
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRuthenium complexes as antimicrobial agents
Fangfei
Li
a,
J. Grant
Collins
*a and
F. Richard
Keene
*bcd
aSchool of Physical, Environmental and Mathematical Sciences, University of New South Wales, Australian Defence Force Academy, Canberra, ACT 2600, Australia. E-mail: g.collins@adfa.edu.au
bCentre for Biodiscovery and Molecular Development of Therapeutics, James Cook University, Townsville, QLD 4811, Australia
cDepartment of Matter & Materials, College of Science, Technology & Engineering, James Cook University, Townsville, QLD 4811, Australia
dDepartment of Chemistry, School of Physical Sciences, University of Adelaide, Adelaide, SA 5005, Australia. E-mail: richard.keene@adelaide.edu.au
First published on 27th February 2015
Abstract
One of the major advances in medical science has been the development of antimicrobials; however, a consequence of their widespread use has been the emergence of drug-resistant populations of microorganisms. There is clearly a need for the development of new antimicrobials – but more importantly, there is the need for the development of new classes of antimicrobials, rather than drugs based upon analogues of known scaffolds. Due to the success of the platinum anticancer agents, there has been considerable interest in the development of therapeutic agents based upon other transition metals – and in particular ruthenium(II/III) complexes, due to their well known interaction with DNA. There have been many studies of the anticancer properties and cellular localisation of a range of ruthenium complexes in eukaryotic cells over the last decade. However, only very recently has there been significant interest in their antimicrobial properties. This review highlights the types of ruthenium complexes that have exhibited significant antimicrobial activity and discusses the relationship between chemical structure and biological processing – including site(s) of intracellular accumulation – of the ruthenium complexes in both bacterial and eukaryotic cells.
 Fangfei Li | Fangfei Li obtained her Bachelor's degree in Chemistry in 2007 and continued two years research work in Ocean University of China before she commenced her PhD under the supervision of Professor Grant Collins at the University of New South Wales Canberra and Professor Richard Keene at James Cook University. Her PhD project centred on antimicrobial activity and pharmacology studies on ruthenium complexes. Fangfei recently completed her PhD and she is currently a postdoctoral fellow at Institute of Microbiology, Chinese Academy of Science in Beijing. |
 J. Grant Collins | Grant Collins obtained his PhD from the Australian National University. Following post-doctoral work at the School of Biochemistry at the University of New South Wales, he was appointed to a lectureship in the School of Chemistry at the Faculty of the University of New South Wales located at the Australian Defence Force Academy in Canberra in 1986. He is currently a Professor in Bio-inorganic Chemistry. His research involves the development of oligonuclear ruthenium(II) complexes as anticancer and antimicrobial agents, including studies of the interactions of these metal complexes with DNA and RNA. |
 F. Richard Keene | Richard Keene graduated as a PhD from the University of Adelaide in 1973. He was appointed to the staff of James Cook University (Townsville) in 1978, from where he retired in 2012 as a Distinguished Professor. His recent research interests have centred primarily on the stereochemistry of metallosupramolecular assemblies and its effect on their physical properties – in particular intramolecular electron transfer, and the interaction with biological molecules such as nucleic acids. This latter interest has extended over the last decade to the efficacy of oligonuclear metal complexes as anticancer and antimicrobial agents. Richard was awarded a DSc by the University of Adelaide in 1998, and was conferred as an Emeritus Professor on his retirement from JCU. He is currently an Honorary Visiting Research Fellow at the University of Adelaide. |
1. Microbial infection
The struggle to control bacterial and other microbial infectious diseases has persisted throughout human history. Over centuries, epidemics such as cholera and plague (‘bubonic plague’ in lymph nodes, ‘septicemic plague’ in blood vessels, and ‘pneumonic plague’ in lungs) have at times been prevalent and widespread, occasionally resulting in dramatic regional population decreases.1 Pneumonia – described as “the captain of the men of death” in the 19th Century – today still infects 7% of the world's population with four million deaths reported every year.2 Tuberculosis (TB) is another common and often deadly infectious disease in humans, with more than 1.5 million deaths per year worldwide.3One of the major advances in medical science over the last century has been the development of antimicrobials.4 However, a consequence of their widespread use has been the emergence of drug-resistant populations of microorganisms. Infection by such drug-resistant pathogens has become an important cause of morbidity and mortality worldwide once again: in a recent update from the Infectious Diseases Society of America,5Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumanni, Pseudomonas aeruginosa and Enterobacter species were identified as the pathogens of most current concern. In particular, methicillin-resistant S. aureus (MRSA, colloquially known as Golden Staph), vancomycin-resistant Enterococcus (VRE) and fluoroquinolone-resistant P. aeruginosa show rapidly increasing incidence of infection, with treatment failure leading to high mortality rates.5 Furthermore, and despite considerable research, Mycobacterium tuberculosis (the causative agent of the disease tuberculosis) and malaria (particularly caused by the eukaryotic Plasmodium falciparum-induced infection) remain major causes of concern, due in part to persistent antimicrobial resistance, leading to treatment failure and high mortality rates. Although 90% of tuberculosis is asymptomatic, it is estimated that 33% of the world population is infected with the organism:3 while the antibiotic rifampicin and the antibacterial isoniazid can be used to treat TB, there is a worrying increase in the emergence of M. tuberculosis strains resistant to these drugs (MDR-TB), with some strains resistant to all known treatments (XDR-TB). In the case of malaria, it has been estimated that the malaria parasite infects 500 million people annually, particularly in sub-Saharan Africa, where “one child dies every 30 seconds from infection”.6
There is clearly a need for the development of new antimicrobials; but more importantly, there is the need for the development of new classes of antimicrobials, rather than drugs necessarily based upon analogues of known scaffolds.
2. Cellular structure of bacteria
Bacteria are a group of microscopic and single-celled prokaryotic microorganisms. In general, the cocci range from 0.5 to 1.0 μm in diameter, while for bacilli, spirilla and other non-spherical species the width ranges from 0.2 to 2.0 μm and the length ranges from 1 to 20 μm.7,8 As prokaryotes, bacteria have a characteristic cellular organisation (Fig. 1) which is distinct from the structure of eukaryotes.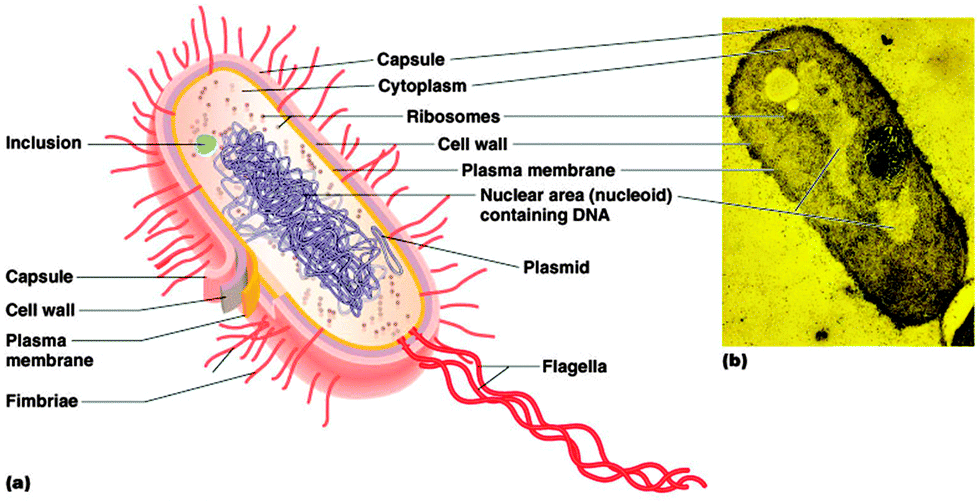 | ||
| Fig. 1 The structure of a typical bacterial cell (a) and an electron microscopy photo of a bacterial cell (b). [Image taken from G. J. Tortora, B. R. Funke and C. L. Case, Microbiology: An Introduction, Pearson Education, Inc., San Francisco, 8th edn, 2004]. | ||
2.1 Genetic material
The genetic information is carried in the bacterial chromosome, which is a single circular double-stranded DNA molecule. Bacterial chromosomal DNA is supercoiled in a non-membrane bound structure known as the nucleoid.9,10 The structural configuration of the DNA is controlled by two enzymes, DNA gyrase and topoisomerase I, which can introduce or relax the supercoils.9Besides chromosomal DNA, a variable number of small circular self-replicating double-stranded DNA molecules called plasmids can also be present in the cytoplasm and carry supplementary information.10,11
2.2 Ribosomes
The only organelle found in bacteria is the ribosome, the site of protein synthesis. Bacterial ribosomes are composed of ribosomal RNA and proteins, with the functional organelle containing two parts, the 30S and 50S subunits (S is the abbreviation of the Svedberg unit, a measure of the rate of sedimentation in centrifugation).12,13 In rapidly growing bacterial cells, most of the ribosomes appear in the form of polysomes, where several ribosomes carry out translation on a single messenger RNA strand.142.3 Cytoplasmic membrane
Like all cells, the bacterial internal structures are surrounded and protected by a cytoplasmic membrane. The cytoplasmic membrane of bacteria consists of a phospholipid bilayer and is similar to the cell membrane of eukaryotes except for the wider variety of fatty acids, a higher content of negatively-charged phospholipids and the absence of sterols within the membrane.15 As a selective permeability barrier, the bacterial cytoplasmic membrane regulates the exchange of substances between the cytoplasm and the outer environment. Many important cellular bio-processes also take place in the cytoplasmic membrane, such as energy production, protein secretion and chromosome segregation.16,17 In addition, under different environmental stresses, the fluidity of the membrane can be modulated by varying the proportions and types of saturated and unsaturated fatty acids in the phospholipids.182.4 Cell wall
Bacteria must survive in some extreme conditions in which eukaryotes are not able to do so. Thus a rigid cell wall outside the cell membrane is essential for the protection of the cell from osmotic pressure, chemical or enzymatic lysis and mechanical damage.17 Due to the distinct structure of the cell wall, bacteria are classified into two groups by the Gram staining procedure – Gram positive and Gram negative bacteria. The cell wall of Gram positive bacteria (which retain the purple colour of the Crystal Violet stain after the procedure) is composed of a thick layer of peptidoglycan with a group of inlaid molecules called teichoic acids.17,19 In contrast, in the absence of teichoic acid molecules, the Gram negative cell wall consists of a thin layer of peptidoglycan but it is covered by an outer membrane.10,17,19 Besides phospholipids, the outer membrane contains LPS (lipopolysaccharide), a unique structure on the surface directed towards the external environment. LPS is a highly negatively-charged amphiphilic molecule and comprises lipid A, the core and the O antigen.20 It is stabilised by divalent cations such as calcium and magnesium. The outer membrane, with the LPS, greatly decreases the permeability to antibacterials and is regarded as one of the major mechanisms of resistance to drugs for many pathogenic Gram negative bacteria.21Bacteria may also have an additional capsule (normally covered with a slimy layer) lying outside of the cell wall for further protection against phagocytosis by host cells. Some other external structures such as flagella, fimbriae and pili may also be present to aid the movement of a bacterium or attachment to a surface.10,15
2.5 Comparison with eukaryotes
The differences in the cellular structure between bacteria and eukaryotes are briefly summarised in Table 1. These differences are the basis for the selective targeting of many antibacterial drugs. It is worth mentioning that from an evolutionary perspective, bacteria are believed to be the origin of the mitochondria in eukaryotic cells.22 A mitochondrion is of similar size to a bacterium, and the mitochondrial ribosome structure is closer to prokaryotes than the ribosomes found in the cytoplasm of eukaryotes. Most importantly, it has been reported that the mitochondrial genomes are evolved from a bacterial ancestor.22| Bacteria | Eukaryotes | |
|---|---|---|
| Cell size | Commonly 1–10 μm | Commonly 10–100 μm |
| Cell type | Usually unicellular | Usually multicellular |
| Genetic material | A non-membrane bound single circular DNA molecule; plasmids | A membrane-bound nucleus is present and contains more than one chromosome |
| Ribosome | 70S, with 30S and 50S subunits | 80S, with 40S and 60S subunits |
| Other organelles | Absent | Mitochondria or chloroplasts, endoplasmic reticulum, golgi apparatus, cytoskeleton, lysosomes |
| Cytoplasmic membrane | Wider variety of fatty acids, higher content of negatively-charged phospholipids, absence of sterols | More uniformed distribution of fatty acids, high percentage of neutral phospholipids, presence of sterols |
| Cell wall | Present | Absent |
| Outer cellular structures | Capsule, flagella, fimbriae, pili | Absent |
3. Antibacterials
Since the discovery of penicillin, the first antibiotic, a great number of compounds have been subsequently developed and clinically used for the treatment of infections by bacteria and other pathogens. However, not long after the beginning of the ‘antibiotic era’, another severe challenge arose – the emergence of bacteria resistant to existing antibiotics as well as other antimicrobials. The pipeline of new antimicrobials is now a particular challenge, and thus there is an urgent need for the development of targets, particularly those with novel structures.3.1 Mechanisms of action of antibacterials
Existing antibacterials are classified into four major groups based upon their intracellular target and their mechanism of action (see Fig. 2).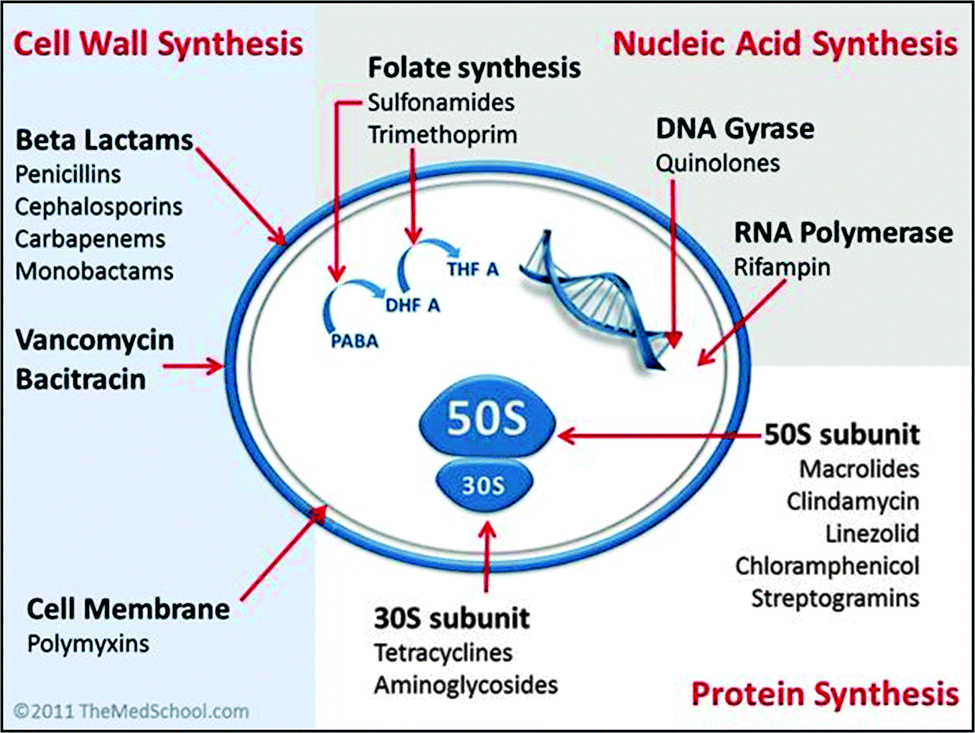 | ||
| Fig. 2 Classification of antibiotics by mechanism of action. [Image by Kendrick Johnson: Creative Commons Attribution-Share Alike 3.0 Unported license]. | ||
(1) Cell wall synthesis inhibition; e.g. penicillin and derivatives, cephalosporins, carbapenems and glycopeptides.23–25 These compounds are more effective against infection by Gram positive bacteria.
