
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhage display selected magnetite interacting Adhirons for shape controlled nanoparticle synthesis†
Andrea E.
Rawlings
a,
Jonathan P.
Bramble
a,
Anna A. S.
Tang
b,
Lori A.
Somner
a,
Amy E.
Monnington
c,
David J.
Cooke
c,
Michael J.
McPherson
b,
Darren C.
Tomlinson
b and
Sarah S.
Staniland
*a
aDepartment, of Chemistry, The University of Sheffield, Sheffield, UK. E-mail: s.s.staniland@sheffield.ac.uk
bFaculty of Biological, Sciences, The University of Leeds, Leeds, UK
cChemical and Biological Sciences, University of Huddersfield, Huddersfield, UK
First published on 30th June 2015
Abstract
Adhirons are robust, well expressing, peptide display scaffold proteins, developed as an effective alternative to traditional antibody binding proteins for highly specific molecular recognition applications. This paper reports for the first time the use of these versatile proteins for material binding, and as tools for controlling material synthesis on the nanoscale. A phage library of Adhirons, each displaying two variable binding loops, was screened to identify specific proteins able to interact with [100] faces of cubic magnetite nanoparticles. The selected variable regions display a strong preference for basic residues such as lysine. Molecular dynamics simulations of amino acid adsorption onto a [100] magnetite surface provides a rationale for these interactions, with the lowest adsorption energy observed with lysine. These proteins direct the shape of the forming nanoparticles towards a cubic morphology in room temperature magnetite precipitation reactions, in stark contrast to the high temperature, harsh reaction conditions currently used to produce cubic nanoparticles. These effects demonstrate the utility of the selected Adhirons as novel magnetite mineralization control agents using ambient aqueous conditions. The approach we outline with artificial protein scaffolds has the potential to develop into a toolkit of novel additives for wider nanomaterial fabrication.
Introduction
Magnetic nanoparticles (MNP) have become an area of intense research interest thanks to their wide ranging applications from high density magnetic data storage, biomedical diagnostics, and therapies.1,2 Current manufacture of MNP with sufficiently high uniformity required for the demands of these applications typically requires high temperatures and expensive reagents.2 Therefore the development of a synthetic methodology is needed that offers mild reaction conditions and a high degree of molecular control over the type of mineral formed, the size, and the morphology. Such alternative approaches for MNP production have looked to nature where magnetotactic bacteria are able to synthesise highly uniform crystals of magnetite, often achieving nanoarchitectures that are unobtainable under any current synthetic method. The bacteria use proteins which are able to strictly regulate the nucleation of the magnetite nanoparticle and then direct its growth along certain crystal faces. Proteins from these bacteria, such as Mms6 and MmsF, can be isolated and shown to exert control over particle production during in vitro magnetite precipitation reactions.3–5 Being able to utilise proteins to regulate mineral formation in synthetic reactions offers a possible route to the precision manufacture of MNP with a narrow size distribution and uniform morphology. However, the use of these proteins limits the synthetic particle production to the characteristics of those found in nature, and the proteins found in the magnetosomes are often difficult and expensive to produce, purify, and therefore utilise in these reactions, making them ill-suited to industrial MNP manufacture.Taking inspiration from these, and similar biomineralisation proteins, the process of biopanning has proven useful in uncovering novel peptides that can interact with specific mineral types.6,7 In this process, diverse libraries of random peptide sequences are expressed on the surface of bacterial cells (cell display) or bacteriophage (phage display) before application to a substrate.8 Through successive rounds of peptide binding, substrate washing, peptide elution and sequence amplification, peptides which show affinity to the substrate are selectively enriched. Binding peptides from these methods have now been identified for a range of materials and have been used for tethering of small molecules and enzymes, patterning surfaces, and also controlling the formation of materials.9,10 This latter use has allowed the synthesis of nanoparticles with characteristics specified by the interacting peptides. Well studied systems include peptides which demonstrate affinity for metals such as gold,11–13 platinum,14–16 palladium17 and titanium18 as well as oxides such as silica,19,20 quartz21 and titania.21,22
Recent advances have seen the development of material binding peptides which are constrained into cysteine bridged loops15,25 or by transposing the binding sequence into an alternative scaffold protein such as a protein cage.26 Constraining the binding peptides appears to alter the binding mode to the material it is selected for, offering different binding affinities and effects by limiting the conformational freedom of the peptide. By constraining peptide binders within a larger protein a number of benefits arise in that the system becomes suitable for large scale biological production, and is also easily amenable to genetic manipulation, thereby allowing the protein to be tailored for different applications. One scaffold protein capable of displaying variable binding loops is the recently developed Adhiron protein (commercially known as Affimers), Fig. 1.27 Based upon a consensus sequence of the phytocystatin fold,28 the Adhiron is engineered as a robust framework for the stable display of two variable nine amino acid long loop regions on the protein surface. Adhirons provide an ideal ‘protein scaffold’ as it is extremely robust and can be produced cost effectively in high quantities in bacterial expression systems. Here we report for the first time the selection by phage display of a suite of magnetite interacting Adhirons, MIA, which feature variable binding loops selected in situ for their affinity to cubic magnetite nanoparticles. These particles have specific planes, denoted [100], to which the MIA bind. These proteins possess the ability to alter the characteristics of magnetite mineralisation products in favour of cubic nanoparticles.
