
A trial proteomics fingerprint analysis of HepaRG cells by FD-LC-MS/MS†
Katsunori
Nakata
ab,
Tomoko
Ichibangase
a,
Ryoichi
Saitoh
b,
Masaki
Ishigai
b and
Kazuhiro
Imai
*a
aLaboratory of Proteomics Analysis, Research Institute of Pharmaceutical Sciences, Musashino University, 1-1-20 Shinmachi, Nishitokyo-shi, Tokyo 202-8585, Japan. E-mail: k-imai@musashino-u.ac.jp; Fax: +81-42-468-9787; Tel: +81-42-468-9787
bResearch Division, Chugai Pharmaceutical Co., Ltd., 1-135 Komakado, Gotemba, Shizuoka 412-8513, Japan
First published on 10th November 2014
Abstract
A proteomics profile analysis was performed on a human hepatocyte carcinoma cell line (HepaRG) by using the FD-LC-MS/MS method. One hundred and fifty-eight proteins were newly identified for the first time of which 10 were found to be specific to human hepatocytes. These proteins are a “proteomics fingerprint” that can be used to characterize HepaRG cells.
HepaRG is a human hepatocellular carcinoma cell line1 composed of hepatocyte- and biliary-like cells. Differentiated HepaRG is the only accepted cell line that is capable of being infected in vitro by the hepatitis B virus (HBV) in a similar way to cultured primary hepatocytes.1 In addition, HepaRG retains a drug metabolic capacity comparable to that of primary human hepatocytes.2 Therefore, HepaRG is regarded as a model for drug metabolism studies.3 Several differential proteomics analyses on HepaRG, including hepatitis B virus-infected vs. non-infected cells4 and on membrane proteins in differentiated vs. non-differentiated cells,5,6 have identified 44,4 108–118 (ref. 5) and 210–307 proteins6 as differentiated proteins. However, there has not been a comprehensive profile analysis of the proteins expressed in HepaRG cells that characterize mammalian hepatocytes from the standpoint of the proteome.
Proteomics analysis is usually performed by using conventional methods, such as two-dimensional polyacrylamide gel electrophoresis (2D-PAGE)7,8 and shotgun proteomics analysis using LC/MS/MS.9–11 However, these methods require pretreatment steps, such as precipitation, cleanup with the extraction column, or enzymatic protein digestion before LC/MS/MS (shotgun proteomics analysis), which tend to remove proteins, thereby resulting in low sensitivity and low reproducibility, and also imprecise information on the expressed proteins.12
In contrast, the fluorogenic derivatization (FD)-LC-MS/MS method12,13 is a quantitative proteomic analysis method that does not require any sample pretreatment procedure; proteins are derivatized by using fluorogenic reagents such as 7-chloro-N-[2-(dimethylamino)ethyl]-2,1,3-benzoxadiazole-4-sulfonamide (DAABD-Cl) and derivatized proteins are separated by HPLC. This is followed by quantification based on the peak height, fractionation of the protein peaks, enzymatic digestion of the isolated proteins, and the final identification of the proteins using HPLC and tandem MS. Because this approach has been used for differential proteomics analysis in many biological samples, such as human breast and colorectal cancer cells, and mouse liver,12–15 FD-LC-MS/MS should be suitable for a profile proteomics analysis of HepaRG cells. Such analysis should identify the expressed proteins that have been lost during the pretreatment steps of conventional proteomics analysis methods. Therefore, the FD-LC-MS/MS method was used in the present study. To separate the DAABD-labeled proteins, we tried using columns of core–shell particles (Aeris WIDEPORE XB, Phenomenex, Torrance, CA, USA)16,17 that have recently become commercially available.