(2) Cell membrane disruption; e.g. the family of polycationic peptide antibiotics called polymyxins.26–28 Polymyxins are used in the treatment of infection by Gram negative bacteria, and are considered a last-line therapy against Gram negative ‘superbugs’.27
(3) Nucleic acid synthesis inhibition; e.g. quinolones, rifampicin and sulphonamides.29–32 The fluoroquinolones are one of a few examples of a broad-spectrum synthetic antimicrobial in clinical use.29
(4) Protein synthesis inhibition; e.g. tetracycline, aminoglycosides, chloramphenicol and macrolides.33–35 A large proportion of clinically-used antibacterials inhibit protein synthesis by targeting the ribosomal-RNA rich surfaces of ribosomes, and in some cases can be effective against tuberculosis.35
3.2 Mechanisms of resistance to antibacterials
The prevalence of drug-resistant bacterial infections makes it crucial to understand the mechanisms of drug resistance developed by microorganisms. Antibacterial resistance is manifested through a variety of biochemical processes that are genetically controlled, developed by either mutation of intrinsic cellular genes or by the acquisition of resistance genes from the other bacteria.36 The biochemical mechanisms of resistance involves drug inactivation or modification by enzymes, target modification, target repair, target overproduction, immunity and bypass, efflux pumps, increased impermeability and tolerance, biofilm formation and other unrecognised mechanisms.36–38 The mechanisms of resistance to the currently-used antibiotics are summarised in Table 2.| Antibiotic | Mechanism of resistance |
|---|---|
| β-Lactams | Antibacterial inactivation (β-lactamase) |
| Modification of penicillin-binding proteins (PBPs) | |
| Efflux | |
| Impermeability | |
| Glycopeptides | Target modification (gene van A) |
| Polymyxins | Modification of target |
| PhoP–PhoQ system | |
| Quinolones | Alteration of target enzymes |
| Impermeability | |
| Rifampicin | Target modification (gene rpoB) |
| Sulfonamides | Target modification (genes sul1 and sul2) |
| Chloramphenicol | Antibacterial inactivation (acetyltransferase) |
| Active efflux | |
| Impermeability | |
| Tetracyclines | Efflux |
| Ribosome protection | |
| Modification of the antibiotic | |
| Aminoglycosides | Impermeability |
| Enzymatic modification (AMEs) |
4. Metal-based antimicrobial agents
Metal- and metalloid-based drugs play an important part in the history of medicinal chemistry. Salvarsan (Fig. 3a–c), an organoarsenic compound for syphilis treatment developed by Paul Ehrlich and his co-workers in the 1900s, was the first successful application of metalloid complexes in chemotherapy.39 After half a century, another breakthrough – the discovery of the anti-tumour properties of cis-diamminedichloridoplatinum(II) (cisplatin, Fig. 3d) and its derivatives – paved the way for the subsequent development of metal-based chemotherapeutic agents.40 A variety of metal complexes with different metal centres (mainly transition metal elements) and ligands of diverse structures were synthesised and studied for their biological activity. Because of the range of coordination geometries, metal complexes provide more stereochemical variability than is possible in organic molecules and often introduce new elements of chirality which may be important for biological molecule recognition and interaction. The metal complexes can also be highly positively-charged: since many biological structures – such as DNA and RNA, several types of phospholipids, and some regions of proteins – are negatively charged, for electrostatic reasons the positive charge of metal complexes could aid the binding with intracellular targets. It has been reported that a range of transition metal complexes (including Ag, Au, Cu, Fe, Ir, Pt, Rh, Ru, Ti, etc.) bind DNA and RNA with a relatively high affinity and many of them have shown anticancer activity.41,42 Furthermore, a number of transition metal complexes display antimicrobial activity. For example, silver nitrate and silver(I) sulfazine are used clinically for treatment for ophthalmia neonatorum and severe burns infections, respectively,43 and silver complexes with oxygen donor ligands exhibit a wide-spectrum antimicrobial activity;44 a series of gold(I) anti-arthritic drugs were shown to exhibit inhibition against Pseudomonas putida;45 Richards et al. found that an iron triple-helicate complex binds bacterial chromosomal DNA and is bactericidal against E. coli and Bacillus subtilis with a moderate minimum inhibitory concentration (MIC).46 Ng et al. demonstrated that some copper(II)- and platinum(II)-based metallointercalator complexes could exhibit good antimicrobial activity against S. aureus.47 Interestingly, some of the copper complexes substantially permeabilised the bacterial membrane while others had little effect.47 Furthermore, a range of metal complexes (Mn, Fe, Co, Ni, Cu and Zn) containing Schiff base ligands have shown modest antimicrobial activity against bacteria and fungi.48 There is also a group of metal complexes containing existing antibacterials as ligands:49,50 compared to their parent organic antibacterial, the metalloantibacterial normally show enhanced antimicrobial activity, especially against drug-resistant bacterial strains.49–51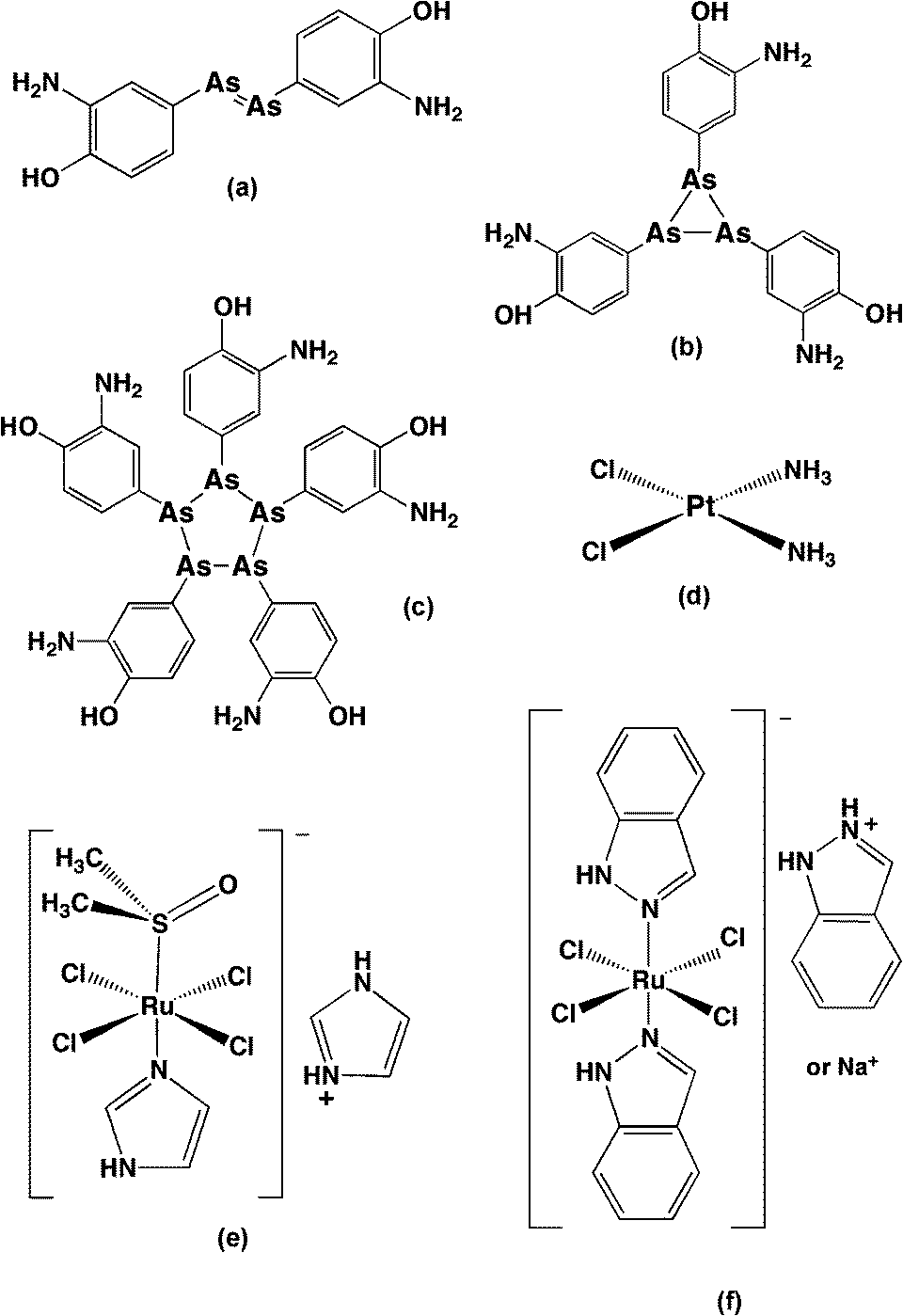 | ||
| Fig. 3 The chemical structures of several metal/metalloid-based therapeutic agents. (a–c) Salvarsan – the initially proposed structure (a), although it has been shown to be a mixture of (b) and (c); cisplatin (d); NAMI-A (e); and KP1019 (f). | ||
4.1 Ruthenium-based antimicrobial agents
Among the transition metal complexes, ruthenium-based complexes have been widely studied and some have displayed significant biological activity.52–63 This can be due to their ability to strongly bind nucleic acids and proteins, ligand exchange kinetics similar to those of their platinum counterparts, the prevalence of two main oxidation states (II and III) and the iron-mimicking property when bound to biological molecules.52–58 In addition, both the commonly accessible oxidation states of ruthenium are octahedral and relatively inert, the synthetic chemistry is very well established (including stereochemical control), and the photophysical properties of many ruthenium(II) complexes facilitate confocal microscopy and flow cytometry studies of cellular accumulation and localisation.57,58 Over the last decade their therapeutic potential as anticancer and antimicrobial agents has been demonstrated.59–63Nucleic acids are generally believed to be a target for many metal-based drugs.64 Consequently, there have been many studies of the interactions of ruthenium complexes with DNA and RNA.53–58,64–67 Complexes with labile ligands (such as KP1019, [Ru(terpy)(bpy)Cl]+ {terpy = 2,2′:6′,2′′-terpyridine} and organometallic arene complexes) can bind DNA coordinatively, predominantly at guanine residues.64,66 Through the addition of extra functionality (e.g. extended arene rings), these coordinating complexes can also bind DNA by hydrogen bonding and hydrophobic interactions.64 Kinetically inert ruthenium complexes {e.g. tris(bidentate) species containing polypyridyl ligands} can bind reversibly to DNA and RNA by intercalation or through association in either the major or minor groove.53–58,65,67 In addition to binding general duplex structures, inert ruthenium complexes have been designed that specifically bind non-duplex structures, such as mismatches, bulge sites, hairpins and quadruplexes.57,58 The tailoring of ruthenium complexes to specific nucleic acid sequences and structures is likely to become more important as the biological significance of non-canonical structures is determined.
