
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent progress in theoretical and computational investigations of Li-ion battery materials and electrolytes
Mahesh Datt
Bhatt
ab and
Colm
O'Dwyer
*ab
aDepartment of Chemistry, University College Cork, Cork, Ireland. E-mail: c.odwyer@ucc.ie; Fax: +353 (0)21 4274097; Tel: +353 (0)21 4902732
bTyndall National Institute, Lee Maltings, Cork, Ireland
First published on 14th January 2015
Abstract
There is an increasing worldwide demand for high energy density batteries. In recent years, rechargeable Li-ion batteries have become important power sources, and their performance gains are driving the adoption of electrical vehicles (EV) as viable alternatives to combustion engines. The exploration of new Li-ion battery materials is an important focus of materials scientists and computational physicists and chemists throughout the world. The practical applications of Li-ion batteries and emerging alternatives may not be limited to portable electronic devices and circumventing hurdles that include range anxiety and safety among others, to their widespread adoption in EV applications in the future requires new electrode materials and a fuller understanding of how the materials and the electrolyte chemistries behave. Since this field is advancing rapidly and attracting an increasing number of researchers, it is crucial to summarise the current progress and the key scientific challenges related to Li-ion batteries from theoretical point of view. Computational prediction of ideal compounds is the focus of several large consortia, and a leading methodology in designing materials and electrolytes optimized for function, including those for Li-ion batteries. In this Perspective, we review the key aspects of Li-ion batteries from theoretical perspectives: the working principles of Li-ion batteries, the cathodes, anodes, and electrolyte solutions that are the current state of the art, and future research directions for advanced Li-ion batteries based on computational materials and electrolyte design.
 Mahesh Datt Bhatt | Mahesh Datt Bhatt received his MSc Degree in Physics (1997) from Tribhuvan University (Nepal) and PhD degree in Engineering (2010) from University of Tsukuba (Japan). Dr Mahesh studied the electronic structure at organic molecule–metal surface interfaces during his doctoral research. He focused on interaction of Li+ with non-aqueous electrolytes in Li-ion batteries and the origin of band-bending in donor–acceptor interfaces in organic solar cells during his postdoctoral positions in Seoul National University (Korea) and University of Warwick (UK) respectively. He has been currently studying ORR/OER kinetics in Li–air batteries using DFT methods in University College Cork (Ireland). |
 Colm O'Dwyer | Colm O'Dwyer received his BS degree in applied physics and PhD degree in physics from the University of Limerick, Ireland in 1999 and 2003, respectively. After postdoctoral research in nanotechnology and nanolithography using ultracold atoms beams at the University Paul Sabatier, Toulouse, France, he conducted work on nanoelectronics/photonics at Tyndall National Institute, Cork, Ireland. From 2008 to 2012, he was a Science Foundation Ireland Stokes Lecturer on nanomaterials at the University of Limerick. He is currently at the Department of Chemistry, University College Cork and at Tyndall National Institute, leading the Applied Nanoscience Group focused on Li-ion and Li–air batteries, energy storage and conversion, electronic and photonics materials, and nanoscience. |
1. Introduction
Due to burning of fossil fuels and biomass, the unusual climate change effects such as global warming, are being exacerbated due to increased CO2 emission. Moreover, the increasing demand of energy worldwide is affecting the prices of fossil fuels. These concerns have led to recommendations on the development and use of alternative energy sources such as solar, wind, tidal, and geothermal energy. These energy sources will require electric energy storage solutions. The most convenient energy storage devices are batteries, which provide the portability of stored chemical energy with the ability to deliver this energy as electrical energy with a high conversion efficiency without gaseous exhaust as with fossil fuels. The development of low cost, safe, rechargeable (secondary) batteries of high voltage, capacity, and rate capability are important for large scale uses and smaller device-based requirements. Among the rechargeable battery technologies (Fig. 11), the current Li-ion battery technology offers the best useable energy and tap density, dominating the worldwide market for mobile electronic devices2 for decades. Li-ion batteries have minimal (unwanted) side reactions when a Li ion intercalates into or desorbs from the cathode/anode materials. Their energy efficiency may be further enhanced by lowering the internal resistance of the battery, and they exhibit limited self-discharge, and no memory effects that limit energy density after many cycles. Li-ion batteries as a result, receive considerable attention at both fundamental and applied research levels.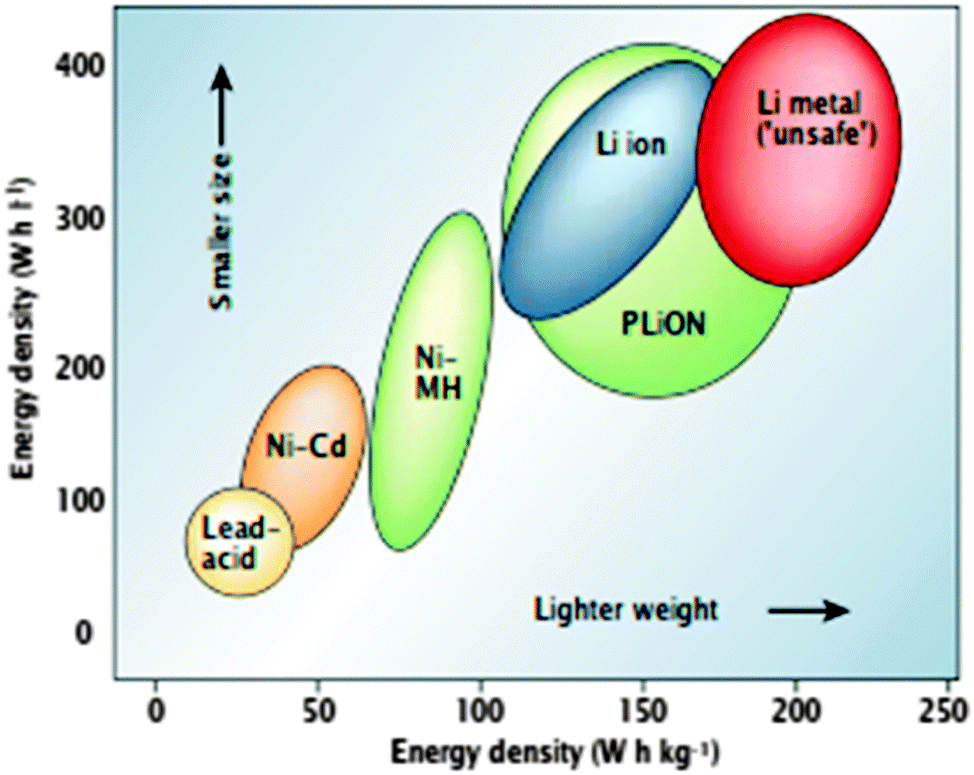 | ||
| Fig. 1 Comparison of the different battery technologies in terms of volumetric and gravimetric energy density. The share of worldwide sales for Ni–Cd, Ni–MeH and Li-ion portable batteries is 23, 14, and 63%, respectively. The use of Pb–acid batteries is restricted mainly to SLI (starting, lighting, and ignition) in automobiles or standby applications, whereas Ni–Cd batteries remain the suitable technologies for high-power applications. Reprinted with permission from ref. 1. Copyright 2001 Nature Publishing Group. | ||
However, alternative forms of transportation, such as plug-in hybrid electric vehicles (PHEV) and all electric vehicles (EV), require significant improvements in many perspectives such as energy density, safety, cost, and durability.3–6 The development of stable novel materials is the key to the success of development of novel and advanced rechargeable batteries. Current research and development has focused on upgrading the energy density of Li-ion batteries. Ideal batteries would possess properties such as long life, light weight, small size, high energy density, safety, low cost, environmental compatibility, and worldwide consumer distribution. However, in real battery options, none yet can fulfil all these challenging requirements. Most practical rechargeable batteries deliver capacities and energy densities far below their theoretical values7,8 due to limited utilization efficiency of the active materials that participate in electrochemical reactions. The major reasons for such limitations include effects that result from slow electrode process kinetics with high polarization and low ionic diffusion or electronic conductivity rates, particularly at the electrolyte–electrode interfaces. Material stability issues caused by a low Li content can also impact on its degree of charging. Therefore, the improvement in existing rechargeable battery systems involves exploring key materials and focusing our attention on the atomic, ionic, or molecular diffusion and transport. Charge transfer, the optimization of surface and interface structure, and the regulation of electrochemical reactions within Li-ion systems may pave the way for improved (i) capacity, and energy and power density, (ii) reactivity, reversibility, and structural stability during charge–discharge cycles, (iii) ionic diffusion and electronic transfer at high charge–discharge rate, and (iv) lower cost, increased safety and environmental compatibility.
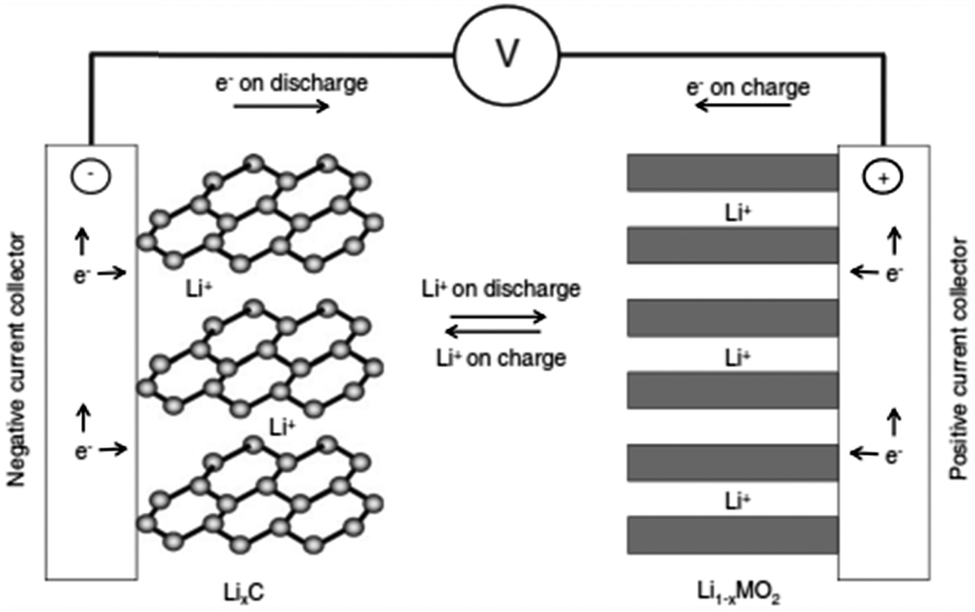
The schematic diagram of the current Li-ion battery based on a carbon based anode (LixC6), cathode (LiCoO2), liquid electrolyte (LiPF6 dissolved in a mixture of EC and DMC or equivalent), and separator is shown in Fig. 2.9 In the foreseeable future, Li-ion batteries will be the most practical solution to a wide range of electrical energy storage applications.10
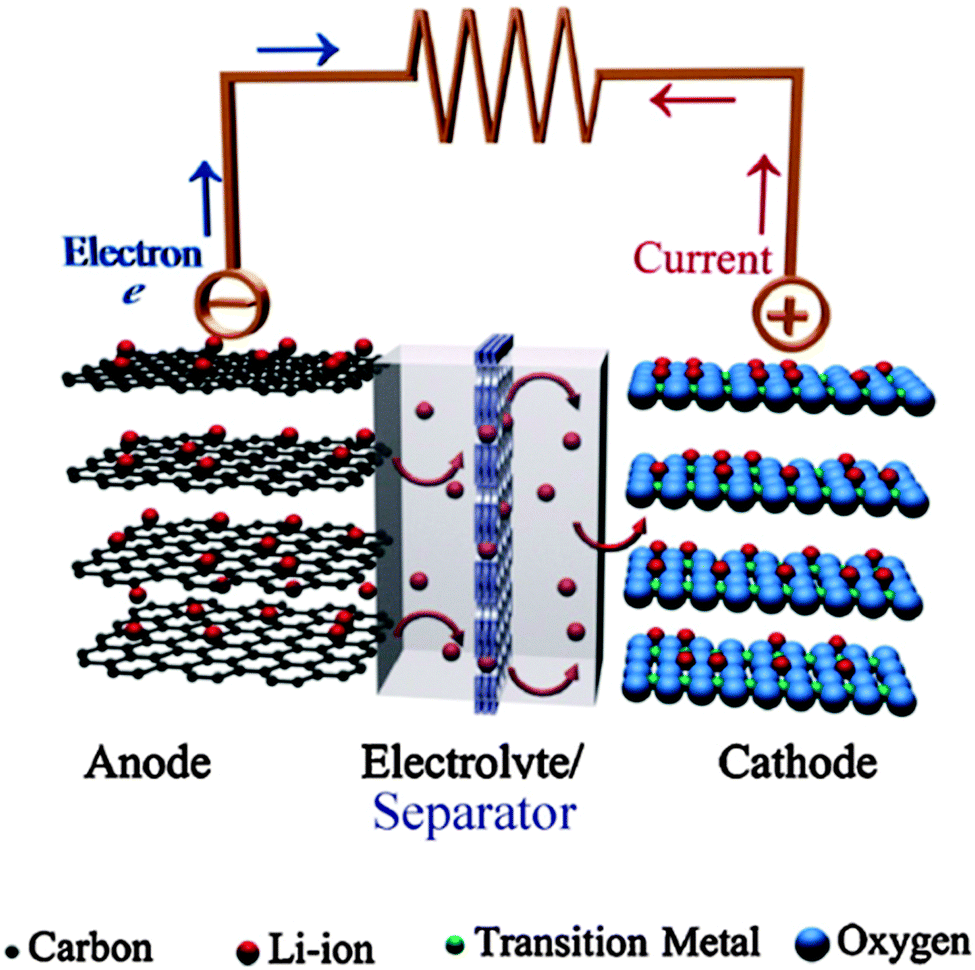 | ||
| Fig. 2 Schematic illustration of a typical lithium-ion battery where graphite and LiCoO2 are used as anode and cathode, respectively. Reprinted from ref. 9. Copyright 2011, with permission of Elsevier. | ||
The power and energy density of a Li-ion battery largely depend on the electrode and electrolyte materials. Fig. 3 summarizes the relationship between electric potential referenced to metallic Li and the discharge capacity density of electrode materials that have been studied.1,11–14
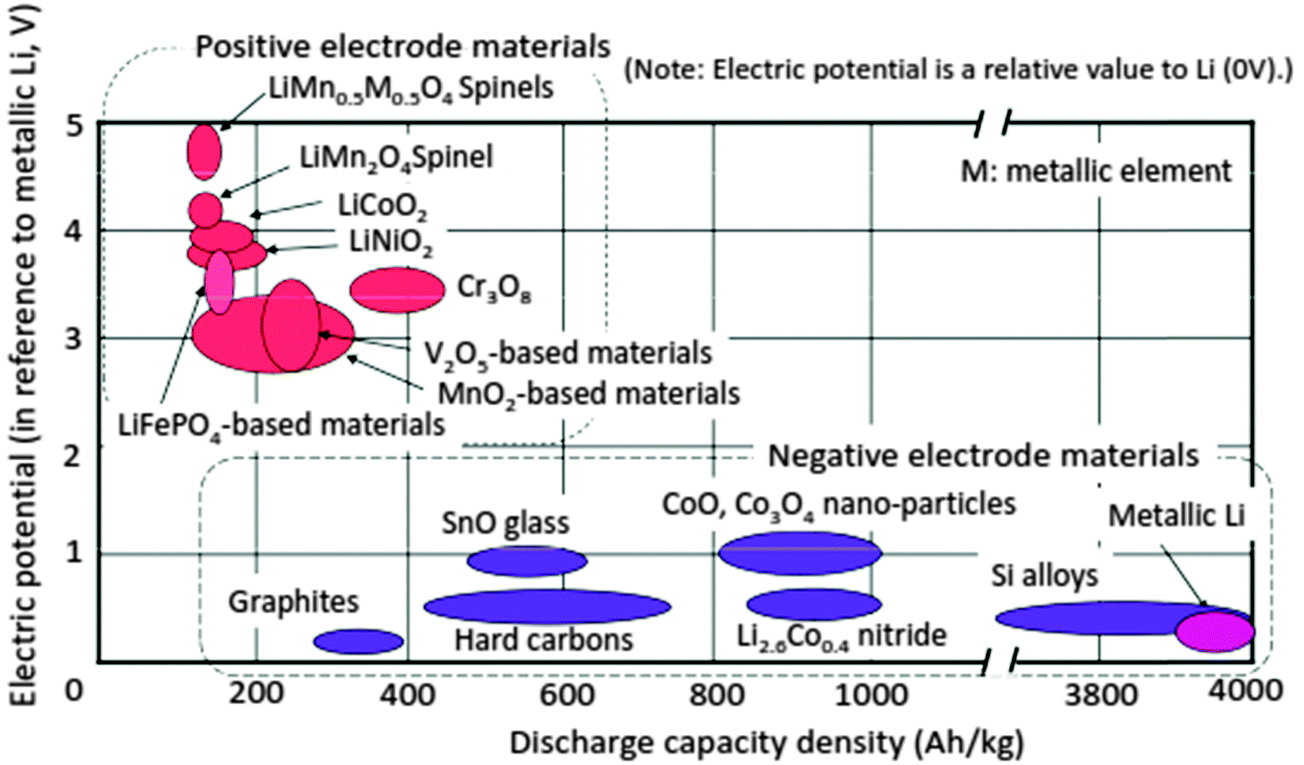 | ||
| Fig. 3 Electric potential vs. charge-capacity density: electrode materials R&D. Reprinted from ref. 14. Copyright 2008, with permission of Elsevier. | ||
The enhancement of current is very important along with the boost of output voltage, for which the internal resistance should be minimum. Fig. 414 summarizes the main factors responsible for internal resistance during a discharge process in a Li-ion battery, which includes the resistance due to Li ion transport (discharge) and the electron conduction resistance inside both the electrodes and electrode current collector interface region.
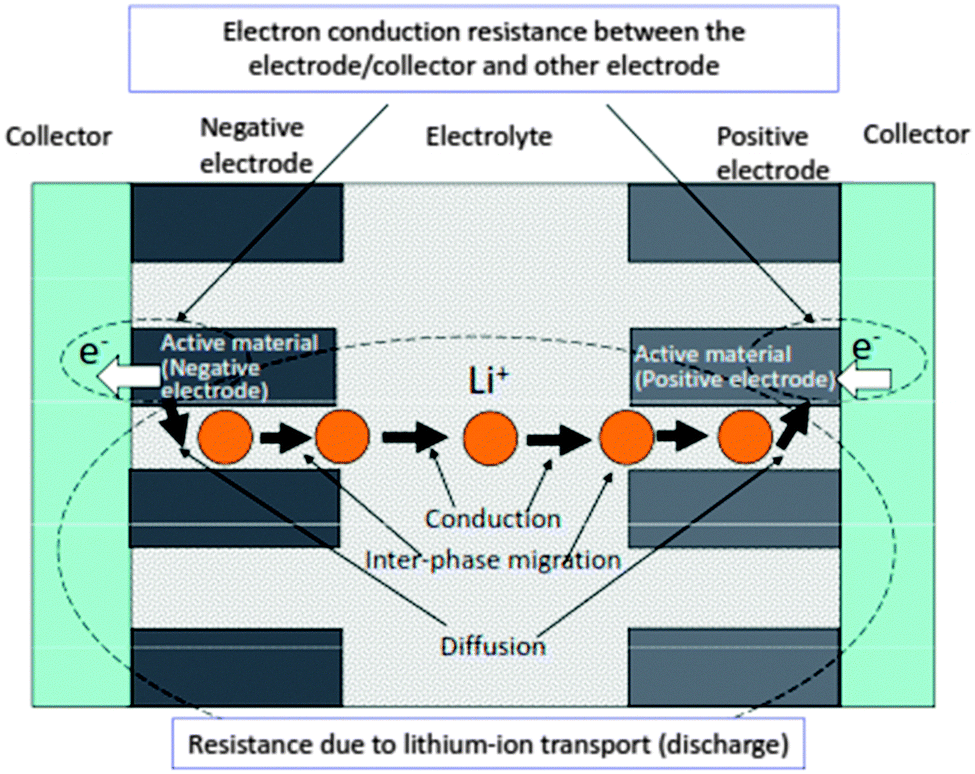 | ||
| Fig. 4 Major factors affecting the internal resistance of discharging Li-ion battery. Reprinted from ref. 14. Copyright 2008, with permission of Elsevier. | ||
Li-ion batteries are the most popular rechargeable batteries, and since the 1970's research efforts have been devoted to understanding and stabilizing the electrochemical performance of a wide variety of active materials for electrodes and electrolytes in rechargeable non-aqueous Li-ion batteries (see Fig. 515).
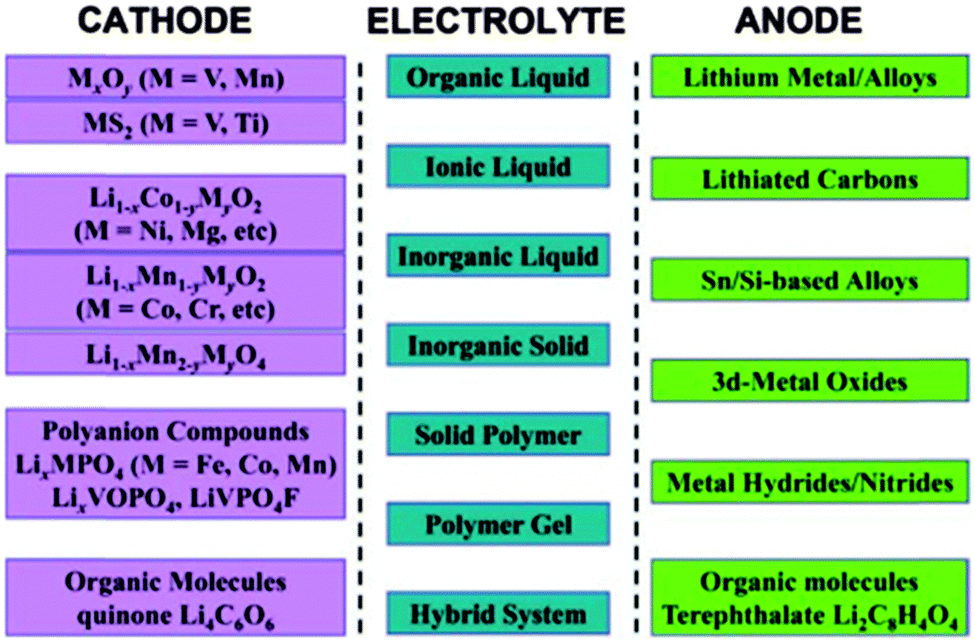 | ||
| Fig. 5 List of representative electrode materials and electrolyte types investigated for rechargeable non-aqueous Li-ion batteries. Reprinted with permission from ref. 15. Copyright 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In Li-ion batteries, LiCoO2 is widely used as a cathode material, while LiMn2O4 is used for some applications which requires higher level of safety. Its insertion potential is lower and less likely to self-discharge. The majority new cathode materials for Li-ion batteries under research and development are transition metal oxides, which tend to provide lower discharge potential as the electric-capacity density increases. Carbon-based materials (usually graphite) are currently used as anode materials in Li-ion batteries. The other variety of carbon-based materials and pure Li metal are currently proposed as alternate anode materials, but many need further improvement with respect to electrode potential and charge–discharge cycle life concerns. Several options are available for electrolytes such as non-aqueous electrolytes (organic and ionic liquids), gelled electrolytes, and solid organic and inorganic electrolyte materials (see Fig. 5). The major electrolytes currently used comprise mixtures of cyclic and linear carbonates. Electrolyte additives such as vinylene and ethylene carbonates or sulfites are also used for some electrolyte solvents to improve cycling performance.
Metal chalcogenides (e.g., TiS and MoS2) and manganese or vanadium oxides have been investigated as the cathode and metallic Li or graphite as the anode, and led to the initial successes for rechargeable Li-ion batteries.16 The introduction of high-capacity lithium-storage materials such as Sn/Si/Ge alloys and transition metal oxides has fostered the development of high-energy batteries.17 Recently, considerable interest has been directed to polyanion-based compounds (LiFePO4 in particular), which potentially allows for lower cost and high safety.
In this review, we start with a brief introduction to Li-ion battery development together with recent advancements, progress, scientific challenges, and the theoretical development of component materials in Section 1. Section 2 provides a brief summary of key working principles of Li-ion batteries focusing on theoretical data for electrochemical reactions that take place at the electrodes. A summary of recent theoretical progress towards enhancing the performance of intercalation compounds as cathodes is discussed in Section 3. Section 4 focuses on theoretical approaches to achieving high performance anode materials, their current development and hurdles associated with electrolytes used in Li-ion batteries is discussed in Section 5. Finally, Section 6 discusses theoretical investigations into the major scientific challenges in current rechargeable Li-ion batteries and possible future research directions, by exploring new materials electrolytes, structures, compositions and reactions through theoretical modelling. These discussions represent the current understanding of existing Li-ion batteries, and in parallel with many informative reviews on experimental findings,16–48 this review concentrates on density functional theory and related methods used to develop a fundamental understanding of electrode reaction mechanisms, which are imperative in gaining critical insights into the rational design of active materials used in Li-ion batteries.
2. Working principles
When a Li-ion based cell discharges, the Li+ ions move from anode (usually graphite) to cathode (usually LiMO2, M = transition metal) and reverse occurs on charging as shown in Fig. 6,49 facilitated by a Li+-containing salt in the electrolyte. The electrochemical reactions take place in a Li-ion battery are:50–53| LiMO2 ⇄ Li1−xMO2 + xLi+ + xe− (at the cathode) | (1) |
| xLi+ + xe− + xC6 ⇆ xLiC6 (at the anode) | (2) |
The overall cell reaction is:
| LiMO2 + C6 ⇄ Li1−xMO2 + LixC6 (x = 0.5, V ∼ 3.7 volts) | (3) |
As metallic Li is not present in this cell, Li-ion batteries are chemically less reactive, safer, and offer longer cycle life than rechargeable lithium batteries using Li metal as anodes.
 | ||
| Fig. 6 Scheme of the electrochemical process in a Li-ion battery. Reprinted with permission from ref. 49. Copyright 2009 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In order to achieve better performance in current rechargeable non-aqueous Li-ion batteries, researchers must design either (i) a cathode with a chemical potential (μC) matched to the HOMO of the electrolyte as well as an anode with a chemical potential (μA) matched to the LUMO of the electrolyte or (ii) a non-aqueous electrolyte (solid or liquid) of high Li+ ion conductivity over the practical ambient temperature range (between −40 °C and 60 °C) that has a potential window that allows a thermodynamically stable open circuit voltage VOC ≥ 4 V. Fig. 7(a)–(c)27 represent the relative energies of the electrolyte window HOMO–LUMO gap Eg, and the electrode chemical potentials μC and μA with no electrode–electrolyte reaction.