Kirkland et al. developed 2.7 μm columns of core–shell particles composed of a 1.7 μm solid core enriched by a 0.50 μm porous layer. Compared with a column with totally porous particles of less than 2 μm, the 2.7 μm columns showed equally efficient separation with much lower column pressure drops18 and were used for the separation of low-molecular-weight compounds, including drugs. A column of this wide-pore type of core–shell material recently became commercially available (Aeris WIDEPORE XB)16,17,19 and has been shown to be highly efficient for the separation of several proteins and monoclonal antibodies. Because there have been no reports on the separation of proteins in bio-samples by a single analysis, we used the Aeris WIDEPORE XB-C8 column (250 × 4.6 mm i.d.; Phenomenex) to achieve efficient separation of the expressed proteins in HepaRG cells. The composition of the eluents was referenced to data taken from the separation of proteins of human hepatocytes on a column of non-porous materials (Presto FF-C18, 250 × 4.6 mm i.d.; Imtakt, Kyoto, Japan).14,15
We investigated the separation efficiency under various flow rates (0.10–0.60 mL min−1) (ESI, Fig. 1†). The number of peaks between the first and act peaks (identified as actin, as shown in the ESI, Fig. 1†) in every chromatogram was compared to evaluate the separation efficiency. The number of peaks was 290, 304, 299, 306, 285, and 269 at flow rates 0.10, 0.15, 0.20, 0.30, 0.40, and 0.60 mL min−1, respectively. The lower flow rates (0.10–0.30 mL min−1) showed higher separation efficiency (290–306 peaks) as compared with the higher flow rates (0.40–0.60 mL min−1) at a column temperature of 60 °C. These results agree with a trend reported by Fekete et al. when using the standard insulin on an Aeris WIDEPORE XB-C18 column.16 Moreover, the peak height increased as the flow rate decreased (ESI, Fig. 1†). This observation suggests that lower flow rates show higher sensitivity than higher flow rates. The same trend was also observed when the column temperature was set at 50 °C. The numbers of peaks were 327, 315, and 311 at flow rates 0.10, 0.15, and 0.20 mL min−1, respectively. Second, as for the column temperatures (30–70 °C) (ESI, Fig. 2†), higher column temperatures (50, 60, and 70 °C) showed higher separation efficiency (311, 299, and 302 peaks, respectively) than the lower column temperatures (208 and 289 peaks for 30 and 40 °C, respectively) at a flow rate of 0.20 mL min−1. Taking these results into consideration and using a single column to separate the HepaRG extract, a flow rate of 0.10 mL min−1 and a column temperature of 50 °C were considered optimal.
In accordance with previous reports,20,21 when the column length was varied (250 mm, 250 + 250 mm, and 250 + 250 + 250 mm) at the flow rate of 0.20 mL min−1 and the column temperature was set at 60 °C, the number of peaks increased as the column length increased (299, 334, and 350 peaks, respectively, data not shown). Furthermore, the number of peaks (350) at the flow rate of 0.20 mL min−1 and at 60 °C was higher than that (328) at the flow rate of 0.10 mL min−1 and a column temperature of 50 °C, with a column length of 750 mm. Finally, for the proteomics profiling analysis of the HepaRG cell extract, a 750 mm connecting column was selected with the column temperature set at 60 °C and the flow rate was changed from 0.4 to 0.2 mL min−1 between 20 and 25 min to shorten the analysis time. The injection volume was changed to 50 μL (20 μg protein) to increase the number of proteins, as shown in Fig. 1.
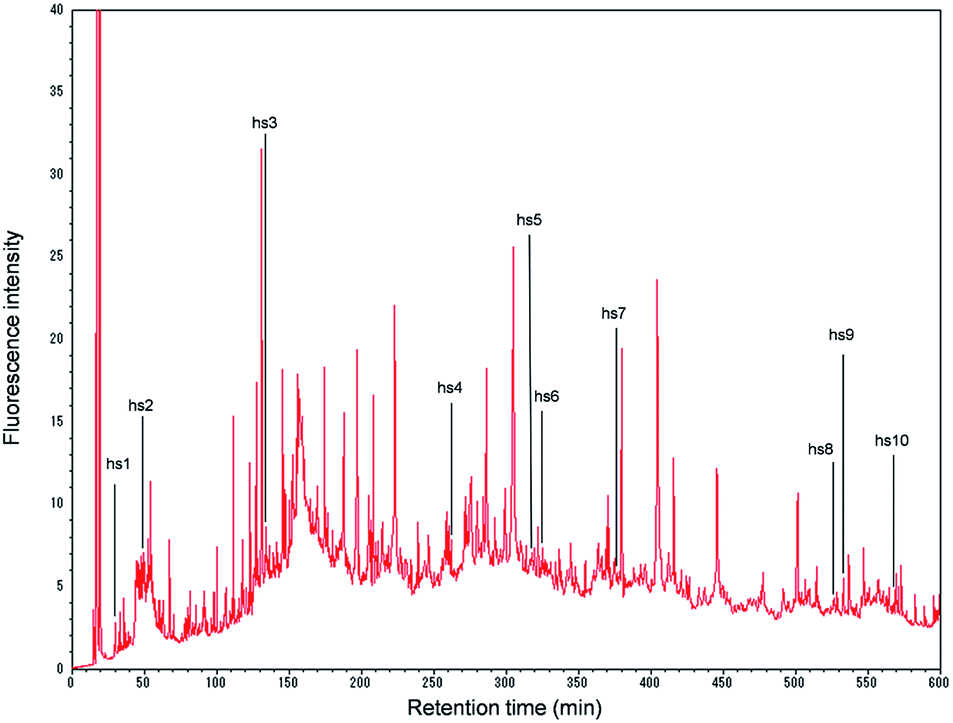 | ||
| Fig. 1 Chromatogram of the derivatized proteins obtained from the HepaRG extract (20 μg proteins) (0–600 min). The abbreviations hs1–hs10 indicate the hepatocyte-specific proteins found in the HepaRG extract. hs1, arginase-1; hs2, haptoglobin; hs3, serotransferrin; hs4, phosphoenolpyruvate carboxykinase [GTP]; hs5, glyoxylate reductase/hydroxypyruvate reductase; hs6, carbamoyl-phosphate synthase [ammonia]; hs7, delta-1-pyrroline-5-carboxylate dehydrogenase; hs8, acyl-coenzyme A synthetase ACSM2B; hs9, apolipoprotein-L2; and hs10, liver carboxylesterase 1. | ||
On the chromatogram, 532 peaks appeared during the 10 h analysis (Fig. 1). When each peak was fractionated, and the isolated proteins were enzymatically digested and subjected to nano-LC/MS/MS, the number of identified proteins was 254.