Although less well explored, the structural properties of metal complexes that allow strong interactions with nucleic acids also provide a basis for targeting proteins and enzymes.68,69 Inert polypyridyl ruthenium complexes and ruthenium–arene complexes have already been shown to bind and inhibit enzymes such as acetylcholinesterase and protein kinases.68–70 For example, the Meggers group have demonstrated that bulky pyridocarbazole ruthenium complexes can inhibit the activity of p21-activated kinase 1, which is implicated in tumourgenesis and metastasis, at nanomolar concentrations.70 As outlined in recent reviews by Meggers and Pandey and co-workers,68,69 metal complexes (particularly octahedral complexes) have a number of features that may make them more suitable than purely organic compounds for the development as enzyme inhibitors. Although no example of a ruthenium complex inhibiting a specific enzyme in live bacteria has been reported to the best of our knowledge, it is likely that proteins and enzymes will become important future targets for the development of ruthenium complexes as antimicrobial agents.
4.1.1.1 Mononuclear complexes. Over 60 years ago, Dwyer and co-workers first investigated the biological activity of polypyridyl metal complexes.59,71,72 The complexes they studied were mononuclear tris(bidentate) inert metal complexes with ligands such as 1,10-phenanthroline and its derivatives (e.g. 3,5,6,8-tetramethyl-1,10-phenanthroline and 5-nitro-1,10-phenanthroline) and 2,2′-bipyridine and its derivatives, coordinated predominantly to ruthenium or iron. The antibacterial activities of these complexes were determined against Gram positive, Gram negative and acid-fast bacteria.59 [Ru(phen)3]2+ (Fig. 4a) was shown to be inactive against all the bacterial strains. However, the introduction of methyl group substituents on the phenanthroline ligands (Fig. 4b) dramatically increased the activity against all bacteria – especially Gram positive bacteria and Mycobacterium tuberculosis – indicating the importance of lipophilicity on antibacterial activity. The substitution of nitro groups also significantly improved the anti-MTB activity. The ruthenium(II) complexes exhibited better activity than quaternary ammonium salts with a lower charge, and cobalt(III) complexes with a higher charge, suggesting the importance of the charge and lipophilicity of the metal complex. Interestingly, they also demonstrated that the bacteria did not easily develop resistance to this class of compound. For example, after sub-culturing Staphylococcus pyogenes var. Phillips (a highly virulent bacterial strain) twenty-five times at 48 hour intervals in the presence of the metal complex, the bacteria only showed a two-fold increase in resistance to [Ru(Me4phen)2(acac)]+ (where acac = acetylacetonato; Fig. 4c). Alternatively, there was a 10
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-fold decrease in the activity of the antibiotic control penicillin.59 Moreover, in vivo bacterial infection treatment studies with mice or guinea-pigs were also conducted. Finally, it was proposed that this class of complexes were suitable for topical application for surface infection treatment rather than injection routes due to the rapid clearance from the blood stream after administration.59,72
000-fold decrease in the activity of the antibiotic control penicillin.59 Moreover, in vivo bacterial infection treatment studies with mice or guinea-pigs were also conducted. Finally, it was proposed that this class of complexes were suitable for topical application for surface infection treatment rather than injection routes due to the rapid clearance from the blood stream after administration.59,72
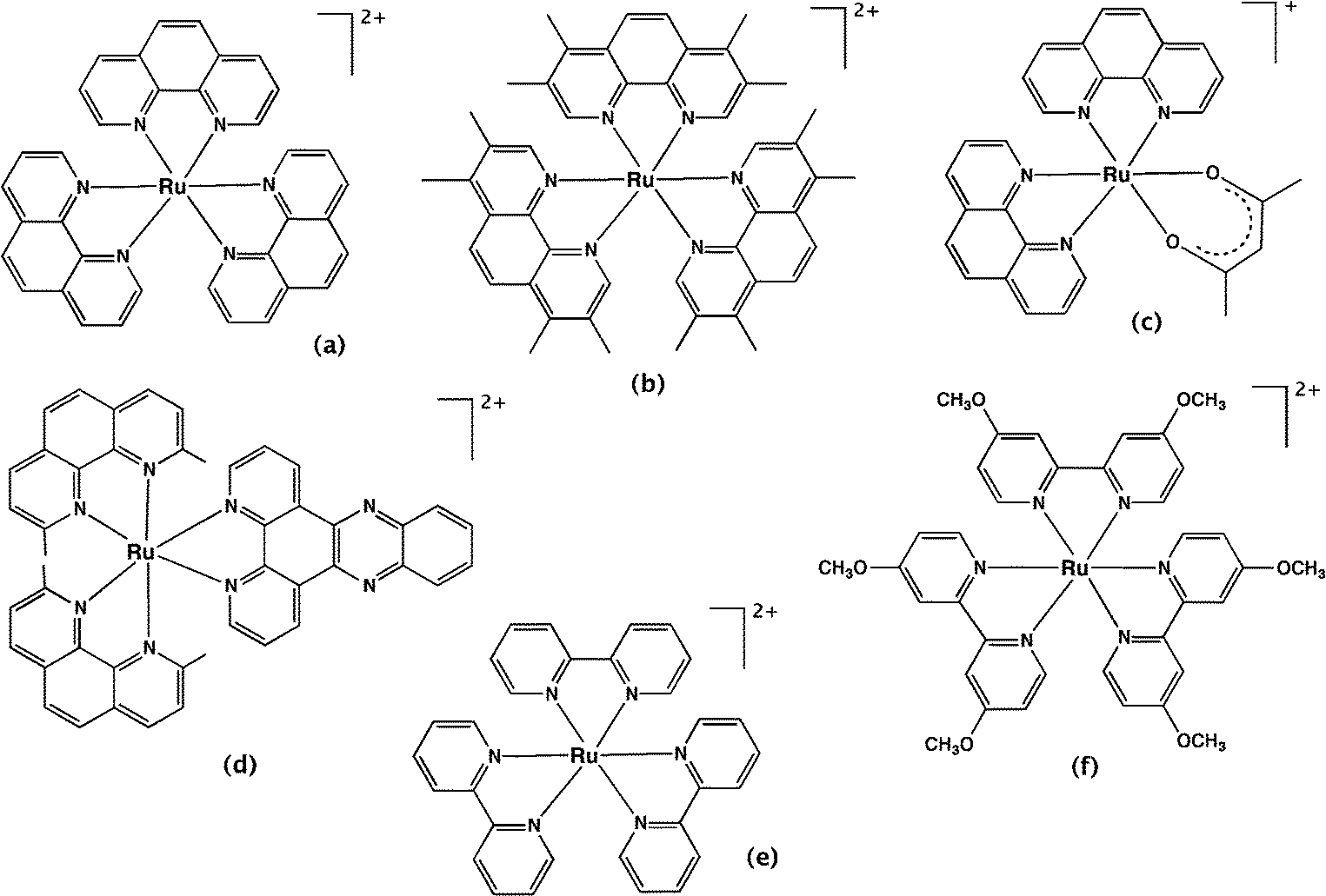 | ||
| Fig. 4 Mononuclear polypyridylruthenium(II) complexes exhibiting antimicrobial activity. (a) [Ru(phen)3]2+; (b) [Ru(Me4phen)3]2+; (c) [Ru(Me4phen)2(acac)]+; (d) [Ru(2,9-Me2phen)2(dppz)]2+; (e) [Ru(bpy)3]2+; (f) [Ru(dmob)3]2+. | ||
Although promising results were published, the mononuclear polypyridyl metal complexes were not further developed as therapeutic agents. This may have been due to the growing interest in their DNA binding ability, or perhaps because of the large number of antibiotics in the pipeline in the 1960s and the vastly lower incidence of drug-resistance at that time. However, recently there has been renewed interest in the antimicrobial activity of polypyridylruthenium(II) complexes. Aldrich-Wright and co-workers reported mononuclear polypyridylruthenium(II) complexes that could bind DNA by intercalation and exhibited significant bactericidal activity against B. subtilis and S. aureus strains, including several methicillin-resistant strains.63 Against the Gram positive strains, some of the complexes exhibited MICs as low as 2 μg ml−1; however, they were inactive against Gram negative bacteria. In addition, the treatment with the most active compound, [Ru(2,9-Me2phen)2(dppz)]2+ (Fig. 4d), increased the survival population of Caenorhabditis elegans that were infected with S. aureus, indicating the relatively lower toxicity against eukaryotic systems.63 Satyanarayana and co-workers also found that a range of mononuclear ruthenium complexes that contained derivatives of either the dppz ligand or 2-phenyl-imidazo-1,10-phenanthroline ligands had moderate activity.73 By contrast, [Ru(L)2bdppz]2+ {where L = 2,2′-bipyridine or 1,10-phenanthroline and bdppz = 9a,13a-dihydro-4,5,9,14-tetraaza-benzotriphenylene-11-yl)-phenyl-methanone} only showed significant antimicrobial activity at 1500 μg ml−1 against S. aureus and E. coli, despite binding DNA with reasonable affinity.74 Although DNA binding is a logical candidate in terms of the responsibility for the antimicrobial activity of the polypyridylruthenium(II) complexes, Lam et al. recently demonstrated that a bis(2,2′-bipyridine)ruthenium(II) complex containing a N-phenyl-substituted diazafluorene ligand significantly increased the production of reactive oxygen species in MRSA.75 The authors suggested that the good activity observed against MRSA (6.25 μg ml−1) could be due to DNA damage caused by the reactive oxygen species.
The antimicrobial activity of polypyridylruthenium(II) complexes upon photo-activation has also been investigated. Another complex, [Ru(dmob)3]Cl2 (dmob = 4,4′-dimethoxy-2,2′-bipyridine, Fig. 4f) exhibited good antimicrobial activity upon irradiation of light (MIC = 12.5 μg ml−1 against S. aureus) and demonstrated considerable promise as a photosensitiser for use in photodynamic antimicrobial chemotherapy.76
The development of the mononuclear polypyridylruthenium(II) complexes as antimicrobial agents was limited due to their comparatively high MIC values compared with antibacterials currently in clinical use; however, their potential against drug-resistant bacterial strains is still promising. Although the mode of action of these ruthenium complexes is not well understood, DNA binding is normally considered the major interaction leading to the antimicrobial activity. Consequently, dinuclear and higher nuclearity complexes with relatively larger size, higher charge and nucleic acid binding affinity were expected to be better candidates as antimicrobial agents.
4.1.1.2 Dinuclear and oligonuclear complexes. Until very recently, few dinuclear or oligonuclear polypyridylruthenium(II) complexes have been studied for their antimicrobial activities. Most of the dinuclear complexes were designed and developed to improve their DNA binding properties compared to the corresponding mononuclear complexes. A selection of the commonly used bridging ligands is shown in Fig. 5.