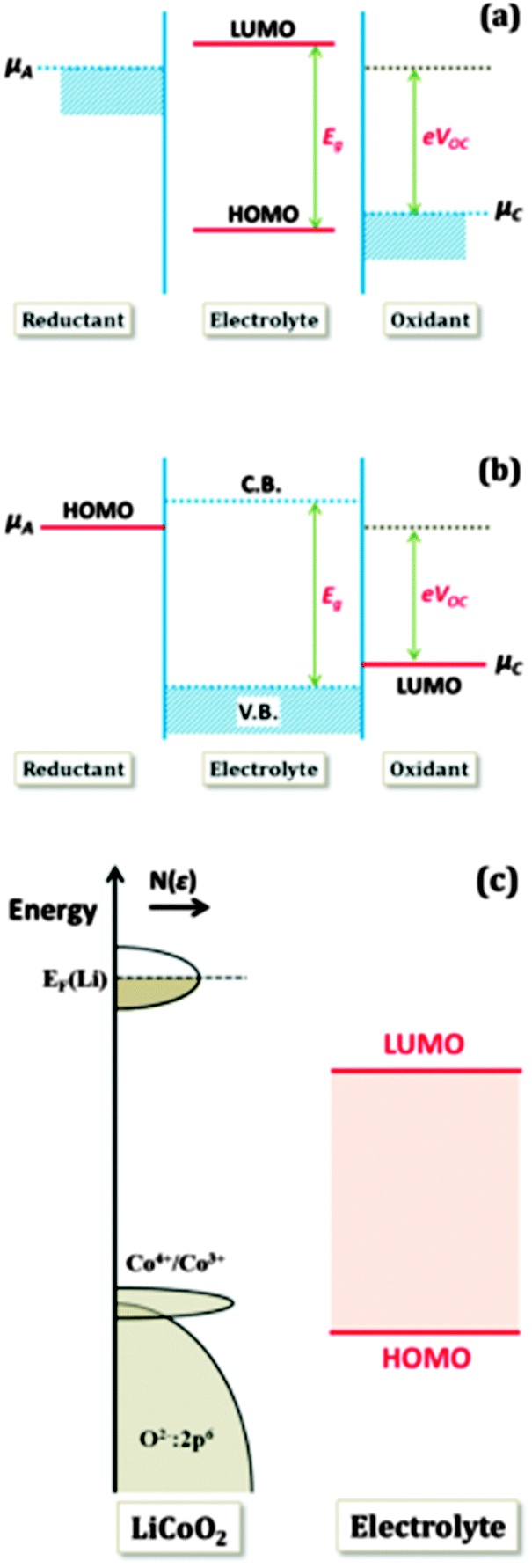 | ||
| Fig. 7 Relative energies of the electrolyte window Eg and the electrode electrochemical potentials μA and μC with no electrode–electrolyte reaction. Reprinted with permission from ref. 27. Copyright 2013 American Chemical Society. | ||
3. Cathode materials
This section mainly focuses on lithium intercalation electrode materials, i.e. where the reaction that occurs at the cathode material is the intercalation of Li ions into the host during the discharge process (spontaneous process), and deintercalation of Li ions from the host during the charge process (non-spontaneous process). Here, the term intercalation is used to describe the interaction process, whether it be true intercalation into a crystal void (van der Walls space) or between atomic planes, or whether it refers to alloying. The typical cathode materials of Li-ion batteries consist of layered lithium transition metal oxides LiMO2 (M = Fe, Mn, Co, Ni, Ti, V, etc.), spinel lithium transition metal oxides, polyanion compounds such as phosphates, silicates, fluorophosphates, fluorosilicates, borates, and graphitic materials such as graphene and graphene oxide. In this section, we review the theoretical studies underlying important properties of these cathode materials such as cell voltages, lithium diffusion, defect chemistry, and surface structures, and highlight the current trends in this field. The crystal structure and the voltage–composition profiles of the most relevant cathode materials for Li-ion batteries are shown in Fig. 8.54 Researchers have full control in designing and classifying new and potential electrode materials that can be explored in detail by DFT methods, since composition and structure are entered as independent variables before they are experimentally prepared, and computations can help guide experiments. In order to design new materials a usual starting point is to analyse the effect of composition modifications for a given structural type, which can be achieved by computation as compared to experiments. Polymorphism in many host compounds has been investigated by first-principles methods: LiCoO2,55–57 LiCoXO4 (X = P, As),58,59 V2O5,60,61 LiFeSiO4,62 TiO2,63,64 LiTiMO4 (M = Ti, V, Cr, Mn, Fe),65 MnO2,66,67 FePO4,68–70etc.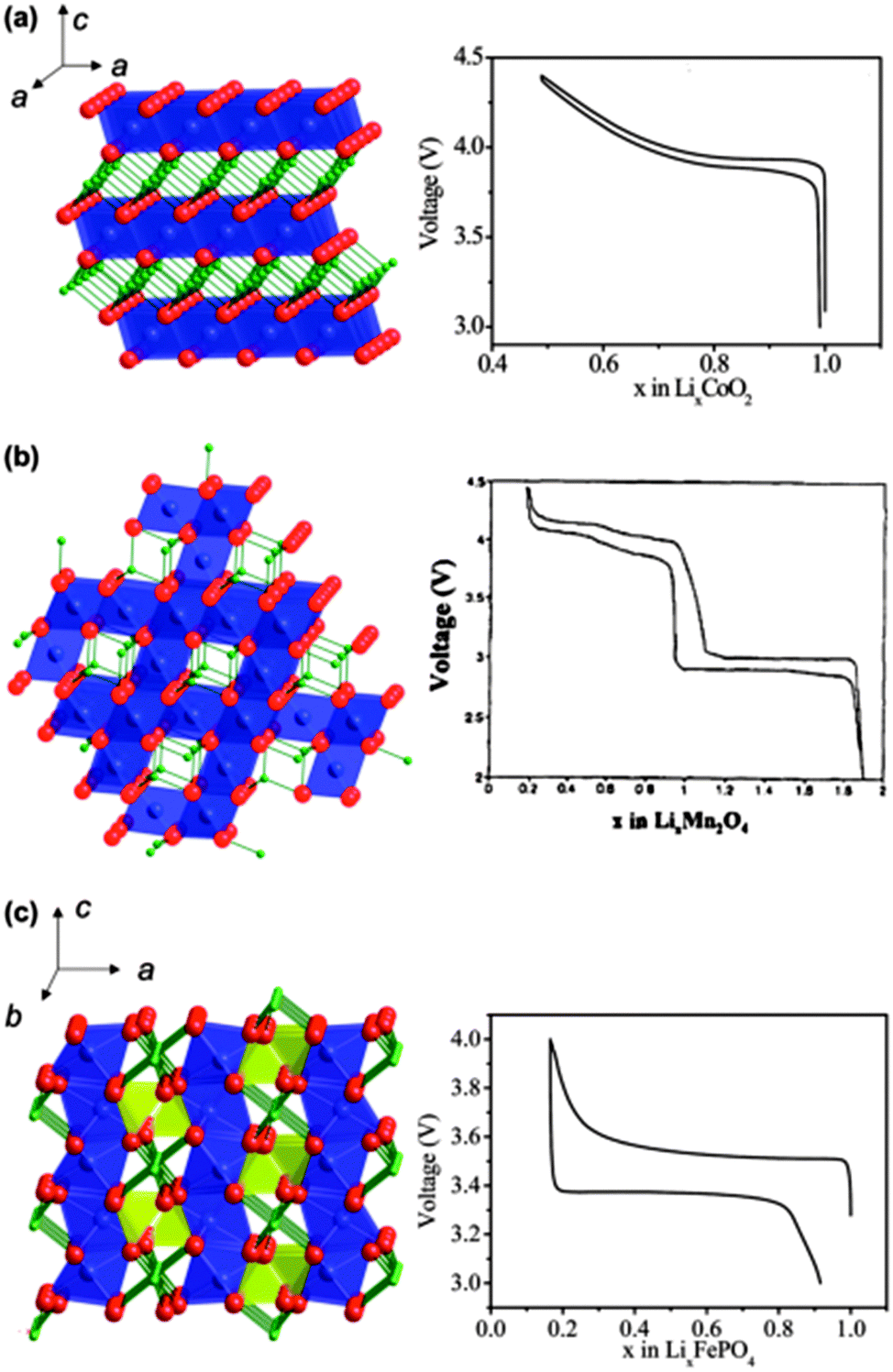 | ||
Fig. 8 Crystalline structures and voltage–composition curves of (a) layered-LiCoO2 (R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m S.G.)—oxygen (red) layers are stacked in ABC sequence, with lithium (green) and cobalt (blue) residing in the octahedral sites of the alternating layers; (b) spinel-LiMn2O4 (Fdm S.G.)—lithium (green) resides in the tetrahedral sites formed by oxygen stacking; and (c) olivine-LiFePO4 (Pnma S.G.)—phosphor (yellow) and oxygen form tetrahedral units linking planes of corner-sharing FeO6 octahedral. Reprinted with permission from ref. 54. Copyright 2009 Royal Society of Chemistry. m S.G.)—oxygen (red) layers are stacked in ABC sequence, with lithium (green) and cobalt (blue) residing in the octahedral sites of the alternating layers; (b) spinel-LiMn2O4 (Fdm S.G.)—lithium (green) resides in the tetrahedral sites formed by oxygen stacking; and (c) olivine-LiFePO4 (Pnma S.G.)—phosphor (yellow) and oxygen form tetrahedral units linking planes of corner-sharing FeO6 octahedral. Reprinted with permission from ref. 54. Copyright 2009 Royal Society of Chemistry. | ||
3.1 Layered lithium transition metal oxides LiMO2 (M = Fe, Mn, Co, Ni, Ti, V, etc.)
The layered lithium transition metal oxides LiMO2 (M = Fe, Mn, Co, Ni, Ti, V, etc.) are currently most extensively studied cathode materials in Li-ion batteries.71 They have been systematically investigated as a family of compounds by DFT methods.72 For example, LiCoO2 is widely used as an active cathode material in commercial Li-ion batteries73,74 and acquires an open circuit voltage of about 4 V with respect to metallic lithium. Such high cell voltages accompanied by high charge storage capacity in typical layered transition metal oxides makes them promising and useful cathode materials. In general, LiMO2 electrodes should meet several design criteria75–77 such as high intercalation voltage and low molar weight. The composition range over which Li can be reversibly intercalated determines the capacity of Li-ion battery. For example, high Li diffusivity is important to satisfy current–density requirements. On other hand, low Li content causes the irreversible structural changes of LiMO2 thereby destroying the part of the capacity that can be used in the next discharge cycle. The structure of layered LiMO2 compounds is typically of rhombohedral symmetry (Rm) with α-NaFeO2 structure as shown in Fig. 9.78 Li+ ions can be inserted into and removed from this structure leaving vacancies in lithiated layers.79
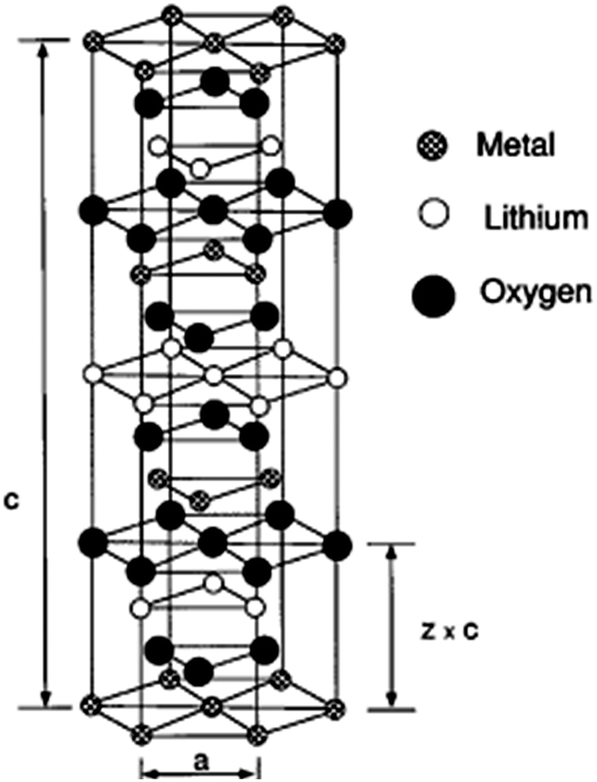 | ||
| Fig. 9 α-NaFeO2-type structure. The oxygen ions form a cubic close-packed array and the metal cations occupy layers of octahedral interstitials. Reprinted with permission from ref. 78. Copyright 1997 American Physical Society. | ||
As Li intercalates into the cathode as positive ion, it is assumed that the compensating electron reduces the metal ion. Therefore, the nature of the transition metal and strength of its MIV/MIII redox couple are expected to be significant variables to determine the intercalation voltage.80 When exposed to electrochemical cycling or high temperature, the original host layer structures are deformed mainly by means of spatial Li-vacancy interactions such as electron count variations and oxygen packing changes.81–83 DFT calculations can provide detailed atomic information that is difficult to acquire or specifically examine in experiments. For LiNiO2, DFT calculations showed that Ni tends to reach a maximum of charge localization. However, Co prefers to achieve a maximum of charge delocalization in LiCoO2.84,85 The theoretical studies based on first-principle calculations successfully predict the average intercalation voltage of lithium transition metal oxides.86,87 Van der Ven et al.83,88 investigated the different phases of the layered LixCoO2 and their results showed that at low Li content, the host structure becomes deformed due to ordering and staging transitions, and this has implications on its charging capability. At high Li content, no phase region occurs. Besides structural deformation and phase transformation, surface properties are also helpful in tailoring the rate performance and understanding parasitic surface reactions in the electrolyte. Kramer et al.89 investigated the surface energies of various low-index surfaces of layered by using DFT methods. Daheron et al.90 proposed that the uncoordinated oxygen atoms existing on the (001) surface of LiCoO2 vary from those in the lattice.
DFT investigations are also helpful in expressing ionic diffusivity in terms of activation barrier along the Li hoping paths during discharge. For example, Kang et al.91 studied the issues related to Li mobility in layered LiMO2 compounds and found that octahedral Li ions migrate through intermediate tetrahedral sites where they experience a strong repulsion from nearby metal (M) ions. The layered LiMO2 doping can cause a small variation in activation barrier, thereby affecting the diffusion rate exponentially. Factors such as local atomic arrangement, size of the tetrahedral site and electrostatic interaction between Li+ ions and face-shared octahedron M+ ions, greatly influence the activation barrier for Li hopping.92,93 There is a major drawback for layered transition metal oxide cathodes in Li-ion batteries when undergoing full discharge and full charge reactions (without depth of discharge control that extends cycle life at the expense of capacity): limiting cycling performance (especially for LiMnO2) at full charge and discharge, which is probably due to its potential failure mechanism94 at high and low Li content causing associated structural phase transitions. For example, LiMnO2 or LiNiO2 doped with transition metals help suppress the Jahn–Teller distortion of Mn3+ or Ni3+ and associated structure transformation. It is important to note that high contents of divalent dopants are especially effective.95 Moreover, the mixed metal LiMO2 compounds are also promising cathode materials for Li-ion batteries. For example, LiMn0.5Ni0.5O2 is a safe and cheaper Li-host material with a high capacity, good cycling stability as demonstrated by theoretical researchers.96,97 Ceder et al.97–100 provided the two lowest energy states as shown in Fig. 10.101 The authors calculated the intercalation potential and Li-site occupancies using both GGA and GGA + U approaches within the flower-like structure as a function of Li as shown in Fig. 11.101
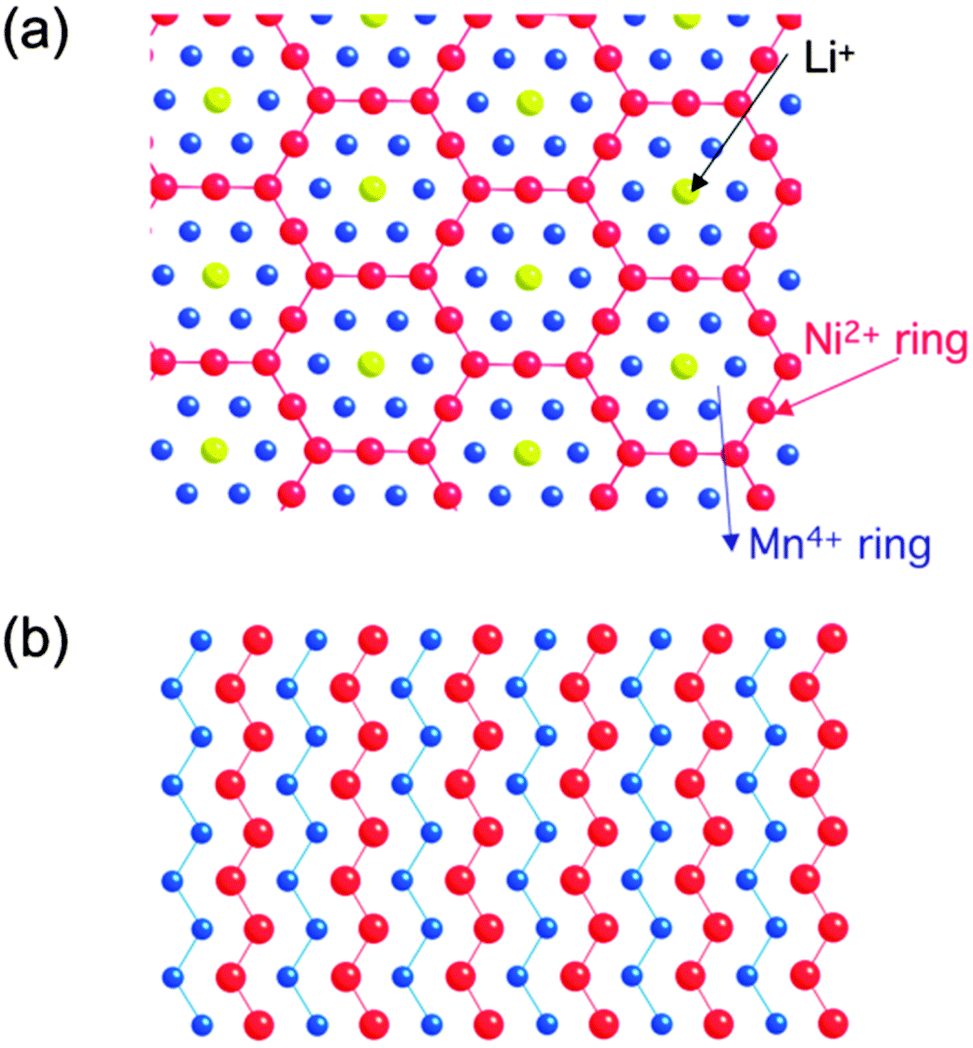 | ||
| Fig. 10 Structural details of LiMn0.5Ni0.5O2 (a) flower-like pattern as proposed by ordering in the transition metal layer between Li, Mn and Ni, and (b) zigzag pattern proposed shows no Li in the transition metal layer. Reprinted with permission from ref. 101. Copyright 2009 Royal Society of Chemistry. | ||
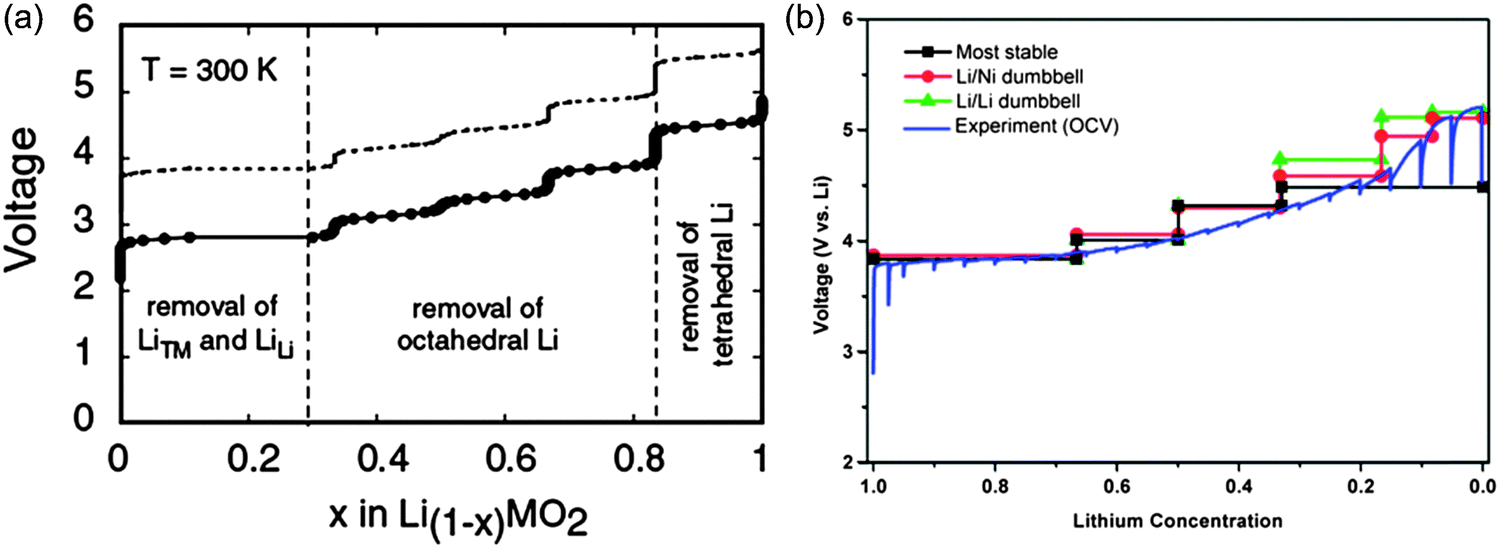 | ||
| Fig. 11 (a) GGA calculated voltage profile of LiMn0.5Ni0.5O2, note the dotted line is obtained by shifting the calculated profile by a constant amount (∼1 V),100 (b) comparison between the calculated voltage curves for different delithiation scenarios and the voltage profile during the first charge of a Li/LixMn0.5Ni0.5O2 cell, charged to 5.3 V at 14 mA g−1 with intermittent OCV stands of 6 h. The calculated curves are obtained with GGA + U, there is no artificial shift of the curves. Reprinted with permission from ref. 102. Copyright 2006 American Chemical Society. | ||
The authors also investigated the phase transformations of layered LiMn0.5Ni0.5O2 at finite temperatures and found the two phase transition temperatures at approximately 550 °C and 620 °C. Their simulation results showed that the Li ions that were part of the flower-like tetrahedral sites early in the charge cycle, then became occupied by Li as tetrahedral Li required high potential to be removed and thus lowering effectively the attainable capacity of the material at practical voltage intervals. As shown in Fig. 11(b), a partially disordered flower-like structure with about 8 to 11% Li–Ni interlayer mixing was found. Such simulation results may help in explaining many of the experimental results for LiMn0.5Ni0.5O2 with and without Li–Ni disorder. Xu et al.103 reported that transition metal migrations mainly occur on the surface layers, thus leading to a surface phase transformation to defect-spinel and large irreversible capacity. Recently, researchers have extended their research to multi-doped materials those with gradient core–shell–surface structures and variable transition metal concentrations, and this research direction is cited as being useful to enhanced thermal stability.104 DFT calculations clearly showed that Mn should ideally be rich at the surface and Ni should be rich in the bulk material to achieve a high energy density. Moreover, there is an increase in electronic charge that is transferred to the anionic band when Li intercalates the MO2 host, with an increase in electron affinity and number of d electrons. The important role of anions can be explained in terms of the charge transfer to the anion that occurs upon Li intercalation.
In layered transition metal oxides, more charge is transferred to the oxide ions than to the metal ions. Early DFT studies included new materials such as Li(Co, Al)O2105 and vanadium oxide cathodes.106 For example, Dianat et al.107 investigated the structural stability during delithiation, the battery voltage, and Li mobility for Al-doped Li–Mn–Ni oxides using DFT and nudged-elastic band methods. The rhombohedral layered structure of LiMn0.5Ni0.5O2 with zigzag and flower arrangements of transition metal atoms as well as the monoclinic structure of Li(Li1/6Ni1/6Mn2/3)O2 were used as base structures. A stabilizing effect of Al-doping was found for all partially lithiated systems that were considered in those studies. The desired battery voltages were found to be enhanced by Al-doping at low temperature. The increase in calculated activation energies for Li suggested lower Li mobility. In this way, the Al-doped Li-rich monoclinic structures are promising cathode material due to comparatively high battery voltage. In addition, Xiao et al.108 studied Li2MnO3-stabilized LiMO2 (M = Mn, Co, Ni) cathode materials. The authors investigated the effect of transition metal substitutions on the oxygen loss in Li-rich layered oxides from the electronic structure calculated with DFT + U approach. Ti substitution increased the band gap due to the higher energy of the non-bonding metal caused by a weaker nuclear attraction compared to Mn. As a result, Ti suppressed oxygen loss. On other hand, Co substitution decreased the band gap due to the fact that the low lying non-bonding metallic band overlaps with the valence band. This particular finding form the calculations suggested that the material became a conductor; as a result, Co facilitated oxygen loss. Thus, a correlation between band gap and oxygen binding energy was confirmed by the authors through examination of other third-period transition metals between Ti and Ni.
There are other layered materials systems that in which Li diffusion and associated materials phase changes strongly influence reversibility during discharge and charge reactions. V2O5 was first reported as an intercalation material in this respect in the late 70's, and its particular layered structure is a ‘model’ system. Wang et al.109 performed DFT calculations for adsorption and diffusion properties of Li atoms on single-layered and bulk V2O5. Their results showed that the diffusion barrier of Li on the single-layered V2O5 (0.20 eV) is decreased compared to that of bulk V2O5 (0.51 eV), indicating the enhancement of Li mobility on the single-layered V2O5. The increased binding energies of Li to single layered V2O5 made them more attractive for promising cathode materials possessing high energy density. However, it has a drawback whereby reversibility is adversely affected due to a phase transition that takes place during the charging–discharging process. Cell reactions between lithium and vanadium oxide produce ternary LixV2O5 phases. Cell reversibility is optimised when these phases are formed instead of bond breaking of the oxide and its change to a V–O polymorph – for small amounts of lithium (∼0.1 mol per V2O5). Jiang et al.110 studied the thermodynamic and kinetic properties of layered LiV3O8 cathode materials by means of DFT and cluster expansion methods to interrogate the mechanism behind reversible lithiation of vanadate bronzes. Their resulting calculations indicated solid solution behaviour from LiV3O8 to Li2.5V3O8 and two-phase co-existence between Li2.5V3O8 and Li4V3O8. Analysis of the lithiation sequence from LiV3O8 to Li2.5V3O8 revealed the mechanism by which Li intercalation proceeds in this material. Calculations of Li migration energies for different Li concentrations and configurations within van der Waals layered positive electrode materials provides insight into the relevant diffusion pathways and their relationship to structural properties.
3.2 Spinel lithium transition metal oxides
Spinel oxides have demonstrated superior reversible Li intercalation capability and longer cycle life,3,71,111 but often with a lesser specific capacity compared to phosphates and layered oxides. Spinel oxides allow high lithium diffusion rates because of the 3D network of interstitial sites112–114 within their crystal structure. Spinels are attractive materials due to their better rate capability for higher power applications. Two main variants of the spinel structure are referred to as normal and inverse. The general formula for a spinel structure can be written as AB2O4, where A is Li metal and B is transition metal. In normal spinel, and the transition metal cations occupy the octahedral sites. In inverse spinel, transition metal cations occupy all tetrahedral sites and half of the octahedral sites and Li+ ions occupy the remaining half of the octahedral sites. In spinel structure, the two metal cations are present in 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio; however, this condition relaxes somewhat since the stoichiometry changes during the cycling of the cathode materials. For instance, the LiM2O4 (M = a transition metal) spinel structures lose Li during the charging process and vacancies are introduced in place of Li. In the completely charged state (i.e. delithiated state), M2O4 structures are also spinel structures, considering vacancies as second cationic species. Bhattacharya et al.115 explored a range of 3d-block transition metal spinels using first-principles calculations to investigate the structural preference at different charge states and its effect on voltage, diffusivity and cycle life of the cathode. The authors considered ternary spinel oxides LixM2O4 across the 3d transition metal series (M = Ti, V, Cr, Mn, Fe, Co, and Ni) in both lithiated (x = 1) and delithiated (x = 0) conditions. They found that for all lithiated spinels, the normal structure is preferred regardless of the metal; the normal structure for all oxides under consideration was found to have a lower size mismatch between octahedral cations compared to the inverse structure, and with delithiation, many of the oxides undergo a change in stability with vanadium in particular showing a tendency to occupy tetrahedral sites. The authors also calculated the average voltage values of lithiation for these spinels. Lastly, they observed that all the normal spinel oxides of the 3d transition metal series have a driving force for a transformation to the non-spinel structure upon delithiation. The O2− ions formed an FCC sub-lattice in spinel, in which the two interstitial sites (octahedral and tetrahedral) are available. The crystalline structure of spinel LiMn2O4 and its corresponding lithium diffusion pathways shown in Fig. 12.116
:1 ratio; however, this condition relaxes somewhat since the stoichiometry changes during the cycling of the cathode materials. For instance, the LiM2O4 (M = a transition metal) spinel structures lose Li during the charging process and vacancies are introduced in place of Li. In the completely charged state (i.e. delithiated state), M2O4 structures are also spinel structures, considering vacancies as second cationic species. Bhattacharya et al.115 explored a range of 3d-block transition metal spinels using first-principles calculations to investigate the structural preference at different charge states and its effect on voltage, diffusivity and cycle life of the cathode. The authors considered ternary spinel oxides LixM2O4 across the 3d transition metal series (M = Ti, V, Cr, Mn, Fe, Co, and Ni) in both lithiated (x = 1) and delithiated (x = 0) conditions. They found that for all lithiated spinels, the normal structure is preferred regardless of the metal; the normal structure for all oxides under consideration was found to have a lower size mismatch between octahedral cations compared to the inverse structure, and with delithiation, many of the oxides undergo a change in stability with vanadium in particular showing a tendency to occupy tetrahedral sites. The authors also calculated the average voltage values of lithiation for these spinels. Lastly, they observed that all the normal spinel oxides of the 3d transition metal series have a driving force for a transformation to the non-spinel structure upon delithiation. The O2− ions formed an FCC sub-lattice in spinel, in which the two interstitial sites (octahedral and tetrahedral) are available. The crystalline structure of spinel LiMn2O4 and its corresponding lithium diffusion pathways shown in Fig. 12.116
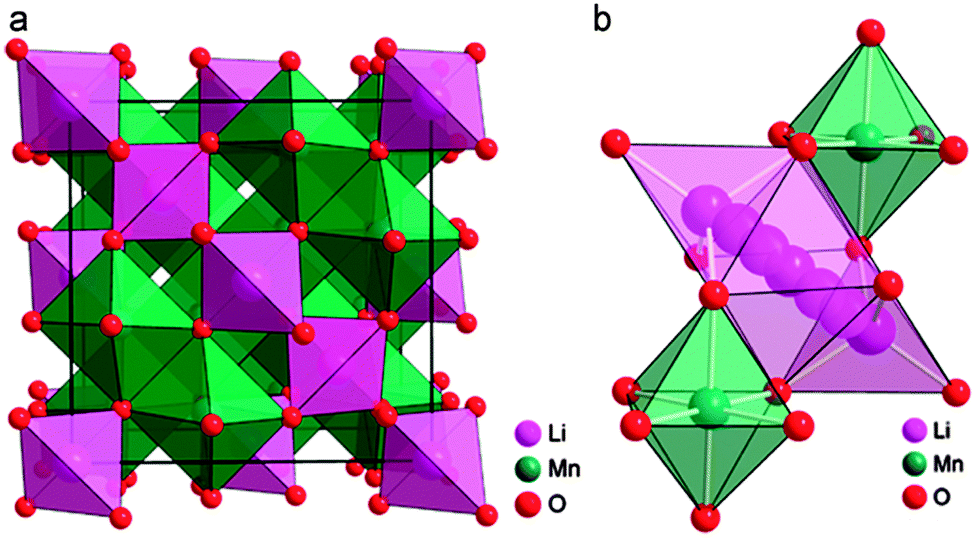 | ||
| Fig. 12 (a) Crystalline structure of spinel LiMn2O4 and (b) its corresponding lithium diffusion pathways. Reprinted with permission from ref. 116. Copyright 2011 Royal Society of Chemistry. | ||
The most studied normal and inverse spinels are LiMn2O4 and LiNiVO4.112,117–126 Previous DFT studies predicted that the spinel phase of LiCo2O4 is stable compared to the delithiated layer structure LiCoO2.71 Due to a relatively low voltage, normal spinel LiTi2O4 is not very useful as a cathode material.114,127–129 Irreversible destabilization and delithiation limit the spinel LiV2O4 as an insertion electrode.130 There are numerous normal and inverse spinels as cathode materials in the literature. There is lack of dedicated reports that investigate the relative stability among them as a function of Li-composition. DFT calculations have been used to understand and predict these and a range of related battery materials in the last two decades.112,131–141
Zhou et al.142 performed first-principles calculations with the local density approximation (LDA) or generalized gradient approximation (GGA) to calculate the redox potentials associated with lithium intercalation in spinel-like transition metal compounds. The authors showed that using a DFT + U approach, the experimental Li intercalation voltages of spinel-like LixM2O4 (M = Mn, Co) can be reproduced accurately. Wagemaker et al.143 studied thermodynamics of spinel LixTiO2 using first-principles calculations. For x < 0.5 in LixTiO2, solid-solution behaviour was found and Li extraction could only occur at higher potentials. The authors calculated a potential of 1.4 V in good agreement with experiment and higher than in the comparable Co and Mn-spinel. Huang et al.144 studied the mechanism of stability of Co-doped spinel λ-MnO2 that is referred to as spinel LixMn2O4 (x = 0) using the first-principle calculation method. The authors found that the total energy and formation enthalpy are markedly decreased due to Co substitution, resulting in a more stable structure of λ-MnxCr2−xO4. The bond order and density of states analysis were given in detail to explain the nature of stability improvement. Their results showed that as the Co dopant content increases, the bond order of Mn–O becomes larger and the density of states (DOS) maximum close to the Fermi level shifted towards lower energy. The nature of Mn–O bonding was found to be ionic and became stronger with increased Co dopant content. Such investigations suggest that Co-doping may enhance the stability of λ-MnO2 and hence improve the electrochemical performance of LixMn2O4. Kebede et al.145 performed first-principles calculations to study the structural and electrochemical properties of Al-doped LiMn2O4 cathode materials and found an increase in lattice parameter for x = 0.05 to 0.1, which is responsible for the increase in first cycle discharge capacity for x = 0.05 and 0.1. As Al content increased towards x = 0.5 in the spinel cathode LiAlxMn2−xO4, the composition became more favourable energetically for Li ion intercalation and consequently provided an increased cell voltage. It should be noted that DFT methods often treat ‘perfect solids’ while defects are always present in real solids in a much more complex form that even deterministic defect inclusions in models and calculation structures. Empirical atomistic simulation methods, with short-range interatomic forces represented by effective pair potentials146 can in certain cases be more useful in accommodating the effects of doping and defects in electrode materials as shown for Li–Mn–Fe–O spinels.147 Such empirical methods often do not provide any information of the electronic structure and redox potentials compared to quantum mechanical methods.