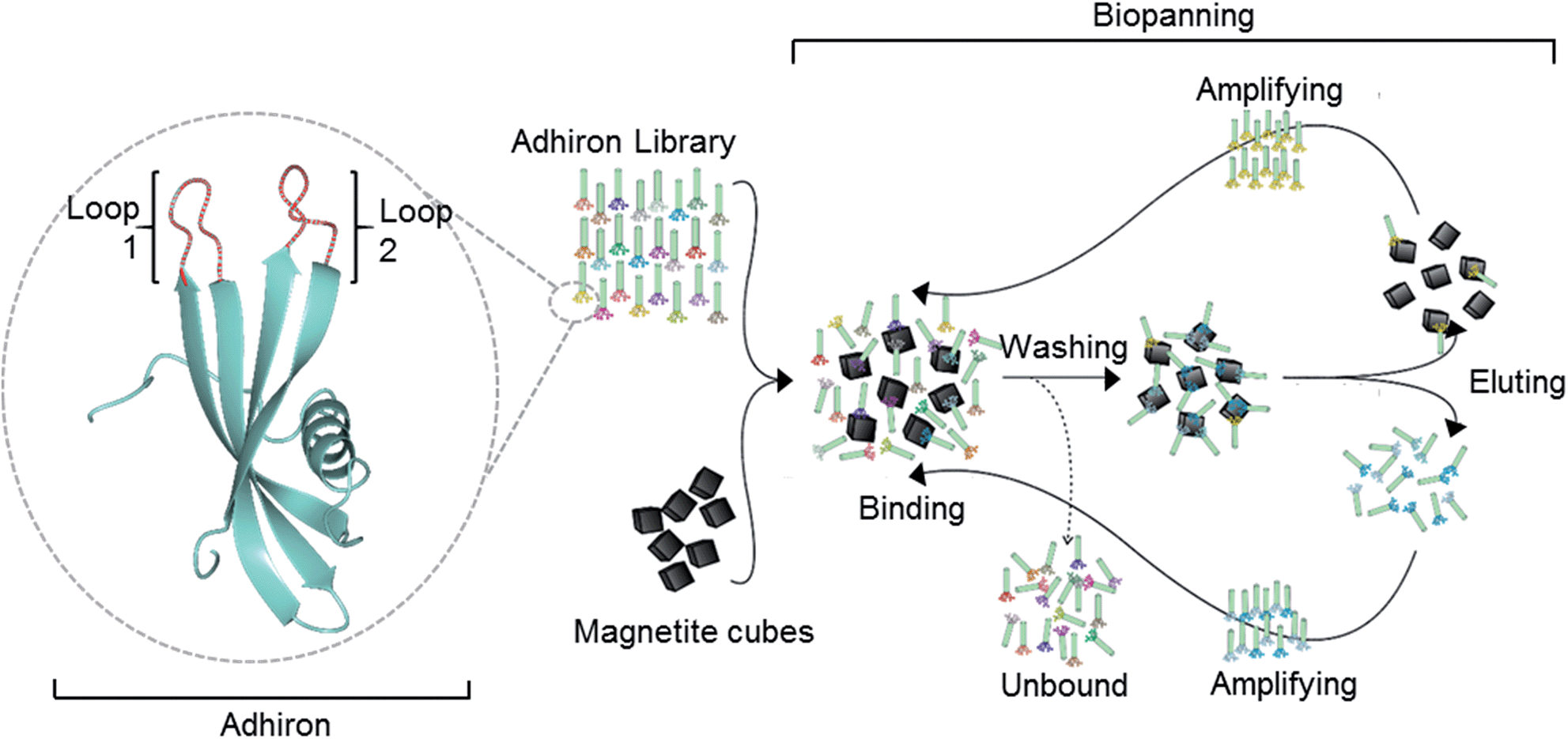 | ||
| Fig. 1 Overview of the magnetite interacting Adhiron selection process. A cartoon representation of the Adhiron scaffold is shown in green with the two variable loop regions depicted in red. Structure model of MIA-1 was generated using the Phyre2 (ref. 23) server and the image produced in CCP4mg.24 | ||
Results
Magnetite interacting Adhirons selected via phage display
The Adhiron scaffold is a well expressing protein that has a melting temperature in excess of 100 °C (ref. 27) making it well suited to exploitation in synthetic mineralisation reactions. The Adhiron library of over 1.3 × 1010 different sequences is fused to a truncated pIII minor coat protein of M13 bacteriophage.27 The phage library was exposed to clean cubic magnetite nanoparticles in the presence of a casein based blocking buffer. We anticipated that the highly charged magnetite surface would show a weak affinity to a wide range of proteins with compatible patches of surface charge, thereby diluting the eluted phage pool with large numbers of weak, non-specific binding sequences which have potentially hindered previous studies.29 By including the blocking protein in excess we aimed to mitigate this by selecting binders which were able to successfully compete for binding to the magnetite rather than the casein.Potentially the strength of association between some of the proteins and magnetite might mean that not all phage would be successfully eluted with the standard pH wash regime (acid then alkali), which has resulted in loss of the highest affinity library members in previous studies.30 However, after each round of selection, in addition to the eluted phage, the remaining MNP with any residual phage attached were also used to directly infect E. coli cells. This approach was adopted to try and maximise the recovery of binding sequences carried through for phage amplification and subsequent selection rounds despite the fact that the phage that remained associated with MNP may be sterically blocked from infecting. After three cycles of selection and amplification the phage pool from each panning round was assayed for magnetite binding using an alkaline phosphatase (AP) linked phage ELISA. The signal intensity increased with each panning round, showing that the phage pool was becoming increasingly enriched with magnetite binding proteins. As shown in Fig. 2, M13 phage alone or with non-magnetite selected Adhirons showed no significant binding to the MNP in the same assay.
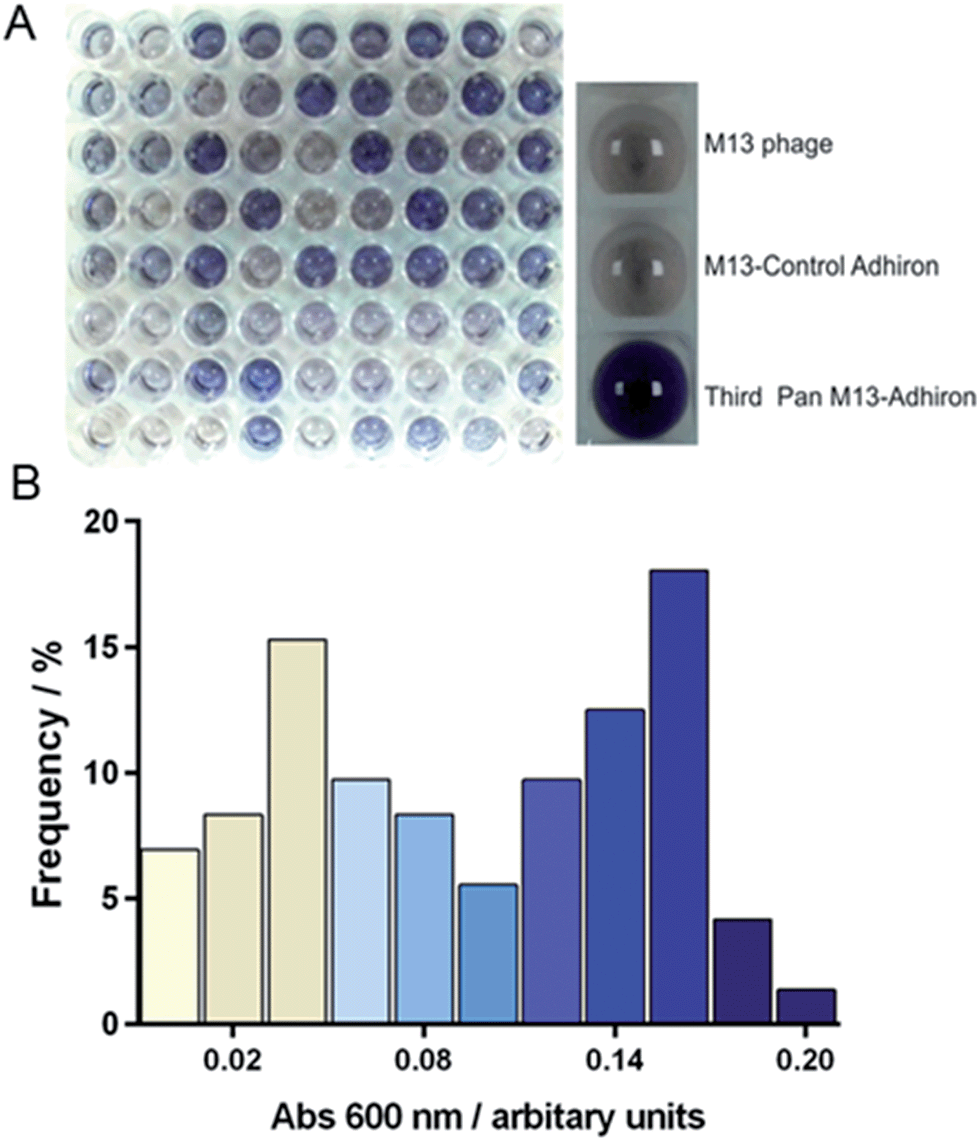 | ||
| Fig. 2 Phage ELISA of magnetite selected Adhirons. (A) Phage ELISA results where a blue colour-change indicates positive binding. The right panel shows ELISA controls of M13 phage, non-magnetite selected phage, and the selected phage pool after the final panning round. The left panel shows the range of binding of 72 clones from the final phage pool. (B) Frequency distribution showing the percentage of clones displaying a certain absorbance in the AP-Phage ELISA. Bar colouring corresponds to that generated in the assay. | ||
After the third and final panning round 72 individual library members were selected. These were amplified and assayed for binding to magnetite nanoparticles using an AP-phage ELISA. From the spectrum of intensities that we observed in the ELISA shown on Fig. 2, we deduced an apparent range of different binding capabilities were present within this subset. Generation of a frequency distribution showed that there were two dominant groupings of lower and higher affinity binders.