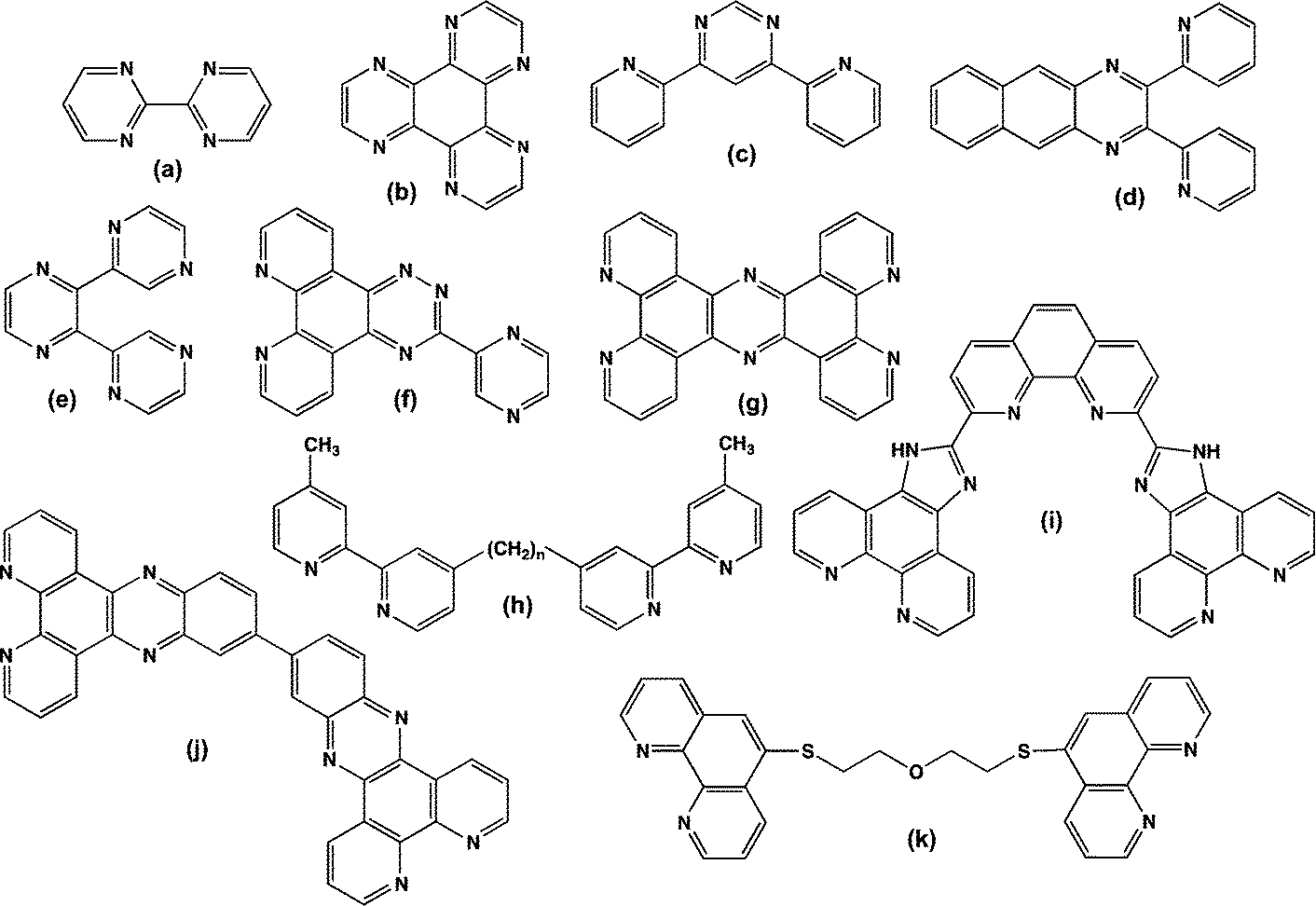 | ||
| Fig. 5 Common di-bidentate bridging ligands for dinuclear complexes: (a) 2,2′-bipyrimidine [bpm], (b) 1,4,5,8,9,12-hexaazatriphenylene [HAT], (c) 4,6-bis(2-pyridyl)pyrimidine [dppm], (d) 2,3-bis(2-pyridyl)benzo[g]quinoxaline [dpb], (e) 2,3-bis(2-pyridyl)pyrazine [2,3-dpp], (f) 3-pyrazin-2-yl)as-triazino[5,6-f]-1,10-phenanthroline [pztp], (g) tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine [tpphz], (h) bis[4(4′-methyl-2,2′-bipyridyl)]-1,n-alkane [bbn] (i) 2,9-bis(2-imidazo[4,5-f][1,10]phenanthroline-1,10-phenanthroline [bipp], (j) 11,11′-bis(dipyrido[3,2-a:2′,3′-c]phenazine [dppz(11,11′)dppz], (k) 1,10-phenanthroline-5-(2 mercaptoethyl ether)-5-1,10-phenanthroline [SOS]. | ||
Aldrich-Wright and co-workers investigated the intercalative dinuclear complex [{Ru(dpq)2}2(μ-phen-x-SOS-x-phen)]4+ (dpq = dipyrido[3,2-d:2′3′-f]quinoxaline; SOS = 2-mercaptoethyl ether; x = 3, 4 or 5) and found the complex had a DNA binding affinity of 6 × 107 M−1, a significant improvement upon the mononuclear analogues [Ru(dpq)2(phen)]2+ (K = 5.4 × 104 M−1), [Ru(dpq)2(phen-4-SOS)]2+ (K = 2.3 × 106 M−1), or [Ru(bpy)2(dpq)]2+ (K = 5.9 × 104 M−1).77,78 Ruthenium complexes containing dppz and tpphz ligands (see Fig. 5) have also been reported to display very high affinity to duplex or quadruplex DNA by intercalation, with low salt concentration dependence.79–83 Lincoln and co-workers developed a bis-intercalating dinuclear complex, ΔΔ-[μ-c4(cpdppz)2-(phen)4Ru2]4+ (cpdppz = 12-cyano-12,13-dihydro-11H-cyclopenta[b]dipyrido-[3,2-a:2′,3′-c]phenazine-12-carbonyl), that bound DNA with extremely high affinity and very slow dissociation kinetics.79,80,82 Additionally, Thomas and co-workers found a series of dinuclear ruthenium complexes containing tpphz bound quadruplex DNA with high affinity, even at high ionic strengths, and exhibited a blue-shifted light-switch effect.83 The beneficial effect of multi-nuclearity on DNA affinity was also well illustrated by the groove-binding dinuclear complexes [{Ru(bpy)2}2(μ-bbn)]4+ (n = 3 or 5) first investigated by Kelly and co-workers.84,85 Comparison of the dinuclear species with mononuclear analogues revealed that the dimetallic complexes had much higher DNA-binding affinities, were more efficient at photosensitising DNA strand breaks, and were less sensitive to ionic strength.84,85 Furthermore Keene, Collins and co-workers used bpm and HAT-bridged species to probe the non-duplex DNA selectivity of bulky dinuclear metal complexes. However, the dinuclear complexes bridged by a rigid planar ligand cannot follow the curvature of the minor groove unless the groove is significantly straightened by bulge-induced bending.86,87 Consequently, these workers investigated the non-duplex DNA binding of a series of complexes [{Ru(phen)2}2(μ-bbn)]4+ (where n = 2, 5, 7, 10, 12 or 16, see Fig. 6), based upon the flexibly-bridged [{Ru(bpy)2}2(μ-bbn)]4+ species described by Kelly et al. In addition, Keene and co-workers resolved the three stereoisomeric forms of the [{Ru(bpy)2}2(μ-bbn)]4+ species, and examined their binding affinity to a number of different oligonucleotide sequences and structures using a variety of techniques.88
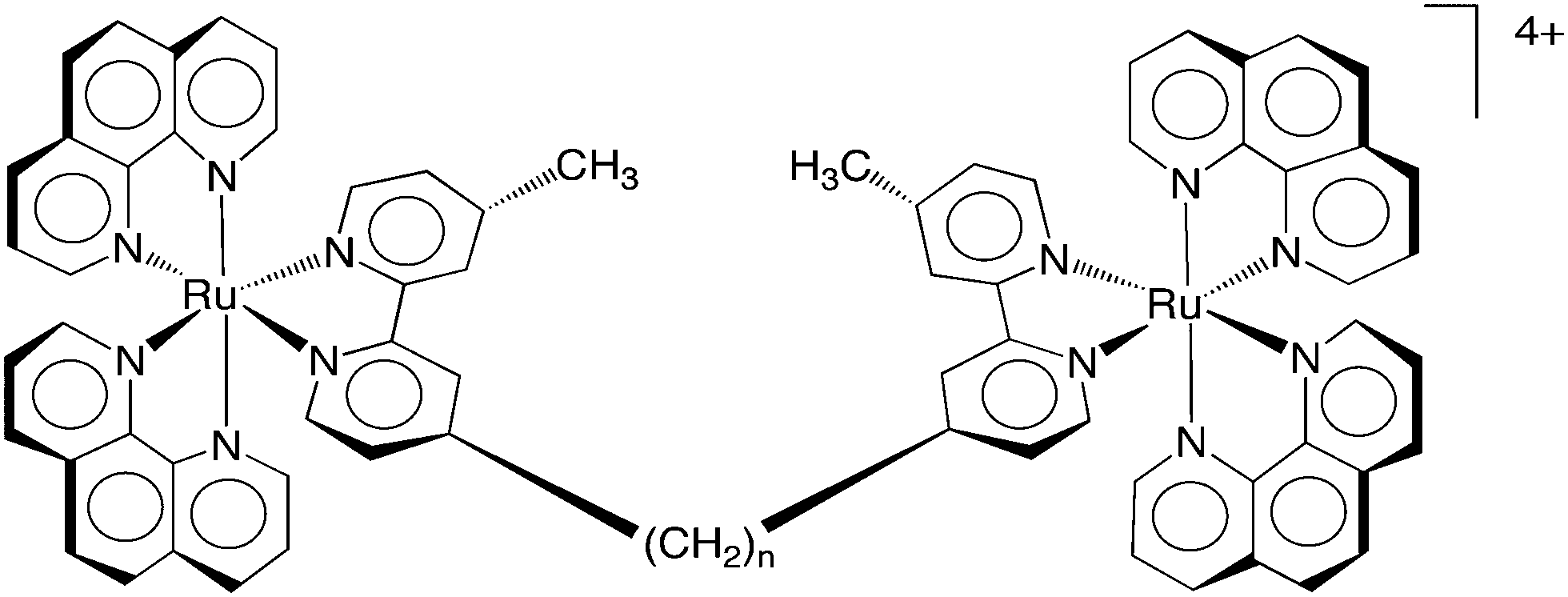 | ||
| Fig. 6 The structure of the dinuclear polypyridylruthenium(II) complexes Rubbn, where n = 2, 5, 7, 10, 12, 14 or 16. | ||
Given their DNA binding ability, the potential of the Rubbn class of complexes as novel antimicrobial agents was subsequently examined. The Rubbn complexes were highly active against a range of pathogenic bacteria, particularly Gram positive strains, e.g. see Table 3.89 In addition, preliminary toxicity experiments indicated the ruthenium complexes were significantly less toxic to eukaryotic cells (see Table 3).89 Furthermore, the Rubbn maintained their activity against drug-resistant bacteria, including strains that are of considerable current concern, e.g. MRSA and VRE. The Rubbn complexes with a longer alkane linking chain (Rubb12, Rubb14 and Rubb16) were the most active.89 Interestingly, the dinuclear complexes with a short linking chain (bb2 and bb5), a rigid polycyclic aromatic linking ligand (bpm and dppm) or those containing an ether or amine in the linking ligand, showed very little or no activity against any of the bacterial strains.89 Only slight differences in activity were observed between the ΔΔ and ΛΛ enantiomers.
