
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exosome isolation: a microfluidic road-map†
A.
Liga
a,
A. D. B.
Vliegenthart
b,
W.
Oosthuyzen
b,
J. W.
Dear
b and
M.
Kersaudy-Kerhoas
*a
aHeriot-Watt University, Institute of Biological Chemistry, Biophysics and Bioengineering, Edinburgh, United Kingdom. E-mail: m.kersaudy-kerhoas@hw.ac.uk
bUniversity of Edinburgh/British Heart Foundation Centre for Cardiovascular Science, Pharmacology, Toxicology and Therapeutics, Queen's Medical Research Institute, Edinburgh, United Kingdom
First published on 28th April 2015
Abstract
Exosomes, first isolated 30 years ago, are nanoscale vesicles shed by most types of cells. The nucleic acid rich content of these nanoparticles, floating in virtually all bodily fluids, has great potential for non-invasive molecular diagnostics and may represent a novel therapeutic delivery system. However, current isolation techniques such as ultracentrifugation are not convenient and do not result in high purity isolation. This represents an interesting challenge for microfluidic technologies, from a cost-effective perspective as well as for enhanced purity capabilities, and point-of-care acquisition and diagnosis. In this frontier review, we present the current challenges, comment the first microfluidic advances in this new field and propose a roadmap for future developments. This review enables biologists and clinicians familiar with exosome enrichment to assess the performance of novel microfluidic devices and, equally, enables microfluidic engineers to educate themselves about this new class of promising biomarker-rich particles and the challenges arising from their clinical use.
1 Introduction
Extracellular vesicles (EVs) are membrane-bound structures that are released by most, if not all, cell types. Due to their “cargo” of protein, RNA and DNA from their cell of origin, EVs offer a minimally-invasive ‘window’ into the intra-cellular world.1,2 In the literature, differing nomenclature and classification of EVs has been used based on size, density, morphology, lipid composition, protein composition, and subcellular origin.3 Exosomes are distinguished from other EV subtypes by their unique intra-cellular biogenesis, in contrast with other EVs, such as microparticles, that are shed directly from the cell membrane.3 Exosomes are reported to measure 20–100 nm and appear cup-shaped when visualized by transmission electron microscopy. Exosomes are first formed as intraluminal vesicles by budding into early endosomes and multi-vesicular bodies (MVBs). Subsequently, the MVBs either fuse with lysosomes or with the plasma membrane and the endosomes are released into the extra-cellular milieu to become exosomes.4 Due to this specific formation process, exosomes contain a cargo that, to some extent, mirrors the cell of origin.The observation that the proteome and transcriptome of exosomes changes according to the cell of origin5,6 and the ability to access exosomes from virtually any biological fluid (including saliva, lymph, urine, milk, blood, synovial and amniotic fluids7) forms the rationale for the use of exosomes as non-invasive biomarkers for disease and toxicity.8–10
Exosomes can also behave as inter-cellular signals by transferring functional mRNA and microRNA between cells in vitro.2,11 The importance of exosomal signalling in vivo remains to be determined. This signalling capacity may enable exosomes to be used as a non-immunogenic RNA delivery system that can cross natural divisions such as the blood–brain barrier.12,13
2. Conventional exosome capturing
Current methods for exosome ‘capturing’ concentrate exosomes, but do not isolate them. Contamination of exosome preparations with non-exosomal proteins and other EV sub-types can lead to spurious biomarker discovery studies and incorrect conclusions about exosome biological activity.14 The isolation of a pure population of exosomes would facilitate studies in exosome biology, in particular regarding their physiological functions and their roles in various pathologies. While next-generation deep sequencing (NGS) provides fast profiling of miRNA or other biomarkers in biological fluids, including from exosomes, liquid biopsy sample preparation is sometimes crucial to reliably differentiate between diseased and healthy patients.15,16 In this case, the isolation of exosomes can improve the sensitivity of biomarker amplification and reduce the number of false-negative results.16 The most common method used for concentrating exosomes is ultracentrifugation, a method that includes differential centrifugation steps reaching speeds of up to 200![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g. However, this procedure is time consuming (4–5 hours),17 requires specialist laboratory equipment that is not available in routine hospital laboratories, and is inefficient with regard to exosome yield (5–25% recovery).18 Variations on this method, such as adding in a sucrose gradient centrifugation step, lead to higher purity of extracted exosomes. However, the more complicated the sample processing the longer the time from sample to exosome preparation. The capital cost of an ultracentrifuge is typically around $50–100k and the running cost is around $3k per year. Additionally it requires training and a dedicated team. Exosomes can be isolated in as little as two hours but isolation time ranges from 2 to 10 hours depending on the biofluid and