819.3635, found 819.3565, 1H NMR (400 Hz, CDCl3) δ (ppm): 8.54 (d, 4H, J = 4.0 Hz), 8.31 (s, 2H), 7.88 (d, 2H, J = 8.0 Hz), 7.61–7.68 (m, 8H), 7.37 (t, 1H, J = 8.0 Hz, 2H), 7.19 (d, 2H, J = 8.0 Hz), 7.16–7.12 (m, 6H), 3.92–4.06 (m, 12H), 4.46 (d, 2H, J = 4.0 Hz),4.35 (d, 2H, J = 4.0 Hz), 2.68 (s, 6H, J =4.0 Hz). 13C NMR (100 MHz, CDCl3), δ (ppm): 53.8, 56.5, 60.3, 99.0, 122.0, 124.9, 125.4, 126.1, 127.8, 128.8, 129.3, 131.8, 132.6, 133.3, 136.5, 148.1, 153.0, 159.6.
:CH3OH = 100:1 as the eluent to give a yellow oil in 85% yield. HRMS (m/z): calcd for C37H36N3O4, [M + H+], 586.2706, found 586.2708. 1H NMR (400 Hz, CDCl3) δ (ppm): 8.55 (d, 2H, J = 4.0 Hz), 8.30 (s, 1H), 7.96 (d, 1H, J = 12.0 Hz), 7.91 (d, 1H, J = 8.0 Hz), 7.86 (d, 1H, J = 8.0 Hz), 7.66–7.74 (m, 4H), 7.58 (d, 1H, J = 8.0 Hz), 7.32–7.40 (m, 2H), 7.13–7.25 (m, 6H), 5.09 (d, 1H, J = 4.0 Hz), 5.00 (d, 1H, J = 8.0 Hz), 4.44 (d, 1H, J = 8.0 Hz), 4.34 (d, 1H, J = 8.0 Hz) 3.99–4.08 (m, 6H), 3.15 (s, 3H),2.80 (s, 3H). 13C NMR (100 MHz, CDCl3), δ (ppm): 53.7, 55.9, 55.6, 60.4, 94.9, 99.0, 116.5, 120.8, 122.0, 122.8, 124.1, 124.9, 125.3, 125.6, 125.7, 125.8, 126.6, 127.9, 129.1, 129.6, 129.8, 130.9, 132.4, 133.1, 134.0, 136.5, 149.0, 152.7, 152.9, 159.8.
:C2H5OH = 1:1 (v/v, 20 mL) of R1 (0.2 g, 0.25 mmol) was added con. HCl (5 mL). The mixture was refluxed for 4 h, after removal of the solvent, the resulting residue was dissolved in water and the aqueous solution was treated with saturated aqueous Na2CO3. The mixture was extracted with CH2Cl2 (3 × 30 mL), and the combined organic layer was washed with brine, and then dried over Na2SO4. After removal of the Na2SO4 by filtration, evaporation of the solvent gave the crude product, and then crude product was purified by alumina column chromatography by using CH2Cl2 as the eluent to give a white solid in 92% yield. HRMS (m/z): calcd for C46H41N6O2, [M + H+], 709.3291, found 709.3293. 1H NMR (400 Hz, CDCl3) δ (ppm): 10.79 (s, 2H, J = 4.0 Hz), 8.41 (d, 2H, J = 4.0 Hz), 7.81 (d, 2H, J = 8.0 Hz), 7.77 (s, 2H), 7.36–7.44 (m, 8H), 7.24 (t, 1H, J = 4.0 Hz), 7.23 (d, 1H, J = 4.0 Hz), 7.17 (s, 1H), 7.14 (s, 1H), 7.00–7.07 (m, 6H), 4.19 (d, 2H, J = 12.0 Hz), 4.06 (d, 2H, J = 12.0 Hz), 3.92–4.00 (m, 8H). 13C NMR (100 MHz, CDCl3), δ (ppm): 58.1, 59.2, 117.4, 122.1, 122.8, 123.7, 125.1, 127.6, 128.2, 129.3, 134.3, 136.8, 148.8, 153.3, 158.1.