a and IC50 values of four dinuclear Rubbn complexes (ΔΔ-Rubb7, ΔΔ-Rubb10, ΔΔ-Rubb12 and ΔΔ-Rubb16) against S. aureus, E. coli, red blood cells and THP-1b cells. The data was taken from ref. 89
| Bacteria | Eukaryotic cells | |||
|---|---|---|---|---|
| S. aureus | E. coli | Red blood | THP-1 | |
| Ruthenium complex | MIC [μg ml−1] | MIC [μg ml−1] | HC50 [μg ml−1] | IC50 [μg ml−1] |
| a HC50: concentration needed to induce 50% haemolysis. b THP-1 cells are a human monocytic leukemia cell line and a good model for nucleated eukaryotic cells. | ||||
| ΔΔ-Rubb7 | 16 | 16 | >1024 | 400 |
| ΔΔ-Rubb10 | 4 | 4 | 410 | 300 |
| ΔΔ-Rubb12 | 1 | 2 | 160 | 135 |
| ΔΔ-Rubb16 | 1 | 4 | 22 | 78 |
Cellular uptake studies provided a rationale for the observed relative differences in activity for the ruthenium complexes.90 Rubb16 exhibited the highest level of cellular uptake, followed by Rubb12 and then Rubb7, consistent with the trend in lipophilicity of the dinuclear complexes (logP = −1.9, −2.7 and −3.4 for Rubb16, Rubb12 and Rubb7, respectively).90 However, this correlation was not observed for the mononuclear complexes [Ru(phen)2(bb7)]2+ and [Ru(Me4phen)3]2+. Consistent with the observed MIC/MBC (MBC = minimum bactericidal concentration) values for each ruthenium complex the uptake into Gram negative bacteria was significantly less than that into Gram positive species.90 Furthermore, it was shown that the dinuclear Rubbn complexes enter bacterial cells in an energy-independent manner, and significantly depolarise and permeabilise the cellular membrane.91 Interestingly, while [Ru(Me4phen)3]2+ also depolarised the bacterial cells, there was no sign of membrane permeabilisation, again indicating a significant difference in the biological processing of this complex and the dinuclear complexes.91 It was proposed that [Ru(Me4phen)3]2+ has a different mode of cellular entry and/or different intracellular target compared to the dinuclear ruthenium complexes. Although the Rubbn complexes can permeabilise membranes, cellular localisation studies also showed that the most active compound Rubb16 preferentially binds RNA in live bacteria, accumulating at ribosomes and condensing the ribosomes when they existed as polysomes (see Fig. 7).92 The specific targeting and condensation of polysomes would halt translation, thereby interrupting protein synthesis in actively growing bacterial cells.
 | ||
| Fig. 7 Left-hand side: fluorescence microscopy image of Rubb16 localisation in E. coli at 4 μg ml−1, showing condensation of polysomes. Scale bar = 5 μm. Right-hand side: Rubb16 localisation with the image re-processed to enhance the luminescence of the Rubb16 bound to all ribosomes. Adapted from ref. 92. | ||
Consistent with the importance of lipophilicity and cellular uptake, a dinuclear ruthenium helicate complex based upon the bidentate ligand pyridyl-1,2,3-triazole – which has the same cationic charge as the Rubbn complexes but is less lipophilic – showed extremely modest antimicrobial activity.93 The authors postulated that the lack of activity was probably due to poor intracellular uptake, and proposed that increasing the hydrophobicity of the ruthenium helicate could lead to better antimicrobial activity.
Given the good antimicrobial activity exhibited by the dinuclear Rubbn complexes, the antimicrobial activities of the corresponding tri- and tetra-nuclear complexes (see Fig. 8) were also examined.94 Additionally, due to the modular nature of the synthesis of these complexes, it was possible to synthesise both linear and non-linear tetranuclear complexes. All the tri- and tetra-nuclear complexes exhibited good antimicrobial activity, with the linear Rubb12-tri, Rubb16-tri, Rubb12-tetra and Rubb16-tetra the most active compounds – up to four-times more active than the dinuclear counterparts. While the trinuclear complexes were the most lipophilic based upon logP values, the linear tetranuclear complexes were generally more active. Interestingly, although the non-linear tetranuclear complexes were slightly more lipophilic they were consistently less active than their linear counterparts.94 Although the level of cellular accumulation of the tri- and tetra-nuclear complexes in Gram negative bacteria was equal to or greater than in Gram positive species, considerably lower activity was observed against the Gram negative species. This suggested that some Gram negative species, particularly P. aeruginosa, have inherent resistance to inert polypyridyl ruthenium complexes.
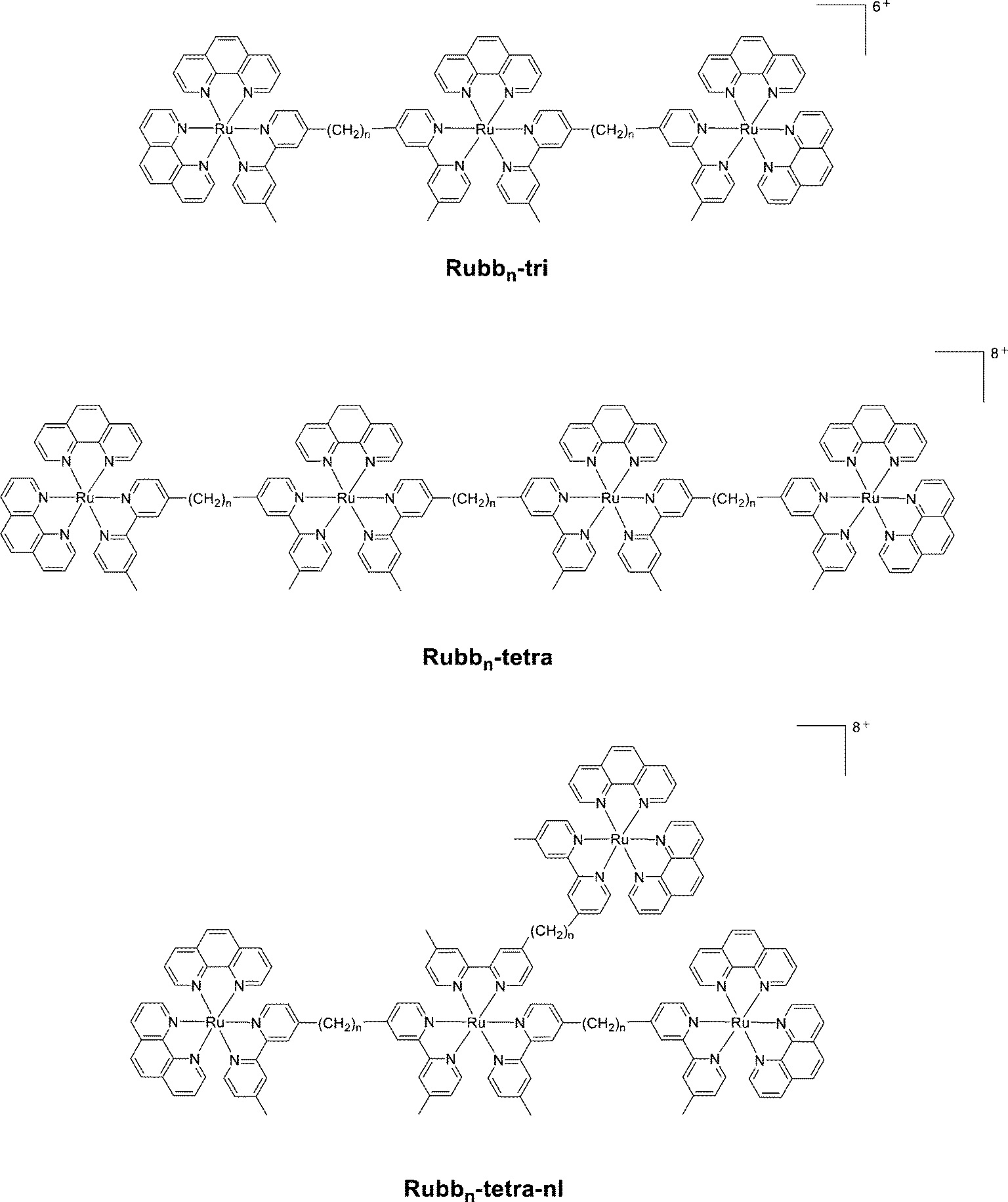 | ||
| Fig. 8 The structure of the tri- (Rubbn-tri) and tetra-nuclear (Rubbn-tetra) polypyridylruthenium(II) complexes. | ||
Considerable diversity in the cellular localisation of mononuclear complexes has been observed, even within the same basic structure. For example, Lincoln and Nordén have reported the remarkable cellular control by the length of an alkyl chain in a dppz-based complex (dppz = dipyrido[3,2-a:2′,3′-c]phenazine) – the least lipophilic species (those with the shortest alkyl chains) were found to stain nuclear DNA; the most lipophilic complexes preferably stained cellular membranes, whereas those derivatives of [Ru(phen)2(dppz)]2+ of intermediate lipophilicity selectively stained the RNA-rich nucleoli.62 Given that lipophilicity appears to play an important role in cellular localisation, appending various moieties to a ruthenium complex could modify the localisation site – and hence the biological activity of the metal complex. For example, Puckett and Barton have demonstrated that conjugating an octaarginine moiety to a dppz complex of ruthenium increased its cellular and nuclear uptake.96 However, while the cellular uptake significantly increased, for incubation at 5 μM complete exclusion from the nucleus was observed. It was concluded that the uptake mechanism had been altered by the addition of the arginine peptide – passive diffusion to endocytosis – with the ruthenium conjugate being trapped in endosomes. Interestingly, addition of fluorescein to the ruthenium–octaarginine complex redirected the conjugate to the nucleus, with nuclear fluorescence and strong nucleoli staining observed.96
In an important early study with dinuclear complexes, Onfelt et al. demonstrated that the ruthenium complex ΔΔ-[μ-c4(cpdppz)2-(phen)4Ru2]4+ can be used as a nuclear stain in live cells, although electroporation of the V79 Chinese hamster cells was required for the ruthenium complex to bind nuclear DNA.82 The DNA imaging potential of another dinuclear polypyridylruthenium(II) complex has been demonstrated by Thomas and co-workers – [{Ru(phen)2}2(tpphz)]4+ (see Fig. 9).58,61 Interestingly, the more lipophilic complex 4,7-diphenyl-1,10-phenanthroline analogue [{Ru(DIP)2}2(μ-tpphz)]4+ localised in the endoplasmic reticulum.97 More recently, Thomas and co-workers reported a study of the cellular uptake and localisation of iridium(III)–ruthenium(II) dinuclear complexes bridged by the tpphz ligand.98 The water soluble cyclometalated complexes [Ir(ppy)2(tpphz)Ru(bpy)2]3+ and [Ir(F2ppy)2(tpphz)Ru(bpy)2]3+ {ppy = 2-phenyl-pyridine and F2ppy = 2-(4-fluorophenyl)pyridine} were rapidly internalised in HeLa cells and localised in the nucleus. This study is particularly significant in that nuclear localisation was maintained even though the [Ir(III)–Ru(II)]3+ complexes are more lipophilic (and hence possess superior uptake) than the corresponding [Ru(II)–Ru(II)]4+ complexes. As noted by the authors,98 generally the addition of hydrophobic groups to improve cellular uptake leads to localisation in hydrophobic regions (e.g. membrane structures) rather than in the nucleus – as observed for [{Ru(DIP)2}2(μ-tpphz)]4+.97
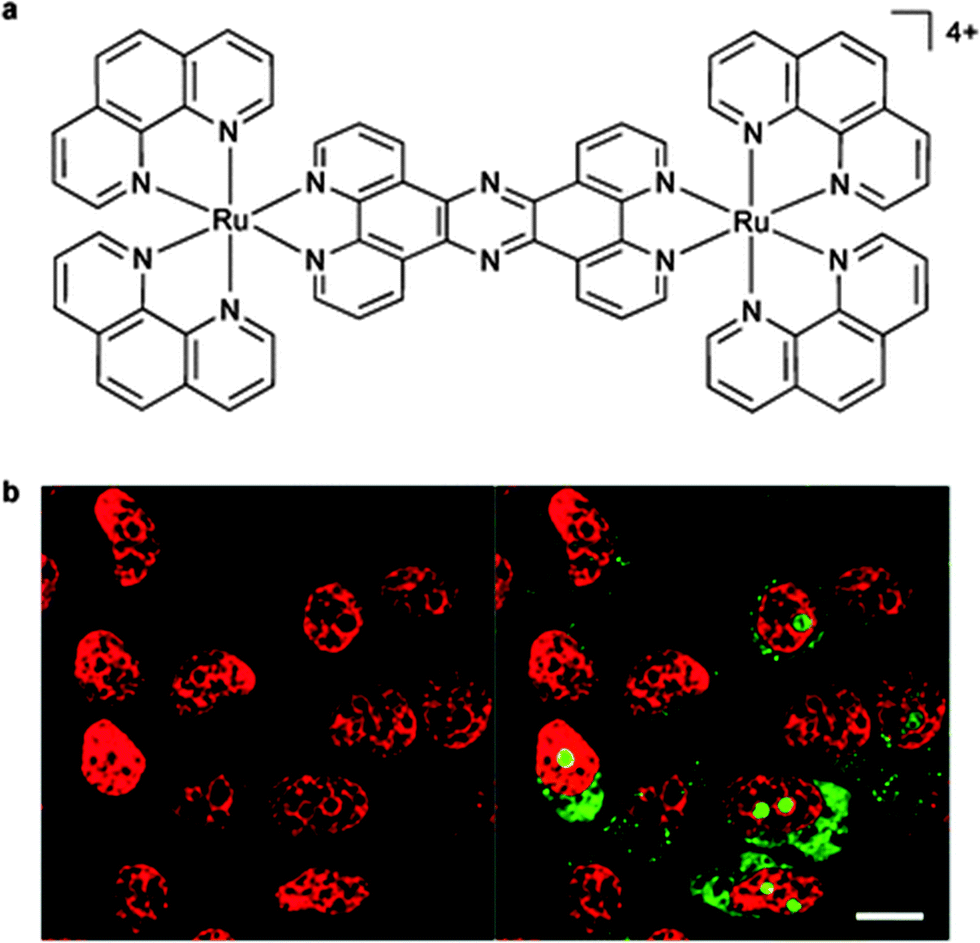 | ||
| Fig. 9 The structure of [(Ru(phen)2)2(tpphz)]4+ (a); and (b) the nuclear DNA staining of the dinuclear complex in MCF-7 cells, as evident by the red luminescence, with co-staining by the general nucleic acid stain SYTO-9 shown in green (highlighting the nucleoli). Adapted with permission from ref. 58. | ||
By contrast, the [{Ru(phen)2}2(μ-bbn)]4+ dinuclear complexes were predominantly taken up by passive diffusion through the cell membrane in L1210 murine leukaemia cells, with a minor contribution from an active structure-specific, non-endocytotic mechanism.99 Confocal microscopy was used to show that the complexes with n = 12, 14 and 16 accumulated exclusively in the mitochondria.99 However, a study of the intracellular localisation of the [{Ru(phen)2}2(μ-bbn)]4+ complexes with organ (liver and kidney) cells lines showed a high degree of selectivity for the nucleus of the eukaryotic cells (see Fig. 10).100 Additional co-localisation experiments with SYTO 9, a general nucleic acid stain, indicated that the ruthenium complexes showed a considerable preference for the RNA-rich nucleolus. However, while the ruthenium complexes exhibited a preference for the nucleoli, significant general DNA binding within the nucleus was also observed when the ruthenium complex was incubated at 50 μM. No significant differences were observed in the intracellular localisation between the ΔΔ and ΛΛ enantiomers of the dinuclear complex. Interestingly, despite targeting the RNA-rich regions in both bacteria (ribosomes) and organ cells (nucleoli), the Rubbn complexes do exhibit a considerable degree of selective toxicity towards bacteria.100 As shown in Table 4, Rubb12 and Rubb12-tetra are significantly more toxic to the Gram positive bacterium S. aureus and the Gram negative species E. coli compared to three eukaryotic cell lines when assayed over similar time frames.