protocol. It may be difficult for standard hospital laboratories and resource-poor countries to acquire and maintain such equipment, thus denying exosome-based diagnostics to a substantial part of the population. A promising method for EV selective concentration relies on antibody-coated magnetic beads. This technique results in higher recovery and purity – at least two-fold more exosome markers and proteins measured in the exosome concentrate compared to ultracentrifugation.19 The use of magnetic beads also facilitates the use of standard analysis methods, such as flow cytometry, immuno-blot and electron microscopy on exosome-bead complexes.20 However, it is important to highlight that the magnetic beads only attach to exosomes that contain the targeted protein that might not be present in all the exosomes in the sample resulting in a biased analysis. Recently, commercial precipitation kits like ExoQuick™ and Total Exosome Isolation™ precipitation solution have become available. The advantages of these kits are that they are easy to use with only 1 or 2 steps and do not require any expensive equipment or advanced technical know-how. The disadvantage is that they commonly require a lengthy overnight incubation step and their mode-of-action has not been disclosed. The purity of the acquired exosomes has been reported to be inferior compared to OptiPrep density gradient centrifugation, as demonstrated by lower enrichment of exosomal marker proteins and more contaminating proteins such as extra-cellular argonaute 2 complexes.21 Chemicals originating from the precipitation kit may contaminate the prep and these commonly undisclosed chemicals may affect the apparent biological activity of the exosomes. Microfiltration techniques and other size exclusion methods can suffer from clogging and vesicle trapping issues and often entail shear stress-induced damages. This paper focuses on the potential of microfluidic devices to concentrate, and even isolate, exosomes, describing the most recent technological developments and highlighting pro and cons of each of these methods that promise to deliver rapid and easy isolation. In this review we have chosen to focus on the diagnostics rather therapeutic use of exosomes, as microfluidic solutions will be most beneficial, or at least of immediate benefit, in the diagnostic area.
000 × g. However, this procedure is time consuming (4–5 hours),17 requires specialist laboratory equipment that is not available in routine hospital laboratories, and is inefficient with regard to exosome yield (5–25% recovery).18 Variations on this method, such as adding in a sucrose gradient centrifugation step, lead to higher purity of extracted exosomes. However, the more complicated the sample processing the longer the time from sample to exosome preparation. The capital cost of an ultracentrifuge is typically around $50–100k and the running cost is around $3k per year. Additionally it requires training and a dedicated team. Exosomes can be isolated in as little as two hours but isolation time ranges from 2 to 10 hours depending on the biofluid and protocol. It may be difficult for standard hospital laboratories and resource-poor countries to acquire and maintain such equipment, thus denying exosome-based diagnostics to a substantial part of the population. A promising method for EV selective concentration relies on antibody-coated magnetic beads. This technique results in higher recovery and purity – at least two-fold more exosome markers and proteins measured in the exosome concentrate compared to ultracentrifugation.19 The use of magnetic beads also facilitates the use of standard analysis methods, such as flow cytometry, immuno-blot and electron microscopy on exosome-bead complexes.20 However, it is important to highlight that the magnetic beads only attach to exosomes that contain the targeted protein that might not be present in all the exosomes in the sample resulting in a biased analysis. Recently, commercial precipitation kits like ExoQuick™ and Total Exosome Isolation™ precipitation solution have become available. The advantages of these kits are that they are easy to use with only 1 or 2 steps and do not require any expensive equipment or advanced technical know-how. The disadvantage is that they commonly require a lengthy overnight incubation step and their mode-of-action has not been disclosed. The purity of the acquired exosomes has been reported to be inferior compared to OptiPrep density gradient centrifugation, as demonstrated by lower enrichment of exosomal marker proteins and more contaminating proteins such as extra-cellular argonaute 2 complexes.21 Chemicals originating from the precipitation kit may contaminate the prep and these commonly undisclosed chemicals may affect the apparent biological activity of the exosomes. Microfiltration techniques and other size exclusion methods can suffer from clogging and vesicle trapping issues and often entail shear stress-induced damages. This paper focuses on the potential of microfluidic devices to concentrate, and even isolate, exosomes, describing the most recent technological developments and highlighting pro and cons of each of these methods that promise to deliver rapid and easy isolation. In this review we have chosen to focus on the diagnostics rather therapeutic use of exosomes, as microfluidic solutions will be most beneficial, or at least of immediate benefit, in the diagnostic area.