DNA sequencing reveals a preference for basic residues
The phagemid vectors bearing the Adhiron coding sequences of the 48 clones ranked highest in terms of signal intensity in the phage ELISA were extracted and sequenced. Alignment of the sequences revealed that approximately one third contained only the first of the variable binding loops, with the second loop completely absent. Previous detailed sequencing of the Adhiron library revealed that few library members were missing this binding loop,27 suggesting that the panning process has selectively enriched binding sequences which lack the second loop. We surmise that loop 1 may be the primary site of the magnetite interaction and the lack of a neighbouring second loop may promote improved binding. The remaining two thirds of sequences showed the expected two binding loops. We analysed the binding loop sequences with a weighted Kullback–Leibler logo plot, shown in Fig. 3.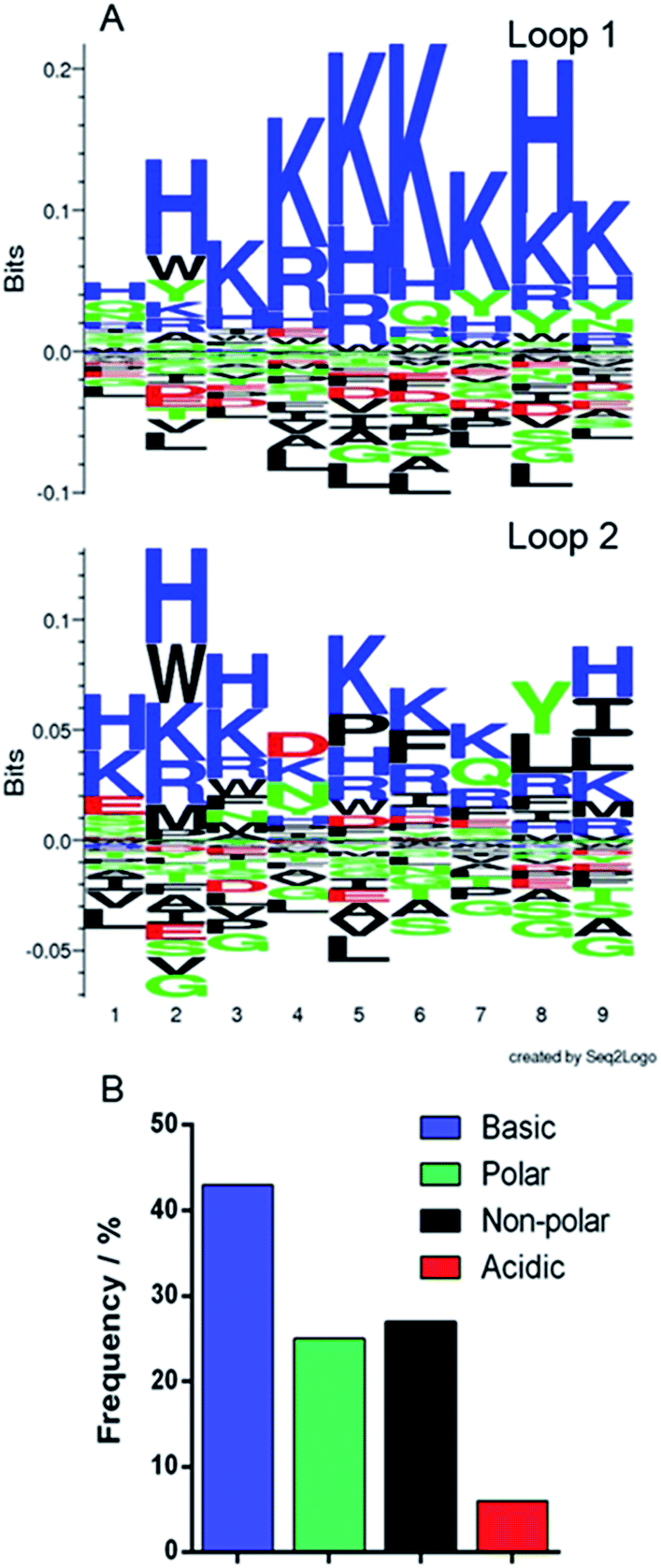 | ||
| Fig. 3 Sequence analysis of selected binding loops. (A) Seq2logo Kullback–Leibler plots31 of the amino acid sequences from the two binding loops. Residues in the positive area of the graph are enriched in the loops, and those in the negative area are depleted. (B) Frequency distribution of residue type in both binding loops. | ||
This revealed a strong preference for basic residues and in particular a predominance of lysine (26.4% of residues) and histidine (9.3%) in loop 1, and an almost total depletion of acidic amino acids. This is in contrast to the starting library which featured an approximately even distribution of amino acids.27 In loop 2 the sequences show a very similar but less stringent preference for basic residues with some hydrophobic and acidic residues reasonably well represented. This also suggests that loop 1 may be the principal magnetite interacting loop due to its more highly selective amino acid preference. The average isoelectric point, pI, of the loops also illustrates this with loop 1 being 10.12 and loop 2 being overall less basic, 8.74. The preference for basic residues (typically arginine) has been previously observed in cell display selected iron oxide binding peptides.32
Purification of magnetite interacting Adhirons
The coding regions of both the highest ranked (600 nm intensity) and median ranked MIA from the phage ELISA, were cloned into expression vectors encoding a C-terminal StrepII tag33 with a final terminating cysteine residue for later gold surface attachment. This plasmid allows high level expression of the selected MIA proteins, referred to from here on in as MIA-1 and MIA-2. A control Adhiron was also introduced into the expression vector. This sequence was not selected during the MNP panning rounds and should not therefore show specific interaction with magnetite. The sequences are provided in Table 1.Both MIA-1 and MIA-2, and the control Adhiron, were produced in E. coli and purified initially with Streptactin resin using the attached StrepII tag. SDS-PAGE analysis demonstrated high yield and good purity, Fig. 4A. However, the UV absorbance of MIA-1 and MIA-2 revealed a 260 nm/280 nm ratio of approximately 1.8. A ratio of greater than 1 is a common indicator that the protein sample also comprises DNA/RNA.34 This contamination appeared to be specific to MIA as the control-Adhiron was unaffected (ratio of 0.8) despite an identical purification procedure. We suspect that the high numbers of lysine residues in the variable loop regions were binding to endogenous cellular nucleic acids. Poly-lysine is a known binder of DNA35 and is well characterised.36 Intriguingly this indicates that Adhirons could potentially be raised against specific nucleic acid sequences for molecular biology applications. To mitigate this we applied the proteins to a Heparin resin which mimics the sugar phosphate backbone of DNA and has been used previously for DNA binding protein purification.37 After recovery from the Heparin resin via sodium chloride elution the UV absorbance profile matched that of the control-Adhiron, confirming that nucleic acids had been successfully removed.