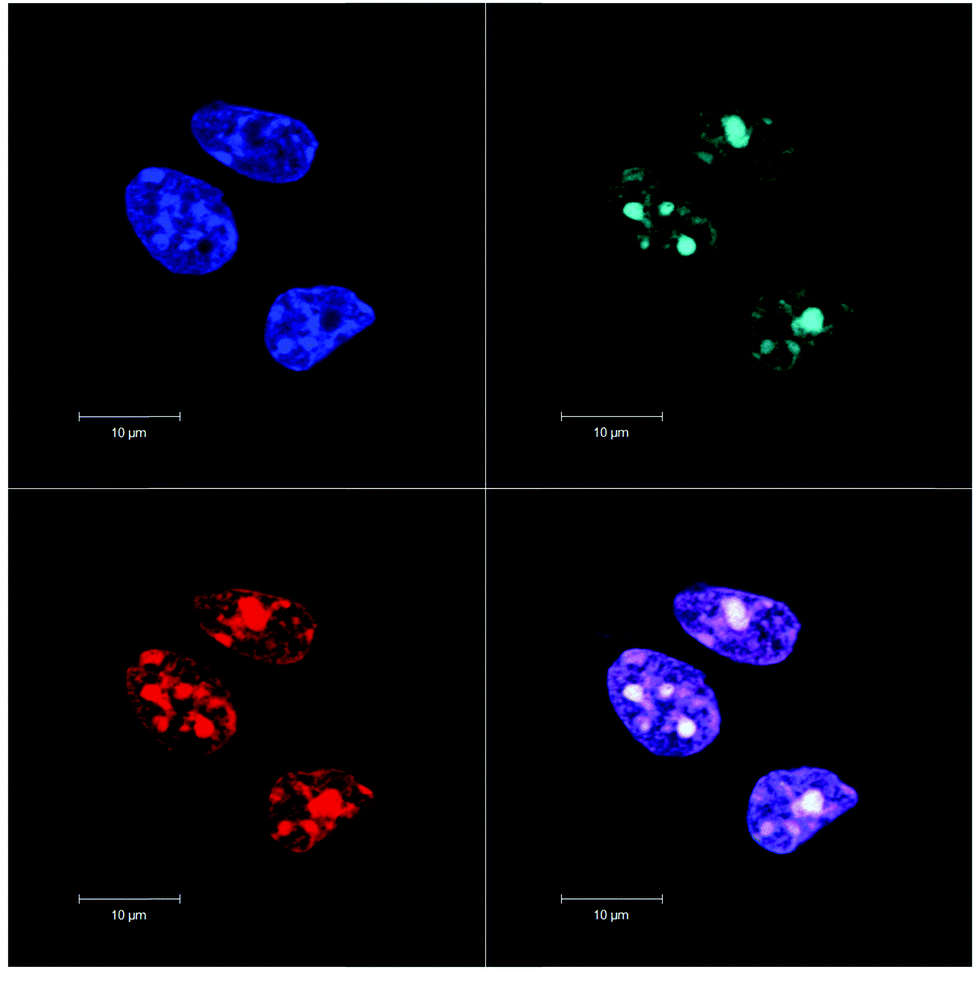 | ||
| Fig. 10 Rubb12 localisation in BHK cells at 10 μM, stained by DAPI (blue), SYTO 9 (light blue), ΔΔ-Rubb12 (red) and merged (bottom right), where white is co-localisation of SYTO 9 and ΔΔ-Rubb12 and magenta is co-localisation of DAPI and ΔΔ-Rubb12. Scale bar = 10 μm. Adapted from ref. 100. | ||
| BHK | HEK-293 | Hep-G2 | S. aureus | E. coli | |
|---|---|---|---|---|---|
| a BHK = baby hamster kidney, HEK-93 = human embryonic kidney and Hep-G2 = liver carcinoma. | |||||
| Rubb12 | 70.5 | 50.9 | 61.7 | 0.6 | 2.5 |
| Rubb12-tetra | 27.7 | 21.7 | 33.8 | 0.3 | 1.2 |
4.2 Ruthenium complexes with labile ligands
Ruthenium complexes were initially developed as lower toxic alternatives to the platinum anticancer complexes. The activities of amminechloridoruthenium(III) complexes, such as cis-[RuCl2(NH3)4]Cl, trans-[RuCl4(Im)2](HIm) (Im = imidazole), and fac-[RuCl3(NH3)3] were explored initially and this class of complexes showed good anticancer activity.101In vivo studies with tumour-bearing animals demonstrated that trans-[RuCl4(Im)2](HIm) exhibited good activity against platinum-resistant colorectal tumours. Another class of ruthenium complexes with labile ligands, based on chloridodimethylsulfoxideruthenium(II), were shown to be significantly less toxic than the platinum complexes;101 the dimethylsulfoxide ligands of these complexes improved their selectivity towards tumour metastases, although they also reduced their activity. The most successful examples of ruthenium complexes with labile ligands as anticancer agents are imidazolium trans-imidazoledimethysulfoxidetetrachloridoruthenate (NAMI-A) and indazolium bis-indazoletetrachloridoruthenate (KP1019) (see Fig. 3e, f), which have entered clinical trials.102 Interestingly, although in general the anticancer activity of metal complexes is due to their DNA binding property, NAMI-A acts as an inhibitor of the metastatic potential of tumours and the activity is not related with DNA binding.101A family of Schiff base ruthenium(III) complexes, [RuX(η3-Schiff)(Eph3)2] (Eph3 = triphenylphosphine/arsine, X = Cl or Br), containing labile ligands showed better antibacterial activity than their parent ligands against B. subtilis and E. coli.103 However, only moderate inhibition was observed using the disc diffusion method at relatively high concentrations. The antimicrobial activity of a series of ruthenium complexes with PTA (1,3,5-triaza-7-phosphaadamantane) ligands and various labile ligands (Cl−, Br−, I− or SCN−) were also tested. The complexes with Cl− or SCN− exhibited antifungal activity while those with Br− or I− were found to be inactive.104,105 More encouragingly, chiral ruthenium(II) salen complexes containing DMSO ligands showed good activity (MIC = 12 to 25 μg ml−1) against Gram positive bacteria, but were inactive against Gram negative species.106 Interestingly, the S enantiomer exhibited better activity than the R enantiomer. In another approach, Kamatchi et al. synthesised several organometallic ruthenium(II) complexes [Ru(HL)(CH3CN)(CO)(EPh3)2] (where HL = 4-oxo-4H-pyran-2,6-dicarboxylic acid and E = As or P) and examined their antimicrobial activity.107 However, while the ruthenium complexes were more active than their parent ligands, they only exhibited moderate activity (MIC ≥ 25 μg ml−1) against a range of bacteria.