3. Microfluidic technologies for exosome isolation
The advent of microfluidic technologies has allowed the development of new exosome manipulation techniques which, though still at an early stage of development (most of these were developed between 2012 and 2015), are proving themselves extremely convenient in terms of reagent volumes, product purity and isolation time (cf. ESI† Table for detailed comparison of techniques). The techniques developed so far for microfluidic-based exosomal purification can be classified in three categories including (i) trapping exosomes with an immune-affinity approach, (ii) sieving (e.g. nanoporous membranes) or (iii) trapping exosomes on porous structures (e.g. nanowire-on-micropillars). Sieving approach differs from the others by the possibility to start from whole blood without previous treatment. However, all methods require further off-chip additional steps for sample preparation, such as plasma extraction, or reagent mixing.223.1 Immunological separation
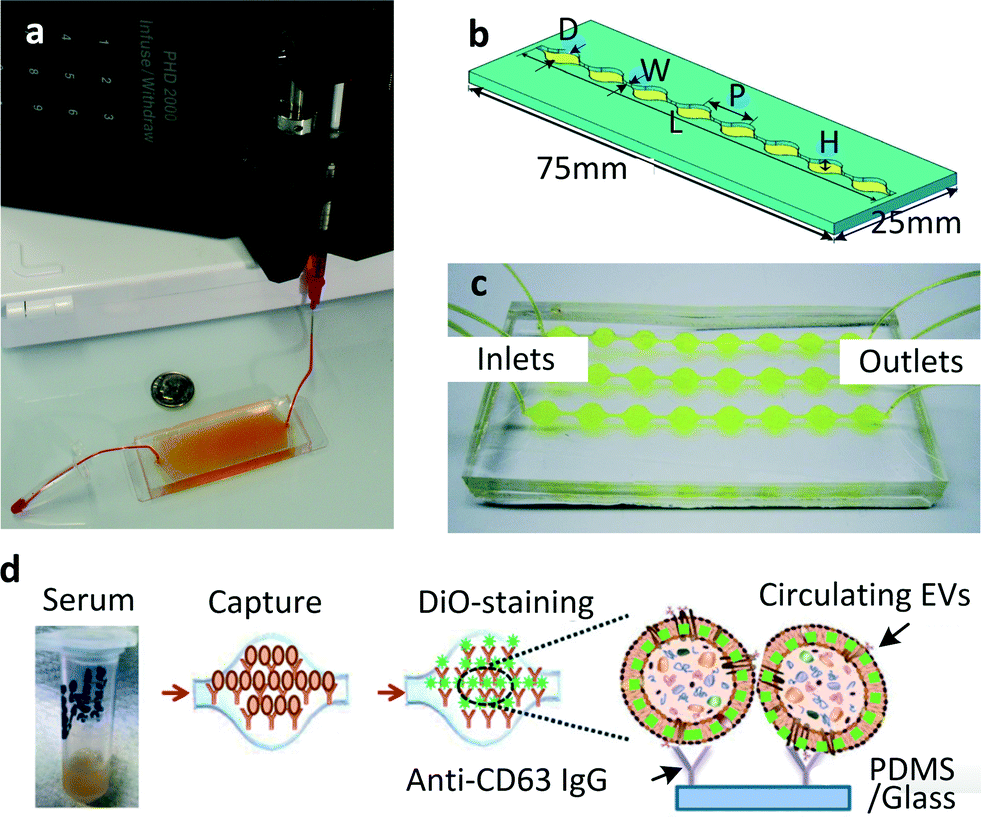 | ||
| Fig. 1 (a) The first chip realised for microfluidic exosome isolation featured a 19 mm × 20 μm × 4.5 cm chamber with herringbone groves 10 μm deep to enhance contact between microparticles and chip surface. The device is pictured here is filled with supernatant fluid and attached to a syringe pump. Reproduced from ref. 23 with the permission of the Royal Society of Chemistry. (b) A modified version of the same scheme: single channel Exochip, the device has the same dimensions of a standard glass-side (W = 0.75 mm, L = 73 mm, P = 9 mm, H = 100 μm). Up to 12 channels could fit within the dimensions of a microtiter-plate. (c) Experimental setup using a 3 channels Exochip (d) Exochip working scheme: exosomes counting is made possible through a simple 3 steps procedure involving: (i) blood serum infusion within the chip (pre-coated with exosome-specific capture antibodies, anti-CD63), (ii) staining with specifically functionalised (Di-O) fluorescent dye, (iii) on chip analysis through conventional counting methods. Reproduced from ref. 22 with the permission of the Royal Society of Chemistry. | ||
In conclusion, it is important to highlight that immunological methodologies are the only ones, so far developed, which can be directed towards the isolation of a pure exosome population. Other methods relying on physical properties (size, density, surface charge) lead to higher percentages of contaminants (similar micro-particles with different origin, proteins).