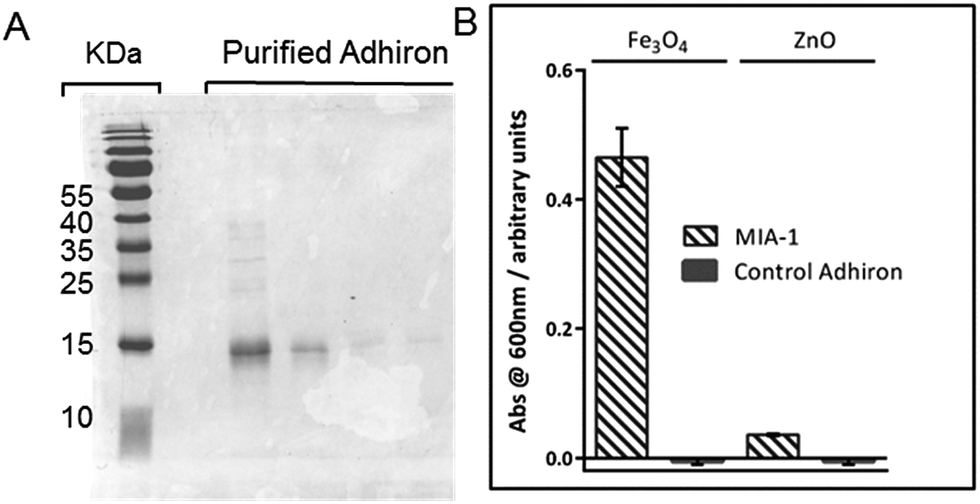 | ||
| Fig. 4 (A) Coomassie stained SDS-PAGE of purified Adhiron. (B) Binding assay of purified Adhirons with magnetite nanoparticle or zinc oxide nanoparticles. | ||
Assessing the specificity of interaction
To ascertain if the purified protein were capable of discriminating between different metal oxides we adapted the AP-Phage ELISA used earlier. In this modified assay the phage were substituted for the purified Adhiron and a primary antibody used which recognised the StrepII sequence of the purification tag. Equal amounts of magnetite and zinc oxide nanoparticles were screened for binding. The assay clearly showed that MIA-1 was able to bind to magnetite as demonstrated by the intense blue colour change, Fig. 4B. MIA-1 displayed a much weaker binding interaction with zinc oxide with a reduced absorbance at 600 nm. In both cases the control Adhiron recorded no colour change during the course of the assay indicating no binding had occurred to either material.Probing the interaction of MIA with magnetite nanoparticles
The interaction of MIA-1 as well as the control-Adhiron with both gold surfaces and magnetite nanoparticles was investigated with QCM, Fig. 5. For comparison we have also included MNP interaction in the absence of any Adhiron. During phase A the proteins are injected into the system and the negative frequency shift (Δf) corresponds to the formation of a protein monolayer via cysteine attachment to the gold surface. At B, the samples are rinsed in ultra-pure (MilliQ) water causing a step change in the resonance position. After returning to PBS some of the adsorbed protein has been removed by the rinsing step, explained by the slight increase in the resonant frequency. Uncoated magnetite nanoparticles are injected into the system during D. A further decrease in Δf would indicate an increase in mass on the surface due to adsorption of nanoparticles to the MIA. If MIA do not interact with MNP we would expect to see no change in frequency with addition of particles.
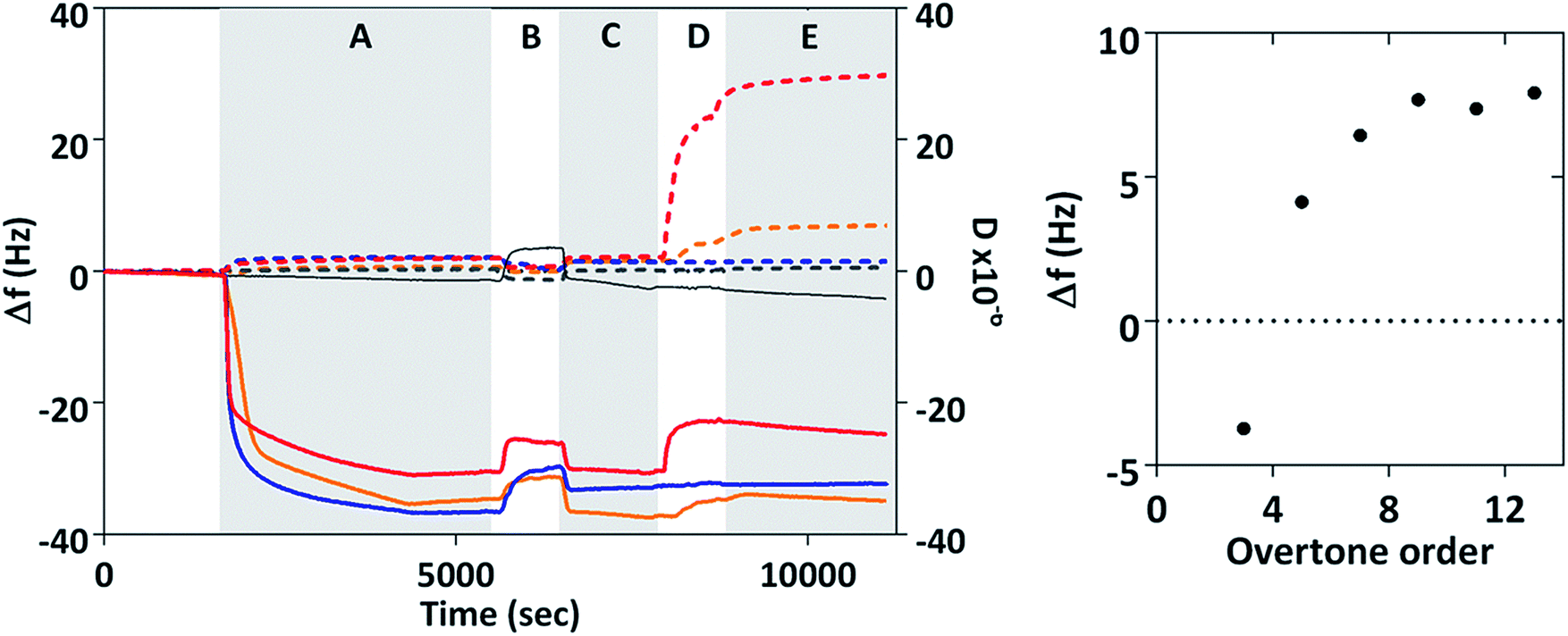 | ||
| Fig. 5 QCM analysis of MIA-nanoparticle interaction. Change in frequency, Δf, and dissipation, D. Dotted lines represent dissipation and solid lines frequency. Red lines are MIA-1, gold lines Mia-2, blue lines control Adhiron, and grey lines blank experiments with no Adhiron. Phase A is injection of protein into the system, B and C are rinses with ultrapure water and PBS respectively. MNP are injected into the system during D, and Phase E is final rinsing with PBS. Time averaged frequency changes for MIA-1 at different overtone orders are also shown. | ||
However, in our experiments we observed a positive frequency shift for both MIA and no frequency change for the control Adhiron. The absence of a frequency change for the control Adhiron is suggestive of non-binding and is consistent with our ELISA results (Fig. 4B). The positive frequency shifts of the MIA-MNP experiment are clearly different to the lack of interaction observed with the control Adhiron which implies both MIA do indeed interact with the applied MNP. Interestingly, when MIA are applied to the QCM sensor surface with DNA occupying the binding site then we see no frequency shift with the addition of MNP (ESI Fig. 5†). Taken together these results show that the positive Δf in Fig. 5 is due to an interaction between the binding loops and the nanoparticles.