A series of dinuclear polypyridylruthenium(II) complexes containing labile chlorido ligands, [{Ru(tpy)Cl}2(μ-bbn)]2+ {Cl–Rubbn; where tpy = 2,2′:6′,2′′-terpyridine and bbn = bis[4(4′-methyl-2,2′-bipyridyl)]-1,n-alkane – see Fig. 11} was examined for antimicrobial activity.108 These labile dinuclear complexes showed good activity, with MIC values for the Cl–Rubb12 complex of 1 μg ml−1 against two Gram positive bacteria (including MRSA) and 2 and 8 μg ml−1 against several Gram negative species.108 Interestingly, the toxicity of the Cl–Rubbn complexes to bacteria increased with increasing methylene groups in the linking ligand up to n = 12, but then decreased for the Cl–Rubb16 complex. It was proposed that, compared to the corresponding inert complexes [{Ru(phen)2}2(μ-bbn)]4+ (Rubbn), the inclusion of the chlorido group on each metal centre increased the cellular uptake but decreased the ability of the ruthenium complexes to kill bacteria.108
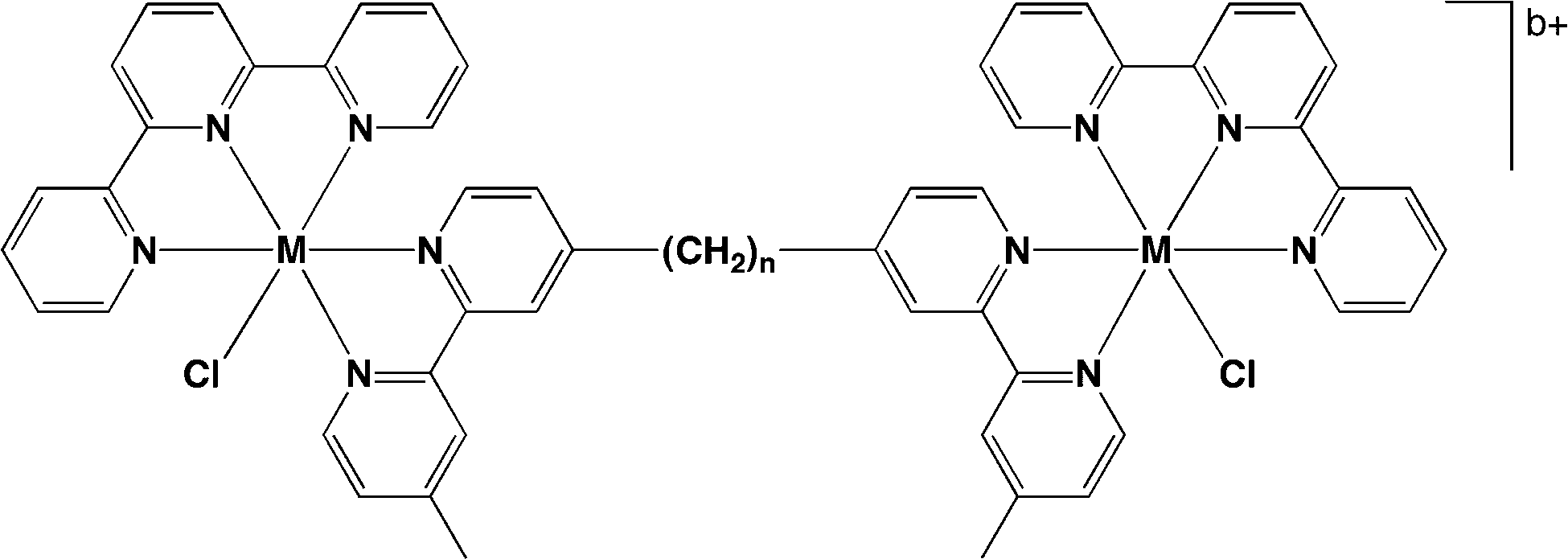 | ||
| Fig. 11 Structure of chlorido-containing dinuclear metal complexes Cl–Rubbn (M = Ru, b = 2). Iridium analogues Cl–Irbbn (M = Ir, b = 4) were also prepared and exhibited good antimicrobial activity.108 | ||
5. Conclusions and future perspectives
It is clear that new antimicrobial agents are needed, but paradoxically, there are fewer drugs in the development “pipeline” than a decade ago. Although research into new organic-based antimicrobial agents continues, there is now growing interest in metal-based drugs. Due to the shifting of interest away from platinum towards ruthenium for the development of novel anticancer compounds, there is now also considerable focus developing on ruthenium(II) complexes as antimicrobial agents. In general, ruthenium(II) complexes have shown good activity towards Gram positive bacteria (e.g. S. aureus and MRSA), but lower activity to Gram negative species (e.g. E. coli and P. aeruginosa). As would be expected, the antimicrobial activity of a ruthenium complex is a function of lipophilicity, charge and charge separation. However, for an antimicrobial to have clinical potential it must also be relatively non-toxic to humans and animals. Simply increasing lipophilicity and charge simultaneously is likely to produce ruthenium complexes that are more active in both bacteria and eukaryotic cells. Given the modular design and general ease with which the structure of the ruthenium complexes can be modified, the aim now becomes preparing new ruthenium complexes that exhibit high levels of selective toxicity towards bacteria.While polycations would generally be toxic to cells, differences in the membrane composition between bacteria and eukaryotic cells can provide a degree of selectivity. Ruthenium complexes could selectively target bacterial cells due to the greater presence of negatively-charged components (phospholipids, such as phosphatidyl-glycerol, teichoic acids and lipopolysaccharides) in the bacterial membrane and cell wall.109 In contrast, the high content of zwitterionic phosphatidylcholine in the outer membrane leaflet of healthy eukaryotic cells confers an overall neutral charge on these cells that results in a greatly reduced capacity for electrostatic interactions. However, and more generally, the relative hydrophilicity and high charge of some of the ruthenium complexes would suggest that the metal complex could not freely diffuse across the bacterial membrane. Hence, there is a need to understand the interaction of the ruthenium complexes with lipid bilayers. Liposomes are a good model for biological membranes and have been extensively used to study drug interactions with phospholipid bilayers,110 and there are a variety of well established procedures to examine the interactions, e.g. inductively-coupled plasma spectroscopy, differential scanning calorimetry and NMR spectroscopy.110
DNA binding is generally suggested or implied as the possible intra-cellular target for ruthenium complexes, particularly inert polypyridyl species, with significant antimicrobial activity. However, toxicity is a major concern for the DNA-targeting metal complexes. Due to the lack of variation in the structure of DNA between bacteria and eukaryotic cells, DNA is unlikely to provide the selectivity required for development of a clinically-useful antimicrobial drug. RNA is more structurally rich than DNA: RNA contains a larger proportion of non-duplex type structures, forming complex three-dimensional structures comprising of loops, bulges, pseudo knots and turns. In addition, viral and bacterial RNA often have ‘unusual’ sequences and folds that could be specifically targeted.111 Consequently, RNA may provide a better target than DNA for the development of new ruthenium-based antimicrobials. In support of this notion, studies have demonstrated that a variety of mono- and di-nuclear polypyridylruthenium complexes preferentially target RNA over DNA, localising in nucleoli in eukaryotic cells and ribosomes in bacteria. Ribosomes are a particularly attractive target for ruthenium complexes, given the significant differences between eukaryotic and bacterial ribosomes. As noted earlier, a variety of organic-based drugs target bacterial ribosomes.
While a variety of ruthenium complexes have demonstrated good in vitro antimicrobial activity, and in some cases significantly less toxicity to eukaryotic cells,63,89,100 the clinical potential is related to the in vivo activity. Before an in vivo study using mice infected by bacteria can be carried out, it is necessary to determine the toxicity of the ruthenium complexes. Pharmacokinetic studies, where the concentration of the metal complex in serum and various organs are determined as a function of time after administration, are also required. However, once an understanding of pharmacokinetics is obtained, it is likely that ruthenium(II) complexes can be designed that have selective toxicity for bacteria and good residence time in human serum after administration. As previously noted, the lipophilicity, charge and charge separation can be relatively easily controlled; and due to the rigid octahedral geometry of the ruthenium complexes, optimising binding affinity at cellular target sites may be easier than for purely organic-based drugs. Furthermore, it seems more likely that a completely new mechanism of antimicrobial activity will be obtained with a ruthenium(II) complex, compared to organic molecules which have been the major class of compounds studied since the development of penicillin.112
References
- L. Seifert, M. Harbeck, A. Thomas, N. Hoke, L. Zöller, I. Wiechmann, G. Grupe, H. C. Scholz and J. M. Riehm, PLoS One, 2013, 8, e75742 CAS
.
- O. Ruuskanen, E. Lahti, L. C. Jennings and D. R. Murdoch, Lancet, 2011, 377, 1264 CrossRef
- WHO Tuberculosis Factsheet, World Health Organization, March 2010.
- The terms ‘antimicrobials’, ‘antibacterials’ and ‘antibiotics’ are sometimes incorrectly used interchangeably, and the differences should be noted. Antimicrobial is a generic term for any compound with a direct action on microorganisms and used for treatment or prevention of infections. Accordingly, antimicrobials will include antibacterials, antivirals, antifungals, antiprotozoals, and antiparasitics. By strict definition, the term antibiotic refers to substances produced by microorganisms that act against other microorganisms – which therefore does not include antimicrobial substances that are synthetic, semi-synthetic or derived from plant or animal sources. The term antibacterial describes such substances that act against bacteria. Consequently, all antibiotics and antibacterials are antimicrobials – but not all antimicrobials are antibiotics or antibacterials.
- H. W. Boucher, G. H. Talbot, J. S. Bradley, J. E. Edwards, D. Gilbert, L. B. Rice, M. Scheld, B. Spellberg and J. Bartlett, Clin. Infect. Dis., 2009, 48, 1 CrossRef PubMed
- N. Okamoto and G. I. McFadden, Future Microbiol., 2008, 3, 391 CrossRef CAS
-
L. M. Elson and I. E. Alcamo, The Microbiology Coloring Book, Addison-Wesley Longman, New York, 1995 Search PubMed
-
A. R. Freitas Jr. and R. C. Merkle, Kinematic Self-Replicating Machines, Landes Bioscience, Georgetown, TX, 2004 Search PubMed
-
M. Schaechter, N. C. Engleberg, B. I. Eisenstein and G. Medoff, Mechanisms of Microbial Disease, Lippincott Williams & Wilkins, Baltimore, USA, 3rd edn, 1999 Search PubMed
-
R. Goering, H. Dockrell, M. Zuckerman, I. Roitt and P. Chiodini, Mims' Medical Microbiology, Saunders, Edinburgh, UK, 5th edn, 2012 Search PubMed
-
G. Lipps, Plasmids: Current Research and Future Trends, Caister Academic Press, Norfolk, UK, 2008 Search PubMed
- I. D. Algranati, N. S. Gonzalez and E. G. Bade, Proc. Natl. Acad. Sci. U. S. A., 1969, 62, 574 CrossRef CAS
- V. Ramakrishnan, Biochem. Soc. Trans., 2008, 36, 567 CrossRef CAS PubMed
- L. A. Phillips and R. M. Franklin, Cold Spring Harbor Symp. Quant. Biol., 1969, 34, 243 CrossRef CAS
- R. M. Epand and R. F. Epand, Biochim. Biophys. Acta, 2009, 1788, 289 CrossRef CAS PubMed
- P. L. Graumann and H. J. D. Soufo, BioEssays, 2004, 26, 1209 CrossRef CAS PubMed
-
E. Goldman and L. H. Green, Practical Handbook of Microbiology, CRC Press, Taylor & Francis Group, Boca Raton, 2nd edn, 2008 Search PubMed
- C. W. M. Haest, A. J. Verkleij, J. De Gier, R. Scheek, P. H. J. Ververgaert and L. L. M. Van Deenen, Biochim. Biophys. Acta, 1974, 356, 17 CrossRef CAS
- G. M. de Tejada, S. Sanchez-Gomez, I. Razquin-Olazaran, I. Kowalski, Y. Kaconis, L. Heinbockel, J. Andra, T. Schurholz, M. Hornef, A. Dupont, P. Garidel, K. Lohner, T. Gutsmann, S. A. David and K. Brandenburg, Curr. Drug Targets, 2012, 13, 1121 CrossRef
- C. Erridge, E. Bennett-Guerrero and I. R. Poxton, Microbes Infect., 2002, 4, 837 CrossRef CAS