3.2 Sieving
Davies et al.28 developed a different approach to exosome collection, by sieving EVs directly from whole blood through a membrane and driving filtration either by pressure or electrophoresis (cf.Fig. 2). The pressure-driven solution leads to a shorter separation time while the electric field produces a higher purity of the extracted vesicles, proteins being less affected by electric fields than phospholipidic vesicles, due to their lower negative charge.29 In the authors' opinion, the non-selectivity of this device with respect to vesicle type can be seen as an advantage compared to the over-specific collection offered by immune-affinity related methods, which can lead to biased data analysis. A significant drawback is the low exosome recovery (unspecified in the text but shown to be next to 2% in a graph) while the device seems to perform well regarding separation time. The device reached a saturation point after 3–4 μL of filtrate was extracted when pressure driven filtration was used. However this volume was enough to assess the exosomal cargo by western blot and RT-PCR. When electro-driven filtration was used, the average yield was 79 ng RNA per 100 μg of protein from a 100 μL sample, compared to 187 ng of RNA per 100 μg of proteins from a 5 mL sample obtained by centrifugation.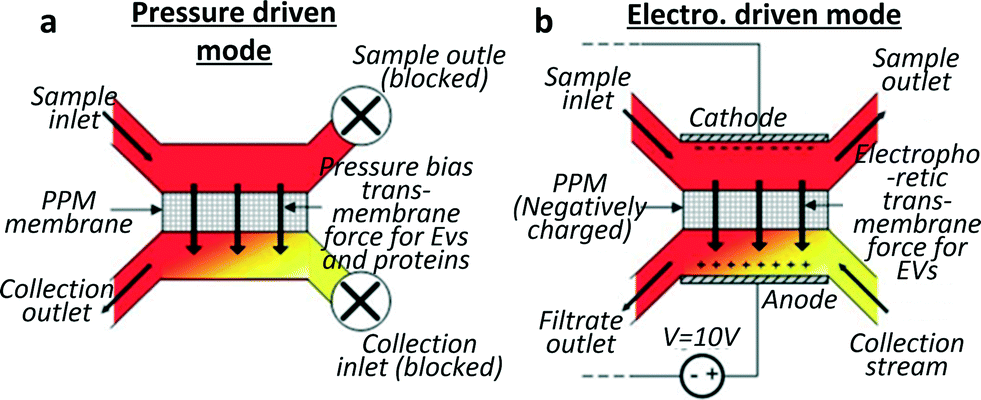 | ||
| Fig. 2 Schematics for two sieving modes for exosome filtration. The membrane is realised by in situ photopatterning of a porous polymer monolith. Sieving can be performed by (a) applying pressure on an on-chip dead end filter or (b) by applying an electric field to drive the exosomes through the porous membrane in a hybrid solution of cross flow and electrophoresis sorting. Vesicles damage through the device was shown to be undetectable by analysing haemolysis and DNA was not contaminated by intracellular proteins. Adapted from ref. 28 with the permission of the Royal Society of Chemistry. | ||
3.3. Trapping on porous structures
Wang et al.7 devised a third approach consisting of trapping EVs through a porous microstructure. They designed a ciliated micropillar structure forming a microporous silicon nano-wire which was able to selectively trap particles in the range of 40–100 nm (Fig. 3). According to Wang et al. this method allows the selective collection of intact phospholipidic “exosome-like” vesicles. Though the trapping step is relatively fast (~10 min), to proceed with imaging and characterisation, it is necessary to dissolve the silicon nanowire in PBS buffer overnight, thus making the recovery procedure time consuming. This device was tested with a solution containing 83 nm and 120 nm lipid vesicles and 500 nm nanoparticles. The highest retention (60%) was obtained for 83 nm vesicles when up to 30 μL of sample was injected (based on fluorescent intensity measured with a plate reader, starting concentration undisclosed), followed by 120 nm vesicles (45%), while the retention of 500 nm bead was only 10%. The retention rate of 83 and 120 nm lipid vesicles decreased when more sample volume was injected, probably due to a saturation effect. The device was not validated with clinical samples, and no analysis of cargo protein or RNA was performed.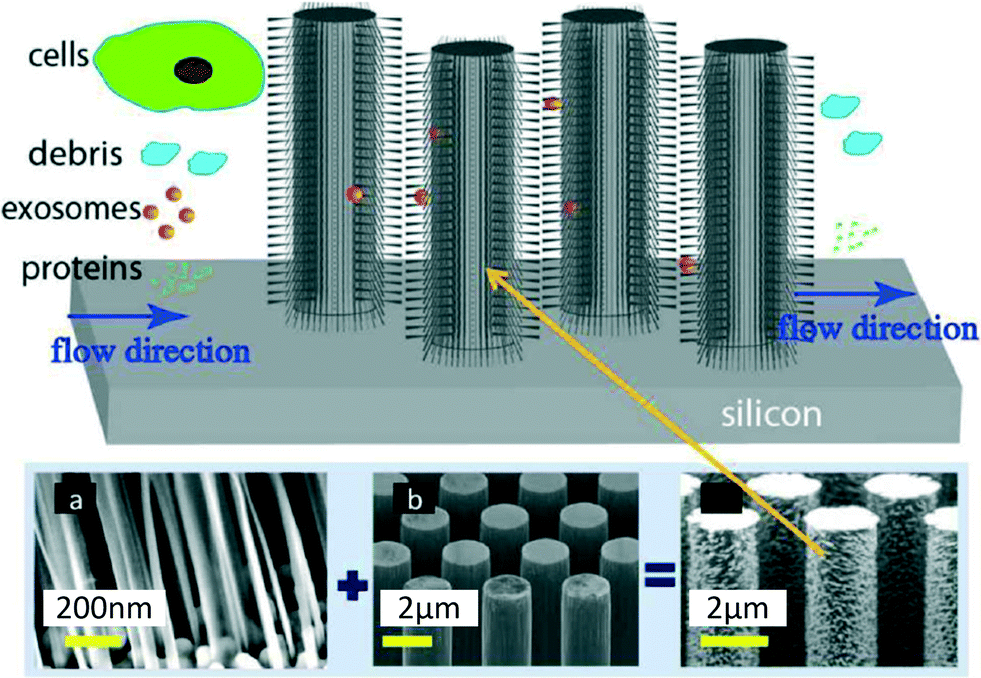 | ||
| Fig. 3 Ciliated structures for exosome isolation and capture by Wang et al. The hierarchical geometry of the device does not allow cells larger than 1 μm to access the wired area, due to the distance between the pillars (b) being too narrow (900 nm). Smaller cell debris can enter the micropillar area but are excluded by the ciliated nano structure (a), which forms pores with diameters ranging between 30 and 200 nm, in order to selectively trap exosomes and small EVs. Smaller proteins and molecules are free to pass through the device without being captured. Reproduced from ref. 7 with the permission of the Royal Society of Chemistry. | ||