Binding of colloidal particles and bacteria39 in QCM have been shown to give rise to this effect which can be explained with a coupled-resonance model40 whereby changes to the stiffness of the bond upon binding affect Δf, rather than simply the mass of the adsorbed material. In those cases the positive frequency shifts scale inversely with order of the overtone. We recorded the frequency shift caused by addition of MNP at different overtones and time averaged them after adsorption of MNP had ceased (phase E in Fig. 5). The average frequency shift was then plotted according to overtone order, n. Δf clearly varies from negative at n = 3 to positive for all higher overtone orders which is representative of a coupled resonance model39 showing interaction between magnetite nanoparticles and surface bound MIA. Interestingly MIA-1 and MIA-2 appear to have different binding affinities, with MIA-1 displaying the larger resonance shift.
Modelling interactions between amino acids and the [100] magnetite surface
To complement our studies and to gain an insight into the interactions at the atomistic level we have performed a series of molecular dynamics (MD) simulations using the DL_POLY classic code41 to further understand the interaction of a magnetite surface with amino acids. In these simulations a selection of individual amino acids with capped termini were brought from an infinite distance away towards a magnetite [100] surface in order to calculate the adsorption energy in water. The [100] planes are found on cubic nanoparticles, which were the dominant nanoparticle form used in the Adhiron selection process. The MD simulations illustrate that the lysine sidechain is able to form strong hydrogen bonds with the surface as well as through the peptide carbonyl whereas glutamic acid is only able to interact through the side chain oxygen as shown in Fig. 6.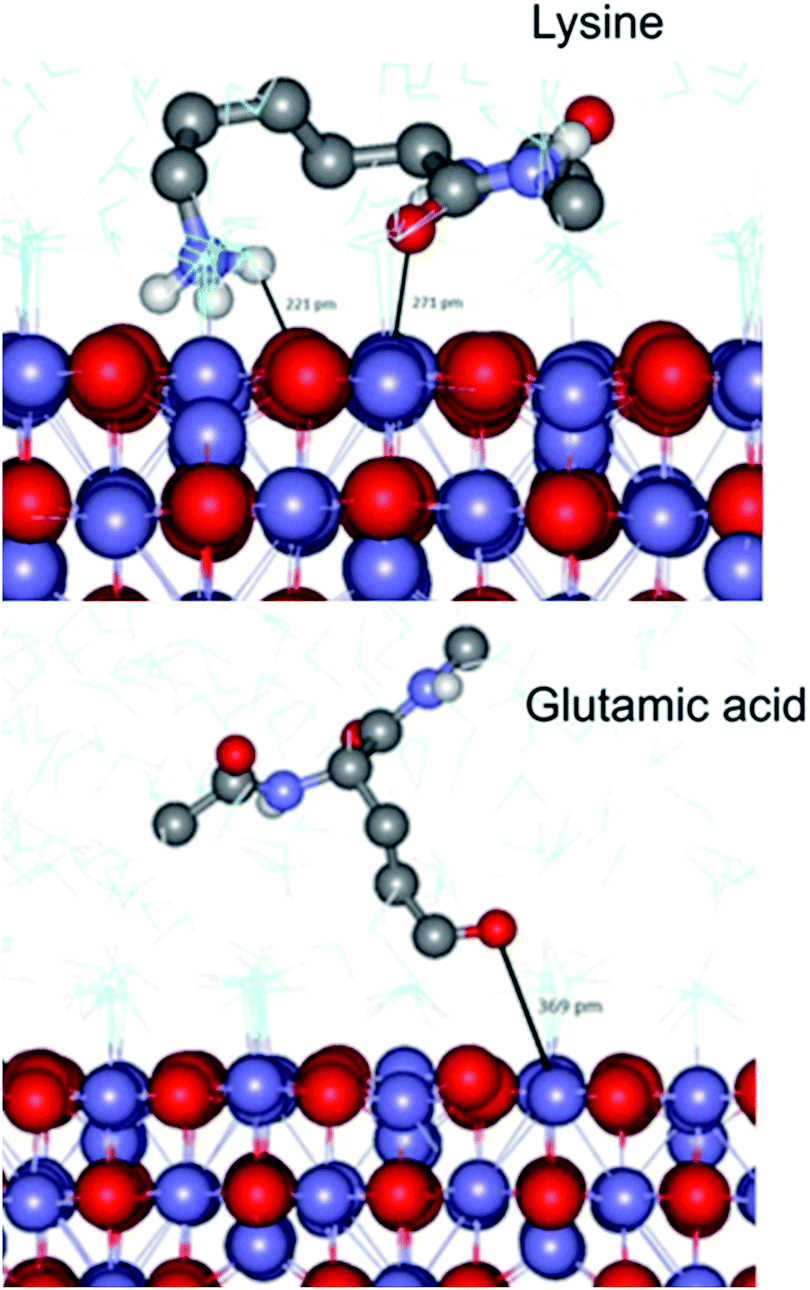 | ||
| Fig. 6 Simulated adsorption of amino acids on magnetite. A snap shot from the MD simulation of capped-lysine in the upper image, and capped-glutamic acid in the lower, on the [100] surface of magnetite. Hydrogen atoms bonded to carbon have been removed for clarity. | ||
In both cases however, the zwitter-ion is considered, which indicates a likely pH dependence upon these adsorption interactions. The simulations reveal a clear trend with lysine showing the lowest adsorption energy (most negative), and acidic amino acids proving the least likely to interact with the surface, listed in Table 2. These data match extremely well with the amino acid preferences we observe in the variable loop regions of MIA.