Conclusions
In conclusion, a series of in situ generated Zn2+ complexes of BINOL–DPA sensors R1–Zn(ii), R2–Zn(ii), R3–Zn(ii) have been presented in this paper. The fluorescence properties of these ensembles were investigated, only the R1–Zn(ii) ensemble showed high sensitivity and selectivity for PPi with a detection limit of 95 nM, and it can successfully be applied in the imaging of PPi in living cells. Moreover, the R1–Zn(ii) ensemble could also realize the real-time detection of PPi by the naked eye through precipitate experiments.Acknowledgements
This work was financially supported by the National Program on Key Basic Research Project of China (973 Program, 2012CB720603 and 2013CB328900), the National Science Foundation of China (no. 21232005, 21321061, J1310008 and J1103315), and the Specialized Research Fund for the Doctoral Program of Higher Education in China (20120181130006).Notes and references
- (a) R.-K. Pathak, K. Tabbasum, A. Rai, D. Panda and C.-P. Rao, Anal. Chem., 2008, 84, 5117 CrossRef PubMed; (b) S. K. Kim, D. H. Lee, J. I. Hong and J. Yoon, Acc. Chem. Res., 2009, 42, 23 CrossRef CAS PubMed; (c) J. J. Deng, P. Yu, L. F. Yang and L. Q. Mao, Anal. Chem., 2013, 85, 2516 CrossRef CAS PubMed.
- (a) X.-J. Zhao, L. He and C.-Z. Huang, Talanta, 2012, 101, 59 CrossRef CAS PubMed; (b) A. Nonaka and Y. Kubo, Org. Biomol. Chem., 2008, 6, 3621 RSC; (c) J.-H. Wen, Z.-R. Geng, Y.-X. Yin, Z. Zhang and Z.-L. Wang, Dalton Trans., 2011, 40, 1984 RSC.
- (a) S.-J. Kim and J.-I. Hong, Tetrahedron Lett., 2009, 50, 1951 CrossRef CAS PubMed; (b) P. Das, A. Ghosh, M.-K. Kesharwani, V. Ramu, B. Ganguly and A. Das, Eur. J. Inorg. Chem., 2011, 20, 3050 CrossRef.
- (a) X.-L. Feng, Y.-X. An, Z.-Y. Yao, C. Li and G.-Q. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 614 CrossRef CAS PubMed; (b) J. Gao, T. Riis-Johannessen, R. Scopelliti, X.-H. Qian and K. Severin, Dalton Trans., 2010, 39, 7114 RSC; (c) S. Kim, M.-S. Eom, S.-K. Kim, S.-H. Seo and M.-S. Han, Chem. Commun., 2013, 49, 152 RSC; (d) K. Ghosh and I. Saha, Org. Biomol. Chem., 2012, 10, 9383 RSC.
- (a) D. Lee, A. Jo, S.-B. Park and J.-I. Hong, Tetrahedron, 2012, 53, 5528 CrossRef CAS PubMed; (b) W.-H. Zhu, X.-M. Huang, Z.-Q. Guo, X.-U. Wu, H.-H. Yu and H. Tian, Chem. Commun., 2012, 48, 1784 RSC; (c) P. Das, A. Ghosh, M. Kesharwani, V. Ramu, B. Ganguly and A. Das, Eur. J. Inorg. Chem., 2011, 20, 3050 CrossRef.
- (a) C.-R. Lohani, J.-M. Kim, S.-Y. Chung, J. Yoon and K.-H. Lee, Analyst, 2010, 135, 2079 RSC; (b) X.-M. Huang, Z.-Q. Guo, W.-H. Zhu, Y.-S. Xie and H. Tian, Chem. Commun., 2008, 5143 RSC.
- (a) L.-G. Pathberiya, N. Barlow, T. Nguyen, B. Graham and K.-L. Tuck, Tetrahedron, 2012, 68, 9435 CrossRef CAS PubMed; (b) R. Josesh, J.-P. Chinta and C.-P. Rao, Inorg. Chem., 2011, 50, 7050 CrossRef PubMed; (c) F.-H. Huang, C. Cheng and G.-Q. Teng, J. Org. Chem., 2012, 77, 11405 CrossRef CAS PubMed.
- (a) J.-H. Lee, A.-R. Jeong, J.-H. Jung, C.-M. Park and J.-I. Hong, J. Org. Chem., 2011, 76, 417 CrossRef CAS PubMed; (b) H.-T. Ngo, X.-J. Liu and K.-A. Jolliffe, Chem. Soc. Rev., 2012, 41, 4928 RSC.
- (a) B.-K. Datta, S. Mukherjee, C. Kar, A. Kojima and G. Das, Anal. Chem., 2013, 85, 8369 CrossRef CAS PubMed; (b) R. Joseph, J.-P. Chinta and C.-P. Rao, J. Org. Chem., 2010, 75, 3387 CrossRef CAS PubMed.
- (a) A. Ojida, Y. Mito-oka, K. Sada and I. Hamachi, J. Am. Chem. Soc., 2004, 126, 2454 CrossRef CAS PubMed; (b) A. Ojida, S. Park, Y. Mito-oka and I. Hamachi, Tetrahedron Lett., 2002, 43, 6193 CrossRef CAS.