4. Road map for microfluidic exosomal isolation
The increased interest in the role of exosomes in biomarker discovery and diagnostics has highlighted the pressing need for new technologies to isolate exosomes in biofluids rapidly and accurately with minimal sample preparation. Here we discuss 5 areas of consideration for future developments.4.1. From research to clinical setting: increased speed and lower cost
Ultracentrifugation is currently used in the research setting, but not yet regularly used in the clinical setting. If an exosome-based diagnostic was to be used in real-life clinical medicine, one could envisage that in a non-urgent situation, such as the diagnosis of cancer, the use of a centralised laboratory with an ultracentrifugation unit could be feasible, providing the extraction is reproducible. However if standardised performance is demonstrated using microfluidic chips, it would open the door for faster, reliable, cost-effective exosome-based diagnostics for emergencies in acute care settings (e.g., myocardial infections and drug toxicity) as well as non-emergency illnesses (cancer diagnostics). Furthermore, this could allow a more accurate exosomes analysis, since studies have indicated that there is a significant loss in exosomes number from biofluids such as urine, even when frozen at −80 °C30 and within 2 h.31 If exosomes are to be used for acute care, the device would ideally allow the biologist or clinician to rapidly and confidently isolate exosomes from biofluids within a 30 minute time frame after sample collection.4.2. Future directions, technological considerations and potential challenges for the development of microfluidic technologies
All prototypes presented in this review have used a single isolation strategy and none are free from issues. Future microfluidic devices might therefore benefit from hybrid approaches using two or more isolation techniques, for example, a first separation based on size could eliminate the largest blood cells from a blood samples, followed by an immunological separation to access a specific exosome population. The challenge, as with all microfluidic development, is to provide a solution to a real clinical question. Two routes are likely to emerge: the first will explore the exosome biology in the research area, and will require refined isolation. For this route, the advantage of microfluidics will be to provide cheaper alternative to ultracentrifugation with low capital investment. The second route will focus on the development of tools to facilitate liquid biopsy preparation for next generation sequencing. The challenge will lie in the rapid extraction from complex matrices. To fit in a clinical work flow the devices will need to be more robust and cost-effective as their current counterparts. The challenges in the development will lie in (i) reducing the cost of the devices, a consideration which will need to be taken into account at the design stage, and which might involve alleviating the need for structures below 80 μm, at which point photolithography might be replaceable by rapid-prototyping and high volume manufacturing processes. (ii) Secondly, achieving the right throughput will be crucial and might necessitate stacking several microfluidic units on the same device.4.3. Achieving specificity and reproducibility
As discussed above, in some cases, devices will need to reliably provide an exosome preparation specific to an organ of origin. If a device has on-chip technologies such as protein specific markers for sorting exosomes from specific organs it allows the clinician access to a relevant exosome population specifically to his site of interest. Current methods of isolation are not delivering on reproducibility of exosome recovery, arguably due to variations in sample handling. Ideally, microfluidic chips should be tested by different laboratories to assess reproducibility and consistency and demonstrate standardised performance.4.5. Characterisation and biological validation