The proteins were classified using DAVID (http://david.abcc.ncifcrf.gov/) software, according to the Gene Ontology terms for their cellular components (ESI, Table 1†).22 The results showed that the proteins originated from the mitochondrion (n = 73; 28.7%), endoplasmic reticulum (n = 36; 14.2%), cytoskeleton (n = 35; 13.8%), ribosome (n = 20; 7.9%), nucleolus (n = 17; 6.7%), and cytosol (n = 78; 30.7%).
As summarized in the ESI, Table 2,† 158 expressed proteins were shown for the first time that have not been found in previous papers in which HepaRG cells were analyzed for hepatitis B virus infections vs. non-infections by the 2D-PAGE proteomics method4 or for membrane proteins expressed on differentiated vs. non-differentiated cells by the shotgun proteomics method.5,6 These proteins may be proteins that were removed by precipitation or other cleanup procedures employed in the shotgun or 2D-PAGE analysis methods. The number of proteins recovered in the present experiment that were localized in the mitochondrion, endoplasmic reticulum, cytoskeleton, ribosome, nucleolus, and cytosol was 49, 22, 15, 13, 11, and 34, respectively. Division of these numbers by the identified proteins in each component (ESI, Table 1†) gives the percentage of recovered proteins for each component as 67.1, 61.1, 42.9, 65.0, 64.7, and 43.6% respectively, showing that the FD-LC-MS/MS method identifies more proteins in each component than the conventional methods. Furthermore, it should be stressed that the present method clearly identifies the proteins based on protein isolation, and the identity is not deduced from the tremendous variety of peptide fragments from large amounts of mixed proteins, as in the case of shotgun proteomics analysis.
Of the 254 proteins identified in the present experiment, 10 proteins were found to be specific to human hepatocytes, according to the classification by Slany et al.8 (Fig. 1 and ESI, Table 1†): hs1, arginase-1; hs2, haptoglobin; hs3, serotransferrin; hs4, phosphoenolpyruvate carboxykinase [GTP]; hs5, glyoxylate reductase/hydroxypyruvate reductase; hs6, carbamoyl-phosphate synthase [ammonia]; hs7, delta-1-pyrroline-5-carboxylate dehydrogenase; hs8, acyl-coenzyme A synthetase ACSM2B; hs9, apolipoprotein-L2; and hs10, liver carboxylesterase 1. These proteins may be suitable for characterizing HepaRG cells (in what might be called a “proteomics fingerprint”) based on the proteomics profile analysis of human cell lines. Further accumulation of these data for many cells should facilitate the construction of a comprehensive “proteomics fingerprint” for mammalian cells.
In the ESI, Table 1,† a single protein name is provided in general, but two or three proteins names are also listed for each isolated peak. This is because some of the peaks were not completely separated from the neighboring protein peaks. Therefore, an effort to achieve more efficient separation would be required for proteomics profile analysis; for example, by using a longer column with a wider inner diameter and a longer analysis time such as 20–30 h, as suggested previously using a column of non-porous particles.21 If more efficient separation can be achieved, the FD-LC-MS/MS approach should be a more powerful method of analyzing the proteomic profiles of various cell species and types. Although, as mentioned above, the present data were insufficient to precisely characterize the HepaRG cells; this trial study is a milestone in obtaining a “proteomics fingerprint” of mammalian cells.
In summary, the present trial study showed that the FD-LC-MS/MS method is applicable to characterizing cells in a profile proteomics analysis of HepaRG cells, because the 10 proteins specific to human hepatocytes were identified in the cells. Although only one single trial study has been achieved, if further profile data for other cells are accumulated, it should allow us to develop a “proteomics fingerprint” for mammalian cells.