Thus, DFT calculations can provide for a variation in lattice parameters and modification in structure on Li intercalation/deintercalation within a given host material, and thus can be useful in examining or predicting likely behaviour for spinels and other compounds where the Li concentration, lattice behaviour and associated potentials largely influence the reversible intercalation/accommodation of cations. Li intercalation into octahedral sites of LiMn2O4 for example, occurs at 3 V via a two phase mechanism involving a transition from cubic LiMn2O4 to tetragonal LiMn2O4, which causes change of 5.6% in volume of unit cell.112,148 In addition, severe structural rearrangements are a major obstacle to topotactically remove Li ions from a material. DFT calculations can provide a better understanding of the phase transformation and accompanying volume changes149 and whether or not topotactic insertion and removal without conversion to polymorphs or bond breaking occurs. Such theoretical investigations for 3d transition metal oxide spinels, among many other materials that accommodate Li is similar fashion, are important for understanding the stability of structures and Li diffusion process for Li-ion batteries. More theoretical work based on DFT methods should be devoted in the future to understand the relationship between the relative stability of the structure of cathode materials and performance and safety in Li-ion batteries.
3.3 Polyanion compounds
Polyanion compounds LixMy(XO4)z (M = Fe, Mn, Co, Ni; X = P, S, Si, Mo, W, etc.) are considered very promising cathode materials for future industrial applications of rechargeable Li-ion batteries. These compounds include olivine phosphates, silicates, fluorophosphates, fluorosulphates, borates as well as anion and metal substitutions.30,150,151(i) Orthophosphates or monophosphates, if O/P = 4.
(ii) Oxyphosphates, if O/P > 4.
(iii) Condensed phosphates, if 2.5 < O/P < 4.
The condensed phosphates are further subdivided into linear phosphates, cyclophosphates, and ultraphosphates.
Ong et al.176 performed hybrid density functional theory calculations based on a Heyd–Scuseria–Ernzerhof (HSE06) functional177–179 to investigate the polaron migration and phase separation in olivine LiMnPO4 and LiFePO4. The olivine LiMPO4 compounds have an orthorhombic Pnma space group where transition metal (M) ions are six-fold coordinated by oxygen ions forming layers of edge-sharing octahedral as shown in Fig. 13.176
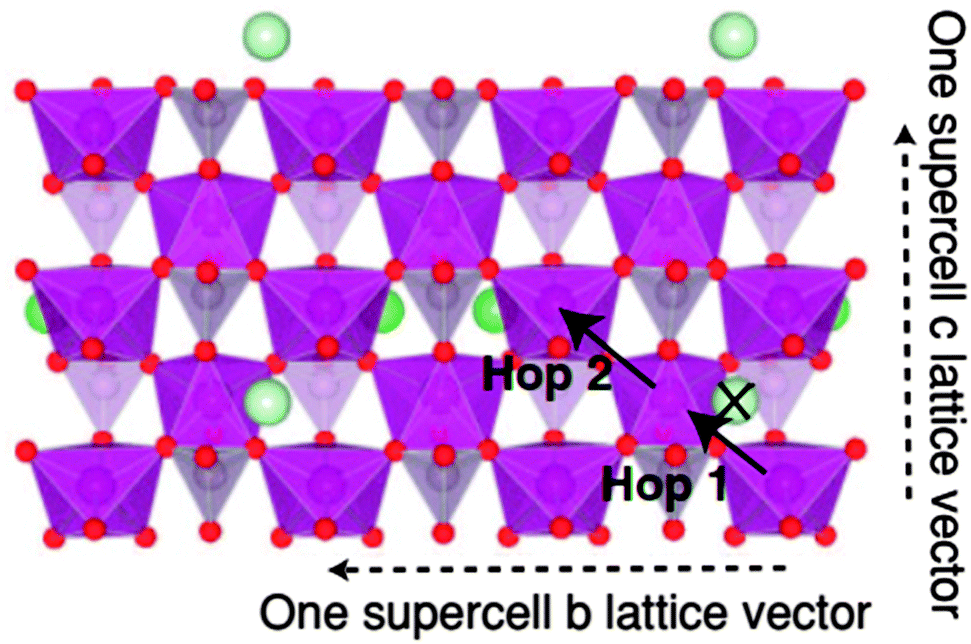 | ||
| Fig. 13 Single layer of an olivine LiMPO4 supercell viewed in projection along the (100) direction, showing polaron hops considered in polaron investigations.176 Reprinted with permission from ref. 176. Copyright 2011 American Physical Society. | ||
The authors computed the formation energies of LixMPO4 (x = 0.25, 0.50, 0.75) to study the phase separation behavior of Mn and Fe olivines as shown in Fig. 14.176 Their results showed an intrinsic difference in the electronic structures and kinetics of LiMnPO4 and LiFePO4. They found free hole and electron polaron migration barriers the Mn olivine, 133 and 63 meV higher than those in the Fe olivine respectively. However, in presence of Li ions or vacancies, these values became 100–120 meV. The computed formation energies were found approximately equal for both Mn and Fe olivines. LiMnPO4 has much lower conductivity than LiFePO4, however, Drezen et al.180 found that a reduction in particle size from 270 to 140 nm significantly improves the rate capability of LiMnPO4 as an electrode and even better performance was subsequently reported by Martha et al.181 with carbon-coated 30 nm particles. Such theoretical investigations were also illustrated in Kang et al.,159,182 Maxisch et al.,183 Yamada et al.,184 and Yonemura et al.185
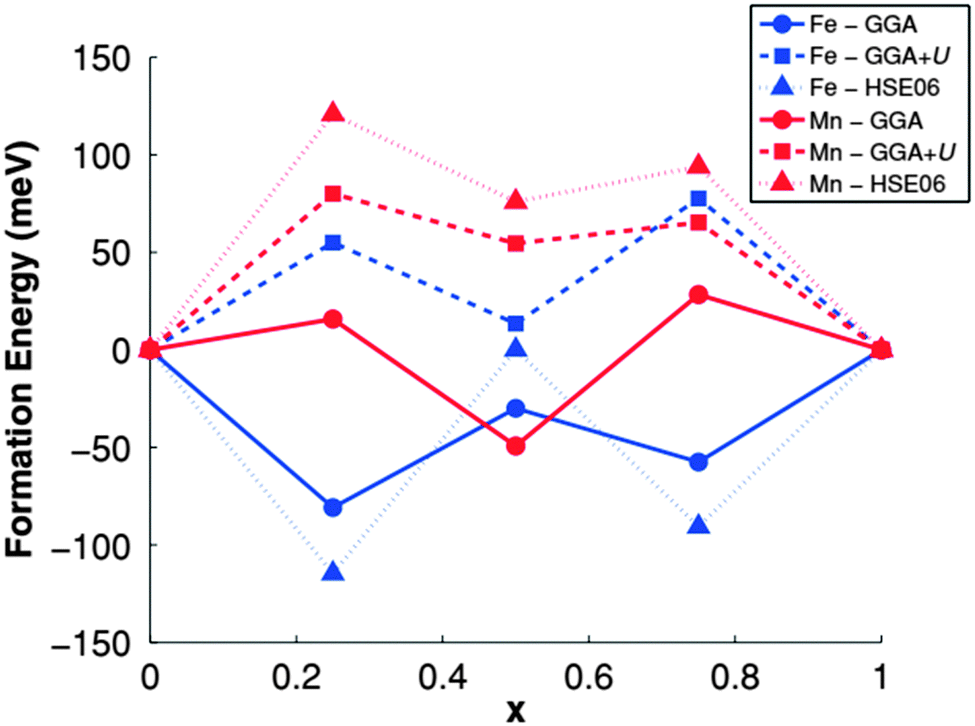 | ||
| Fig. 14 Formation energies of LixMPO4 using different functional. Reprinted with permission from ref. 176. Copyright 2011 American Physical Society. | ||
Varanasi et al.186 performed a first-principles study aimed at identifying structures to enhance the electrochemical potential of Al-substituted olivine phosphates LiFePO4 and LiCoPO4. The Al-substitution reduced the electrochemical potential, which is due to the sharing of charge transferred from Li by the oxygen and Co/Fe in olivine phosphates because of the strong covalency between oxygen and Co/Fe. Zhou et al.142 performed first-principles studies to calculate the redox potentials for olivine LixMPO4 (M = Mn, Fe, Co, Ni) using LDA/GGA and DFT + U approaches. The LDA/GGA approach gave underestimated values of redox potentials, while DFT + U approaches can reproduce experimental results. Zhu et al.187 performed DFT calculations to investigate the average voltage of lithiation/delithiation for Li-ion battery materials across 7 categories and 18 series (see reference for definitions), including LiMO2, LiMn2O4, LiMPO4, Li2MSiO4 and graphite. The average voltage of lithiation/delithiation in the relevant electrode materials was obtained by comparing the total energy difference before and after an electrochemical reaction consistent with experimental results. Such calculation methods could be applied as an easy and effective tool for predicting the potential performance of new lithiation/delithiation materials. Feng et al.188 performed ab initio calculations for ferrotoroidic olivine Li4MnFeCoNiP4O16 and found that this compound possesses ferrotoroidic characteristics and ferromagnetic configuration with a magnetic moment of 1.56 μB per formula unit. The ferrotoroidicity of this compound makes it a potential candidate as a magnetoelectric material. Their DFT + U calculations revealed that the indirect band gap is ∼1.25 eV but the electronic conductivity in Li4MnFeCoNiP4O16 is not defined by this band gap. The valence state was found to be Mn2+, Fe3+, Co2+, and Ni2+ for Mn, Fe, Co, and Ni ions respectively. Conclusively, more focused DFT calculations are required to address the relationship between physical characteristics and electrochemical potentials in complex phosphate materials as cathode materials in Li-ion batteries.
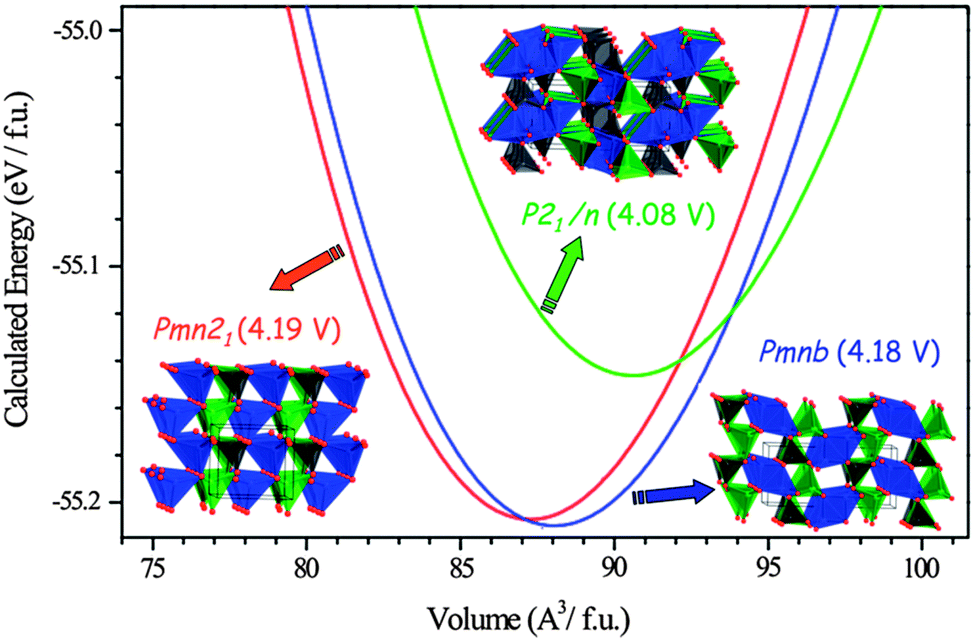 | ||
| Fig. 15 Calculated total energy versus volume curves of Li2MnSiO4 polymorphs; Pmn21 (red), Pmnb (blue) and P21/n (green). DFT (GGA + U, Ueffect = 4 eV) data were fitted to the Murnaghan equation of state. Calculated average voltage for the 2 electron process is given in parentheses. Reprinted with permission from ref. 193. Copyright 2008 American Chemical Society. | ||
Larsson et al.200 and Dompablo et al.201 both reported that the main drawback of this family of cathode materials is a strong driving force for the transition metal (M) ions to change their coordination together with randomized Li-site and M-site occupations upon Li extraction. It is difficult to obtain idealized two-step Li ion intercalation/deintercalation as well as changes in Li diffusion pathways because of structure transformation as stated in Armstrong et al.202 The authors used a combination of both experimental and theoretical methods to demonstrate that it is important to implement structure modification corresponding to the identified pathways to reduce the activation barrier. Fig. 16202 shows both P21/n and Pmn21 structures of Li2FeSiO4 and Li diffusion pathways.
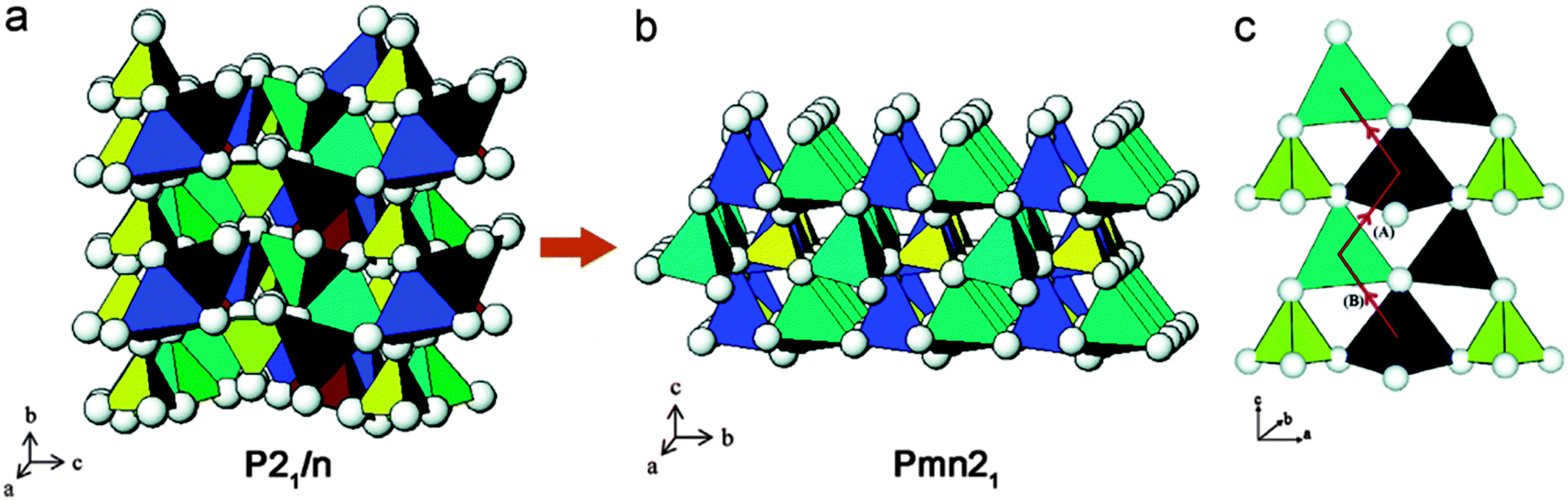 | ||
| Fig. 16 (a) Crystal structure of as-prepared Li2FeSiO4, space group P21/n, (b) crystalline structure of cycled Li2FeSiO4, space group Pmn21, and (c) pathways for Li ion migration. Reprinted with permission from ref. 202. Copyright 2011 American Chemical Society. | ||
In order to improve electrochemical performance, one of the most promising materials Li2MnxFe1−xSiO4 has been explored to achieve reversible exchange of up to two Li ions per formula unit. Kokalj et al.203 developed computational studies of LixMn0.5Fe0.5SiO4 and found that 0.7 V is required to extract more than one Li ion per formula unit, which is lower than the pure Fe counterpart. Additionally, the Mn/Fe structure could not be collapsed if the potential is controlled in such a way that less than 1.5 Li ions per formula unit is exchanged during the electrochemical cycling. However, Larsson et al.204 reported that the structural distortion and high voltage would limit the feasibility of this design with a decrease in the ratio of Mn substitution up to 12.5%. There are many previous studies involving the replacement of O by N,205 SiO4 by AsO4206 or BO3207 or VO4,208 and doping trivalent Al and Ga on the Si site.199 For example, Liivat et al.208 performed DFT calculations to study the substitution of SiO44− for VO43− poly-anions in the orthosilicate Li-ion cathode material Li2FeSiO4 to enhance the electron transfer between the transition metal ions and thereby obtain an increase in capacity due to the potential redox activity of the orthovanadate polyanion. The authors considered five different model structures for Li2FeXO4 (Si, P, V) and revealed that VO43− substitution destabilizes the tetrahedral structures towards olivine- or spinel-type structures. Moreover, Li et al.209 performed DFT calculations to study the feasibility of vanadium substitution into Li2FeSiO4 to allow more than one electron to be involved in the reaction, so as to increase the capacity of cathode material. The authors calculated electronic structure of Li2Fe0.5V0.5SiO4 upon delithiation and found that vanadium substitution into Li2FeSiO4 may be thermodynamically possible allowing more than one Li ion extraction and hence significantly enhance the capacity of the Li2FeSiO4 cathode material as well as improving the electronic conductivity of Li2FeSiO4 with lower band gap.
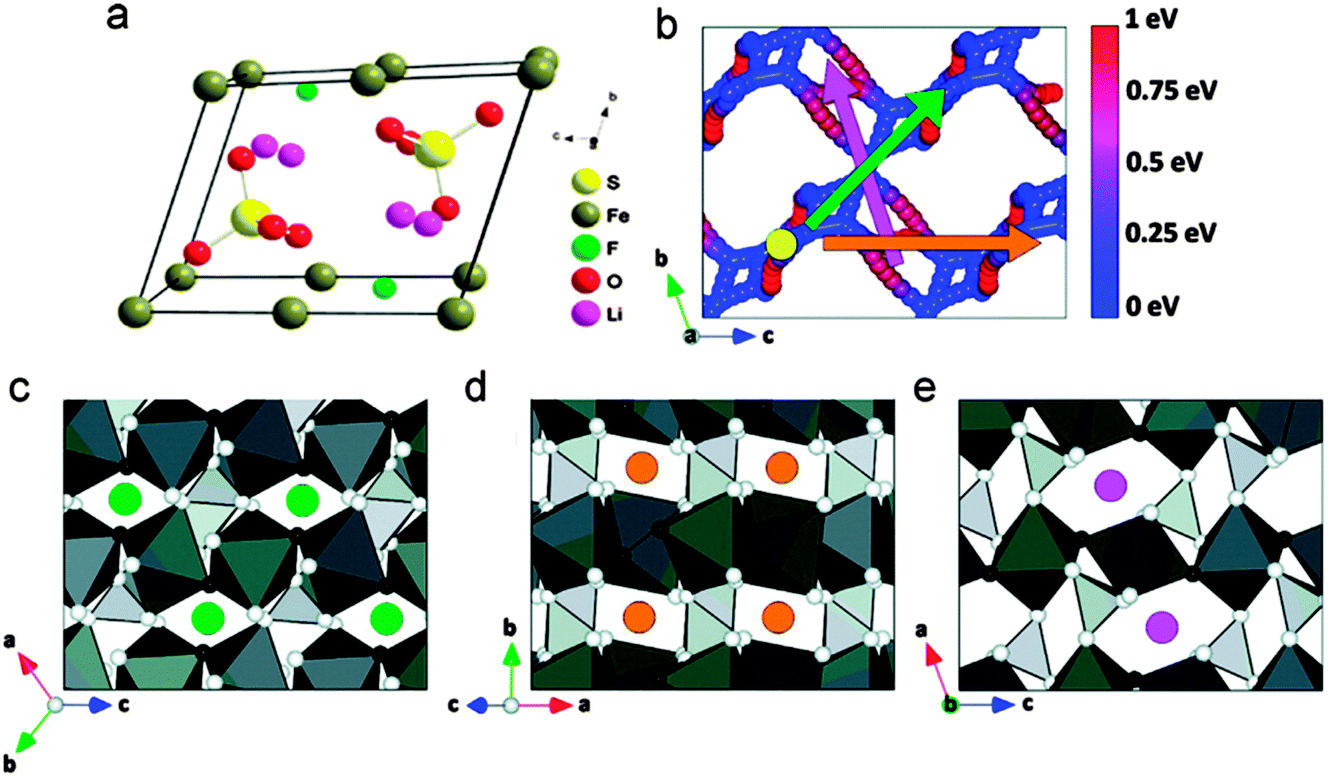 | ||
| Fig. 17 Lithium diffusion paths through a 2 × 2 × 2 supercell of FeSO4F: (a) crystalline structure of LiMnSO4F, (b) direct view of lithium diffusion paths through a 2 × 2 × 2 supercell of FeSO4F, corresponding to (c)–(e) by color. The host structure is removed and the color sphere indicates the energy difference between a point on the path and the lowest-energy lithium site, (c) the green circles mark diffusion channels in the (111) direction, with an activation barrier of 208 meV, (d) the orange circles mark diffusion channels in the (101) direction, with an activation barrier of 700 meV, (e) the pink circles mark diffusion channels in the (010) direction with an activation barrier of 700 meV (dark grey octahedra represent iron, light grey tetrahedra represent sulfur, white spheres represent oxygen, and black spheres represent fluorine). Reprinted with permission from ref. 218. Copyright 2011 American Chemical Society. | ||
Chung et al.220 investigated the degenerate energy states of Li in both tavorite (LiFeSO4F) and triplite (FeSO4F) polymorphs and found that the voltage difference is mainly due to the different stabilities of FeSO4F, likely because of Fe3+–Fe3+ repulsion in the edge-sharing geometry of the FeSO4F structure. Moreover, Yahia et al.221 reported that the voltage enhancement is due to the electrostatic repulsions induced by the configuration of F atoms around Fe cations using combined DFT + U approach and crystallographic analyses.
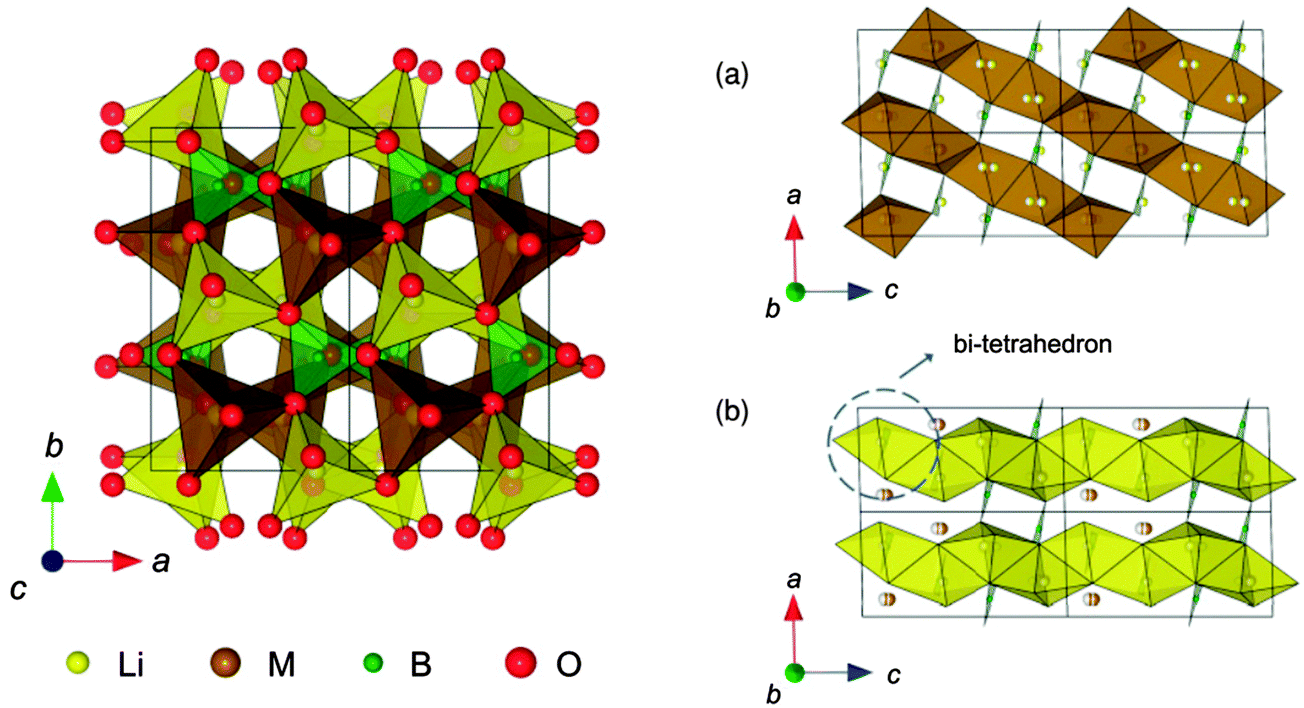 | ||
| Fig. 18 The structure of LiMBO3 (M = Mn, Fe, and Co): (a) MO chains with edge-sharing along the [−101] direction, and (b) edge-shared chains of LiO4 parallel to the [001] direction. Reprinted with permission from ref. 223. Copyright 2011 American Physical Society. | ||
The authors found small volume changes of LixMBO3 (M = Mn, Fe, and Co) with delithiation and these changes might facilitate intercalation or deintercalation with high reversibility. Their calculated DOS (see Fig. 19223) showed that LixMBO3 can be polaronic conductors similar to olivine phosphates. In addition, they found that the high probability of antisite defects of LixMBO3 may be disadvantageous for high power capability. As LiMBO3 are promising cathode materials in Li-ion batteries, more theoretical work based on DFT methods should be devoted to address the current challenges in LiMBO3 cathode materials.
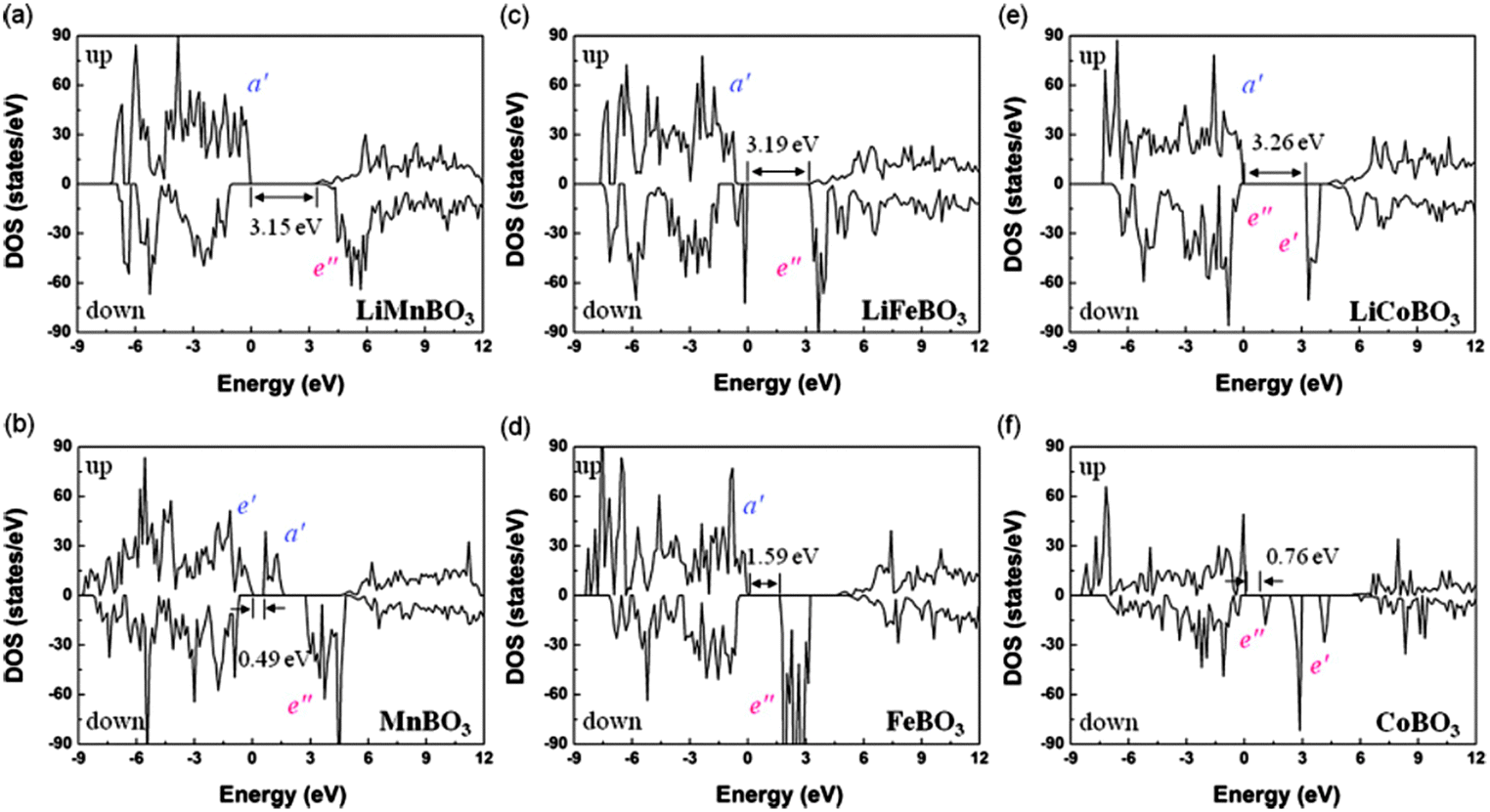 | ||
| Fig. 19 DOS of LixMBO3 (x = 1, 0; M = Mn, Fe, Co). Highest occupied bands, lowest unoccupied bands, and bandgaps are presented. Reprinted with permission from ref. 223. Copyright 2011 American Physical Society. | ||
Conclusively, DFT calculations could provide the following information about Li-ion battery cathode materials: (i) rate capacity can be known by calculating Li diffusion pathways and corresponding activation energies, (ii) the reaction mechanism can interpreted by calculating the phase diagrams and lithiated/delithiated voltage profiles, and (iii) cyclability can be predicted by calculating structural stability before and after Li intercalation. In addition, surface and interface chemical reactions and the processes that occur in electrolytes can also be assessed in exquisite detail. As such, DFT calculations have the potential to provide detailed understanding on major drawbacks of some cathode materials and predict possible mitigating solutions while filtering new materials.