At present, the methods of quantifying exact or ideal exosomal yield are still very semi-quantitative. There is an unmet need for quantitative and qualitative assessment of exosomes and the definition of clear biological controls.32 We have demonstrated that nanoparticle tracking analysis is a promising method for quantifying exosomes in biofluids, such as urine31 and it would therefore be interesting to compare the yield of current and future devices using this method. However, there is no “one fits all” solution for the characterisation of exosomes. The characterisation required will depend on the clinical application. Spiking-in labelled exosomes offers a reliable method to determine the yield, either via fluorescence measuring or RT-PCR. Furthermore, it is recommended to assess not only the presence of the selected markers (yield) but also the absence of contaminants (specificity).325. Conclusion
Through highly precise fluid control and high surface to volume ratios, microfluidics has the power to differentiate, capture, enrich and isolate particles of very similar sizes and shapes. Despite being in its infancy, several microfluidic developments have demonstrated effective exosome separation, leading to robust clinical diagnostics. However none of the first prototypes are free from issues, in particular they often require prior sample preparation based on macro-scale procedures. Among the aforementioned methods, immunological separation leads to high specificity, and can be performed in a fairly short time in most cases (~1.5 h). Trapping exosomes in porous structure, such as ciliated micro-pillars, seemingly leads to high purity exosomes recovery, but the recovery procedure is currently time consuming (over 1 day). Sieving is a particularly interesting method as it can be performed directly on whole blood, without any treatment before the separation, but it is not exempt from drawbacks, such as low recovery and the possibility of damage to vesicles due to shear stress. Some isolation techniques demonstrated for microvesicles (typically of diameter above 100 nm) might also be suitable for exosome recovery.33,34 For example, Rho et al. have demonstrated a rapid and sensitive microvesicle detection in packed red blood cell units using filter-assisted filtration, magnetic labelling and target-specific detection via miniaturised nuclear magnetic resonance system.33 Detection and characterisation are an essential part of the capture-to-diagnosis path, and some microfluidic researchers have focused all, or part, of their efforts towards the development of detection techniques which could be the subject of another focused review.25,35–37 Some of these techniques propose the direct (preparation free) detection of exosomal RNA from raw cell medium, which could be potentially applicable to bodily fluids.37 The applications of microfluidic technology to bodily fluids for exosome isolation have not been described extensively with few devices using clinical samples, and microfluidic engineers should seek to establish strong collaboration with biologists and clinicians to demonstrate the full potential of the technology. While current ultracentrifugation techniques require significant capital and maintenance costs, microfluidic-based exosome isolation techniques are promising and cost-effective, and could be deployed in a large number of clinics and hospitals, as well as in resource-scarce settings.Acknowledgements
AL is funded by a James Watt Scholarship. JWD acknowledges the support of NHS Research Scotland via NHS Lothian. MKK acknowledges the Royal Academy of Engineering for fellowship funding. The authors would like to thank the anonymous reviewers for valuable insights that enhanced the original manuscript.Notes and references
- T. Pisitkun, R. F. Shen and M. A. Knepper, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(36), 13368–13373 CrossRef CAS PubMed.