Notes and references
- P. Gripon, S. Rumin, S. Urban, J. Le Seyec, D. Glaise, I. Cannie, C. Guyomard, J. Lucas, C. Trepo and C. Guguen-Guillouzo, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15655–15660 CrossRef CAS PubMed.
- C. Aninat, A. Piton, D. Glaise, T. Le Charpentier, S. Langouet, F. Morel, C. Guguen-Guillouzo and A. Guillouzo, Drug Metab. Dispos., 2006, 34, 75–83 CrossRef CAS PubMed.
- S. Antherieu, C. Chesne, R. Li, C. Guguen-Guillouzo and A. Guillouzo, Toxicol. In Vitro, 2012, 26, 1278–1285 CrossRef CAS PubMed.
- R. Narayan, B. Gangadharan, O. Hantz, R. Antrobus, A. García, R. a. Dwek and N. Zitzmann, J. Proteome Res., 2009, 8, 118–122 CrossRef CAS PubMed.
- I. Sokolowska, C. Dorobantu, A. G. Woods, A. Macovei, N. Branza-Nichita and C. C. Darie, Proteome Sci., 2012, 10, 47 CrossRef CAS PubMed.
- C. Petrareanu, A. Macovei, I. Sokolowska, A. G. Woods, C. Lazar, G. L. Radu, C. C. Darie and N. Branza-Nichita, PLoS One, 2013, 8, e71859 CAS.
- G. L. Corthals, V. C. Wasinger, D. F. Hochstrasser and J. C. Sanchez, Electrophoresis, 2000, 21, 1104–1115 CrossRef CAS.
- A. Slany, V. J. Haudek, H. Zwickl, N. C. Gundacker, M. Grusch, T. S. Weiss, K. Seir, C. Rodgarkia-Dara, C. Hellerbrand and C. Gerner, J. Proteome Res., 2010, 9, 6–21 CrossRef CAS PubMed.
- C.-L. Lee, H.-H. Hsiao, C.-W. Lin, S.-P. Wu, S.-Y. Huang, C.-Y. Wu, A. H.-J. Wang and K.-H. Khoo, Proteomics, 2003, 3, 2472–2486 CrossRef CAS PubMed.
- Y. Zhang, B. R. Fonslow, B. Shan, M. C. Baek and J. R. Yates 3rd, Chem. Rev., 2013, 113, 2343–2394 CrossRef CAS PubMed.
- S. R. Langley, J. Dwyer, I. Drozdov, X. Yin and M. Mayr, Cardiovasc. Res., 2013, 97, 612–622 CrossRef CAS PubMed.
- K. Imai, A. Koshiyama and K. Nakata, Biomed. Chromatogr., 2011, 25, 59–64 CrossRef CAS PubMed.
- T. Ichibangase and K. Imai, Biol. Pharm. Bull., 2012, 35, 1393–1400 CAS.
- K. Nakata, R. Saitoh, J. Amano, A. Koshiyama, T. Ichibangase, N. Murao, K. Ohta, Y. Aso, M. Ishigai and K. Imai, Cytokines, 2012, 59, 317–323 CrossRef CAS PubMed.
- K. Nakata, R. Saitoh, J. Amano, T. Ichibangase, M. Ishigai and K. Imai, Biomed. Chromatogr., 2014, 28, 742–750 CrossRef CAS PubMed.
- S. Fekete, R. Berky, J. Fekete, J.-L. Veuthey and D. Guillarme, J. Chromatogr. A, 2012, 1236, 177–188 CrossRef CAS PubMed.
- S. Fekete, R. Berky, J. Fekete, J.-L. Veuthey and D. Guillarme, J. Chromatogr. A, 2012, 1252, 90–103 CrossRef CAS PubMed.
- J. J. Kirkland, T. J. Langlois and J. J. Destefano, Am. Lab., 2007, 39, 18–21 CAS.
- C. Sanchez and T. Farkas, Am. Lab., 2012, 44, 11–14 CAS.
- A. Koshiyama, T. Ichibangase, K. Moriya, K. Koike, I. Yazawa and K. Imai, J. Chromatogr. A, 2011, 1218, 3447–3452 CrossRef CAS PubMed.
- T. Ichibangase, K. Nakata and K. Imai, Biomed. Chromatogr., 2014, 28, 862–867 CrossRef CAS PubMed.
- D. W. Huang, B. T. Sherman and R. A. Lempicki, Nat. Protoc., 2009, 4, 44–57 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of experimental and FD-LC-MS/MS data. See DOI: 10.1039/c4an01434k |
| This journal is © The Royal Society of Chemistry 2015 |