4. Anode materials
To date, graphite-based anode materials are widely used in commercial Li-ion batteries due to their advantages of a long cycle life, low cost, and abundance. However, these materials have disadvantages such as (relatively) low gravimetric and volumetric specific capacity (approx. 372 mA h g−1 and 833 mA h cm−3 respectively).224 The development of new anode materials for Li-ion batteries has included carbon nanotubes, graphenes, Li-carboxylates, Li–M (M = Si, Ge, Sn, etc.) alloys, and lithium transition metal oxides such as oxides of Ni, Co, Fe, Mn, Cu, and Ti.224–229 Carbon nanotubes and graphenes are expected to have excellent lithium storage capability. Carboxylates are found to be good anode materials due to their high cyclability and thermal stability. Many issues related to their crystal structures and lithiation mechanisms are still unanswered. Alloys, especially Si-based alloy (Li4.4Si), have garnered considerable interest due to extremely high theoretical capacity of Si (∼4200 mA h g−1), and driven by the plethora of nanoscale silicon research for other reasons. Transition metal oxides such as Fe2O3, Fe3O4, Co3O4 and NiO have high theoretical capacity, for example, 920 mA h g−1 for Fe3O4. However, low conductivity, severe side reactions inside the cell, and volume expansion problems limit the practical applications of these new materials. Therefore, in order to explore their properties, more theoretical investigations based on DFT methods are now devoted to these new anode materials in order to understand reversible (de)alloying behavior and to predict metal–metalloid alloys that may improve capacity and rate performance as anodes for Li-ion batteries.4.1 Graphene
Challenges arose for graphite anodes in Li-ion batteries from continuous formation of a solid electrolyte interface (SEI), which results from poor interfacial properties. In order to improve the Li intercalation and interfacial characteristics during operation, graphene was considered as an alternative anode material230–232 in Li-ion batteries due to its high surface area, high electrical conductivity and robust mechanical integrity. The high capacity of about 540 mA h g−1 was reported for graphene compared to graphite anode.233 As the diffusion time of Li+ ions is directly proportional to the square of the diffusion length, the current rates can be enhanced due to reduced dimensions of graphene.234 The success of both the applications of graphene as anode material or artificial SEI layer strongly depends on the rate of Li diffusion, through and along the graphene sheet. However, there is still a lot of confusion as to why this benefit exists since mixed composites of graphite and graphene are nominally similar save for their preparation i.e., exfoliation of the graphitic sheets within the composite. Accessing intercalation sites should thus be possible with graphite if graphene-like chemo-mechanical exfoliation techniques are used to render these potential sites more accessible, even within the polymer-containing composite matrix material.Graphene is a single layer of graphite and can be considered as a promising material as Li-battery anode material due to its unusual electronic,235,236 mechanical,237,238 and transport239 properties. It can be grown by graphite exfoliation,240 Si sublimation from SiC substrates241 or by chemical vapour deposition242,243 on metal substrates. DFT calculations have been performed to determine the electronic structure of graphene with charge transfer and band structure calculations developed to understand the interaction between Li and graphene.244,245 Graphene may uptake Li+ on both sides of its unique geometric sheet-like structure.
Bhardwaj et al.246 reported that an initial capacity of 1400 mA h g−1 and reversible capacity of 800 mA h g−1 are observed for oxidized graphene nanoribbons (GNRs), which was obtained by unzipping pristine multi-walled CNTs probably due to abundant edges in GNRs. Wu et al.247 performed DFT calculations for N atom-doped graphene as an anode material and reported no much improvement in Li intercalation. Reddy et al.248 studied the reversible Li intercalation properties of N-doped graphene (NG) and attributed its performance to the large number of surface defects caused by N-doping. Das et al.249 studied the effect of both boron and nitrogen doping on graphene by calculating the energy barrier through the defected doped graphene sheets using DFT methods. They found that the height of the barrier depends strongly on the concentration and type of dopants suggesting that a boron doped divacancy emerges, improving its performance as a candidate anode material. Moreover, Ma et al.250 investigated the performance of differently doped N atoms (including graphitic, pyridinic, and pyrrolic N atoms) on graphene, and reported that pyridinic N-doping is the most suitable for Li storage.
Wang et al.251 demonstrated boron-doped graphene under limited conditions and found a Li storage capacity of 2271 mA h g−1 as well as formation of Li6BC5 compound after Li adsorption. Yang et al.252 reported that Li+ intercalation may cause structural changes. Uthaisar et al.253 have investigated Li diffusion in graphene and quasi-1D graphene nanoribbons, and found that the presence of zigzag and armchair edges affects both Li adsorption and Li diffusion. Li tends to migrate to the edges of graphene for lower energy barriers as shown in Fig. 20.253 Their calculated results showed that narrower graphene nanoribbons are promising Li-ion battery anode materials in agreement with experimental results.246
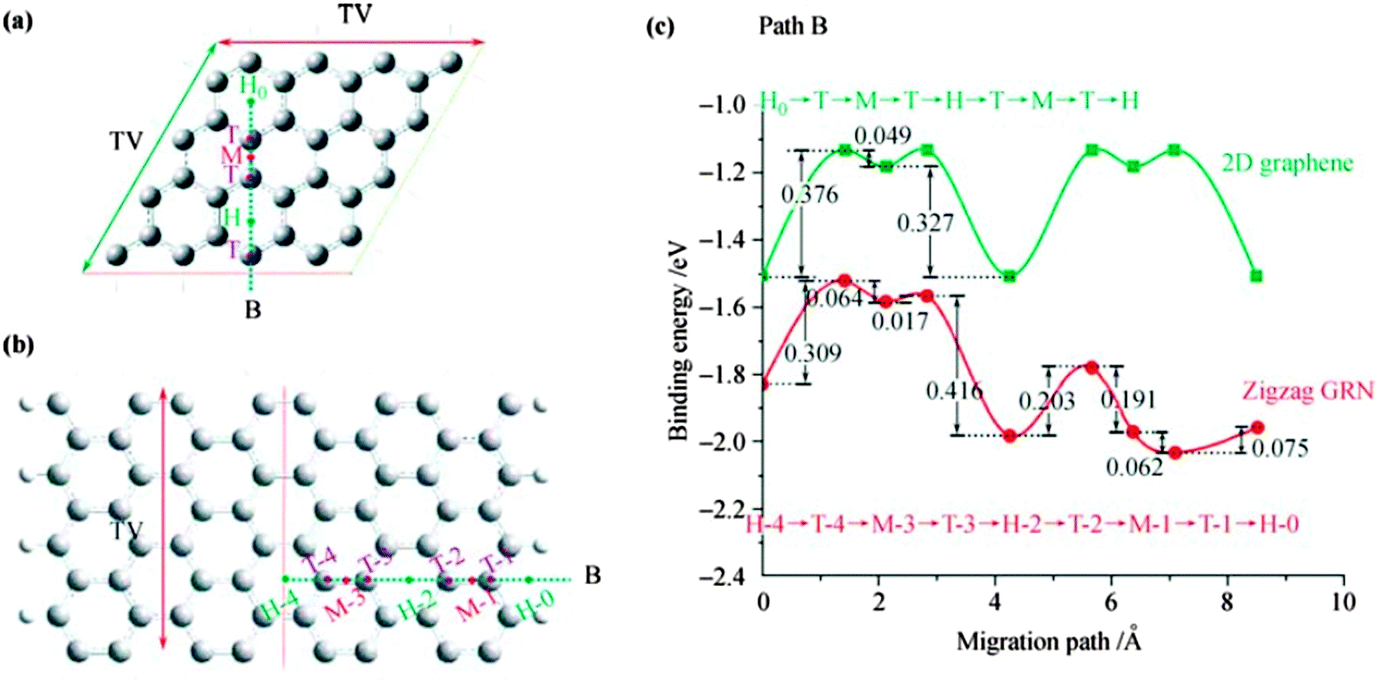 | ||
| Fig. 20 Schematic representation of grapheme: (a) and zigzag graphite nanoribbons, (b) cells and B migration paths, and (c) energy barriers for path B in graphene and the zigzag GNR. H, M, and T represent the Li adsorption positions on top of the hexagon, on the middle of the bond, and on top of a C atom, respectively. Reprinted with permission from ref. 253. Copyright 2010 American Chemical Society. | ||
As there is lack of theoretical investigations based on DFT methods on graphene (single and bilayers, nitrogen and boron doped, nanoribbons and nanosheets) as anode materials in Li-ion batteries, further investigations are still needed to fully understand the lithium storage mechanism.
4.2 Li–M (M = Si, Ge, Sn) alloys
Lithium can form well-defined intermetallic phases (LixM) with numerous metals M (M = Mg, Ca, Al, Si, Ge, Sn, Pb, As, Sb, Bi, Pt, Ag, Au, Zn, Cd, Hg, etc.) at room temperature if the metal is polarized to a sufficiently negative potential in a Li+ ion containing organic electrolyte solvent.254 As the LixM alloys are reversible i.e. can be dealloyed, researchers have shown considerable interest for their use as anode materials in rechargeable Li-ion batteries.255–257 Among all above metal anode materials, attractive high energy density alloy anode materials include Si, Ge, and Sn, which have high theoretical capacities, for example, 4200 mA h g−1 for Li4.4Si, 1600 mA h g−1 for Li4.4Ge, and 900 mA h g−1 for Li4.4Sn.258,259 Li–M alloys possess the higher energy storage capacity due to their safe thermodynamic potential. However, these materials have poor cyclability due to the larger volume change during lithiation–delithiation process, for an instance, up to 400% for Si, 370% for Ge, and 300% for Sn.260–262 In order to overcome these problems, one of the successful strategies for designing alloy anodes for Li-ion batteries is to synthesize them into nanoparticles or nanotubes or nanowires.255,263–283 The reason is that nano-electrodes possess high surface to volume ratio, which enhances the battery performance. The energetics and kinetics of Li insertion/extraction in nano-electrode surfaces can be understood better by availing of DFT calculations probe the effects of Li ion transport inside the cell. It is well known that Si has the highest specific capacity compared to Ge and Sn, so it is a primary choice for next-generation anode materials in Li-ion batteries. Peng et al.284 performed DFT calculations for Li transport properties in Si surfaces with a six layers slab for the Si(100) surface and found that surface incorporation of a Li atom is responsible for retarding fast Li transport. There is lack of research on Li-transport properties in Ge and Sn surfaces. Bulk Ge and Sn possess better atomic transport for Li as compared to Si, for example, diffusivities of Li in Ge and Sn are found to be 10−12 to 10−10 and 10−8 to 10−7 cm2 s−1 respectively275,285 compared to 10−14 to 10−13 cm2 s−1 in Si. Therefore, these excellent Li transport properties in Ge and Sn surfaces have lead to investigations into the differences in Li intercalation behaviors in Ge and Sn compared to Si surfaces.Shenoy et al.286 performed a first-principles study on volume change in Li–Si phase and the relationship between fracture energy and Li concentration. Si alloys with inserted Li ion transforming finally into a well-known crystalline phase Li15Si4,287–289 accompanied by an amorphization of crystalline phase.290 The inserted Li atom prefers the tetrahedral site and the charge transfer from Li ion leads to an isolated state of Li impurities, resulting in weakening of nearby Si–Si bonds291 due to decrease in Young's modulus, decrease in strength, and a brittle-to-ductile transition.292,293 Chou et al.294 performed DFT calculations to investigate the structure, stability, Li diffusion, Li–Li interaction, and influence on host lattice M (M = Si, Ge, and Sn). The authors computed the structure, energetics, electronic, and mechanical properties of Li–M alloys, and analyzed structural parameters in terms of Li content for both crystalline and amorphous phases. They further and estimated the relative stabilities of the alloy based on their mixing enthalpies. The authors also analyzed the bulk modulus and elastic constants of alloys to determine the relationship between the structural and mechanical properties. Such investigation supports the understanding of the nature of Li incorporation and alloying with Si, Ge, and Sn, which may provide a framework for the comparative study of various Li–M alloys. Li et al.295 established a quantitative correlation for calculating the bulk modulus of Li–M binary alloys on the basis of a newly proposed metallic electronegativity scale. The authors simulated the cell parameters by generating various stable lithium silicide such as LiSi, Li12Si7, Li7Si3, Li13Si4, Li15Si4, Li21Si5, and Li22Si5296 and calculated bulk moduli of Li–Si alloys with increasing Li concentration along with other DFT results for comparison,290,291,293,296 signifying that the bulk modulus of Li–Si alloy strongly depends on Li intercalation and shows significant decrease with increase in Li fraction. For Li diffusion inside Si nanowires, the Li surface diffusion has a much higher chance to occur than surface-to-core diffusion (see Fig. 21297).
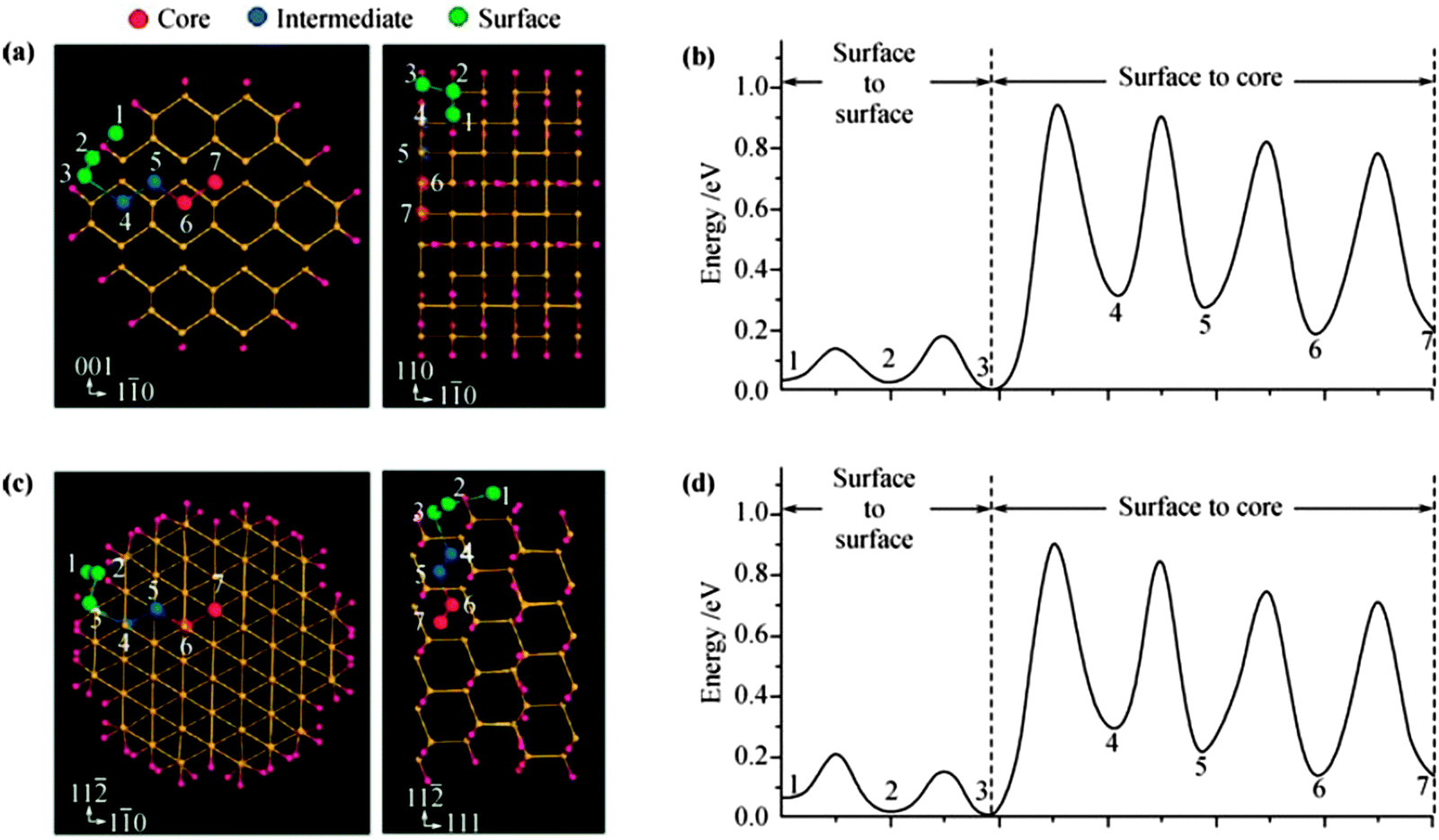 | ||
| Fig. 21 Li diffusion in 1.5 nm (110) and (111) SiNWs. (a), (c) and the diffusion barriers along the pathways are shown in panels (b) and (d), respectively. Reprinted with permission from ref. 297. Copyright 2010 American Chemical Society. | ||
In Si nanostructures, Li prefers to reside in the interstitial tetrahedral sites at the core and the migration is mediated by the hexagonal sites as saddle points as shown in Fig. 22.284,298
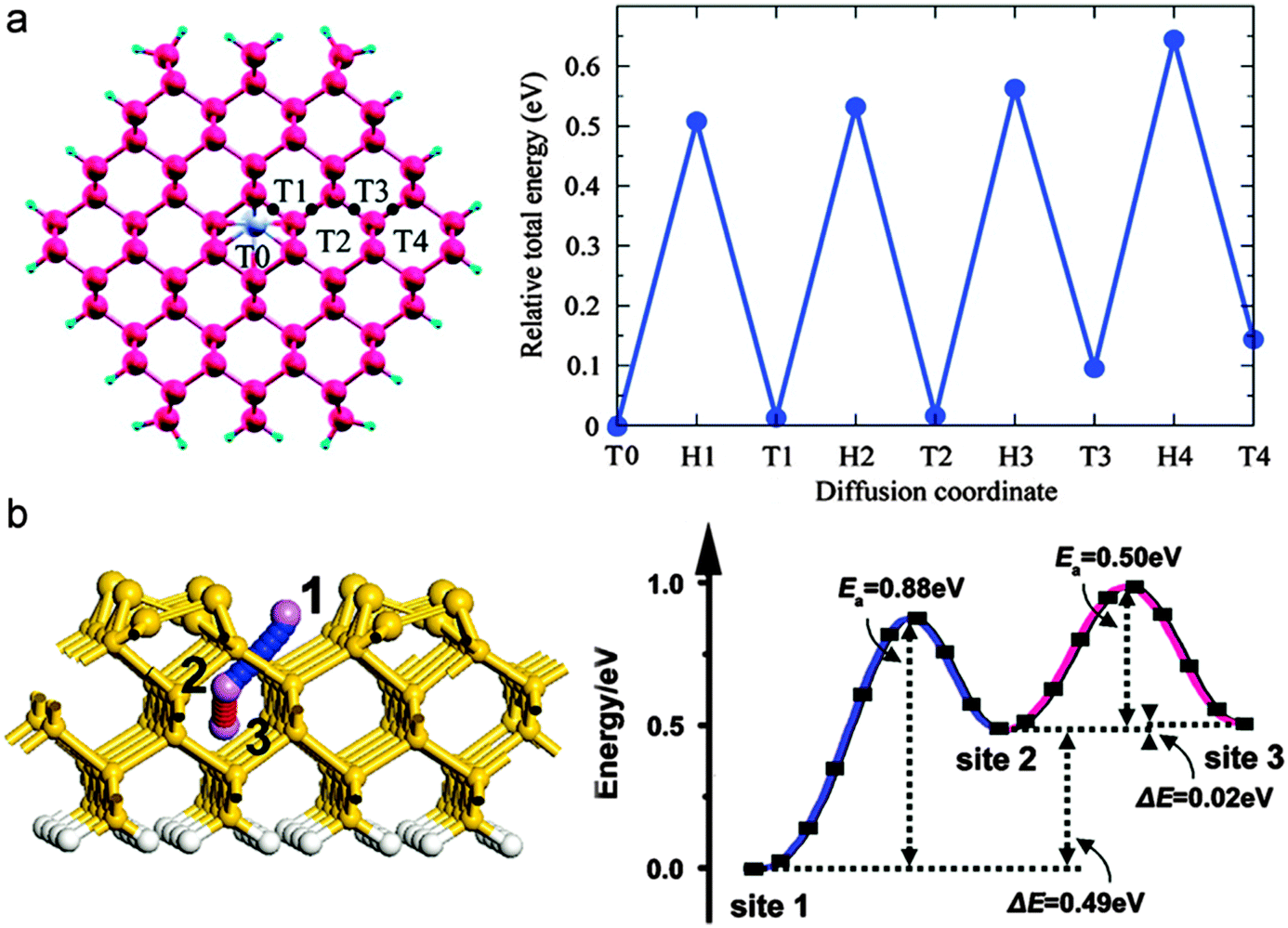 | ||
| Fig. 22 (a) Li diffusion pathway from the center tetrahedral site toward surface in Si(110) nanowire and corresponding energy change. Reprinted with permission from ref. 298. Copyright 2010 American Chemical Society, and (b) Li transport pathway through Si(100) nanofilm and corresponding energy profile. Reprinted with permission from ref. 284. Copyright 2010 American Physical Society. | ||
The Li–Sn alloys possess various crystal structures such as LiSn, Li2Sn5, Li7Sn3, Li5Sn2, Li13Sn5, and Li7Sn2.299 The authors used the optimized cell volume of Li–Sn phases in their calculations to compute bulk moduli of Li–Sn alloys compared to other DFT results294,296,300 and found elastic softening with increasing Li content. Jung et al.301 performed DFT calculations to investigate the Li intercalation behavior at (100) and (111) surfaces of Ge and Sn by considering thick 14-layer slab models for the surfaces. The authors found 4 and 12× faster Li diffusion in Ge and Sn respectively, along the 〈100〉 direction compared to diffusion along the 〈111〉 direction. Some retardation of fast Li transport was observed at the Sn surfaces, but negligible in case of Ge surfaces. The rate-limiting step in Sn may be the subsurface diffusion in both (100) and (111) surfaces. Their calculated results suggest that the Li transport efficiency remains unchanged in the Ge electrode regardless the size of the electrode, but Li transport in the Sn electrode becomes worse due to the surface retardation effect as surface/volume ratio increases for the Sn electrode.
Excessive volume change is big challenge for alloy anodes in Li-ion batteries. In order to tackle such issues, scientists have developed alloy materials on the nanoscale. Chan and Chelikowsky performed ab initio calculations to discuss Li diffusion in Si nanostructures298 and found that Li diffusion barrier is ultimately affected by nanocrystal size. The diffusion barrier of Li+ ions moving into the core is lower than that moving towards the surface, indicating the fact that charging of Si nanostructures is easier than discharging, as in case of Si nanowires due to high surface/volume ratio.301 In actuality, surface sites, specifically (110) on Si nanowires, were found to be energetically more stable than the core and intermediate sites.302 Thus, the Li binding energy gradually increases to bulk values with the increase in the diameter of Si nanowire. Previous DFT investigations reported that Ge and Sn have the same problems as Si. Li–Sn system was investigated by using both experimental and DFT methods.303 Kumar et al.304 proposed that Sn can be filled into carbon nanotubes to overcome the issue of volume expansion by benefiting from elastic deformation accommodation offered by the carbon. The DFT investigations have been accomplished for modified morphologies of C, Si, and Sn such as core–shell structures, nanowires, and nanotubes to improve the electrochemical and mechanical properties as promising new anode materials.305–307
4.3 Lithium transition metal oxide anodes
Lithium transition metal oxides are usually used as active cathode materials in Li-ion batteries. However, researchers have been focused on determining whether lithium transition metal oxides may be used as active anode materials in Li-ion batteries at low voltages. Theoretical studies have been devoted to the progress of these materials.308,309 The transition metal oxides used as Li-ion battery anode materials adopt a conversion procedure310 in some cases, while allowing classic reversible intercalation mechanisms in other lithiated metal oxides, but a lower voltages. The Li intercalation mechanism can be interpreted as| 2yLi+ + 2ye− + MxOy ↔ yLi2O + xM | (4) |
Here M is transition metal such as Cu, Mn, Fe, Co, and Ni310–314 and many conversion processes following similar mechanisms.
Experimentally, spinel-type lithium titanium oxides ranging from LiTi2O4 to Li4Ti5O12 possess good safety and cyclability, and thus these materials can be used as active anode materials in Li-ion batteries.315,316 These materials have also been studied theoretically by using first-principles methods. Jung et al.317 computed electrode potential curve using Monte Carlo methods. First principle calculations showed that Li4Ti5O12 is insulating, while Li7Ti5O12 is metallic318 and that the Li concentration increased to Li8.5Ti5O12 to improve the energy density of Li-ion battery anode significantly.319
TiO2 anodes have been received more attention in battery research320 since these are used in many applications.321 Many experimental and theoretical investigations showed that TiO2 has great potential to achieve better performance as a Li-ion battery anode322–327 because it acquires minimal volume expansion325,328 and thus it is in principle, safer than graphite.329 The Li insertion into TiO2 can be accompanied by phase transitions.330–332 In actuality, Li insertion into TiO2 at room temperature has two phase equilibria with a tetragonal Li-poor and an orthorhombic Li-rich phase. The Li-rich phase acquires the composition of Li0.5TiO2,333,334 which was predicted by first-principles calculations with good agreement with observed results, suggesting a mechanism for the tetragonal to orthorhombic transformation of Li0.5TiO2 phase.335 Kerisit et al.336 investigated the various polymorphs of TiO2 and found that the main factors controlling the relative stability of the lithiated titania polymorphs are primarily the lithium bonding environment, the configuration of LiOx and TiO6 polyhedrons, and the extent of lattice deformation upon lithiation. The calculated structures of TiO2 polymorphs are shown in Fig. 23.336
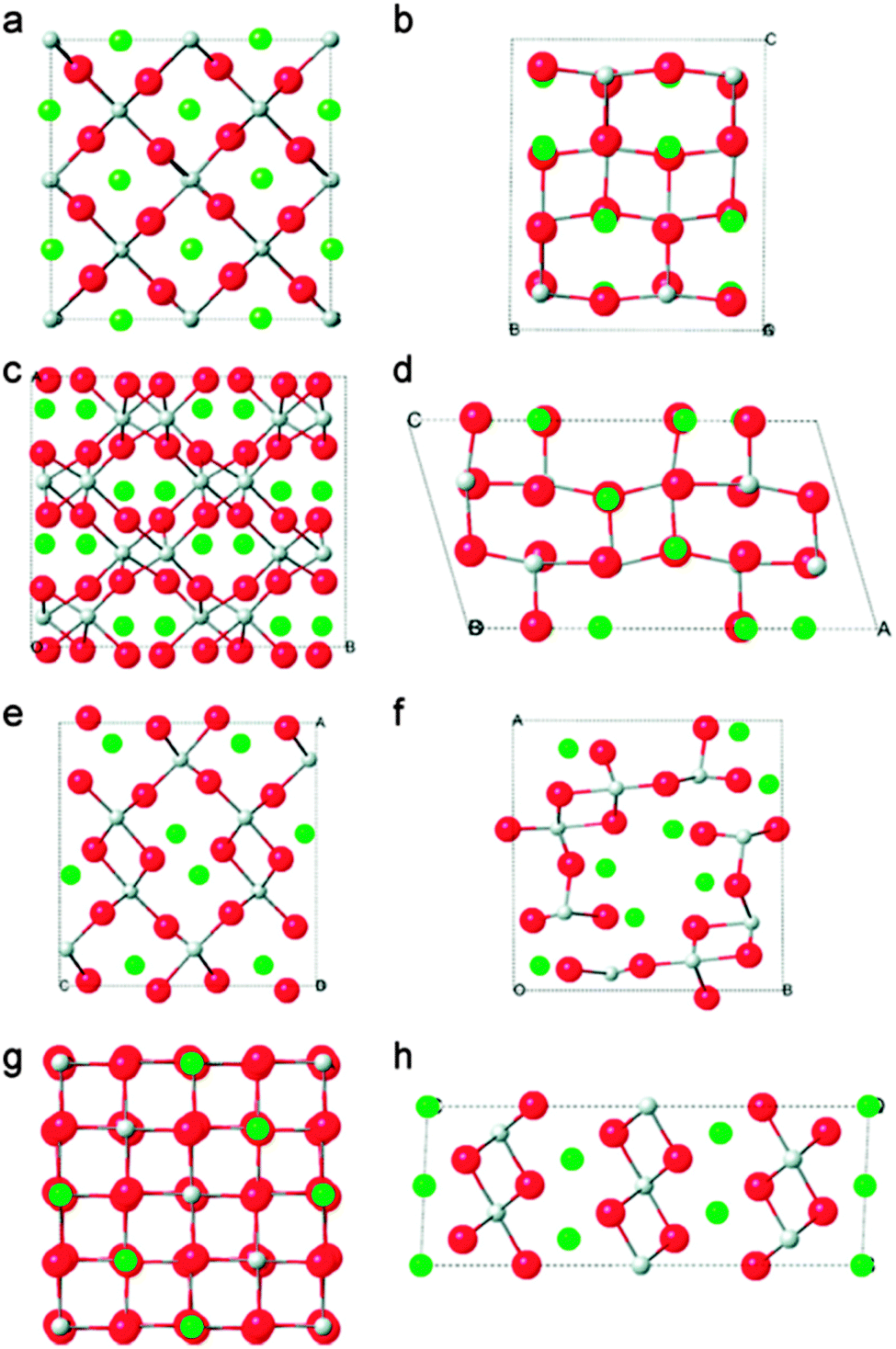 | ||
| Fig. 23 DFT-optimized structures of the eight LiTiO2 polymorphs: (a) rutile, (b) anatase, (c) brookite, (d) TiO2-B, (e) ramsdellite, (f) hollandite, (g) spinel, (h) hexagonal. Reprinted with permission from ref. 336. Copyright 2010 American Chemical Society. | ||
It was observed that conductivity depends upon factors such as Li concentration, size of nanoparticle, crystallographic direction, and titania polymorph in case of TiO2 nanoparticles.337,338 However, the original rutile and anatase structures of TiO2 yield lithium titanate during Li insertion with many polymorph symmetries such as hexagonal, monoclinic, cubic spinel, and cubic rocksalt.339–342 The anatase TiO2 was found to be more energetically stable than rutile for Li insertion.339 Li intercalation into anatase TiO2 has been studied using both experimental and theoretical techniques.333,334,339,343,344 Koudriachova et al.335 investigated a model and kinetics of TiO2 anode material and explained why Li intercalation into rutile TiO2 is not available.