- H. Valadi, K. Ekstrom, A. Bossios, M. Sjostrand, J. J. Lee and J. O. Lotvall, Nat. Cell Biol., 2007, 9(6), 654–659 CrossRef CAS PubMed.
- E. Van Der Pol, A. N. Boing, P. Harrison, A. Sturk and R. Nieuwland, Pharmacol. Rev., 2012, 64(3), 676–705 CrossRef CAS PubMed.
- J. Kowal, M. Tkach and C. Thery, Curr. Opin. Cell Biol., 2014, 29, 116–125 CrossRef CAS PubMed.
- O. G. de Jong, M. C. Verhaar, Y. Chen, P. Vader, H. Gremmels, G. Posthuma, R. M. Schiffelers, M. Gucek and B. W. van Balkom, J. Extracell. Vesicles, 2012, 1, 18396–18418 CAS.
- P. Kucharzewska, H. C. Christianson, J. E. Welch, K. J. Svensson, E. Fredlund, M. Ringner, M. Morgelin, E. Bourseau-Guilmain, J. Bengzon and M. Belting, Proc. Natl. Acad. Sci. U. S. A., 2013, 110(18), 7312–7317 CrossRef CAS PubMed.
- Z. Wang, H.-J. Wu, D. Fine, J. Schmulen, Y. Hu, B. Godin, J. X. J. Zhang and X. Liu, Lab Chip, 2013, 13, 2879–2882 RSC.
- J. Skog, T. Würdinger, S. van Rijn, D. H. Meijer, L. Gainche, M. Sena-Esteves, W. T. Curry Jr, B. S. Carter, A. M. Krichevsky and X. O. Breakefield, Nat. Cell Biol., 2008, 10(12), 1470–1476 CrossRef CAS PubMed.
- L. Musante, D. E. Tataruch and H. Holthofer, Front. Endocrinol., 2014, 5(149), 1–12 Search PubMed.
- X. Yang, Z. Weng, D. L. Mendrick and Q. Shi, Toxicol. Lett., 2014, 225(3), 401–406 CrossRef CAS PubMed.
- J. Wahlgren, L. K. T. De, M. Brisslert, F. Vaziri Sani, E. Telemo, P. Sunnerhagen and H. Valadi, Nucleic Acids Res., 2012, 40(17), 130–142 CrossRef PubMed.
- L. Alvarez-Erviti, Y. Seow, H. Yin, C. Betts, S. Lakhal and M. J. Wood, Nat. Biotechnol., 2011, 29(4), 341–345 CrossRef CAS PubMed.
- S. El Andaloussi, S. Lakhal, I. Mager and M. J. Wood, Adv. Drug Delivery Rev., 2013, 65(3), 391–397 CrossRef CAS PubMed.
- T. F. Hiemstra, P. D. Charles, T. Gracia, S. S. Hester, L. Gatto, R. Al-Lamki, R. A. Floto, Y. Su, J. N. Skepper, K. S. Lilley and F. E. Karet Frankl, J. Am. Soc. Nephrol., 2014, 25(9), 2017–2027 CrossRef CAS PubMed.
- L. Cheng, R. A. Sharples, B. J. Scicluna and A. F. Hill, J. Extracell. Vesicles, 2014, 3, 23743–23757 Search PubMed.
- A. Gallo, M. Tandon, I. Alevizos and G. G. Illei, PLoS One, 2012, 7(3), 30679–30684 Search PubMed.
- F. Momen-Heravi, L. Balaj, S. Alian, P.-Y. Mantel, A. E. Halleck, A. J. Trachtenberg, C. E. Soria, S. Oquin, C. M. Bonebreak, E. Saracoglu, J. Skog and W. P. Kuo, Biol. Chem., 2013, 394(10), 1253–1262 CrossRef CAS PubMed.
- H. G. Lamparski, A. Metha-Damani, J. Y. Yao, S. Patel, D. H. Hsu, C. Ruegg and J. B. Le Pecq, J. Immunol. Methods, 2002, 270(2), 211–226 CrossRef CAS PubMed.