| Amino acid | Adsorption energy (kJ mol−1) | Frequency in binding loop (%) |
|---|---|---|
| Lysine (K) | −52 | 26.4 |
| Arginine (R) | −45 | 7.0 |
| Leucine (L) | −31 | 3.9 |
| Glutamic acid (G) | −3 | 3.3 |
| Aspartic acid (D) | 2 | 2.3 |
MIA-1 can influence particle morphology in synthetic magnetite precipitation reactions
To ascertain the effect that MIA have upon magnetite nanoparticle synthesis we included MIA-1 and MIA-2 in nanoparticle formation reactions. In the room temperature coprecipitation reaction a mixture of ferrous and ferric ions are precipitated by the addition of hydroxide ions. MIA-1 was included at a ratio of 50 μg protein per 10 ml reaction volume. The particles were visualised by transmission electron microscopy (TEM) and compared to nanoparticles prepared in an identical process but without the addition of protein. Grain size analysis of over 500 nanoparticles from each batch revealed little difference in size, with modest improvements in homogeneity in the MIA-1-MNP compared to the control, shown in Fig. 7D. However, we observed that the protein prepared particles had an overall more cubic and angular appearance when compared to the control particles, Fig. 7A and B. This is significant as the MIA were enriched using affinity to magnetite cubes and previous studies of peptide selection to materials indicate that sequences which bind preferentially to a certain crystal face are able to stabilise the formation of that same face during material synthesis.16 We therefore speculate that MIA-1 may show higher affinity for [100] faces and promote the formation of cubic particles. This effect was not observed for nanoparticles produced with either the addition of MIA-2 or the control Adhiron (ESI Fig. 7†). A partial oxidation of ferrous hydroxide reaction was also performed. This is a higher temperature reaction, which typically results in particles with an octahedral morphology. In these reactions we do not observe any effect from addition of MIA-1, with the control particles indistinguishable to those prepared with the protein (ESI Fig. 6†). It is likely that the extended high temperature incubation during these reactions causes denaturation of the Adhiron protein and an accompanying loss of conformational integrity to the binding loops. This indicates that the conformation and structure of the binding loop could be necessary for functional activity of the protein. To test this hypothesis further we prepared MNP by the room temperature coprecipitation method with addition of free flexible peptides comprising the loop sequences of MIA-1. When supplied at an equivalent molar ratio to the amount of MIA-1 we see no increased generation of cubic nanoparticles, and indeed the reaction appears to generate a more heterogeneous range of particles compared to even the Adhiron free particles (ESI Fig. 7†). This suggests that the constraint imposed by the Adhiron scaffold plays an important role in facilitating the activity of the binding loop sequences in cube production.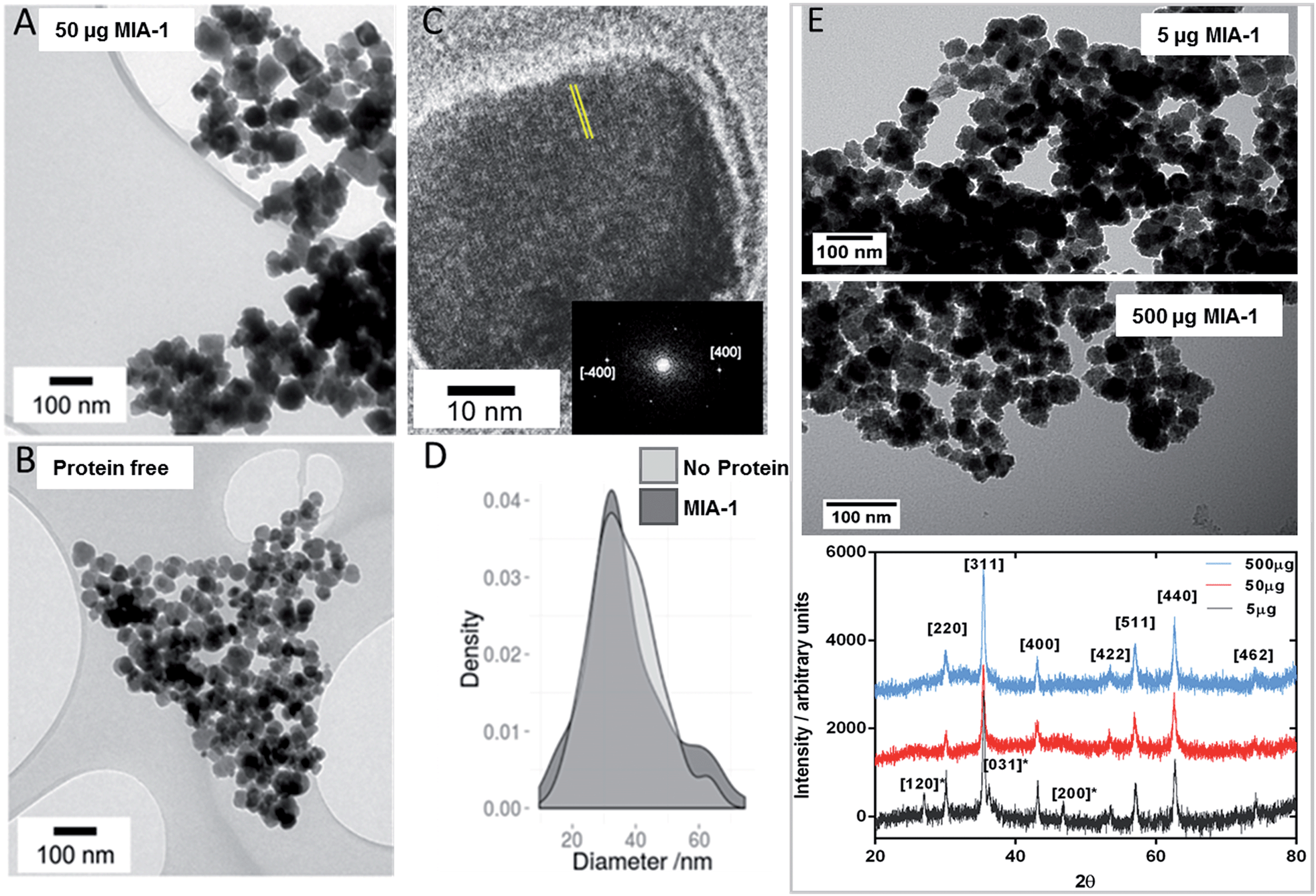 | ||
| Fig. 7 Transmission Electron Microscopy analysis of magnetite nanoparticles. (A) Particles prepared with MIA-1 at 50 μg per 10 ml reaction. (B) Particles prepared in the absence of MIA-1. (C) HRTEM analysis of a representative MIA-1 prepared MNP. A region of the lattice fringe is highlighted with yellow lines and the Fourier transform is shown inset with the [400] peaks identified. (D) Particle size analysis of MNP prepared with and without addition of MIA-1. (E) Transmission Electron Microscopy and X-ray diffraction analysis of magnetite nanoparticles at varying amounts of MIA-1. Powder XRD analysis of particles produced at 5 μg (grey), 50 μg (red) and 500 μg (blue) of MIA-1. Principal magnetite peaks have been assigned. Peaks present in the 5 μg sample assigned to lepidocrosite are denoted with an asterisk. | ||
To confirm the crystal faces present on the nanoparticles synthesised with MIA-1 we performed high resolution TEM (HRTEM). Analysis of the lattice fringes, and the corresponding Fourier transform, revealed a spacing of 0.20 nm which is consistent with the 0.20 nm lattice spacing of the [400] cubic plane of magnetite (Fig. 7C).