Among all polymorphs of TiO2 such as rutile, anatase, brookite, and bronze (B), TiO2(B) has attracted more attention of researchers due to its high energy density and its ability to nanostructure into several architectures. Initially, TiO2(B) lithiates according to the equation:
| xLi+ + xe− + TiO2(B) ↔ LixTiO2(B) | (5) |
In subsequent cycles, LixTiO2(B) is reversibly lithiated/delithiated according to:
| LixTiO2(B) ↔ Lix−yTiO2(B) + yLi+ + ye− | (6) |
TiO2(B) is suggested to be an active anode material due to its unique crystal structure. It has the lowest density and a perovskite-like layered structure unlike other polymorphs of TiO2, allowing faster Li+ diffusion. The mechanism facilitating high rate capabilities is still a subject of debate.345,346 The high surface areas and unique surface energetics have been found to be the key factors in explaining the high capacity and rates for TiO2(B) nanostructures.347 There are three possible Li intercalation sites as labelled by C, A1, and A2 in Fig. 24.348
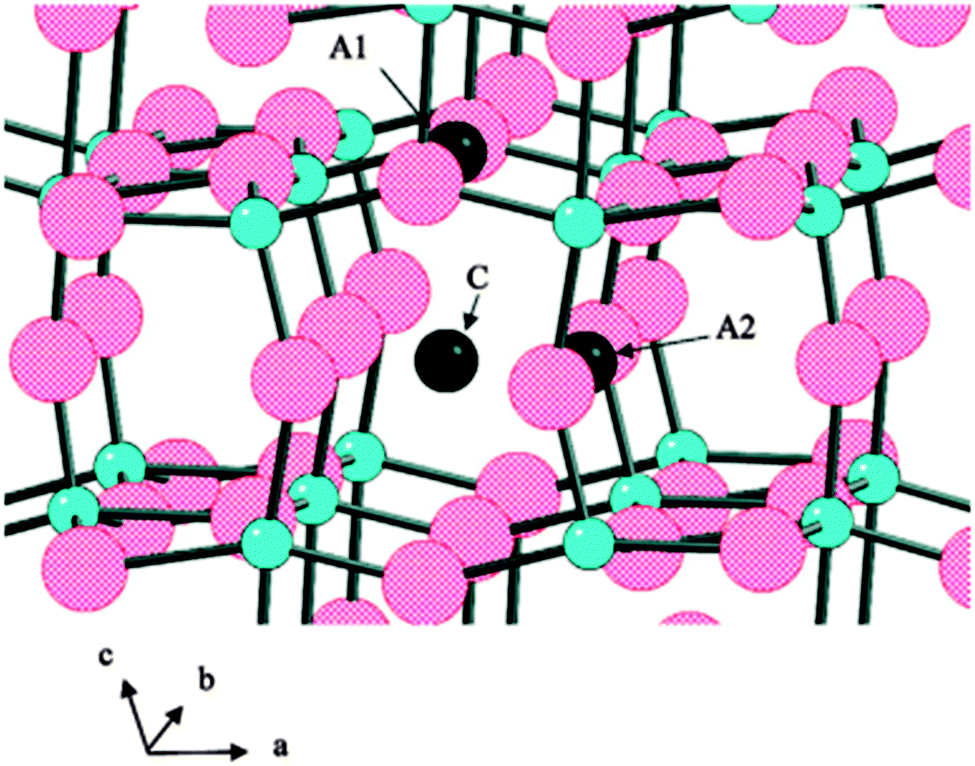 | ||
| Fig. 24 Possible lithium intercalation sites in TiO2-B labeled C, A1, and A2. Large spheres are oxygen atoms (red) and small spheres are Ti atoms (blue). Reprinted with permission from ref. 348. Copyright 2009 American Chemical Society. | ||
Moreover, previous studies regarding the doping of TiO2 with Zr reported its effect on the transformation of anatase TiO2 into rutile and on the photocatalytic activity of doped anatase349–351 due to the fact that Zr4+ ions can be easily incorporated into the anatase lattice by substituting on the Ti side as Zr4+ ions are isovalent with Ti4+ ions.
Finally, many key issues are still unclear for transition metal oxides as Li-ion battery anode materials, and DFT studies could be used effectively to provide insight into the factors that affect the changes in the intercalation potential of transition metal oxides and a meaningful understanding of the relationship between structure and properties to optimize the electrochemical performance of anode materials. Therefore, new novel oxide materials as well as new computational methodologies including DFT are required in the future.
4.4 Carbon nanotubes (CNTs)
It is well known that graphite has been widely used as anode material due to its high electronic conductivity as a consequence of the delocalized π-bonds as well as its appropriate structure for Li+ ion intercalation and diffusion.352 However, the capacity of Li-ion battery graphite anodes is theoretically limited, since Li+ ions can only combine with every second carbon hexagon in the graphite sheets (LiC6) resulting the limited theoretical (372 mA h g−1) and experimental (280–330 mA h g−1) based on the types of graphite used.353 In order to overcome issues of traditional graphite anodes, researchers have focused on an allotrope of graphite known as carbon nanotubes (CNTs) due to an improvement in capacities of Li-batteries because of their structures and properties.354 In general CNTs show three chiralities: armchair, zigzag, and chiral. CNTs with (m,m) and (m,0) lattice vectors are called armchair and zigzag respectively, and otherwise chiral. If the difference between m and n is a multiple of 3 in an (m,n) nanotube, it is metallic, otherwise it is semiconducting.355–359 The chirality of CNTs is directly related to their metallic and semiconducting structures.There have been many scientific investigations of the mechanism of Li storage in CNTs including theoretical research work.354,360–374 Yang et al.360 investigated a surface adsorption mechanism through which Li+ ions are stored in the naked surface of CNTs and intercalation of Li+ ions into raw end-closed CNTs. It is reported that single-walled carbon nanotubes (SWCNTs) and multi-walled carbon nanotubes (MWCNTs) possess high conductivities ∼106 and 105 S m−1 respectively and high tensile strength up to 60 GPa.375–377 Zhao et al.361 studied the intercalation of Li+ ions in both zigzag and armchair SWCNTs using first-principles methods, suggesting the susceptibility of both inside and outside of CNTs capable of Li intercalation to give higher Li density. Senami et al.362 studied the adsorption of Li+ ions on the surface of (12,0) SWCNTs using ab initio methods to investigate more favorable sites for Li adsorption and suggested that inner part of tube is more favorable than outside for a single Li+ ion adsorption and the bonds between Li and CNTs have ionic characters. They assumed that the charge transfer from attached Li+ ions to CNTs depends on the radii of curvature of CNTs. Research on the multiple attachments of Li+ ions on the inside of the CNTs proposed that four Li insertion into SWCNTs is the most stable. Shimoda et al.354 demonstrated chemically etched CNT bundles can achieve a high reversible Li storage capacity, which probably is due to its ability to intercalate Li+ ions in the interstitial spaces of CNT bundles via van der Waals forces. The intercalation and diffusion of Li+ ions in a CNT bundle was investigated by the authors in Song et al.363 Khantha et al.364 investigated the interaction and diffusion of Li+ ions in (5,5) armchair CNTs using DFT methods and analyzed the diffusion of Li+ ions in two directions: radial and axial. It was found that the most favorable positions for Li are along a straight line passing through the center of a six-member carbon ring on one side of the wall and the mid-point of a C–C bond on the opposite side for a Li+ ion moving in radial direction. The Li–Li repulsion associated with Li-tube interaction was demonstrated by Zhao et al.,365 in which the authors found that Li possesses high mobility along the tube axis with low energy barrier and high diffusion barrier along the radial direction.
It is well known that the morphology of CNTs as anode materials such as defects, lengths, diameters, etc. influence their electrochemical response. There are many primary methods to modify the morphologies of CNTs in scalable bulk quantities, two of which are chemical etching and ball-milling. The capacity of CNTs was increased to 1000 mA h g−1 after aggressive ball-milling due to the fact that topological defects were introduced to CNT walls during milling process so that both interior and exterior of CNT can be used to adsorb Li+ ions. Udomvech et al.378 reported the importance of chirality in Li-tube interactions, which may affect the applications of CNTs in Li-batteries. The results revealed that the diffusion of Li is largely impossible across the hexagonal carbon structure371,379 – it remains to be seen what size cation or charge can pass through graphene, possibly protons. Some researchers have assumed that the interaction between Li+ ions and CNTs is somewhat covalent.303,371
Zhao et al.361 also reported that boron- or nitrogen-doping CNTs anodes could improve their performance. Nitrogen has one extra electron and boron is an electron-deficient compared with carbon in carbon-based materials. Therefore, doping of these two elements may cause different Li adsorption and different charge distributions. Accordingly, Li storage capacity of CNTs is directly related to their electronic structure. Mukhopadhyay et al.380 proposed that B-doped MWCNTs have greater Li storage capacity due to higher specific area, larger defect concentration, and higher conductivity. However, due to negative effects on Li adsorption caused by electron rich structure, N-doped CNTs may not be appropriate Li-battery anode materials.304–306 Li et al.381 proposed one electron deficient pyridine-like CNTs in place of N-doped CNTs due to their low diffusion barrier (1.44 eV) compared to pristine CNTs with an enneagon (nonagon) hole.382 Such pyridine-like CNTs have large adsorption energies and high irreversible capacities in practical applications, since Li is adsorbed at the site of pyridine-like structure. The structure of various single-walled nanotubes with defects is shown in Fig. 25.382
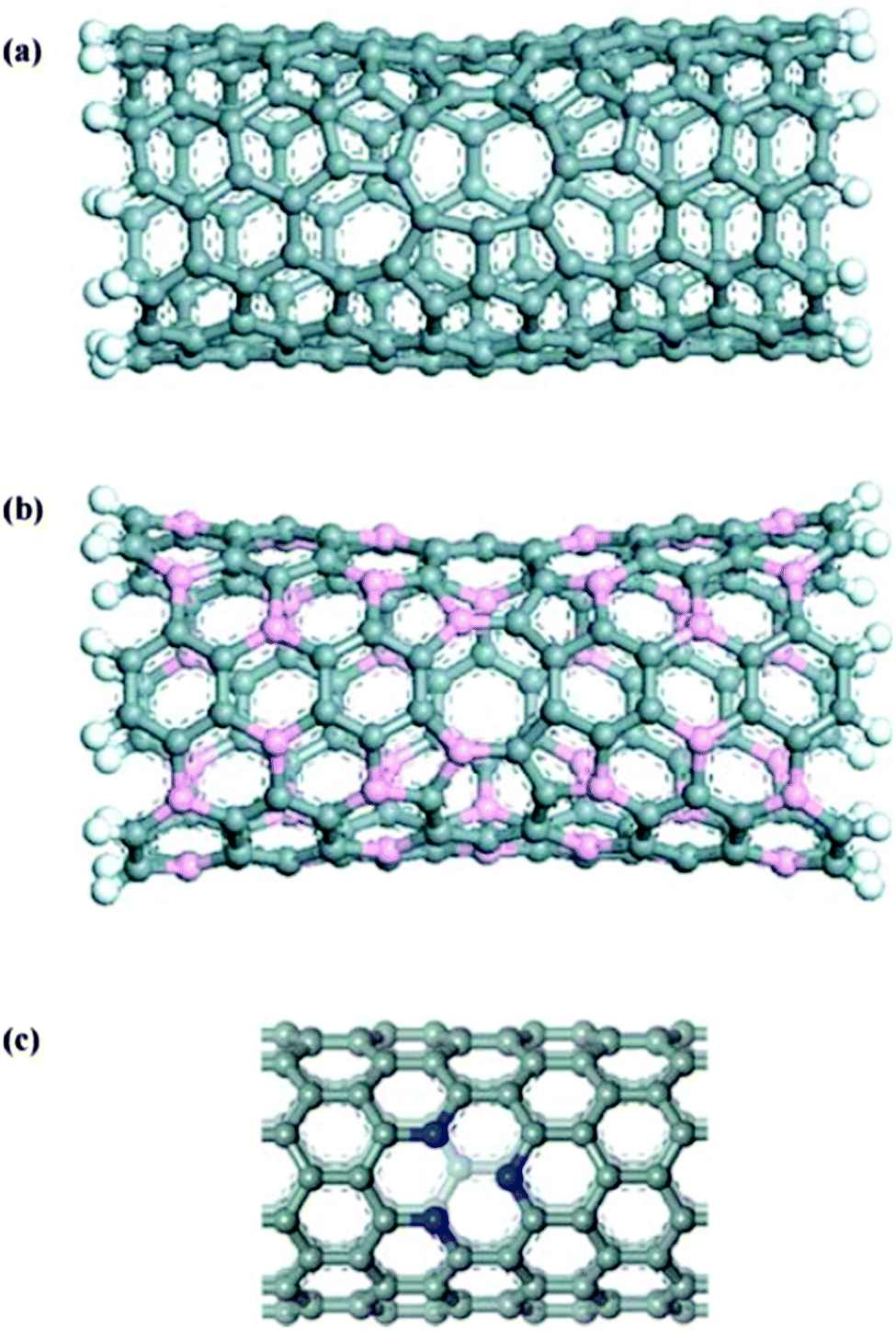 | ||
| Fig. 25 Various single-walled nanotubes with defects: (6,6) carbon nanotube with a enneagon hole (a), (3,3) BC nanotube with an octagonal hole (b), and (10,0) carbon nanotube with a pyridine-like structure (c). Grey, orange, blue, and white balls denote carbon, boron, nitrogen, and hydrogen atoms, respectively. Reprinted from ref. 382. Copyright 2005, with permission of Elsevier. | ||
Finally, there remains a lack of clear understanding regarding the Li storage capacity of CNTs. This is in part caused by issues related to inaccurate weighing of the active materials, capacity calculation based on only the CNT portion of the complete anode, and more succinct differences caused by purity differences in the CNT product used between various labs. These issues of course arise in complex material mixtures of many forms and compositions in Li-ion battery research. Therefore, more theoretical research based on DFT methods can help understand whether such large variations are due to the very nature of the material itself, so as to help deconvolute the response of the materials from the response of the electrode formation in some systems.
4.5 Li-carboxylates
Carboxylates have been reported to be better anode materials in Li-ion batteries with respect to their observed thermal stability, rate capability, and high cyclability. The operating voltages of these anodes are found to be slightly larger than those of graphite or Li metal, thus avoiding SEI formation and solvent breakdown. Such carboxylates can also be used in Li–air batteries due to their redox potentials, Li+ ion transport, and stability properties. These carboxylates are expected to form ordered organic crystals. Some DFT methods are limited in describing the long range interactions in molecular crystals in the sense that the prediction of structure, relative energies, and relevant energy storage properties such polymorphs is quite difficult.54,163 Burkhardt et al.258 performed DFT calculations for a set of 12 known crystalline organic Li-ion electrode materials based on carboxylates. The authors made a comparison of two carboxylate molecular crystals based on molecular length, carboxylate location and π-electron topology. In addition, they compared the computed properties of novel materials such as formal reduction potential, Li+ binding energy, solvation energy, and reaction indices, which can then be used to make general conclusions about relevant organic anode properties such as redox tuning/anode potential, solubility, and stability. In this literature, two carboxylates (see Fig. 26258) were examined by authors in detail to develop a generalized approach for computing approximate redox potentials from the test set of 12 molecules. The authors found several structural observations such as their planarity upon reduction and maximum coordination of Li+ with oxygen atoms as shown in Fig. 27.258 The correlation between calculated and experimental potentials was found to be quite good in all cases, when they considered the contribution of experimental galvanostatic discharge data for cell internal resistance and/or sluggish kinetics. The coordination by multiple carboxylate functionalities in the crystalline state may stabilize the reduced form and these effects were not observed in lower dimensional calculations. Electronic interactions (π–π* stacking) within the crystal may enhance charge delocalization, shifting the reduction potential to more positive values. In such a case, more DFT studies are required to provide a clearer description of these materials by modelling the reduction processes of π-stacked carboxylate dimers. | ||
| Fig. 26 Structural details and melting points for a series of dicarboxylic acids. Dimer geometries were extracted from crystal structure data. Reprinted with permission from ref. 258. Copyright 2013 Royal Society of Chemistry. | ||
 | ||
| Fig. 27 Relative energies for several different Li positions relative to the reduced carboxylates. In both cases, low energy structures reflect maximum coordination of the Li-ion by oxygen. Reprinted with permission from ref. 258. Copyright 2013 Royal Society of Chemistry. | ||
The predicted reductions in this article spanned a potential range between 0.6 V and 1.6 V vs. Li/Li+ as shown in Fig. 28.258
 | ||
| Fig. 28 Calculated potentials (Ecalcd) for the reduction of molecules with (filled circles) and without (empty circles) lithium present. In every case, the electrostatic interactions with lithium shift the reduction potential to more positive values. Additionally, the computed reduction potential shifts positive with increasing molecular length with the exception of 19. The secondary (upper) x-axis uses the correlation established between computed and experimental potentials (vide supra) to give a predicted reduction potential. Reprinted with permission from ref. 258. Copyright 2013 Royal Society of Chemistry. | ||
Compared to graphitic carbon anodes, organic anode materials are found to be more competitive in terms of capacity/energy density in the quest to develop sustainable and inexpensive secondary Li-ion battery anode materials for widespread deployment, by assuming that at least one 2-electron reduction is possible. As solubility is a major consideration for energy storage materials in the case of electrolyte–solvent systems, the dissolution of neutral salt electrode material can be an important issue addressable by considering the molecular nature of these active materials. Therefore, calculated solvation energies for a subset of dissociated anions in a variety of solvents with properties relevant to electrical energy storage applications were determined. Moreover, carboxylic acids have a rich chemistry since carboxylic groups may dehydrate to anhydrides, carboxylic acids can be oxidatively decarboxylated, and reduction can cause their dimerization and subsequent degradation.383–388
Finally, carboxylate anodes may be good choice for anode materials in Li-ion batteries and even in Li–air batteries. Some issues related to their crystal structures and lithiation mechanisms are still unanswered.
Conclusively, DFT calculations are found to be useful for exploring new anode materials. However, it is well known that DFT fails to describe long range van der Waals interactions correctly in all cases (with some exceptions namely vdW-DF2), so the computational investigation of traditional graphite material in Li-ion batteries can be difficult. Using nudged elastic band methods and Monte Carlo simulations, Li+ diffusion in graphite can be examined. Solid electrolyte interface (SEI) film formation is a very important factor that would affect the performance of Li-ion battery anode materials, which is also related to the safety issues and the reversible capacity of anode materials. Regarding the complexity of SEI films, it is quite difficult to be simulated through computational methods and significant effort is still needed to improve the accuracy and predictive power of the calculations, and the true nature of the SEI film formation process.
5. Electrolyte (solvent and salt) materials
The electrolyte is currently the primary obstacle to progress in next-generation Li-ion batteries, often because of unwanted side reactions that in some cases become exacerbated by reactive nanoscale materials. Understanding, controlling and hence eliminating side reactions is a significant task. Currently, the applied electrolytes in non-aqueous Li-ion battery systems can be divided into two categories based on the types of solvent: organic carbonates (ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), diethyl carbonate (DEC)), and ethers (tetrahydrofuran, dioxolane, dimethoxyethane, tetraethylene glycol dimethyl ether), which solvate lithium salts such as LiPF6, LiBF4, LiAsF6, LiN(SO2CF3)2 and LiSO3CF3,389–393 among others.EC and PC exhibit a large permittivity due to their relatively high polarity, but they are very viscous due to strong intermolecular interactions. On the other hand, DMC and DEC have a low permittivity and a low viscosity due to their molecular structure giving a large degree of freedom of the molecule. Alkylcarbonate solvents such as EC form a stable passivating SEI film required for reversible Li intercalation at the graphite electrode at low voltages. Electrolytes for Li-ion batteries mostly constitute of mixture of solvents with high permittivity (for example, EC) and low viscosity (DMC or DEC) in order to simultaneously promote ionic dissociation and ion mobility.
Ethers were also studied as solvents for Li-ion batteries to replace PC due to their low viscosity and low melting point. These solvents seem to be less interesting due to an oxidation potential less than 4 V, especially with traditional cathodes in Li-ion batteries. Electrolytes containing γ-butyrolactone or γ-valerolactone are very promising due to their large electrochemical window, high flash point, high boiling point, low vapour pressure and high conductivity at low temperatures in spite of moderate permittivity and absolute viscosity of 1.75 cP at 25 °C. Sulfones such as ethylmethylsulfone, methoxy-methylsulfone or tetramethylsulfone are good candidates for high voltage electrolytes as their electrochemical stability in the presence of LiPF6 remains good up to 5 V versus Li/Li+ on a Pt electrode.394–396 However, these solvents can not be used with graphite electrodes as they do not form SEI film onto the graphite surface. Nitrile solvents have low viscosity and good anodic stability (∼6.3 V versus Li/Li+). Nagahama et al.397 reported that the electrochemical window of dinitrile based electrolytes such as sebaconitrile mixed with EC and DMC with LiBF4 salt can reach 6 V at the graphite electrode and shows promise when used with LiFePO4 positive electrodes.
Another developing class of electrolytes are ionic liquids, i.e. low temperature molten salts having specific properties such as low vapour pressure, large electrochemical window and high conductivity.398 Room temperature ionic liquids (RTILs) are liquid at room temperature and contain large organic cations associated with small inorganic or organic anions by strong electrostatic interactions. Moreover, they can be mixed with numerous organic solvents. For example, Chagnes et al.399,400 studied the electrochemical and thermal behaviours of 1-butyl-3-methylimidazolium tetrafluoroborate or 1-butyl-3-methylimidazolium tetrafluorophosphate mixed with BL and found that this mixture has very good thermal stability (>350 °C) and remains liquid at very low temperature (<−110 °C). Zugmann et al.401 studied the properties of LiBOB by adding fluorine atoms and found excellent Al-corrosion-protection properties, excellent cycling behaviour of lithiated carbon anodes and Li Ni0.33Co0.33Mn0.33O2 cathode, no HF formation by hydrolysis, and a good solubility in dipolar aprotic solvent.
Although the detailed composition of the SEI film on the graphite anode depends on the settings and conditions of the electrolyte, the SEI film is basically composed of insoluble inorganic solids such as Li2O, Li2CO3, LiF, etc. and organic solids such as dilithium ethylene dicarbonate (Li2EDC).1,45–47 Solid electrolytes represent one of the most promising candidates, as they potentially allow the construction of lighter and safer all solid-state batteries with design flexibility in the shape and size. Solid electrolytes are classified as gelled (or wet) polymers, solvent free polymers, inorganic crystalline compounds, and inorganic glasses.402 Polymer electrolytes can serve as both electrolytes and separators between the anode and cathode, are of peculiar interest. Significant attention has been focused on poly(ethylene oxide) (PEO) and related polymers that are blended with Li salts. However, PEO-based electrolytes have poor low-temperature conductivity because the segmental chain motion required for ion transport quickly diminishes upon cooling below the glass transition temperature. In addition to PEO-related electrolytes, poly(vinylidene fluoride) (PVDF)-based polymer electrolytes are also attractive candidates with ionic conductivities up to 0.1 S m−1 at room temperature.403–408 When TiO2 or Al2O3 nanoparticles as solid plasticizers in PEO, a solid state polymer electrolyte has been developed.409 The conductivity of these electrolytes increased and the crystallization was prevented. Li-ion doped plastic crystalline matrices are stable over a potential of 5 V and very attractive for battery applications in combination of possible structural vibrations of plastic crystal matrices and conductivities.410 Zhang et al.411 prepared a novel solid-state composite polymer electrolyte based on PEO by using LiClO4 as doping salts and inorganic hybrid poly(cyclotriphosphazene-co-4, 40-sulfonyldiphenol) (PZS) microspheres as filters due to the fact that this electrolyte leads to higher enhancement in ionic conductivity compared to traditional ceramic fillers such as SiO2. Other promising candidates for solid state electrolytes are inorganic materials (brittle superionic glass electrolytes). For inorganic solid electrolytes, lithium superionic conductors (LISICON) are of key importance to achieve all solid-state Li-ion battery. This technology may solve the safety issue of rechargeable Li-ion batteries using non-aqueous electrolytes. In 1978, Alpen et al.412 developed Li14Zn(GeO4)4, a type of LISICON, which attracted attention for its potential application as a solid electrolyte.413 A new solid system based on lithium germanium sulphide and lithium silicon sulphide known as thio-LISICON was subsequently discovered414,415 with a higher electrochemical stability. Many studies have been focused on the binary Li2S–P2S5 system416 since.
Numerous additives can be added to the electrolyte to improve the performance of Li-ion batteries by influencing intercalation SEI formation at anode surfaces and the properties of the electrolyte as a function of operating conditions, among other reasons. However, the use of such additives can be responsible for deleterious effects if they are used at high concentration in some solvents or if they chemically interact with other compounds. The microscopic mechanism responsible for the reductive decomposition and subsequent growth of the SEI is unclear because of the difficulty in situ observation of the electrode–electrolyte interface in the Li-ion batteries. Additives to the electrolytes have large impact on the SEI formation mechanism and its performance. For example, addition of vinylene carbonate (VC) up to 5–10% to the ethylene carbonate (EC) solvent significantly improves the irreversible capacity at the first charging and the cycle life of Li-ion polymer cells.417–419 A proposed mechanism is that VC additives are sacrificially reduced and decomposed on behalf of EC and form the oligomer structures, leading to the improved SEI.417,420 However, the solvation properties of VCs with respect to Li+ as well as the subsequent decomposition process have not yet been established on the atomic scale. An atomistic investigation of the role of VC additives in SEI film formation is therefore crucial for an in-depth understanding of Li-ion battery durability and performance. It is well known from previous reports that Li+ does not co-intercalate with EC into graphite anodes, and the latter undergoes specific decomposition or reduction mechanisms the lead in part to the formation of the SEI film in tandem with salt interactions. However, Li+ can co-intercalate with PC into graphite anode under certain conditions, resulting in the exfoliation of anode structure by PC. In such cases, PC is less suitable electrolyte for graphite anodes. In order to address this issue, some electrolyte additives such as vinylene carbonate (VC), vinyl ethylene carbonate (VEC), vinyl ethylene sulphite (VES), ethylene sulphite (ES), etc. have been explored to investigate their role for PC-based electrolytes regarding the SEI-film formation near the graphite anode and hence enhance the cycling performance of Li-ion batteries.
It is well known that EC has been widely used as a solvent of commercial Li-ion batteries using carbonaceous materials (e.g. graphite) as anodes. However, due to high viscosity and high melting point (∼36 °C) of EC, a certain amount of linear carbonates such as DMC or DEC that have much lower viscosities and lower melting points can be added to EC-based electrolytes for better low temperature response. Therefore, the performance of Li-ion batteries may differ depending on the type of co-solvents. Previous studies reported that EC decomposes in a battery solvent of EC/DMC or EC/DEC binary mixture to contribute to the SEI film formation, and DMC or DEC mainly improves viscosity and conductivity.
It was reported in experiments that the oxidation potential of PC decreases in the presence of electrolyte salts.421 In order to understand how and why anions affect the oxidation stability of a solvent, the initial oxidation reactions of isolated PF6−, BF4−, and ClO4− ions were investigated in Wang et al.422 and the authors found that the oxidation stability of lithium salts is in the order LiPF6 > LiBF4 > LiClO4.404,423,424 Therefore, the influence of the anion and surrounding solvent must be taken into account in modelling when considering the oxidation reaction on the electrode surfaces in Li-ion cells. In literature, LiClO4, LiAsF6, LiBF4, LiTFSI, LiTf, and LiPF6 are the most studied and commercialized salts.