- (a) J.-F. Zhang, S. Kim, J.-H. Han, S.-J. Lee, T. Pradhan, Y.-C. Qian, S.-J. Lee, C.-H. Kang and J.-S. Kim, Org. Lett., 2011, 13, 5294 CrossRef CAS PubMed; (b) A.-J. Moro, P.-J. Cywinski, S. Korsten and G.-J. Mohr, Chem. Commun., 2010, 46, 1085 RSC.
- (a) A.-S. Rao, D. Kim, H. Nam, H. Jo, K.-H. Kim, C. Ban and K.-H. Ahn, Chem. Commun., 2012, 48, 3206 RSC; (b) A.-S. Rao, S.-S. Singha, W. Choi and K.-H. Ahn, Org. Biomol. Chem., 2012, 10, 8410 RSC.
- (a) X. Chen, M.-J. Jou and J. Yoon, Org. Lett., 2009, 11, 2181 CrossRef CAS PubMed; (b) H.-N. Lee, Z. Xu, S.-K. Kim, K.-M.-K. Swamy, Y. Kim, S.-J. Kim and J. Yoon, J. Am. Chem. Soc., 2007, 129, 3828 CrossRef CAS PubMed.
- (a) L. Xue, H.-H. Wang, X.-J. Wang and H. Jiang, Inorg. Chem., 2008, 47, 4310 CrossRef CAS PubMed; (b) B.-A. Wang, S. Friedle and S.-J. Lippard, Inorg. Chem., 2009, 48, 7009 CrossRef PubMed; (c) Z. Xu, D.-R. Spring and J. Yoon, Chem.–Asian. J., 2011, 6, 2114 CrossRef CAS PubMed.
- (a) H.-W. Rhee, C.-R. Lee, S.-H. Cho, M.-R. Song, M. Cashell, H.-E. Choy, Y.-J. Seok and J.-I. Hong, J. Am. Chem. Soc., 2008, 130, 784 CrossRef CAS PubMed; (b) Z.-H. Zeng, A.-A.-J. Torriew, A.-M. Bond and L. Spiccia, Chem.–Eur. J., 2010, 16, 9154 CrossRef CAS PubMed.
- (a) D.-H. Lee, S.-Y. Kim and J.-I. Hong, Angew. Chem., Int. Ed., 2004, 43, 4777 CrossRef CAS PubMed; (b) S.-Y. Kim and J.-I. Hong, Bull. Korean Chem. Soc., 2010, 31, 716 CrossRef CAS; (c) B. Roy, A.-S. Rao and K.-H. Ahn, Org. Biomol. Chem., 2011, 9, 7774 RSC.
- (a) P. Das, S. Bhattacharya, S. Mishra and A. Das, Chem. Commun., 2011, 47, 8118 RSC; (b) D.-H. Lee and J.-I. Hong, Bull. Korean Chem. Soc., 2008, 29, 497 CrossRef CAS.
- S.-J. Yang, G.-Q. Feng and N.-H. Williams, Org. Biomol. Chem., 2012, 10, 5606 CAS.
- G.-K. Walkup, S.-C. Burdette, S.-J. Lippard and R.-Y. Tsien, J. Am. Chem. Soc., 2000, 122, 5644 CrossRef CAS.
- H.-N. Lee, Z. Xu, S.-K. Kim, K.-M.-K. Swamy, Y. Kim, S.-J. Kim and J. Yoon, J. Am. Chem. Soc., 2007, 129, 3828 CrossRef CAS PubMed.
- X. Chen, Z. Huang, S.-Y. Chen, K. Li, X.-Q. Yu and L. Pu, J. Am. Chem. Soc., 2010, 132, 7297 CrossRef CAS PubMed.
- B. Roy, A.-S. Rao and K.-H. Ahn, Org. Biomol. Chem., 2011, 9, 7774 CAS.
- (a) M.-Q. Wang, K. Li, J.-T. Hou, M.-Y. Wu, Z. Huang and X.-Q. Yu, J. Org. Chem., 2012, 77, 8350 CrossRef CAS PubMed; (b) S.-Y. Jiao, L.-L. Peng, K. Li, Y.-M. Xie, M. Z. Ao, X. Wang and X.-Q. Yu, Analyst, 2013, 138, 5762 RSC.
- J.-H. Lee, J. Park, M.-S. Lah, J. Chin and J.-I. Hong, Org. Lett., 2007, 9, 3729 CrossRef CAS PubMed.
- M. Shortreed, R. Kopelman, M. Kuhn and B. Hoyland, Anal. Chem., 1996, 68, 1414 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: NMR and HRMS spectras. See DOI: 10.1039/c4an01615g |
| This journal is © The Royal Society of Chemistry 2015 |