We wanted to understand the effect that different concentrations of MIA-1 had upon nanoparticle formation. We therefore used 5 μg and 500 μg of Adhiron per 10 ml magnetite coprecipitation (compared to the 50 μg we had used previously) and analysed the products by both TEM and powder X-ray diffraction (XRD), Fig. 7E. Addition of 5 μg produced particles, which appear similar to the control (protein free) nanoparticles, Fig. 7B. 500 μg appeared to produce particles with a rough, uneven surface. This may be because the production of iron oxides is especially sensitive to changes in reaction conditions which may have been caused by the high amount of organic material present. We have estimated the amount of Adhiron required to produce an approximate monolayer coverage of the MNP produced in our experiments. Based on our calculations (ESI†) we estimate that at 5 μg the amount of Adhiron is insufficient to interact with the total surface area of particles during formation. 50 μg provides approximate monolayer coverage, and 500 μg provides a 10 fold excess of protein. This estimation matches well with our TEM data which indicates that 50 μg of Adhiron produces the largest effect. XRD analysis, Fig. 7E, confirms that the samples are magnetite, however interestingly the MNP produced in the presence of 5 μg amount of MIA-1 reveals peaks assigned to the iron oxide hydroxide, lepidocrosite γ-FeO(OH). These peaks are not observed in samples produced with higher quantities of protein, suggesting that the addition of MIA-1 helps to prevent the formation of alternative iron oxide materials during the precipitation reaction.
Discussion
Using a phage display methodology we have selected a range of Adhiron proteins for their ability to bind to magnetite nanoparticles. By selecting binding sequences in situ within the confines of a robust scaffold protein we have established a novel approach to material binder development. Recent comparison between free peptide binders and antibody recognition of specific crystal planes shows that a protein-constrained binding loop has greater potential for discriminating between rigid material surface than a flexible peptide due to its fixed spatial structure and molecular rigidity.42 This is supported by other studies which indicate that a peptide will be interact with a material differently depending if it is free and flexible or constrained into a cyclic structure.25The magnetite selected Adhirons appear to have a clear interaction with magnetite nanoparticles in our QCM experiments, confirming that material binding is not dependent upon the presence of the pIII fusion protein, and is not an interaction with the scaffold itself, but a property derived from the variable loops. The positively charged surface of the MIA created by the lysine rich binding loops gives rise to a nucleic acid binding ability. This capability coupled with the proteins adsorption to magnetite nanoparticles gives these binding proteins a potential novel dual function which could have interesting applications.
The MIA we have analysed appear to share many of the properties of poly-lysine, which has been used previously as a magnetite coating material43 and is also a known DNA binding molecule.36 However, the conformationally constrained binding loops and presence of other amino acids offers a route to produce changes in the formed nanoparticles when included in synthetic precipitation reactions. MIA-1 is able to exert shape control in room temperature reactions; tipping the balance of the MNP reaction in favour of the specific cubic crystal morphologies that these sequences were enriched against. In higher temperature reactions or when free peptides are used we do not observe this activity, indicating that the constraint imposed on the binding loops by the protein scaffold may be necessary for function. The presence of MIA-1 also appears to reduce the occurrence of alternative iron oxides in these reactions.
A key advantage of constraining the binding sequences is that with more defined conformation, differences in binding can be more easily interpreted in terms of sequence variability rather than alternative conformation. Our analysis of a large number of binding sequences reveals a high level of consensus with a strong preference for the positively charged residues of lysine, histidine, and arginine. This matches particularly well with our molecular modelling of amino acid side chain interactions with a magnetite [100] surface which indicates that the most negative adsorption energy is observed with the amine group of lysine. The trend is in favour of interaction with positively charged amino acids at neutral pH with the negatively charged aspartate residue the least favoured. However, MIA-1 shows increased magnetite binding compared to MIA-2 in QCM-D and phage ELISA, and also displays morphology control during MNP synthesis. Yet MIA-1 contains fewer basic residues than MIA-2. Clearly the interplay between binding affinity and loop sequence is not a simple correlation to numbers of basic residues (although this does seem to be an important factor), but is most likely due to a subtle range of factors. A recent analysis of specific peptide binding to titania and silica surfaces revealed that positively charged residues are crucial for making the binding interactions, and that the balance of charged residues is important depending on the material used.44 In our case we hypothesise that due to the strong interaction between lysine and magnetite in our modelling studies, and the corresponding dominance of basic residues in the binder pool, lysine residues will be the main binding mediators. Their position and precise local topology within the binding loop are likely to govern how the residues interact with the specific arrangement of charges on the magnetite surface. The nuances of the MIA sequences and their implications for magnetite binding can only be unlocked through further in depth study of the entire binder pool.
Conclusions
Biological systems are able to synthesise materials with exquisite control down to the nanoscale into well-defined morphologies and nanoarchitectures using specialised biomineralisation proteins which have evolved over millennia. Mimicking this ambient, aqueous chemistry synthetically is a current challenge in nanomaterial development, and the use of screening strategies such as phage display and other combinatorial strategies offers a versatile fast-track route for achieving similar results in vitro. The manufacture of MNP with cubic faces currently requires high temperatures and a range of organic reagents. By using these cubic particles as a substrate for phage display we have selected a protein which demonstrates the ability to direct the production of particles with similar cubic morphology, thereby suggesting the potential to replicate an original extreme condition synthesis at room temperature and with mild reaction conditions. This highlights the power of combinatorial approaches in the development of novel nanomaterial green synthesis strategies. By screening for binding sequences within the robust Adhiron scaffold we are able to limit the conformational freedom of the binding loops and focus instead on sequence preference which is dominated by lysine and other basic residues. This is in contrast to other iron oxide screening results which have shown the difficulty of generating significant sequence consensus from flexible peptide libraries.29,32 We believe the Adhiron scaffold represents a promising new approach for precision manufacture of magnetite nanoparticles and that the method we outline could potentially be expanded to other materials for future nanomaterial synthesis.Acknowledgements
This work was supported by funding from the BBSRC (BB/H005412/2) and EPSRC (EP/L000202). Additional funding was from The University of Leeds via the Biomedical and Health Research Centre and The University of Huddersfield. The authors would like to thank J. Galloway for assistance with the QCM-D and figure drawing, C. Tiede for useful discussions during the phage display, and to A. Judge and J. Adams for help in preparing the cube MNP used in the Adhiron selection process. We also extend our thanks to S. Tsokov, C. Hill and the Sheffield Electron Miscroscopy unit for TEM and HRTEM, and to S. Spey for powder XRD data collection.References
- Q. A. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. Vander Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS PubMed.
- Y. Amemiya, A. Arakaki, S. S. Staniland, T. Tanaka and T. Matsunaga, Biomaterials, 2007, 28, 5381–5389 CrossRef CAS PubMed.
- A. Arakaki, J. Webb and T. Matsunaga, J. Biol. Chem., 2003, 278, 8745–8750 CrossRef CAS PubMed.
- A. E. Rawlings, J. P. Bramble, R. Walker, J. Bain, J. M. Galloway and S. S. Staniland, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 16094–16099 CrossRef CAS PubMed.