5.1 Organic carbonates
Organic carbonates are widely used as solvents in state-of-the-art rechargeable Li-ion batteries as a result of their high oxidation potential (high stability) and relatively low viscosity (low activation barrier for Li diffusion).392,425,426 Aurbach et al.427 studied the effects of contaminants (O2, H2O) on the performance of Li-ion batteries and reported the instability of organic carbonates in the presence of O2 in the early 1990s. Many groups reported the multiple cycle of Li-ion cells, and recently also in Li–O2 batteries428–431 due to the assumption that the ability to cycle such cells implied reversible formation of Li2O2 and capacity fading due to side reactions with the electrolyte. Furthermore, Mizuno et al.432 and Freunberger et al.433 independently reported the instability of organic carbonates, which is an important issue still.The common non-aqueous organic solvents are ethylene carbonate (EC), propylene carbonate (PC), dimethyl carbonate (DMC), diethyl carbonate (DEC), and ethyl methyl carbonate (EMC). Among these electrolytes, EC has an important role due to its stability at anode surfaces in Li-ion batteries.434 These batteries have liquid electrolytes consisting of a lithium salt such as LiPF6 or LiBF4 or LiClO4 or LiAsF6 dissolved in organic solvent (EC or PC or DMC). The mixture of two or more electrolytes has been found to be more convenient435 due to the fact that it allows optimization of salient features such as viscosity, chemical stability, ionic diffusion, ionic transport, salt dissociation, dielectric constants etc., leading to enhanced performance and stability in Li-ion batteries under various operating conditions. The transport of Li+ ions in electrolyte is important for understanding the molecular adsorption mechanism. At the same time, the strong influence of Li+ ion can affect the structural and dynamic properties of the surrounding electrolytes. It is well known that some organic electrolytes decompose during the first lithium intercalation into graphite to form a SEI film on the graphite anode surface and SEI film largely determines the performance of the graphite anode in rechargeable batteries.436 Unlike PC, EC does not readily co-intercalate with Li+ into graphite anode, but it helps to build the SEI layer near graphite anode that can inhibit further decomposition of solvent molecules on the anode.1,437 Because of the relatively high melting point of EC, it can allow products that build up a stable SEI.438 Therefore, the mixture of linear carbonates such as DMC or DEC with a high content of EC, are widely used in Li-ion batteries439,440 due to the fact that the high dielectric permittivity of DMC or DEC combined with low viscosity of EC or PC provides the electrolyte with a high ionic conductivity. The understanding of ionic diffusion mechanisms in the solid phases and the prediction of transport coefficients are of crucial importance in achieving optimal battery designs.438,439,441,442 In this context, several theoretical and experimental investigations have focused on ionic diffusion process,443–445 thermal behaviour,446 graphite structural changes447 and side reactions.439,443,448
The influence of SEI film formation on the performance of cathode materials becomes noticeable at elevated temperatures, resulting from pronounced surface-related capacity fading due to the development of higher electrode impedance. Kanamura et al.449,450 showed that the electrochemical oxidation processes of organic solvents, such as PC, are influenced by the type of electrolyte salt used. Similarly, Arakawa et al.451 used GC/MS to investigate the oxidative decomposition products of PC containing different lithium salts and reported the formation of CO2, CO, alkanes, and cis- and trans-2-ethyl-4-methyl-1,3-dioxalane on the surface of the cathode. The understanding of the SEI film formed on the cathode surface is essential in the quest to produce Li-ion batteries that can deliver higher levels of energy while maintaining safety. However, there have been relatively fewer studies dedicated to the understanding of this issue.452–454 SEI films are primarily composed of LiF, Li2O, Li2COG, lithium alkyl-carbonates, and non-conductive polymers. The SEI films are able to transport Li ions, but block electron transfer, and thus play a crucial passivating role in the performance of Li-ion batteries.423,436,455 On other hand, specific electrolyte compositions may also influence the SEI structure and anode performance. For example, enhanced cyclic behaviours of Si electrode have been observed by using fluoroethylene carbonate (FEC).456–461 Moreover, the experimental evidence also showed that FEC-modified SEI films possess smoother surfaces and low Li+ transfer resistance as well.462
Bhatt et al.463 studied electronic structures of Li+ ion–ethylene carbonate using DFT methods in the gas phase. The authors found that the solvation energy and Mulliken charge of Li+ solvated by EC molecule decreases with increase in number of EC molecule. Such analysis favoured the stability of 4-coordinated solvation shell among [Li+(EC)n=1–5] complexes. Moreover, Bhatt et al.464 performed DFT calculations to study the interaction of Li+ ions with EC and PC comparatively. The optimized structures of [Li+(S)n=1–5] (S = EC, PC) complexes were obtained as shown in Fig. 29(i) and (ii).464
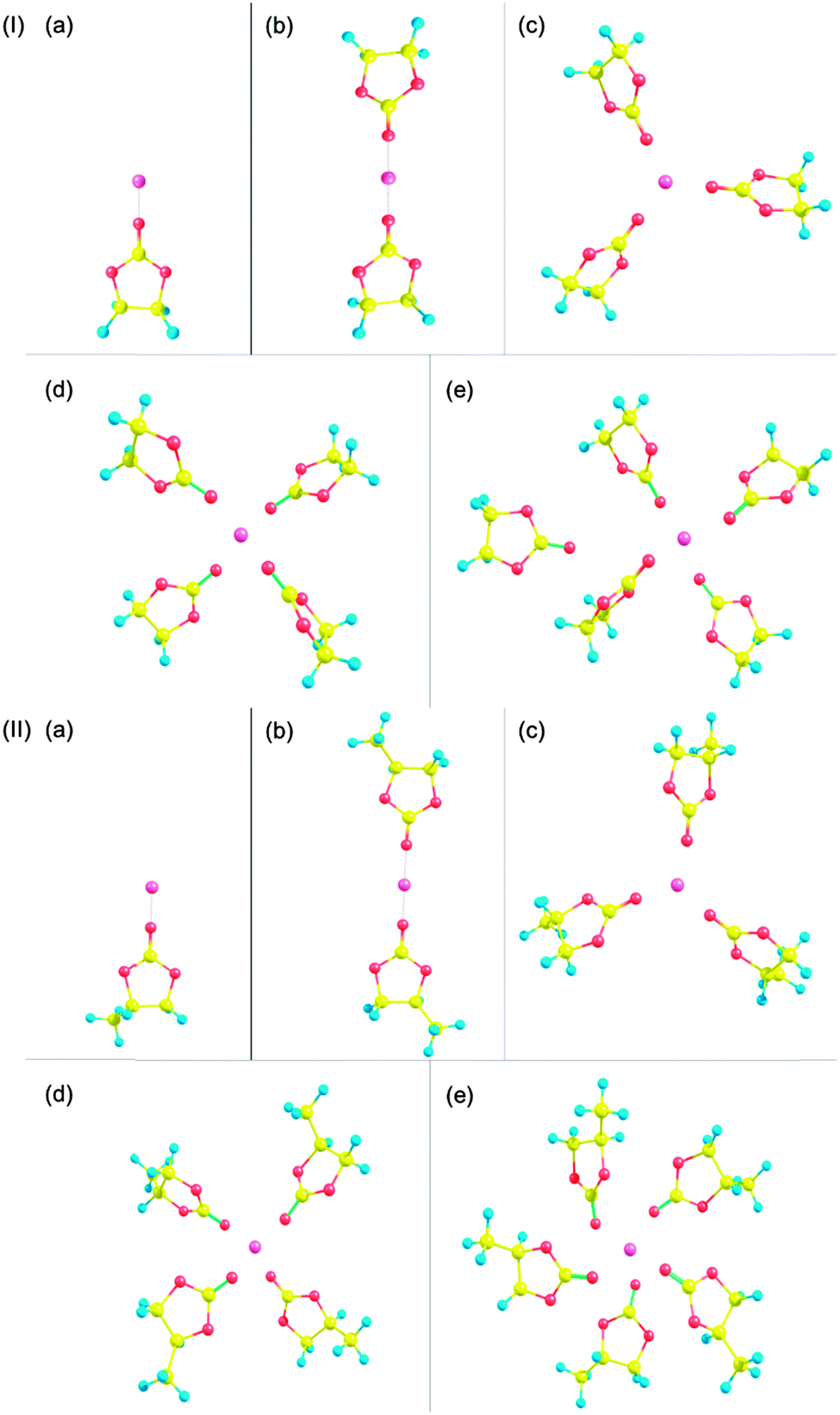 | ||
| Fig. 29 Optimized structures of (i) [Li+(EC)n=1–5] complexes and (ii) [Li+(PC)n=1–5] complexes. Reprinted with permission from ref. 464. Copyright Springer-Verlag Berlin Heidelberg 2012. | ||
The authors calculated the solvation energy, desolvation energy, electron affinity and charge on Li+ solvated by EC and PC as a function of solvation number as shown in Fig. 30(a)–(d).464 They calculated Gibb's free energies and heats of formation for the coordination of EC and PC to Li+ as shown in Table 1.464 From Table 1, it was found that although the heats of formation (ΔH) for [Li+(EC)n=1–5] complexes are exothermic, its Gibb's free energy of formation (ΔG) for [Li+(EC)n=5] complex is positive, favouring the stability of [Li+(EC)n=4] complex. This calculated result was found consistent with conclusion from Raman spectroscopic data465 and classical molecular dynamics simulations.466
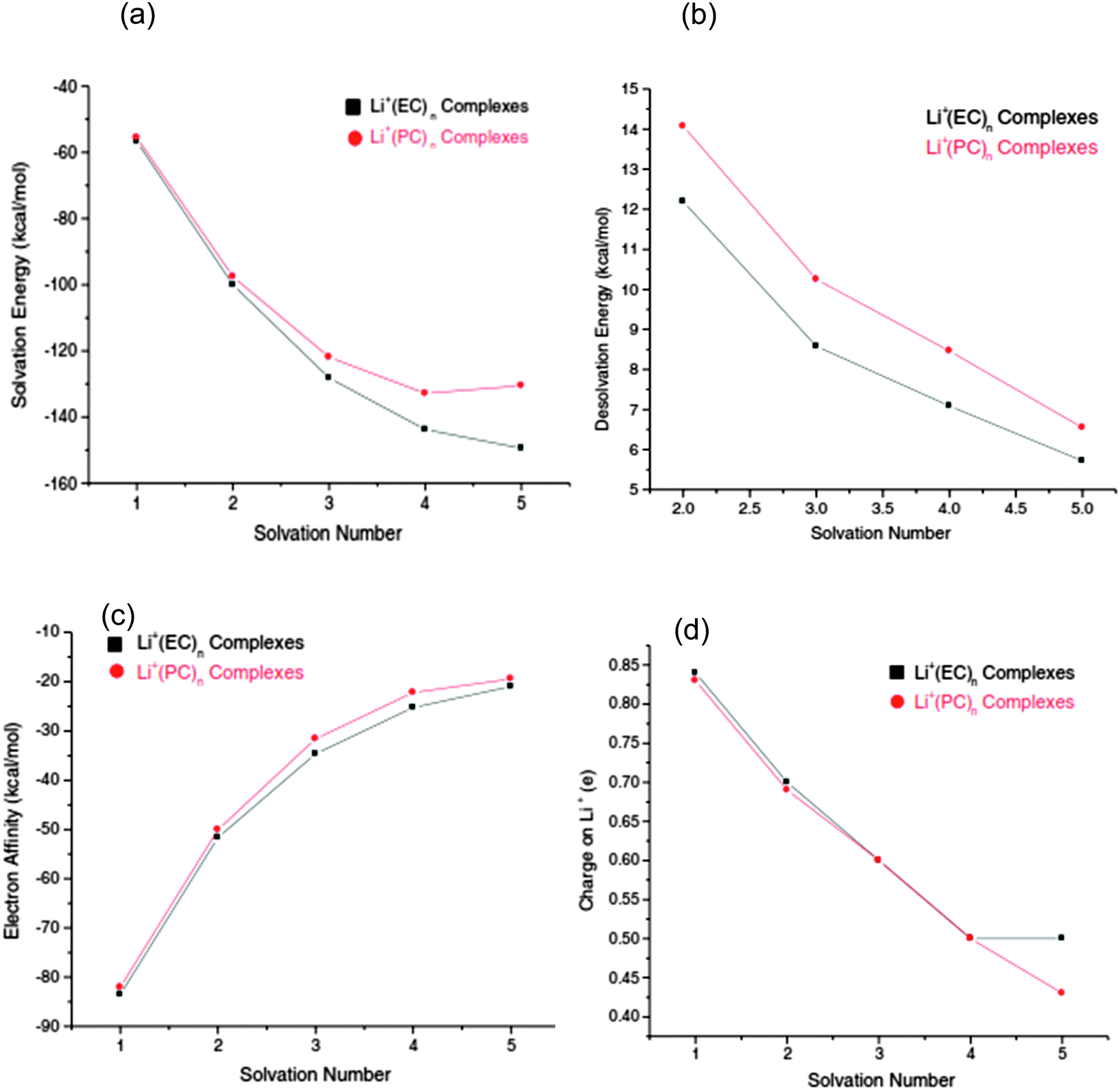 | ||
| Fig. 30 (a) Solvation energy, (b) desolvation energy, (c) electron affinity, (d) Mulliken charge on Li+ solvated by EC and PC, as a function of solvation number. Reprinted with permission from ref. 464. Copyright Springer-Verlag Berlin Heidelberg 2012. | ||
| Reaction | ΔG (kcal mol−1) | ΔH (kcal mol−1) |
|---|---|---|
| Li+ + EC = Li+(EC)1 | −43.5 | −49.7 |
| Li+(EC)1 + EC = Li+(EC)2 | −30.6 | −39.3 |
| Li+(EC)2 + EC = Li+(EC)3 | −12.9 | −23.8 |
| Li+(EC)3 + EC = Li+(EC)4 | −5.4 | −13.3 |
| Li+(EC)4 + EC = Li+(EC)5 | 9.6 | −5.4 |
| Li+ + PC = Li+(PC)1 | −43.2 | −49.5 |
| Li+(PC)1 + PC = Li+(PC)2 | −28.4 | −36.7 |
| Li+(PC)2 + PC = Li+(PC)3 | −11.6 | −19.8 |
| Li+(PC)3 + PC = Li+(PC)4 | 2.2 | −11.2 |
| Li+(PC)4 + PC = Li+(PC)5 | 7.8 | −4.6 |
Bhatt et al.467 performed DFT calculations for intercalation of Li+ cations and PF6− anions with non-aqueous electrolytes in the gas phase in terms of coordination of Li+ and PF6− with solvents. The authors found that EC coordinates with Li+ and PF6− ions most strongly and reaches the anode and cathode more easily because of its highest dielectric constant among all solvent molecules, resulting in its preferential reduction on the anode and oxidation on the cathode. For EC and PC, Li+(S)4 was found to be the most stable. However, for DMC, DEC, and EMC, the formation of PF6−(S)n=1–3 was not favourable. The optimized structures of PF6−(S) complexes (S = EC, PC, DMC, DEC, EMC) are shown in Fig. 31(a)–(e).467 The calculated infrared (IR) spectra of Li+(S) and PF6−(S) compared to those of S (S = EC, PC, DMC, DEC, EMC) are shown in Fig. 32(a) and (b).467 From Fig. 32(b), it is worth noting that very small change in IR frequency was found for PF6−(S) complexes (S = EC, PC, DMC, DEC, EMC). Further computational investigations468 performed using DFT investigated the electronic structure of EC in the gas phase together with ab initio molecular dynamics simulations for its liquid phase at T = 450 K to avoid freezing. The authors found that the stability of the primary solvation shell [Li+(EC)4] that contains four strongly bound EC molecules in a tetrahedral arrangement both in gas and liquid phases (see Fig. 33468). The ab initio molecular dynamics simulation provided the diffusion and conduction of Li+ in liquid EC regarding the SEI film formation mechanism.
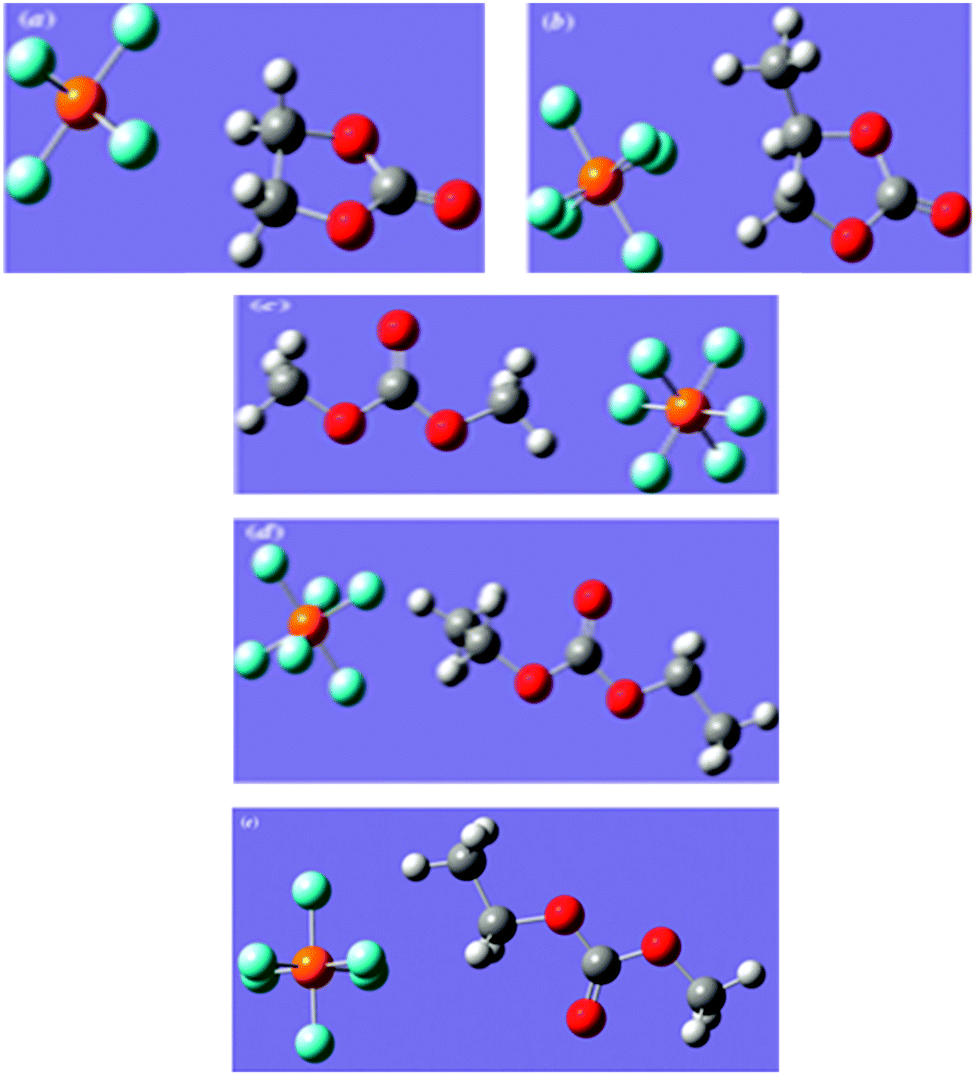 | ||
| Fig. 31 Optimized structures of PF6−(S) complexes (S = EC, PC, DMC, DEC, EMC). Reprinted with permission from ref. 467. Copyright 2011 NRC Research Press. | ||
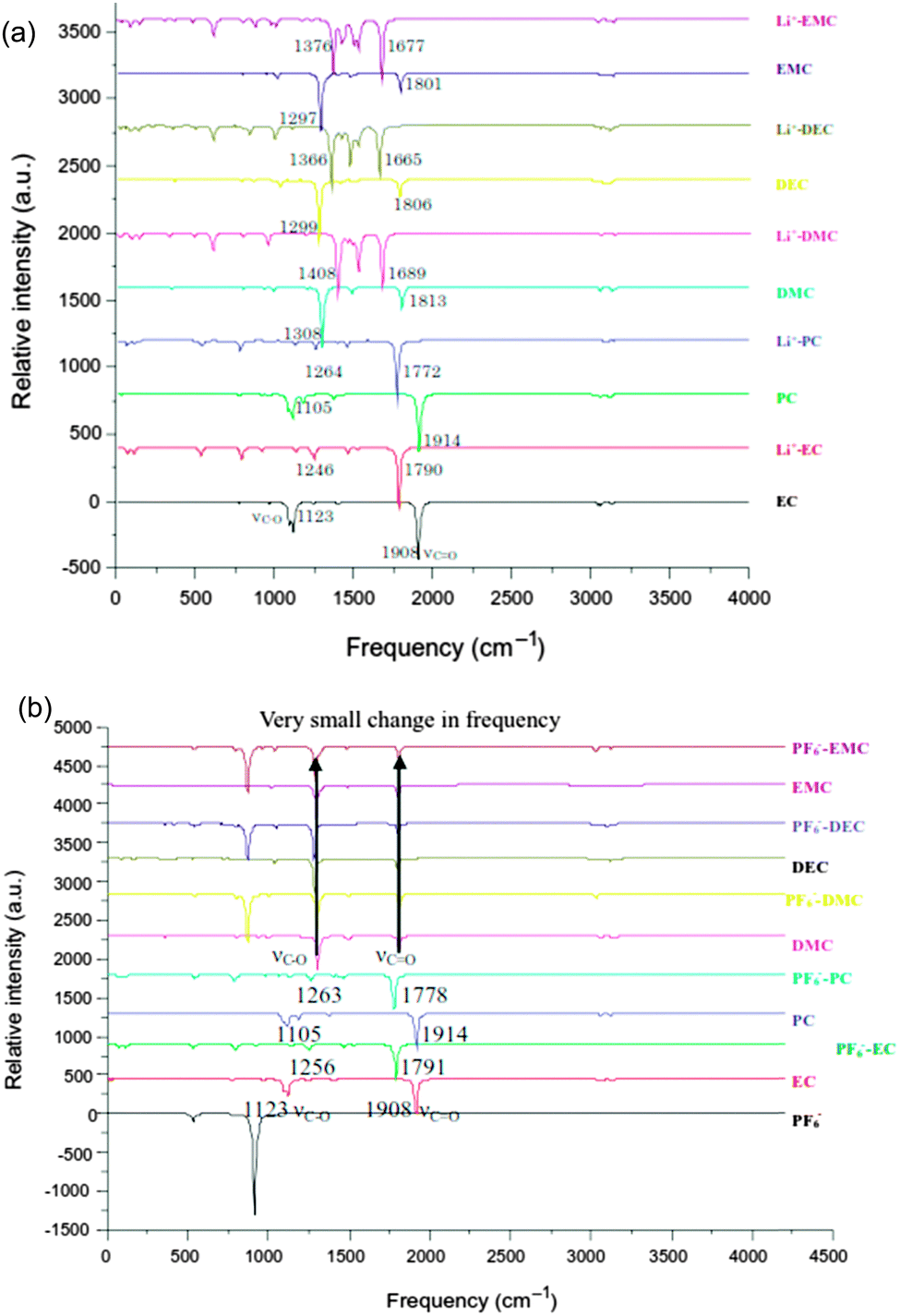 | ||
| Fig. 32 Calculated infrared (IR) spectra of (a) Li+(S) and (b) PF6−(S) compared to those of S (S = EC, PC, DMC, DEC, EMC). Reprinted with permission from ref. 467. Copyright 2011 NRC Research Press. | ||
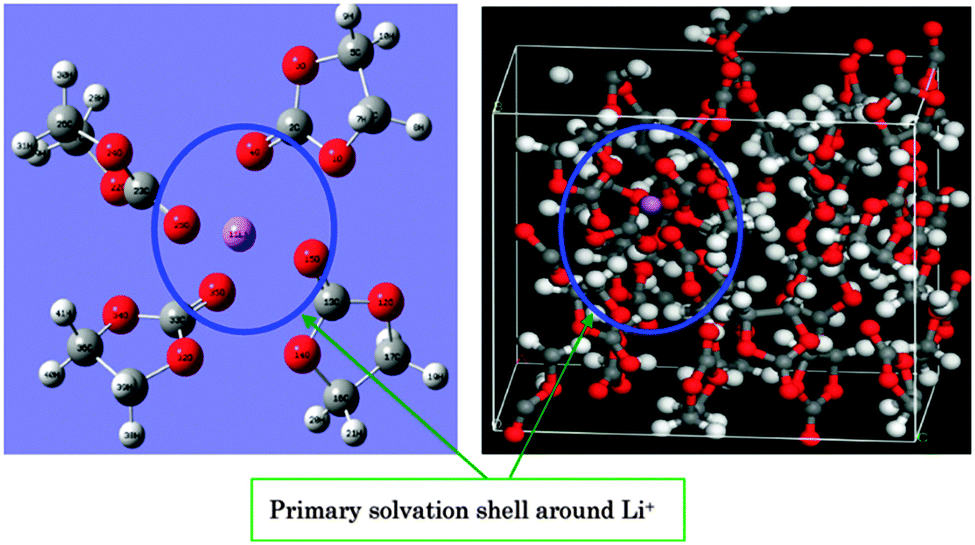 | ||
| Fig. 33 Comparison of the structures of the first EC solvation shell around Li+ in the gas phase (calculated by Gaussian) and in the liquid phase (calculated by VASP). Reprinted with permission from ref. 468. Copyright 2011 IOP Publishing Ltd. | ||
Similarly, Leggesse et al.469 investigated oxidative decomposition of PC by using DFT methods in presence of lithium salts LiClO4, LiBF4, LiPF6, and LiAsF6. The solvent effect was accounted by using the implicit solvation model in DFT. The authors found that the shortening of the original carbonyl C–O bond and a lengthening of the adjacent ethereal C–O bonds of PC, which occurs as a result of oxidation, leads to the formation of acetone radical and CO2 as primary oxidative decomposition product. Their thermodynamic and kinetic data showed that the major oxidative decomposition products of PC are independent of the type of lithium salt. The decomposition rate constants of PC are strongly affected by the lithium salt type. On the basis of rate constants using transition state theory, the authors found the order of gas volume generation as [PC–ClO4]− > [PC–BF4]− > [PC–AsF6]− > [PC–PF6]−. Tasaki et al.470 performed DFT calculations for the reduction decompositions of solvents EC, PC, DMC, DEC, and EMC including a typical electrolyte additive VC both in the gas phase and solution using the polarizable conductor calculation model. In the gas phase, the first electron reduction for the cyclic and linear carbonates was found to be exothermic and endothermic, respectively, while the second electron reduction was exothermic for all compounds. On other hand, in solution, both first and second electron reductions were exothermic for all compounds.
Although experimental investigations have revealed many important details of the electrochemical decomposition reactions, there are still some key issues to be elucidated. Quantum chemical calculations can provide a reliable approach to study the reduction reaction pathways.471–481 Theoretical studies can accurately predict reaction energies, energy barriers and intermediate structures, which are practically immeasurable by experiments under complex battery operation conditions. Computational results can be used for verifying reaction pathway, checking the feasibility of reaction steps, and eventually finding the kinetically and thermodynamically favourable mechanisms for electrolyte decomposition. Ma et al.482 proposed complete decomposition mechanisms and other organic solvents in bulk electrolyte solutions in their previous work.474–476 In this study, the authors investigated the reductive decomposition mechanisms of EC and FEC in very low-lithiated surfaces modelled with a Si cluster and found that Li+ ions are bounded to the Si surface and form adsorption sites for EC/FEC bindings. The Li location in this ultra-low lithiated surface was found to be very different than observed in the LixSy surfaces. Their study showed that the Si cluster can store either positive or negative charge in the reduction process. However, the charge on the surface/cluster depends on the degree (mole fraction) of lithiation. This study supports the stability of Si cluster rather than Si surface for decomposition of EC and FEC. Moreover, Han et al.483 performed DFT investigations for reductive ring opening reactions of Li+-coordinated EC and VC. The authors have also explored the ring opening of Li+–EC and Li+–VC by reaction with a nucleophilic (CH3O−) thermodynamically. Their calculated results revealed that the possible reaction products are CH3OCH2CH2OCO2Li (O2–C3 cleavage in Fig. 34483) Li+–EC + CH3O−, and CH3OCHCHOCO2Li (C1–O2 cleavage) Li+–VC + CH3O−.
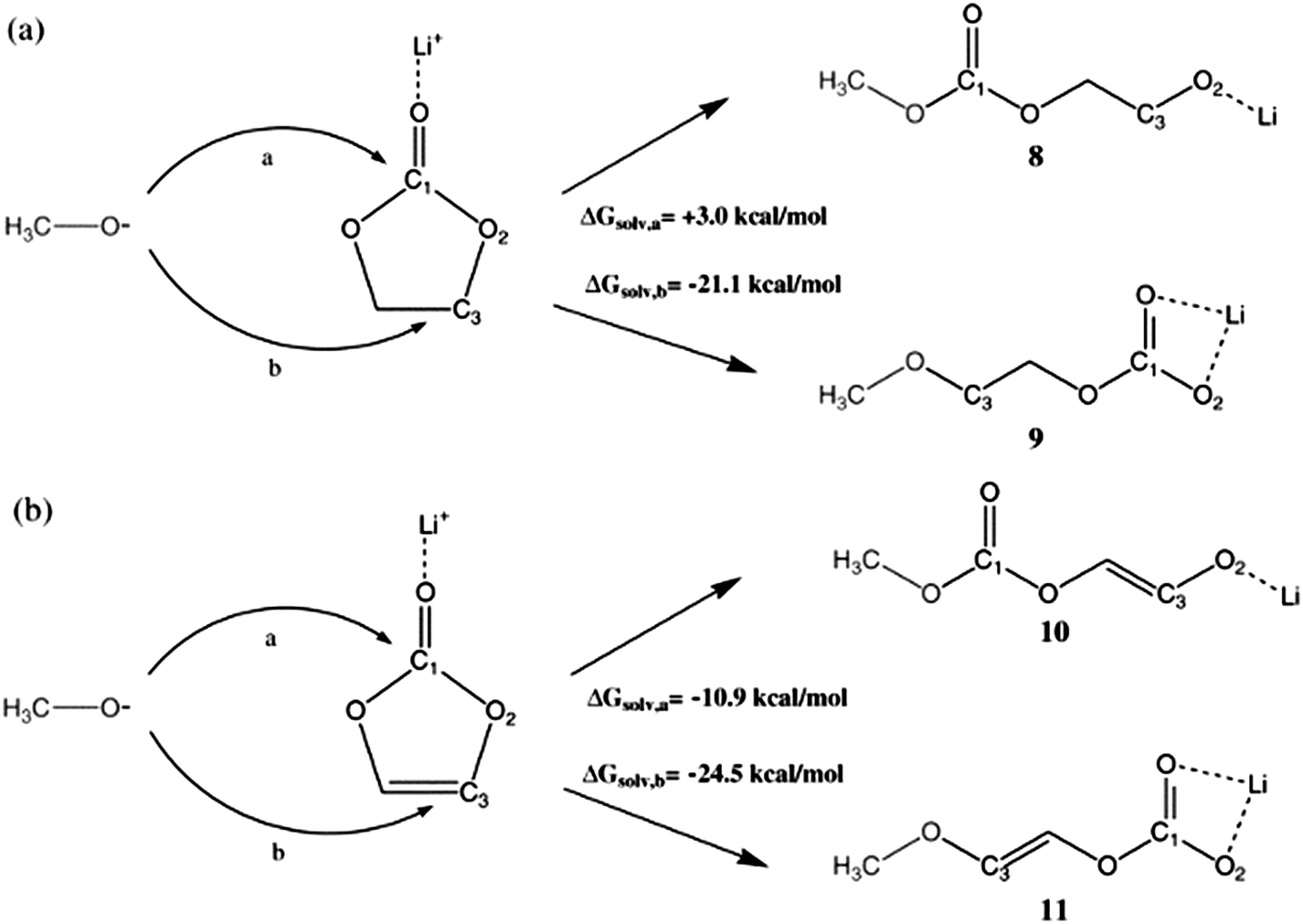 | ||
| Fig. 34 Structures of reactants and products and reaction free energies for the reactions (a) CH3O− + Li+–EC and (b) CH3O− + Li+–VC. Reprinted with permission from ref. 483. Copyright 2005 KCS Publications. | ||
Wang et al.484 performed DFT calculations to study the electroreductive decompositions of PC and VC in Li-ion battery electrolyte solutions using Li+(PC)n (n = 2, 3) and (PC)nLi+(VC) (n = 1, 2) cluster models in order to understand the experimentally observed irreversible capacity of anodes when PC alone is used as a solvent versus the reported reversible capacity found in the presence of small amounts of VC in PC-based electrolyte solutions. Their calculated results indicated that PC solvates Li+ more strongly than EC and VC do, which implies that the co-intercalation of PC with Li ion into graphite layers is preferred. However, PC is more difficult to be reduced than EC and VC. On other hand, the reaction kinetics for the reductive decomposition of PC is very similar to that of EC. The authors proposed the graphite intercalation compound (GIC) model, which can also interpret the VC role in PC-based electrolyte solutions. Nevertheless, the stability difference between GIC from PC and PC–VC mixture needs further investigation. Yu et al.485 applied static and dynamic hybrid functional DFT calculations to study the interactions of 1 and 2 excess electrons with EC liquid and clusters. The excess electron was found to be localized on a single EC molecule in all cases and the EC dimeric radical anion exhibited a reduced barrier associated with the breaking of the ethylene carbon–oxygen covalent bond compared to EC−. In ab initio molecular dynamics simulations of EC− solvated in liquid EC, large fluctuations in the carbonyl surfaces yielded products similar to those predicted using non-hybrid DFT functional. Vollmer et al.475 performed quantum mechanical calculations based on DFT methods to study the reduction mechanisms of EC, PC and VEC in electrolyte solutions. The authors investigated the direct 2-electron reduction of these species and no barrier to reaction was found for the formation of Li2CO3 and 1,4-butadiene from VEC. In contrast, they found the reaction barrier of 0.5 V for EC and PC. The formation of Li2CO3 when VEC was reduced may explain why it acts as a good passivation agent in Li-ion batteries. Li et al.466 performed ab initio calculations to study the pair interactions between Li+ ion and ClO4− ion, LiClO4, and polar aprotic solvents: EC, PC and their mixtures. The authors reported limited molecular association in pure EC solution. In EC–PC mixtures, the tendency was for EC to substitute PC in the first solvation shell of the cation, and is consistent with previous experimental studies. Therefore, the effective radius of the complex ion solvent was postulated to be smaller for the EC–PC mixture than in the case of pure PC, leading to higher ionic conductivities. The authors then investigated the potential energy surface for ion pair association of Li+ and ClO4− ions with a self-consistent polarizable continuum model and found that the minimum corresponding to the ion pair association is shifted toward larger values of the ion–ion separation in solution. Wang et al.486 performed DFT calculations to study the associations of lithium alkyl (vinylene, divinylene, ethylene, and propylene) dicarbonates, resulting from the reductive decomposition of organic carbonates and playing a crucial role on the formation of SEI layers in rechargeable Li-ion batteries. The authors found that lithium alkyl dicarbonates can associate through intermolecular O⋯Li⋯O interactions. For their dimers, the cage-like isomer was found the most stable structure and closed pseudo-planar structures turned out to be the global minima for trimers as well as for tetramers. O⋯Li⋯O interactions were characterized with atoms-in-molecules (AIM) and natural bond overpopulation (NBO) analysis and it was found that Li⋯O behave as ionic interactions. It was also found that the partial charges of Li+ ions are decreased in the range of 0.04 e to 0.035 e when O⋯Li⋯O interactions occur or when a Li atom is being shared by more adjacent oxygen atoms, whereas the overall NBO bond orders of Li are considerably increased. The calculated energetics and IR spectra of lithium alkyl dicarbonates association indicated that lithium alkyl dicarbonates exist on the anode surface, forming 2D n-mers and even 3D ones rather than monomers.