- B. J. Tauro, D. W. Greening, R. A. Mathias, H. Ki, S. Mathivanan, A. M. Scott and R. J. Simpson, Methods, 2012, 56(2), 293–304 CrossRef CAS PubMed.
- C. Thery, A. Clayton, S. Amigorena and G. Raposo, Curr. Protoc. Cell Biol., 2006, 1–29 Search PubMed , unit 3.22 pag.
- J. Van Deun, P. Mestdagh, R. Sormunen, V. Cocquyt, K. Vermaelen, J. Vandesompele, M. Bracke, O. De Wever and A. Hendrix, J. Extracell. Vesicles, 2014, 3, 24858–24862 Search PubMed.
- S. S. Kanwar, C. J. Dunlay, D. M. Simeone and S. Nagrath, Lab Chip, 2014, 14, 1891–1900 RSC.
- C. Chen, J. Skog, C.-H. Hsu, R. T. Lessard, L. Balaj, T. Wurdinger, B. S. Carter, X. O. Breakefield, M. Toner and D. Irimia, Lab Chip, 2010, 10, 505–511 RSC.
- M. He, J. Crow, M. Roth, Y. Zeng and A. K. Godwin, Lab Chip, 2014, 14, 3773–3780 RSC.
- H. Im, H. Shao, Y. I. Park, V. M. Peterson, C. M. Castro, R. Weissleder and H. Lee, Nat. Biotechnol., 2014, 32(5), 490–495 CrossRef CAS PubMed.
- R. Vaidyanathan, M. Naghibosadat, S. Rauf, D. Korbie, L. G. Carrascosa, M. J. Shiddiky and M. Trau, Anal. Chem., 2014, 86(22), 11125–11132 CrossRef CAS PubMed.
- J. S. Dudani, D. G. Gossett, H. T. K. Tse, R. J. Lamm, R. P. Kulkarni and D. Di Carlo, Biomicrofluidics, 2015, 9(1), 014112 CrossRef PubMed.
- R. T. Davies, J. Kim, S. C. Jang, E.-J. Choi, Y. S. Gho and J. Park, Lab Chip, 2012, 12, 5202–5210 RSC.
- D. L. Sparks and M. C. Phillips, J. Lipid Res., 1992, 33, 123–130 CAS.
- H. Zhou, P. S. T. Yuen, T. Pisitkun, P. A. Gonzales, H. Yasuda, J. W. Dear, P. Gross, M. A. Knepper and R. A. Star, Kidney Int., 2006, 69(8), 1471–1476 CAS.
- W. Oosthuyzen, N. E. L. Sime, J. R. Ivy, E. J. Turtle, J. M. Street, J. Pound, L. E. Bath, D. J. Webb, C. D. Gregory, M. A. Bailey and J. W. Dear, J. Physiol., 2013, 591(23), 5833–5842 CrossRef CAS PubMed.
- K. W. Witwer, E. I. Buzàs, L. T. Bemis, A. Bora, C. Lässer, J. Lötvall, E. N. Nolte-'t Hoen, M. G. Piper, S. Sivaraman, J. Skog, C. Théry, M. H. Wauben and F. Hochberg, J. Extracell. Vesicles, 2013, 2, 20360 Search PubMed.
- J. Rho, J. Chung, H. Im, M. Liong, H. Shao, C. M. Castro, R. Weissleder and H. Lee, ACS Nano, 2013, 7(12), 11227–11233 CrossRef CAS PubMed.
- S. M. Santana, M. A. Antonyak, R. A. Cerione and B. J. Kirby, Biomed. Microdevices, 2014, 16, 869–877 CrossRef CAS PubMed.
- H. Shao, J. Chung, L. Balaj, A. Charest, D. D. Bigner, B. S. Carter, F. H. Hochberg, X. O. Breakefield, R. Weissleder and H. Lee, Nat. Med., 2012, 18(12), 1835–1840 CrossRef CAS PubMed.
- F. Wei, J. Yang and D. T. W. Wong, Biosens. Bioelectron., 2013, 44, 115–121 CrossRef CAS PubMed.
- D. Taller, K. Richards, Z. Slouka, S. Senapati, R. Hill, D. B. Go and H.-C. Chang, Lab Chip, 2015, 15(7), 1656–1666 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5lc00240k |
| This journal is © The Royal Society of Chemistry 2015 |