- D. S. Snyder and T. J. Mclntosh, Biochemistry, 2000, 39, 11777 CrossRef CAS PubMed
- M. W. Gray, G. Burger and B. F. Lang, Genome Biol., 2001, 2, 1018.1 CrossRef
-
A. de J. Sosa, D. K. Byarugaba, C. Amabile, P.-R. Hsueh, S. Kariuki and I. N. Okeke, Antimicrobial Resistance in Developing Countries, Springer, New York, USA, 2009 Search PubMed
- M. C. McManus, Am. J. Health-Syst. Pharm., 1997, 54, 1420 CAS
- J. Cremniter, J.-L. Mainardi, N. Josseaume, J.-C. Quincampoix, L. Dubost, J.-E. Hugonnet, A. Marie, L. Gutmann, L. B. Rice and M. Arthur, J. Biol. Chem., 2006, 281, 32254 CrossRef CAS PubMed
- L. Zhang, P. Dhillon, H. Yan, S. Farmer and R. E. W. Hancock, Antimicrob. Agents Chemother., 2000, 44, 3317 CrossRef CAS
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2010, 53, 1898 CrossRef CAS PubMed
- J. D. F. Hale and R. E. W. Hancock, Expert Rev. Anti-Infect. Ther., 2007, 5, 951 CrossRef CAS PubMed
- G. Cheng, H. Hao, M. Dai, Z. Liu and Z. Yuan, Eur. J. Med. Chem., 2013, 66, 555 CrossRef CAS PubMed
- E. A. Capmbell, N. Korzheva, A. Mustaev, K. Murakami, S. Nair, A. Goldfarb and S. A. Darst, Cell, 2001, 104, 901 CrossRef
- A. Feklistov, V. Mekler, Q. Jiang, L. F. Westblade, H. Irschik, R. Jansen, A. Mustaev, S. A. Darst and R. H. Ebright, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14820 CrossRef CAS PubMed
- G. M. Brown, J. Biol. Chem., 1962, 237, 536 CAS
- S. Yoshizawa, D. Fourmy and J. D. Puglisi, EMBO J., 1998, 17, 6437 CrossRef CAS PubMed
- J. Poehlsgaard and S. Douthwaite, Nat. Rev. Microbiol., 2005, 3, 870 CrossRef CAS PubMed
- R. E. Stanley, G. Blaha, R. L. Grodzicki, M. D. Strickler and T. A. Steitz, Nat. Struct. Mol. Biol., 2010, 17, 289 CAS
-
L. B. Rice and R. A. Bonomo, in Manual of Clinical Microbiology, ed. J. Versalovic, ASM Press, Washington, DC, 10th edn, 2011, vol. 1, pp. 1082–1114 Search PubMed
- M. C. McManus, Am. J. Health-Syst. Pharm., 1997, 54, 1420 CAS
- L. L. Silver and K. A. Bostian, Antimicrob. Agents Chemother., 1993, 37, 377 CrossRef CAS
- R. I. Aminov, Front. Microbiol., 2010, 1, 134 Search PubMed
- E. Wong and C. M. Giandomenico, Chem. Rev., 1999, 99, 2451 CrossRef CAS PubMed
- A. C. Komor and J. K. Barton, Chem. Commun., 2013, 49, 3617 RSC
- K. B. Garbutcheon-Singh, M. P. Grant, B. W. Harper, A. M. Krause-Heuer, M. Manohar, N. Orkey and J. R. Aldrich-Wright, Curr. Top. Med. Chem., 2011, 11, 521 CrossRef CAS
- S. Rafique, M. Idrees, A. Nasim, H. Akbar and A. Athar, Biotechnol. Mol. Biol. Rev., 2010, 5, 38 CAS
-
V. Bobbarala, A Search for Antibacterial Agents, InTech Press, Rijeka, Croatia, 2012 Search PubMed
- M. D. Rhodes, P. J. Sadler, M. D. Scawen and S. Silver, J. Inorg. Biochem., 1992, 46, 129 CrossRef CAS
- A. D. Richards, A. Rodger, M. J. Hannon and A. Bolhuis, Int. J. Antimicrob. Agents, 2009, 33, 469 CrossRef CAS PubMed
- N. S. Ng, P. Leverett, D. E. Hibbs, Q. Yang, J. C. Bulanadi, M. J. Wu and J. R. Aldrich-Wright, Dalton Trans., 2013, 42, 3196 RSC
- M. A. Neelakantan, M. Esakkiammal, S. S. Mariappan, J. Dharmaraja and T. Jeyakumar, Indian J. Pharm. Sci., 2010, 72, 216 CrossRef CAS PubMed
- B. S. Sekhon, J. Pharm. Educ. Res., 2010, 1, 1 Search PubMed
- L. J. Ming, Med. Res. Rev., 2003, 23, 697 CrossRef CAS PubMed
- N. Metzler-Nolte, Top. Organomet. Chem., 2010, 32, 195 CrossRef CAS
- C. S. Allardyce and P. J. Dyson, Platinum Met. Rev., 2001, 45, 62 CAS
- B. Nordén, P. Lincoln, B. Akerman and E. Tuite, Met. Ions Biol. Syst., 1996, 33, 177 Search PubMed
- N. W. Luedtke, J. S. Hwang, E. Nava, D. Gut, M. Kol and Y. Tor, Nucleic Acids Res., 2003, 31, 5732 CrossRef CAS PubMed
- C. Metcalfe and J. A. Thomas, Chem. Soc. Rev., 2003, 32, 215 RSC
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565 RSC
- F. R. Keene, J. A. Smith and J. G. Collins, Coord. Chem. Rev., 2009, 253, 2021 CrossRef CAS PubMed
- M. R. Gill and J. A. Thomas, Chem. Soc. Rev., 2012, 41, 3179 RSC
-
F. P. Dwyer and D. P. Mellor, Chelating Agents and Metal Chelates, Academic Press, New York, 1964 Search PubMed
- C. A. Puckett and J. K. Barton, Biochemistry, 2008, 47, 11711 CrossRef CAS PubMed
- M. R. Gill, J. Garcia-Lara, S. J. Foster, C. Smythe, G. Battaglia and J. A. Thomas, Nat. Chem., 2009, 1, 662 CrossRef CAS PubMed
- M. Matson, F. R. Svensson, B. Nordén and P. Lincoln, J. Phys. Chem. B, 2011, 115, 1706 CrossRef CAS PubMed
- A. Bolhuis, L. Hand, J. E. Marshall, A. D. Richards, A. Rodger and J. Aldrich-Wright, Eur. J. Pharm. Sci., 2011, 42, 313 CrossRef CAS PubMed
- A. M. Pizarro and P. J. Sadler, Biochimie, 2009, 91, 1198 CrossRef CAS PubMed
- H.-K. Liu and P. J. Sadler, Acc. Chem. Res., 2011, 5, 349 CrossRef PubMed
- V. Brabec and O. Nováková, Drug Resist. Updates, 2006, 9, 111 CrossRef CAS PubMed
- C. Moucheron, New J. Chem., 2009, 33, 235 RSC
- E. Meggers, Chem. Commun., 2009, 1001 RSC
- S. K. Singh and D. S. Pandey, RSC Adv., 2014, 4, 1819 RSC
- J. Maksimoska, L. Feng, K. Harms, C. Yi, J. Kissil, R. Marmorstein and E. Meggers, J. Am. Chem. Soc., 2008, 130, 15764 CrossRef CAS PubMed
- F. P. Dwyer, I. K. Reid, A. Shulman, G. M. Laycock and S. Dixson, Aust. J. Exp. Biol. Med. Sci., 1969, 47, 203 CrossRef CAS
- F. P. Dwyer, E. C. Gyarfas, W. P. Rogers and J. H. Koch, Nature, 1952, 170, 190 CrossRef CAS
- C. S. Devi, D. A. Kumar, S. S. Singh, N. Gabra, N. Deepika, Y. P. Kumar and S. Satyanarayana, Eur. J. Med. Chem., 2013, 64, 410 CrossRef PubMed
- K. A. Kumar, K. L. Reddy, S. Vidhisha and S. Satyanarayana, Appl. Organomet. Chem., 2009, 23, 409 CrossRef CAS
- P.-L. Lam, G.-L. Lu, K.-M. Hon, K.-W. Lee, C.-L. Ho, X. Wang, J. C.-O. Tang, K.-H. Lam, R. S.-M. Wong, S. H.-L. Kok, Z.-X. Bian, H. Li, K. K.-H. Lee, R. Gambari, C.-H. Chui and W.-Y. Wong, Dalton Trans., 2014, 43, 3949 RSC
- R. F. Donnelly, N. C. Fletcher, P. J. McCauge, J. Donnelly, P. A. McCarron and M. M. Tunney, Lett. Drug Des. Discovery, 2007, 4, 175 CrossRef CAS
- J. Aldrich-Wright, C. Brodie, E. C. Glazer, N. W. Luedtke, L. Elson-Schwaband and Y. Tor, Chem. Commun., 2004, 1018 RSC
- S. Delaney, M. Pascaly, P. K. Bhattacharya, K. Han and J. K. Barton, Inorg. Chem., 2002, 41, 1966 CrossRef CAS PubMed
- P. Lincoln and B. Nordén, Chem. Commun., 1996, 2145 RSC
- P. Nordell and P. Lincoln, J. Am. Chem. Soc., 2005, 127, 9670 CrossRef CAS PubMed
- S. A. Tysoe, R. Kopelman and D. Schelzig, Inorg. Chem., 1999, 38, 5196 CrossRef CAS
- B. Onfelt, L. Gostring, P. Lincoln, B. Nordén and A. Onfelt, Mutagenesis, 2002, 17, 317 CrossRef CAS
- C. Rajput, R. Rutkaite, L. Swanson, I. Haq and J. A. Thomas, Chem. – Eur. J., 2006, 12, 4611 CrossRef CAS PubMed
- F. O'Reilly, J. Kelly and A. Kirsch-De Mesmaeker, Chem. Commun., 1996, 1013 RSC
- F. M. O'Reilly and J. M. Kelly, J. Phys. Chem. B, 2000, 104, 7206 CrossRef
- J. A. Smith, J. L. Morgan, A. G. Turley, J. G. Collins and F. R. Keene, Dalton Trans., 2006, 3179 RSC
- D. P. Buck, C. B. Spillane, J. G. Collins and F. R. Keene, Mol. BioSyst., 2008, 4, 851 RSC
- J. L. Morgan, C. B. Spillane, J. A. Smith, D. P. Buck, J. G. Collins and F. R. Keene, Dalton Trans., 2007, 4333 RSC
- F. Li, Y. Mulyana, M. Feterl, J. Warner, J. G. Collins and F. R. Keene, Dalton Trans., 2011, 40, 5032 RSC
- F. Li, M. Feterl, Y. Mulyana, J. M. Warner, J. G. Collins and F. R. Keene, J. Antimicrob. Chemother., 2012, 67, 2686 CrossRef CAS PubMed
- F. Li, M. Feterl, J. M. Warner, F. R. Keene and J. G. Collins, J. Antimicrob. Chemother., 2013, 68, 2825 CrossRef CAS PubMed
- F. Li, E. J. Harry, A. L. Bottomley, M. D. Edstein, G. W. Birrell, C. E. Woodward, F. R. Keene and J. G. Collins, Chem. Sci., 2014, 5, 685 RSC
- S. V. Kumar, W. K. C. Lo, H. J. L. Brooks and J. D. Crowley, Inorg. Chim. Acta, 2015, 425, 1 CrossRef CAS PubMed
- A. K. Gorle, M. Feterl, J. M. Warner, L. Wallace, F. R. Keene and J. G. Collins, Dalton Trans., 2014, 43, 16713 RSC
- C. A. Puckett, R. J. Ernst and J. K. Barton, Dalton Trans., 2010, 39, 1159 RSC
- C. A. Puckett and J. K. Barton, J. Am. Chem. Soc., 2009, 131, 8738 CrossRef CAS PubMed
- M. R. Gill, D. Cecchin, M. H. Walker, R. S. Mulla, G. Battaglia, C. Smythe and J. A. Thomas, Chem. Sci., 2013, 4, 4512 RSC
- A. Wang, M. R. Gill, D. Turton, H. Adams, T. M. Roseveare, C. Smythe, X. Su and J. A. Thomas, Chem. – Eur. J., 2014, 20, 14004 CrossRef PubMed
- M. J. Pisani, P. D. Fromm, Y. Mulyana, R. J. Clarke, H. Korner, K. Heimann, J. G. Collins and F. R. Keene, ChemMedChem, 2011, 6, 848 CrossRef CAS PubMed
- X. Li, A. K. Gorle, T. D. Ainsworth, K. Heimann, C. E. Woodward, J. G. Collins and F. R. Keene, Dalton Trans., 2015, 44, 3594 RSC
- K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Young, Chem. Rev., 2009, 109, 3859 CrossRef CAS PubMed
- A. Bergamo and G. Sava, Dalton Trans., 2007, 1267 RSC
- N. Thilagavath, A. Manimaran and C. Jayabalakrishnan, J. Coord. Chem., 2010, 63, 1252 CrossRef
- A. I. Ramos, T. M. Braga and S. S. Braga, Mini-Rev. Med. Chem., 2012, 12, 227 CrossRef CAS
- C. S. Allardyce, P. J. Dyson, D. J. Ellis, P. A. Salter and R. Scopelliti, J. Organomet. Chem., 2003, 668, 35 CrossRef CAS
- N. H. Khan, N. Pandya, R. I. Kureshy, S. H. R. Abdi, S. Agrawal, H. C. Bajaj, J. Pandya and A. Gupte, Spectrochim. Acta, Part A, 2009, 74, 113 CrossRef PubMed
- T. S. Kamatchi, P. Kalaivani, P. Poornima, V. V. Padma, F. R. Fronczek and K. Natarajan, RSC Adv., 2014, 4, 2004 RSC
- M. Pandrala, F. Li, M. Feterl, Y. Mulyana, J. M. Warner, L. Wallace, F. R. Keene and J. G. Collins, Dalton Trans., 2013, 42, 4686 RSC
- A. J. Mason, A. Marquette and B. Bechinger, Biophys. J., 2007, 93, 4289 CrossRef CAS PubMed
- Q. Liu, Y. Qu, R. Van Antwerpen and N. Farrell, Biochemistry, 2006, 45, 4248 CrossRef CAS PubMed
- J. Petruska, N. Arnheim and M. F. Goodman, Nucleic Acids Res., 1996, 24, 1992 CrossRef CAS PubMed
- A. Fleming, Br. J. Exp. Pathol., 1929, 10, 226 CAS
| This journal is © The Royal Society of Chemistry 2015 |