- C. Tamerler, S. Dincer, D. Heidel, N. Karaguer and M. Sarikaya, Biomicroelectromechanical Systems (Biomems), 2003, vol. 773, pp. 101–110 Search PubMed.
- U. O. Seker and H. V. Demir, Molecules, 2011, 16, 1426–1451 CrossRef CAS PubMed.
- T. Bratkovic, Cell. Mol. Life Sci., 2010, 67, 749–767 CrossRef CAS PubMed.
- K. I. Sano and K. Shiba, J. Am. Chem. Soc., 2003, 125, 14234–14235 CrossRef CAS PubMed.
- C. R. So, J. L. Kulp, E. E. Oren, H. Zareie, C. Tamerler, J. S. Evans and M. Sarikaya, ACS Nano, 2009, 3, 1525–1531 CrossRef CAS PubMed.
- S. Brown, Nat. Biotechnol., 1997, 15, 269–272 CrossRef CAS PubMed.
- J. M. Slocik, M. O. Stone and R. R. Naik, Small, 2005, 1, 1048–1052 CrossRef CAS PubMed.
- J. Kim, Y. Rheem, B. Yoo, Y. Chong, K. N. Bozhilov, D. Kim, M. J. Sadowsky, H. G. Hur and N. V. Myung, in Acta Biomater, Acta Materialia Inc. Published by Elsevier Ltd, England, 2010, vol. 6, pp. 2681–2689 Search PubMed.
- Y. Li, G. P. Whyburn and Y. Huang, J. Am. Chem. Soc., 2009, 131, 15998–15999 CrossRef CAS PubMed.
- U. O. Seker, B. Wilson, S. Dincer, I. W. Kim, E. E. Oren, J. S. Evans, C. Tamerler and M. Sarikaya, Langmuir, 2007, 23, 7895–7900 CrossRef CAS PubMed.
- C. Y. Chiu, Y. J. Li, L. Y. Ruan, X. C. Ye, C. B. Murray and Y. Huang, Nat. Chem., 2011, 3, 393–399 CrossRef CAS PubMed.
- D. B. Pacardo, M. Sethi, S. E. Jones, R. R. Naik and M. R. Knecht, ACS Nano, 2009, 3, 1288–1296 CrossRef CAS PubMed.
- Y. Liu, J. Mao, B. Zhou, W. Wei and S. Gong, J. Mater. Sci.: Mater. Med., 2010, 21, 1103–1107 CrossRef CAS PubMed.
- R. R. Naik, L. L. Brott, S. J. Clarson and M. O. Stone, J. Nanosci. Nanotechnol., 2002, 2, 95–100 CrossRef CAS PubMed.
- S. R. Whaley, D. S. English, E. L. Hu, P. F. Barbara and A. M. Belcher, Nature, 2000, 405, 665–668 CrossRef CAS PubMed.
- H. Chen, X. Su, K. G. Neoh and W. S. Choe, Anal. Chem., 2006, 78, 4872–4879 CrossRef CAS PubMed.
- Y. Fang, N. Poulsen, M. B. Dickerson, Y. Cai, S. E. Jones, R. R. Naik, N. Kröger and K. H. Sandhage, J. Mater. Chem., 2008, 18, 3871–3875 RSC.
- L. A. Kelley and M. J. Sternberg, Nat. Protoc., 2009, 4, 363–371 CrossRef CAS PubMed.
- S. McNicholas, E. Potterton, K. S. Wilson and M. E. Noble, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 386–394 CrossRef CAS PubMed.
- M. Hnilova, E. E. Oren, U. O. S. Seker, B. R. Wilson, S. Collino, J. S. Evans, C. Tamerler and M. Sarikaya, 2008.
- D. W. M. T. Klem, D. J. Solis, A. M. Belcher, M. Young and T. Douglas, Adv. Funct. Mater., 2005, 15 CrossRef.
- C. Tiede, A. A. Tang, S. E. Deacon, U. Mandal, J. E. Nettleship, R. L. Owen, S. E. George, D. J. Harrison, R. J. Owens, D. C. Tomlinson and M. J. McPherson, Protein Eng., Des. Sel., 2014, 27, 145–155 CrossRef CAS PubMed.
- H. Kondo, K. Abe, Y. Emori and S. Arai, FEBS Lett., 1991, 278, 87–90 CrossRef CAS PubMed.
- J. Baumgartner, M. A. Carillo, K. M. Eckes, P. Werner and D. Faivre, Langmuir, 2014, 30, 2129–2136 CrossRef CAS PubMed.
- R. R. Naik, S. E. Jones, C. J. Murray, J. C. McAuliffe, R. A. Vaia and M. O. Stone, Adv. Funct. Mater., 2004, 14, 25–30 CrossRef CAS.
- M. C. F. Thomsen and M. Nielsen, Nucleic Acids Res., 2012, 40, W281–W287 CrossRef CAS PubMed.
- S. Brown, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8651–8655 CrossRef CAS.
- T. G. Schmidt, J. Koepke, R. Frank and A. Skerra, J. Mol. Biol., 1996, 255, 753–766 CrossRef CAS PubMed.
- J. A. Glasel, Biotechniques, 1995, 18, 62–63 CAS.
- D. E. Olins, A. L. Olins and P. Vonhippe, J. Mol. Biol., 1967, 24, 157 CrossRef CAS PubMed.
- G. Liu, M. Molas, G. A. Grossmann, M. Pasumarthy, J. C. Perales, M. J. Cooper and R. W. Hanson, J. Biol. Chem., 2001, 276, 34379–34387 CrossRef CAS PubMed.
- T. Middleton and B. Sugden, J. Virol., 1992, 66, 489–495 CAS.
- K. A. Marx, Biomacromolecules, 2003, 4, 1099–1120 CrossRef CAS PubMed.
- A. Pomorska, D. Shchukin, R. Hammond, M. Cooper, G. Grundmeier and D. Johannsmann, Anal. Chem., 2010, 82, 2237–2242 CrossRef CAS PubMed.
- G. L. Dybwad, J. Appl. Phys., 1985, 58, 2789–2790 CrossRef CAS.
- W. Smith and T. R. Forester, J. Mol. Graphics, 1996, 14, 136–141 CrossRef CAS PubMed.
- A. Artzy-Schnirman, E. Abu-Shah, M. Dishon, H. Soifer, Y. Sivan, Y. Reiter, I. Benhar and U. Sivan, J. Pept. Sci., 2014, 20, 446–450 CrossRef CAS PubMed.
- C. Riggio, M. P. Calatayud, C. Hoskins, J. Pinkernelle, B. Sanz, T. E. Torres, M. R. Ibarra, L. J. Wang, G. Keilhoff, G. F. Goya, V. Raffa and A. Cuschieri, Int. J. Nanomed., 2012, 7, 3155–3166 CAS.
- B. Z. Megumi Fukuta, M. Uenuma, N. Okamoto, Y. Uraoka, I. Yamashita and H. Watanabe, Colloids Surf., B, 2014, 118C Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods, supporting experimental data, and details of the molecular dynamics study. See DOI: 10.1039/c5sc01472g |
| This journal is © The Royal Society of Chemistry 2015 |