5.2 Ethers
Ethers are attractive electrolyte solvents for Li-ion batteries in the sense that these are capable of operating with a Li metal anode, stable to high oxidation potential greater than 4.5 V vs. Li/Li+, safe, cheap, and have low volatility in case of high molecular weights, for example, tetraglyme.390,487 Other important ethers are triglyme409 and dimethoxyethane389,410,411,487 as shown Fig. 35. Choi and Freunberger reported that ethers are more stable than organic carbonates407,488 towards O2 reduction and formation of Li2O2 at approximately 2.7 V at least on the first discharge, but ether electrolyte still decomposed to yield Li2CO3, HCO2Li, and CH3CO2Li.412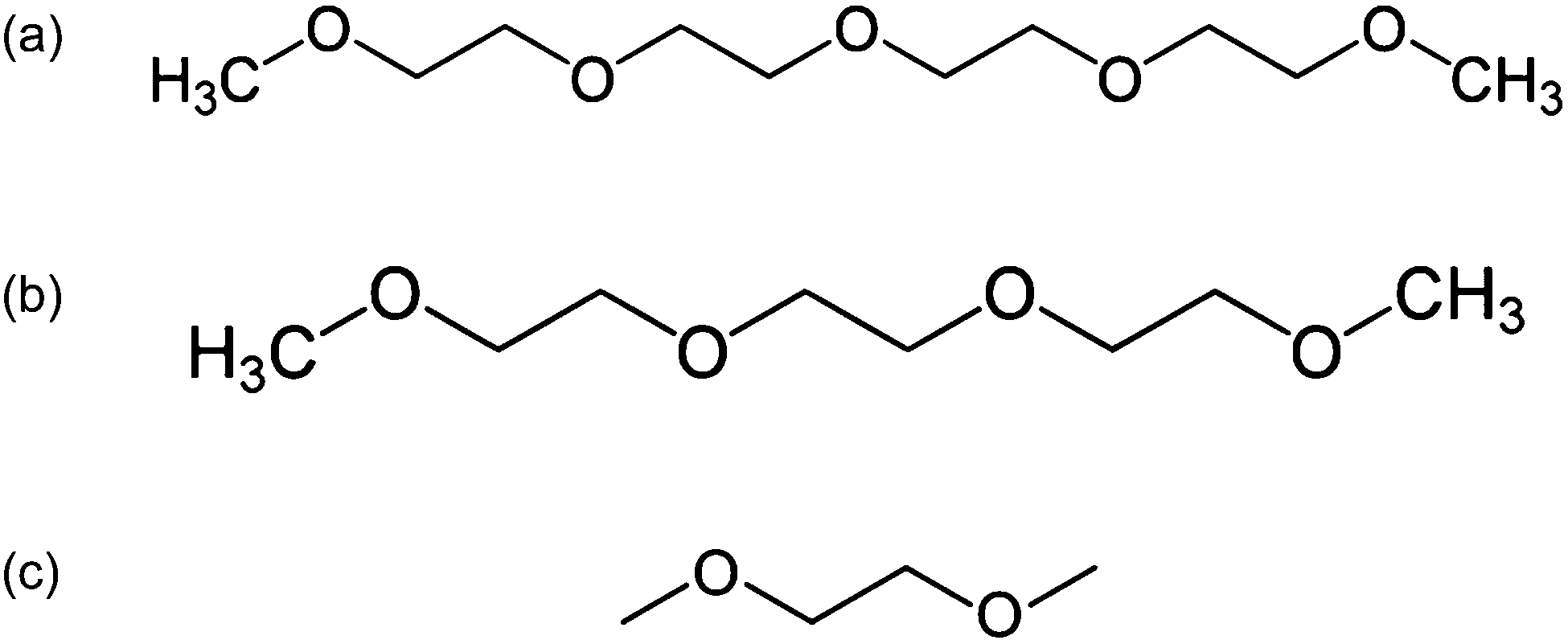 | ||
| Fig. 35 Chemical structures of (a) tetraglyme, (b) triglyme, and (c) dimethoxyethane. | ||
5.3 Ionic liquids
Hydrophobic (room temperature ionic) liquids are attractive electrolyte solvents for Li-ion batteries mainly due to their unique properties such as hydrophobic nature, low flammability, low vapour pressure, wide potential window, and high thermal stability. Due to their hydrophobic properties, ionic liquids can better protect the Li metal anode from moisture compared to other aprotic solvents. The hydrophobicity and negligible vapour pressure make ionic liquids a promising electrolyte for Li-ion battery system.489–491 Some researchers have already considered ionic liquids as electrolyte systems in Li-ion batteries,256,492 for example, Kuboki et al.256 utilized 1-alkyl-3-methylimidazolium bis(trifluoromethylsulfonyl) imide (EMITFSI) in Li-ion batteries. Allen et al.493 also investigated the oxygen electrode rechargeability in a room temperature ionic liquid EMITFSI and found that the nature of the electrode affects the reaction mechanism. For example, gold showed the ability for high efficiency recharging of the oxygen without electrode passivation. Therefore, ionic liquids can be considered as stable electrolytes for Li-ion batteries with Li metal and oxygen electrodes for future research.494 Mizuno et al.495 reported very low capacity [200 A h kg−1 (total electrode mass)] of Li-ion cells with ionic liquid electrolytes. Ong et al.496 performed combined DFT and MD simulations to investigate the cathodic and anodic limits of six room-temperature ionic liquids formed from a combination of two common cations, BMIM and P13, and three common anions, PF6, BF4 and TFSI. Their calculated results for the cathodic and anodic limits for ionic liquids and individual ions are shown in Fig. 36(a) and (b).496 Their calculated results revealed that TFSI anion is less stable than the P13 cation against reduction and BMIM cation is less stable against oxidation than the PF6 anion in P13–PF6.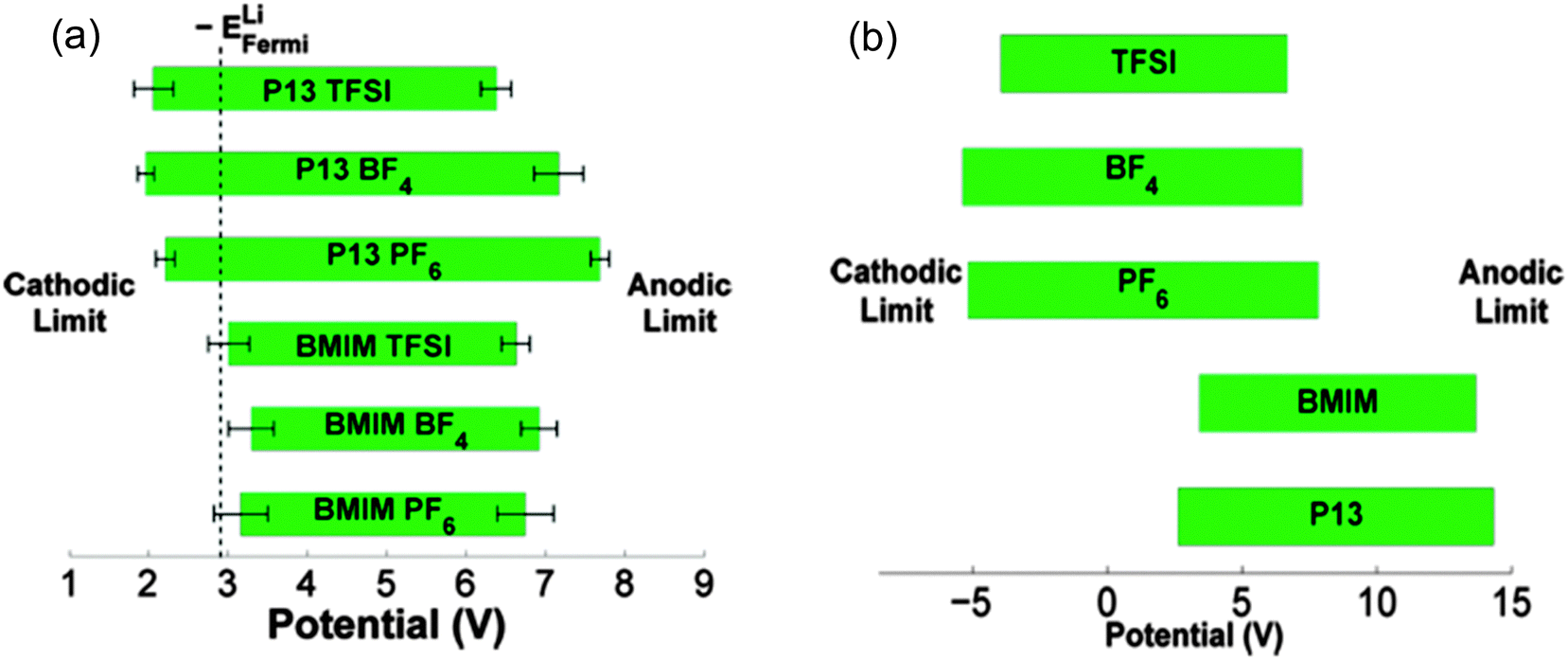 | ||
| Fig. 36 Calculated cathodic and anodic limits for (a) ionic liquids and (b) individual ions. Reprinted with permission from ref. 496. Copyright 2011 American Chemical Society. | ||
Angenendt et al.497 performed DFT calculations for three different popular imidazolium based ionic liquids EMI–BF4, EMI–PF6, and EMI–TFSI using large cluster models to understand the structure and ion–ion interactions in these ionic liquids. The optimized structures of these ionic liquids are shown in Fig. 37(a)–(c).497 Moreover, Angenendt et al.498 performed DFT calculations for triplet species from combinations of ionic liquids and lithium salts due to the fact that the charge, stability and size of the triplets have a large impact on the total ionic conductivity, the Li ion mobility, and also the Li-ion delivery at the electrode.
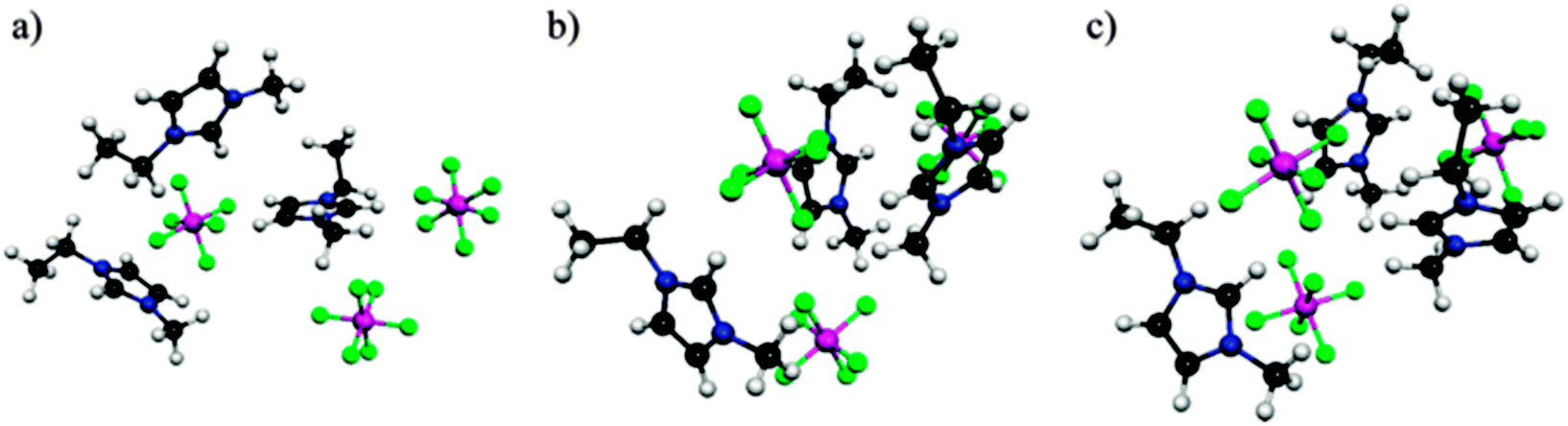 | ||
| Fig. 37 Optimized structures of (a) EMI–BF4, (b) EMI–PF6, and (c) EMI–TFSI. Reprinted with permission from ref. 497. Copyright 2010 American Chemical Society. | ||
In fact, current Li-ion batteries utilizing ionic liquid electrolytes have lower discharge capacity compared to carbonate-based electrolytes due to their high viscosity and hence inferior wetting of the cathode in some cases. Therefore, further modifications in ionic liquids to optimize their high electrochemical stability as well as lowering their viscosity, may facilitate better wetting and ionic diffusion. Moreover, further computational research work is necessary to fully investigate the mechanisms leading to side-reactions and electrolyte decomposition with ionic liquid electrolytes in Li-ion batteries.
5.4 Solid state electrolytes
Abraham and Jiang499 first reported about the use of polymer based electrolyte in the rechargeable non-aqueous Li-ion batteries. Kumar et al.500 demonstrated a totally solid-state fabricated from glass- and polymer-ceramic materials as the electrolytes and found a good rechargeability in Li-ion battery systems at a wide range of temperatures due to unique properties of these electrolytes such as high stability on exposure to moisture, a wide electrochemical window, and excellent thermal stability. The low electrochemical performance due to low ion conductivity of solid state electrolytes needs to be improved.Ogata et al.501 performed DFT and MD simulations to study the microscopic mechanisms of Li-ion transfer through the boundary between the SEI formed on the graphite anode and liquid electrolyte in the Li-ion battery. The authors considered Li2EDC, EC, and LiPF6 as the boundary components and found that the enhanced stability of the Li-ions at the boundary where EDC2− and EC contact with each other is thereby found in the computations without salt, which acts to impede the Li-ion transfer through the boundary. The ground-state structures of EDC2−, LiPF6 and EC molecules with normalized distributions of dihedral angles at T = 825 K without salt are shown in Fig. 38.501
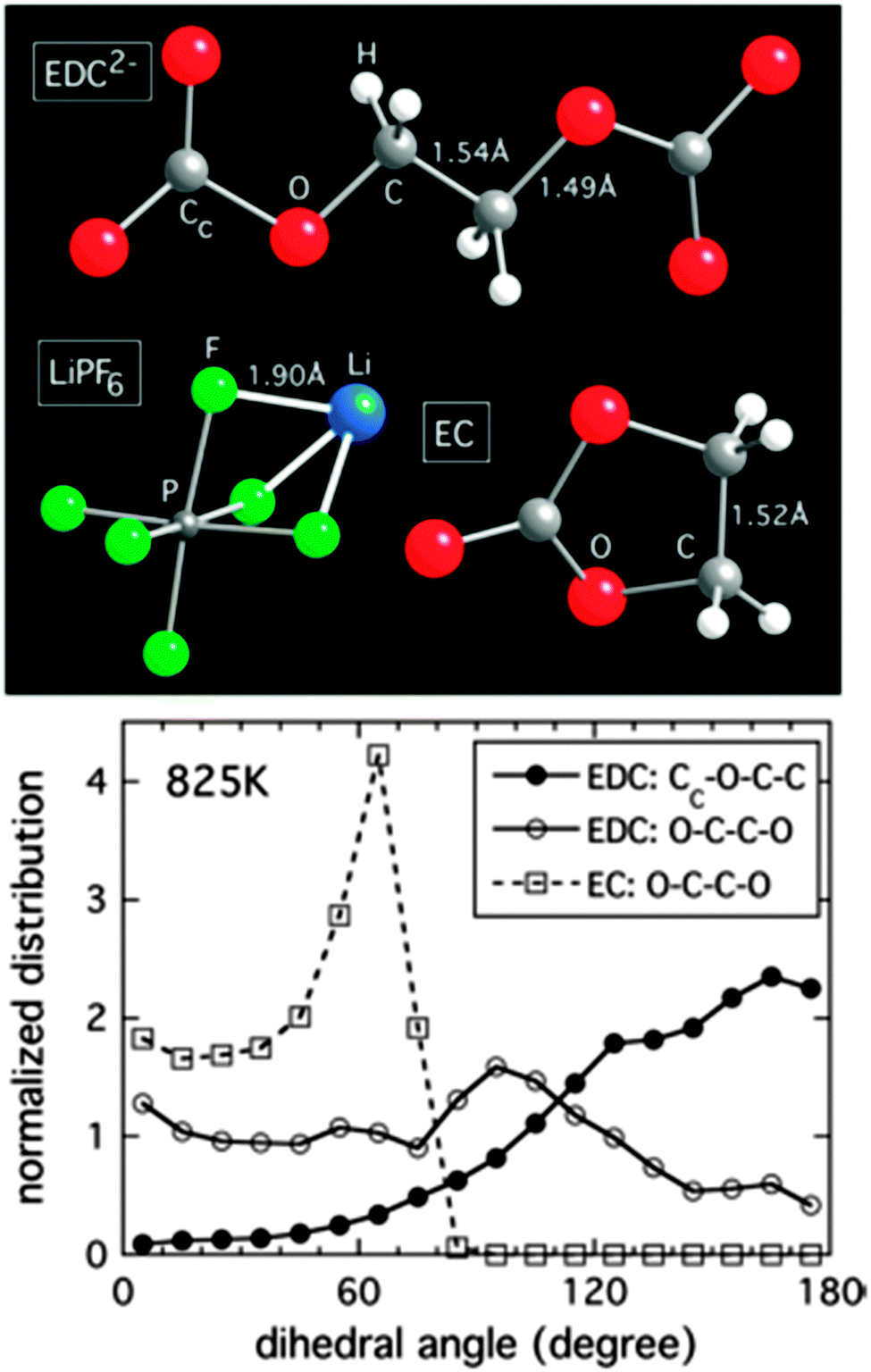 | ||
| Fig. 38 Ground-state structures of EDC2−, LiPF6 and EC molecules with normalized distributions of dihedral angles at T = 825 K without salt. Reprinted with permission from ref. 501. Copyright 2013 American Chemical Society. | ||
Moreover, Wang et al.502 reported structural and elastic properties of a new ultrafast lithium ion conductor Li10GeP2S12 (LGPS) and other two compounds of the thio-LISICON family Li3P7S11 and Li4P2S7 (see Fig. 39502) using first-principles calculations. The authors calculated six elastic constants C11, C12, C13, C33, C44, and C66 satisfying the Born's criteria of lattice stability and found that the bulk modulus of LGPS is larger than that of the Li3P7S11 and Li4P2S7, indicating difficulty in battery assembly processes if Li3P7S11 and Li4P2S7 are replaced with LGPS. The calculated B/G ratio showed that the LGPS is ductile, favouring the mechanical processing of LGPS in solid-state electrolytes in all solid-state Li-ion batteries. Their calculated results also showed that the VDW interaction is very important to the mechanical properties of LGPS. Balbuena et al.503 performed both DFT and classical MD simulations to study the ionic and molecular transport for the design of polymer electrolyte membranes. The authors considered two systems: (a) ionic transport in PEO compared to that in a PZS membrane targeted to be a good ionic carrier but an inefficient water carrier and (b) transport of oxygen and protons through hydrated NAFION in the vicinity of a catalyst phase. The authors found that Li+ has much higher diffusion coefficient in PZS than in PEO electrolyte membranes, with the consequence relevance to lithium–water battery technology and NAFION membrane used in low-temperature fuel cells interacts strongly with the catalytic metal nanoparticles directing the side chain towards the catalyst surface. The formation of water clusters surrounding the polymer hydrophilic sites revealed how the connectivity of these clusters influences proton transport and that of other molecular species.
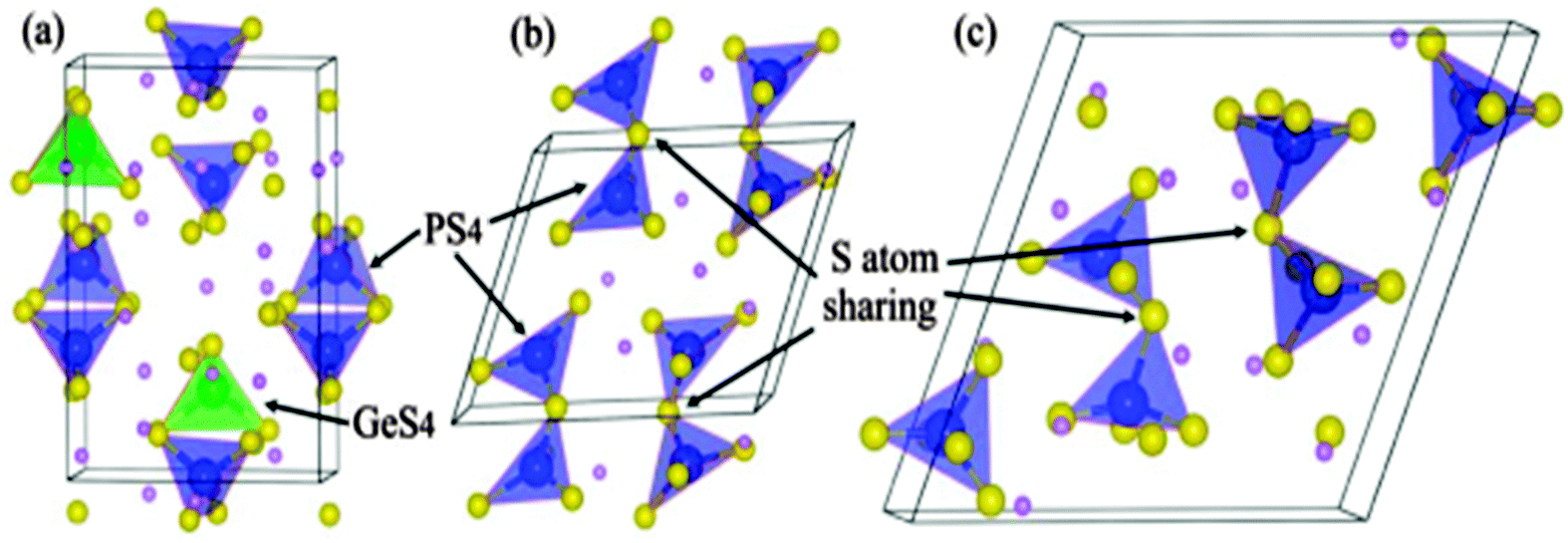 | ||
| Fig. 39 Schematic view of the atomic structures of (a) LGPS, (b) Li4P2S7, and (c) Li7P3S11. The PS4 and GeS4 tetrahedrons and the S atom shared PS tetrahedrons are marked with arrows. Reprinted with permission from ref. 502. Copyright 2014 The Electrochemical Society. | ||
Johansson et al.504 performed ab initio calculations for Li ion-tetra-, penta- and hexa-glyme [CH3O(CH2CH2O)nCH3, n = 4–6] complexes by modelling of amorphous Li salt-PEO polymer electrolyte. The authors found 23 different stable complexes with coordination numbers of Li ranging from 4 to 6. The calculated results showed the total and binding energies to increase with the glyme length. Lopez et al.505 investigated first the potential applicability of Li3−xNaxPO4 as a lithium ion solid electrolyte using DFT methods. Their calculated results revealed that for small values of x (ca. x < 1.5), Na ions could be accommodated in the β-Li3PO4 network and compounds with greater Na contents could be stable within crystal structures possessing octahedral sites. The crystal structure of Li2NaPO4 is shown in Fig. 40.505
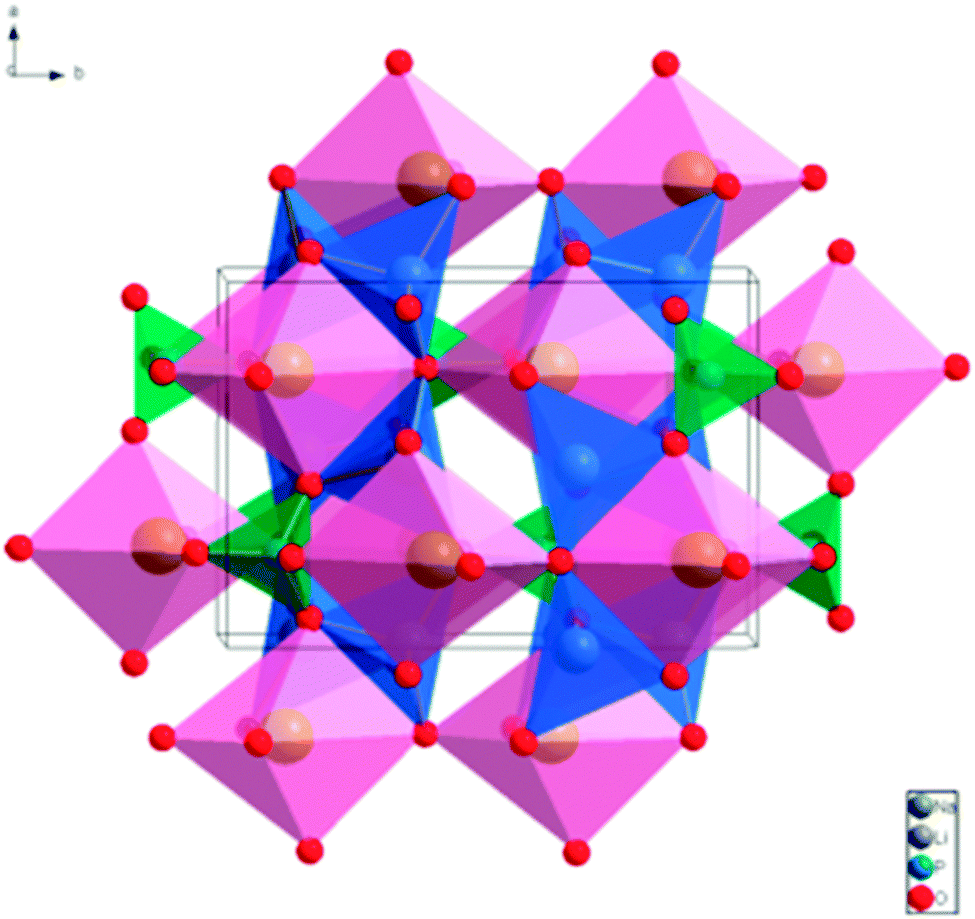 | ||
| Fig. 40 Structure of the Li2NaPO4 unit cell in the ab plane, showing PO4 tetrahedra (green), LiO4 tetrahedra (blue), and NaO6 distorted octahedral (pink). Reprinted with permission from ref. 505. Copyright 2014 American Chemical Society. | ||
5.5 Electrolyte additives
The performance of Li-ion batteries can be improved with the use of the additives in non-aqueous electrolytes by increasing the solubility of phases in the electrolyte due to an increase in the oxidation kinetics and charging rate. In Li–air (Li–O2) batteries by comparison, O2 reduction products (Li2O, Li2O2) are insoluble in non-aqueous electrolytes that precipitate on the surface of the porous carbon electrode.2,9,506 Re-oxidation of Li2O2 and its mechanism and factors that affect the large charge overpotential is still a matter of debate, but the oxygen evolution reaction is severely limited by irreversible reactions the crystal morphology and electrolyte decomposition mechanisms that occur during discharge. Additives can also increase the discharge rate due to increase in the O2 solubility in the electrolyte. Yang et al.507–511 demonstrated the synthesis and characterization of a group of boron-based anion receptors and found that when these additives are used with non-aqueous electrolytes, these make some insoluble compounds such as LiF, Li2O2, etc. soluble due to the strong interaction between those inorganic compounds and the boron centers. Such a boron-based Lewis acid additive, for example, tris(pentafluorophenyl) borane (TPFPB) has great potential to increase the solubility of the solid Li2O2 formed during the reduction of O2 in non-aqueous electrolytes, thus keeping more surface of porous carbon electrode active and in turn increase the capacity and discharge rate. Xu et al.489 reported on the positive effects of TPFPB on the oxidation of solid Li2O2 and Li2O in their Li-O2 batteries.It is well known that a Li-ion battery with a graphite anode and an electrolyte EC can be cycled, while the charging of a similar battery in an electrolyte PC gives rise to exfoliation of graphite.240,512–514 Balbuena and co-workers performed theoretical calculations on the reductive decomposition intermediates of EC and PC in absence of graphite to examine the difference in the interaction of Li+ ions with EC and PC420,484 and confirmed the latter is not suitable due to its tendency to co-intercalate into graphite during the first charge process, which leads to the destruction of the graphite structure.515–518 Therefore, many methods have been developed to solve this issue. One effective method is the use of film-forming electrolyte additives which are reduced predominantly on the graphite anode surface during the first charging process. Such additives are vinylene carbonate (VC), vinyl ethylene carbonate (VEC), vinyl ethylene sulphite (VES), ethylene sulphite (ES), etc. These additives suppress both solvent and salt anion reduction. Han and Lee investigated possible reaction products and the thermodynamic stability of Li+–EC and Li+–VC by nucleophilic addition reaction with CH3O−.519 In this sense, the electrolyte additives play an important role in the protection of the structure of the graphite anode from exfoliation/destruction by PC, and have been used to enhance the stability in modern nanomaterial-based anodes.520,521 Bhatt et al.522 investigated the electronic structures of ternary graphite intercalation compounds (GICs), Li+(S)n=1–4C14 (S = EC, PC) to determine the best choice of a range of carbonate and sulphite additives for PC-based electrolytes that promote stable SEI film formation at a graphite anode in Li-ion batteries. The authors found that the higher desolvation energy of PC limits Li+ intercalation into graphite compared to solvated Li+ in EC. Li+(PC)3 clusters are found to be unstable with graphite intercalation compounds and become structurally deformed, preventing decomposition mechanisms and associated SEI formation in favour of co-intercalation. Their calculated results revealed that the reduction decomposition of PC and electrolyte additives is such that the first electron reduction energies scales as ES > VES > VEC > PC, whereas the second electron reduction follows ES > VES > VEC > VC > PC. Thus, the reactivity of additives under consideration was found as ES > VES > VEC > VC. The authors concluded that role of certain additives is found to be supportive, particularly sulphites, in PC-based electrolytes for SEI film formation and stable cycling at graphite carbon-based Li-ion battery anodes without exfoliation or degradation of the anode structure. The optimized structures of propylene carbonate (PC), vinylene carbonate (VC), vinyl ethylene carbonate (VEC), vinyl ethylene sulfite (VES), ethylene sulfite (ES) including their open carbonate anions are shown in Fig. 41.522 Similarly, the optimized structures of Li+(PC)3 and Li+(PC)3A complexes (A = ES) are shown in Fig. 42.522
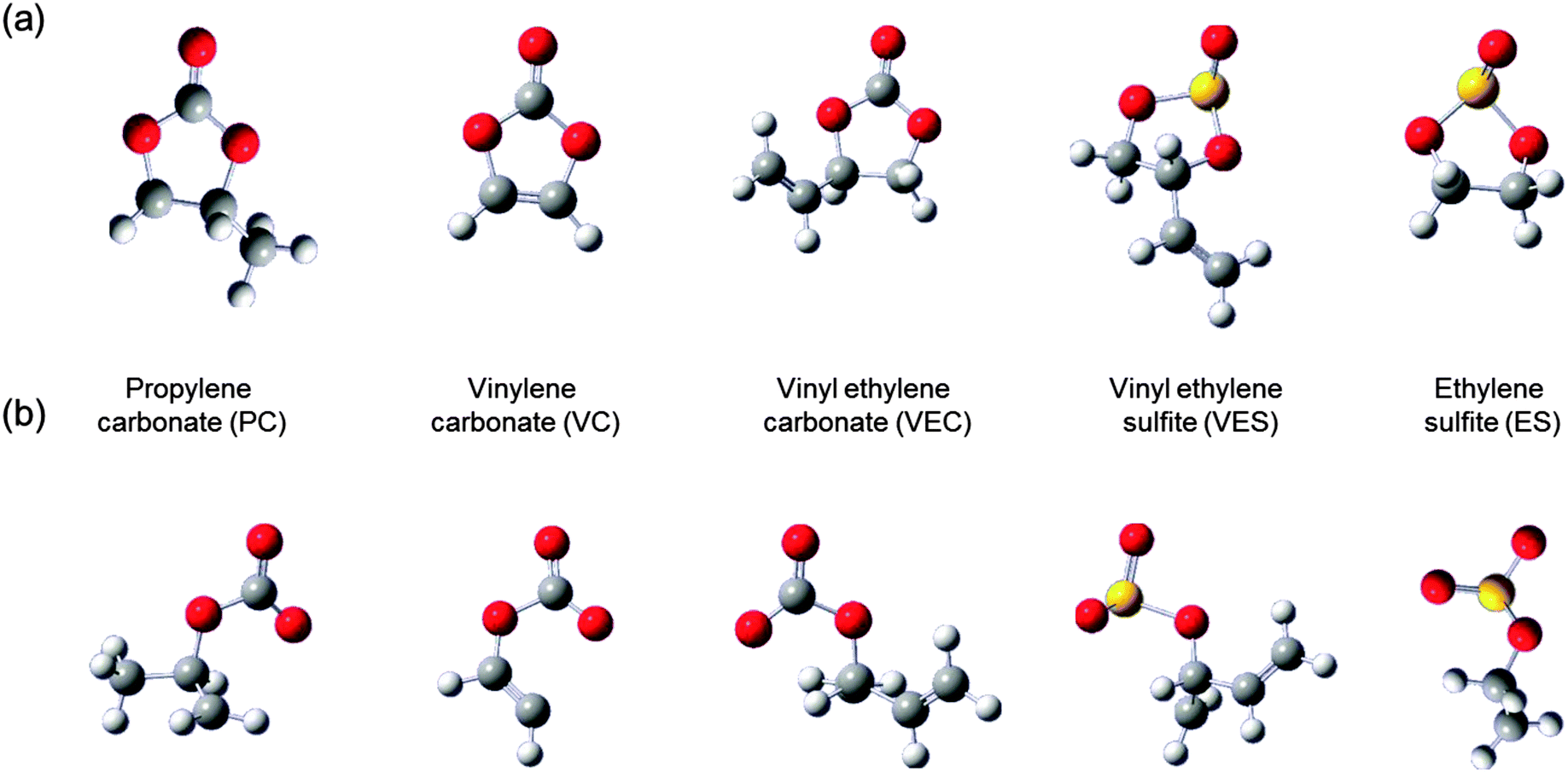 | ||
| Fig. 41 Optimized structures of propylene carbonate (PC), vinylene carbonate (VC), vinyl ethylene carbonate (VEC), vinyl ethylene sulfite (VES), ethylene sulfite (ES) including their open carbonate anions. Red atoms are oxygen, yellow atoms are sulfur, grey atoms are carbon, and white atoms are hydrogen. Reprinted with permission from ref. 522. Copyright 2014 The Electrochemical Society. | ||
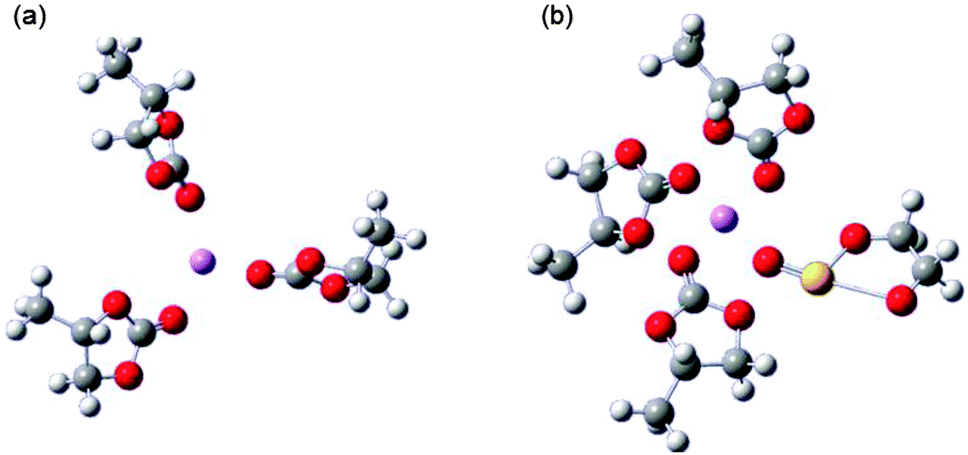 | ||
| Fig. 42 Optimized structures of (a) Li+(PC)3 and (b) Li+(PC)3A complexes (A = ES). Reprinted with permission from ref. 522. Copyright 2014 The Electrochemical Society. | ||
The IR spectra of Li+(PC)3A complex (A = VC, VEC, VES, ES), together with isolated PC and each of the additive molecules, are shown in Fig. 43(a) and (b) respectively.522 Similarly, the IR spectra of PC with all additives (that includes the increasing amount of PC) are reproduced in Fig. 44.522
 | ||
| Fig. 43 IR spectra of Li+(PC)3A complex (A = VC, VEC, VES, ES); and isolated PC and additive molecules. Reprinted with permission from ref. 522. Copyright 2014 The Electrochemical Society. | ||
 | ||
| Fig. 44 IR spectra of PC as a function of the PC:additive ratio for additives (a) VC, (b) VEC, (c) VES, and (d) ES. Reprinted with permission from ref. 522. Copyright 2014 The Electrochemical Society. | ||
The theoretical IR vibrational characteristics of PC with and without PC (Fig. 41) indicated the bonding within the solvated complexes and characteristically, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O frequency was found to be 1818.30, 1804.02, and 1799.26 cm−1 for PC:VC ratios of 1:1, 2:1, and 3:1 respectively (Fig. 42), while the corresponding values for PC:VEC contents are found to be 1799.26, 1804.02, and 1813.54 cm−1. Such variations in IR response clearly shows that other three additives VEC, VES, and ES were found to be superior to VC for PC-based electrolytes consistent with the first and second electron reduction energies.
O frequency was found to be 1818.30, 1804.02, and 1799.26 cm−1 for PC:VC ratios of 1:1, 2:1, and 3:1 respectively (Fig. 42), while the corresponding values for PC:VEC contents are found to be 1799.26, 1804.02, and 1813.54 cm−1. Such variations in IR response clearly shows that other three additives VEC, VES, and ES were found to be superior to VC for PC-based electrolytes consistent with the first and second electron reduction energies.
Moreover, Ushirogata et al.523 investigated the effects of adding vinylene carbonate (VC) to ethylene carbonate (EC) solvent, on the reductive decomposition mechanism. The authors calculated the free energy changes along several EC or VC decomposition pathways under one-electron (1e−) reduction conditions. Two-electron (2e−) reduction and attacks of anion radicals on intact molecules were also examined and found that VC additive preferentially reacts with the EC anion radical to suppress the 2e− reduction of EC and enhance the initial SEI formation, contrary to the conventional scenario in which VC additive is sacrificially reduced and its radical oligomerization becomes the source of the SEI.
Such findings for electrolyte additives may enhance the development of high capacity and high rate rechargeable Li-ion batteries. However, computational approaches to these issues that focus on predictive discharge and charge reactivity for higher voltage materials and associated electrolytes would be very useful particularly those that do not require the formation of an SEI layer but need to the thermodynamically stable at high overvoltages.
5.6 Electrolyte mixtures
It is well known that EC has been proposed a better solvent than PC due to its higher dielectric constant and lower viscosity. These properties enhance the ionic conductivity because they favour salt dissociation and high ionic diffusion rates. Mixtures of EC with liquid solvents such as PC and linear carbonates (DMC or DEC) are used because of their practical advantages as co-solvents. An ionic conductivity of ∼10−3 Ω−1 cm−1 has been reported for mixtures of these solvents.524,525 Many experimental studies reported that the maximum ionic conductivity depends on the ratio of EC/PC while maintaining chemical and electrochemical stability in solution.524,526 Yang et al.527 reported that the SEI composition greatly depends on the types of solvents. With an EC solvent, the passive film mainly consists of an organic compound, (CH2OCO2Li)2. However, for DEC and DMC solutions, the passive film contains C2H5OCO2Li and Li2CO3 respectively. It seems that only EC is decomposed in a binary solvent of EC/DEC or EC/DMC to contribute to the SEI film formation, and that DMC and DEC mainly improve the viscosity and conductivity. Although the reductive decomposition of EC has been extensively investigated by theoretical studies,474,475 little has been reported on the reduction of DMC, DEC, and EMC. Moreover, Wang et al.528 performed DFT calculations to investigate the co-solvation of Li+ and the solvent reductive decomposition in binary mixtures containing EC and one of the three linear carbonates such as DMC, DEC, and EMC. The authors reported that the calculated results may help to understand some aspects of the SEI layer related phenomena at the molecular level.Li et al.466 performed ab initio calculations to study the pair interactions between Li+ ion and ClO4− ion, LiClO4, and polar aprotic solvents: EC, PC and their mixtures. The authors found that in EC–PC mixtures, EC tends to substitute PC in the first shell of the cation in agreement with previous experimental studies. Therefore, with EC substituting PC in the first ionic shell, the effective radius of the ion–solvent complex was found to be reduced and the ionic mobility was enhanced. Blint et al.529,530 performed DFT calculations for liquid electrolytes including different ethers and carbonyl oxygen containing species and their mixtures and the coordination of Li ions. The authors found that a four-coordinated complex would be dominant, which was also supported by Gibb's free energy calculations in Kalssen et al. and Wang et al. They also considered dimethyl ether, diethyl ether, acetone and water and acetaldehyde. Some studies have used acetonitrile531 and γ-butyrolactone532 as co-solvents. Bhatt et al.533 performed DFT calculations to investigate the solvation of Li ions in EC-based binary mixtures of non-aqueous electrolytes and explain it at molecular level. The calculated results favour the stability of EC-based binary mixtures and high EC-content binary mixture systems. The optimized structures of EC-based binary mixtures are shown in Fig. 45.533
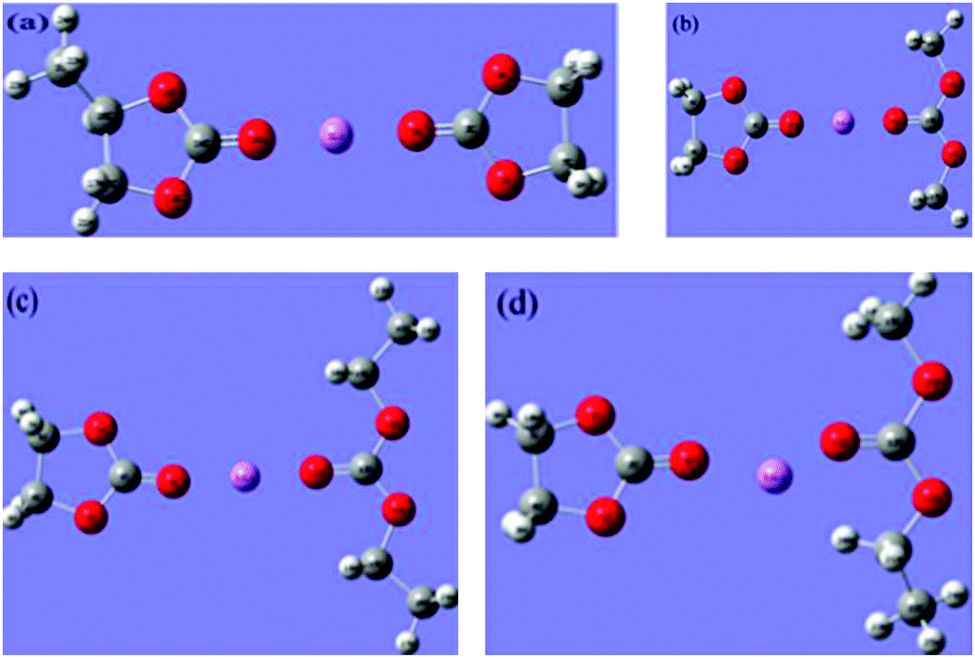 | ||
| Fig. 45 Optimized structures of Li+(EC)x(S) (S = PC, DMC, DEC, and EMC; x = 1–3) complexes. Reprinted from ref. 533. Copyright 2014, with permission of Elsevier. | ||
The compared IR spectra EC-based binary mixtures are shown in Fig. 46.533 From IR spectra, the IR active modes of the solvent showed the significant changes due to the cation–solvent interaction.
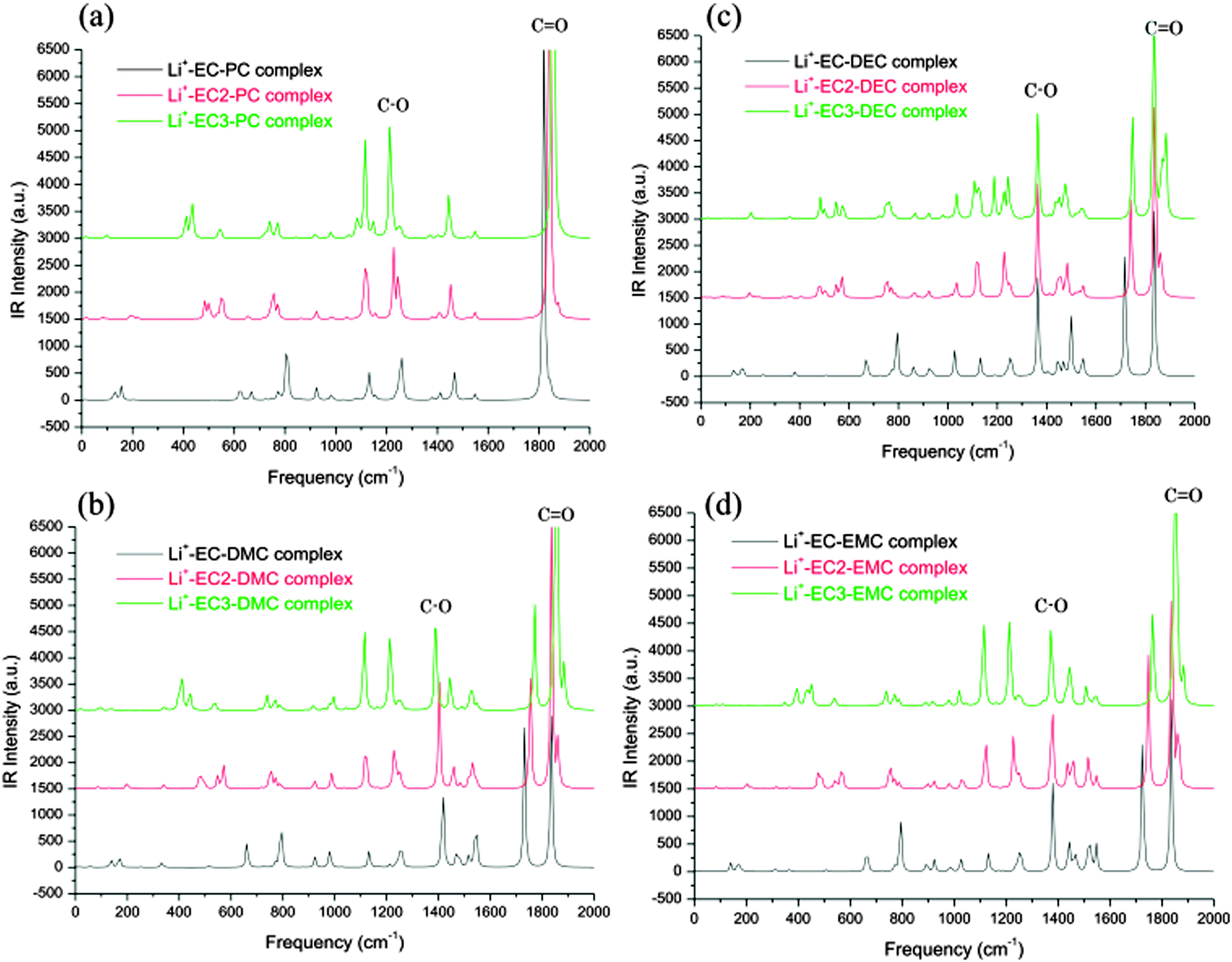 | ||
| Fig. 46 Compared IR spectra of (a) ECx/PC/Li+, (b) ECx/DMC/Li+, (c) ECx/DEC/Li+, and (d) ECx/EMC/Li+ complexes (x = 1–3). Reprinted from ref. 533. Copyright 2014, with permission of Elsevier. | ||
5.7 Lithium salts
Besides the stability of solvents, another important factor is side reactions occurring due to decomposition of lithium salts used in the electrolytes,534 which has not been studied in as much detail as the negative and positive electrode materials. The stability of lithium salts plays a crucial role in the cyclability and capacity of Li-ion batteries. If the side reactions due to lithium salts are not electrochemical reactions, it can be quite difficult to accurately assess and understand the voltage-capacity profiles of the discharge–charge processes.Moreover, the interaction between solvent and lithium salts during the discharge–charge of the cells may severely affect the electrochemical performance of Li-ion batteries. LiPF6 decomposition causes the formation of a thick layer of LiF on the Li2O2.535,536 LiClO4 is found to be more stable against Li2O2,536 but may suffer from lower oxidative stability. LiTFSI and LIFSI both have been proposed as more stable salts for use in Li-ion batteries.537 In such cases, the computational investigations devoted to investigate the interaction between the lithium salts and solvents (as well as the lithium salts and Li2O2 in Li–O2 battery chemistries) may prove invaluable when conducted in tandem with the efforts being conducted on appropriate materials for Li-ion batteries. The complex processes that occur within the electrolyte as a function of the state of charge or discharge are as important as the response to newly predicted materials, structures and compositions to uptake and reversible intercalation with cations.
It is well known that LiPF6 has been widely used as an effective electrolyte salt for Li-ion batteries due to its high conductivity. The understanding of the salt decomposition and its process is foremost in controlling the stability of LiPF6 in electrolyte solutions. Yet the difficulty of probing PF5 in solution hinders the chemical analysis of the reactions involved in the LiPF6 decomposition, although several experimental studies on the reactivity of these species have been reported.538–540 Therefore, computer simulations are very useful tools for examining chemically very unstable compounds.541,542 Tasaki et al.543 performed DFT and MD simulations for decomposition of LiPF6 and the stability of PF5 in EC, DEC, DMC, and GBL and suggested that a stable PF5–solvent adduct is formed in solution and its stability depends on the solvent. The structures of PF5 solvation in various solvents are shown in Fig. 47.543 The authors calculated the enthalpy of decomposition using thermodynamic cycle as shown in Fig. 48.543 The solvation free energy of each solute as a function of dielectric constant of the solvent obtained by SCRT–DFT calculations is shown in Fig. 49.543 The stability of PF5 stayed virtually unchanged, whereas the other solutes became more stable as the polarity of the solvent increases with the stability increase of LiF being the largest.
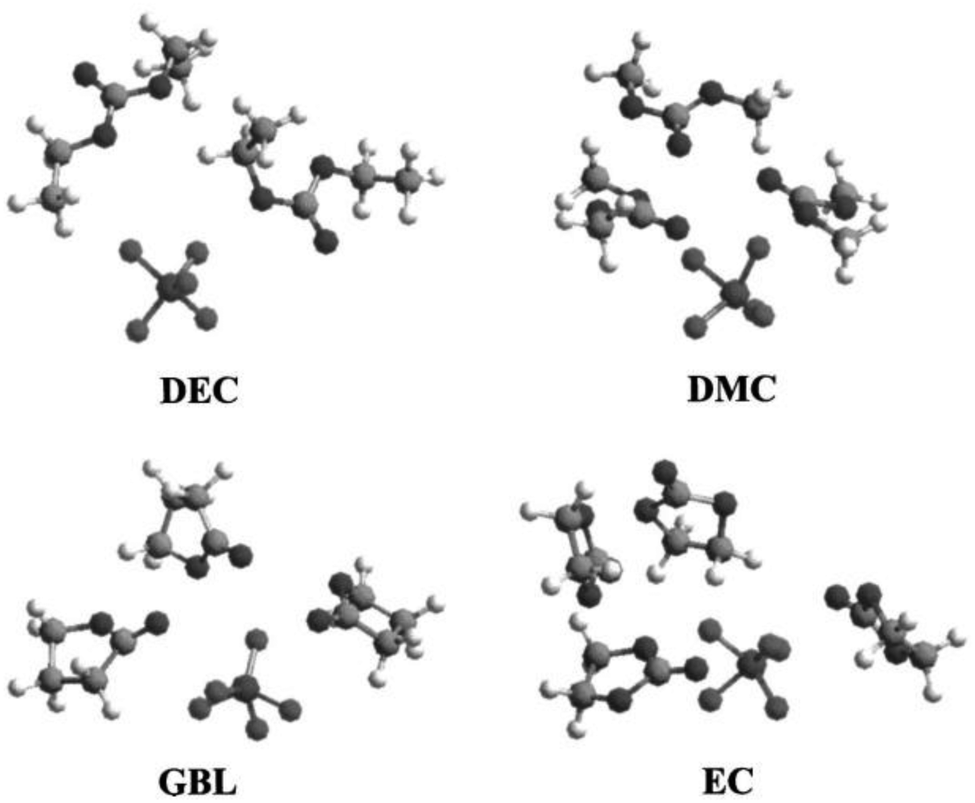 | ||
| Fig. 47 Structures of PF5 solvation in various solvents with the first-solvation shell. Reprinted with permission from ref. 543. Copyright 2003 The Electrochemical Society. | ||
 | ||
| Fig. 48 The thermodynamic cycle for the LiPF6 decomposition. Reprinted with permission from ref. 543. Copyright 2003 The Electrochemical Society. | ||
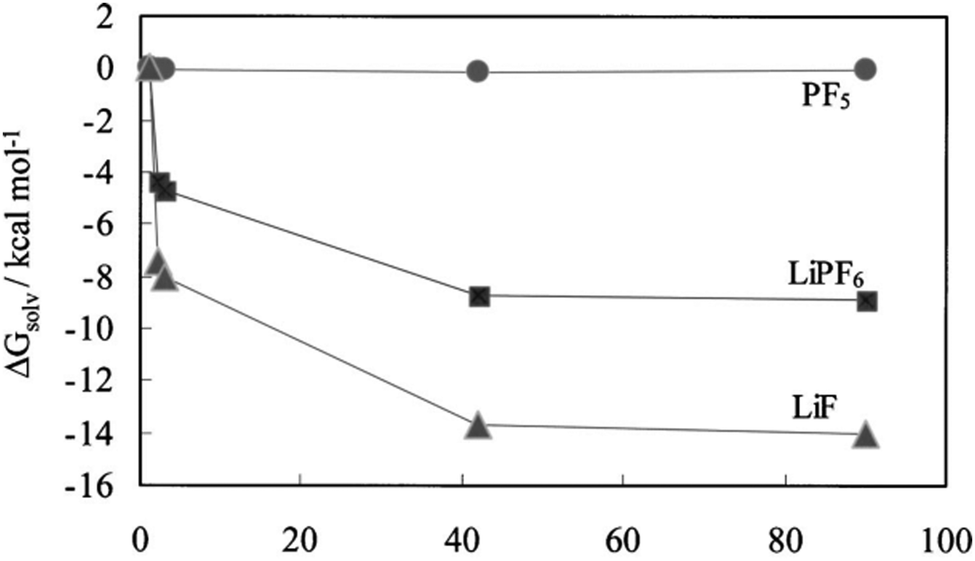 | ||
| Fig. 49 The solvation free energy of each solute as a function of dielectric constant of the solvent obtained by SCRT–DFT calculations. Reprinted with permission from ref. 543. Copyright 2003 The Electrochemical Society. | ||
Moreover, Han et al.544 investigated a relatively new salt lithium difluoro (oxalate) borate (LiDFOB) as a battery electrolyte usage using DFT methods and provided an insight into the vibrational modes of its Raman spectra to aid in the interpretation of the experimental results. The authors considered the two different forms of anion contact ion pair (CIP) coordination in the (G1)2:LiDFOB and (Et–G1)1.5:LiDFOB crystalline solvates as shown in Fig. 50.544 The stable ion pairs obtained for LiDFOB from DFT calculations are shown in Fig. 51.544
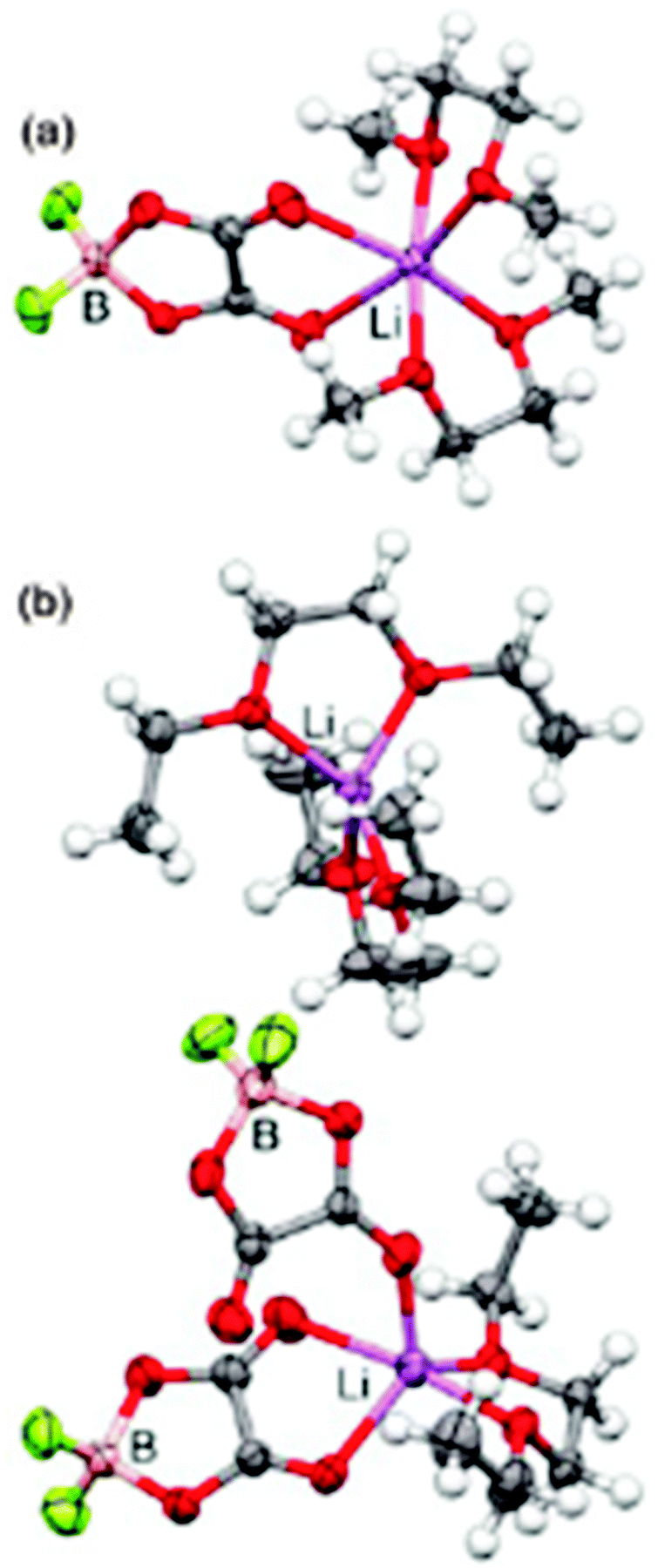 | ||
| Fig. 50 The two different forms of anion contact ion pair (CIP) coordination in the (G1)2:LiDFOB and (Et–G1)1.5:LiDFOB crystalline solvates. Reprinted with permission from ref. 544. Copyright 2013 American Chemical Society. | ||
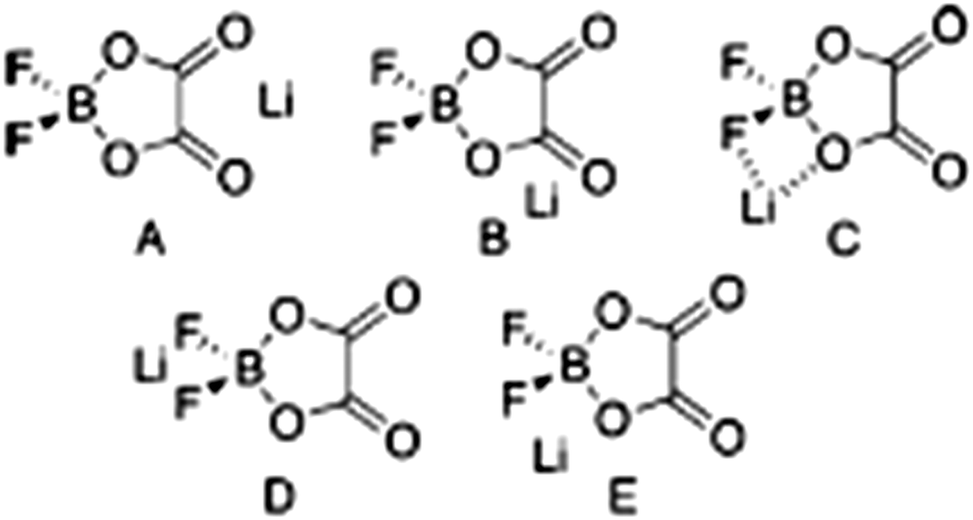 | ||
| Fig. 51 Stable ion pairs obtained for LiDFOB from DFT calculations. Reprinted with permission from ref. 544. Copyright 2013 American Chemical Society. | ||
Consequently, improved techniques to model the complex properties of liquid electrolytes would further advance the field, as would a better understanding of intercalation dynamics in systems with mixed redox couples. Moreover, modelling conversion reactions that involve large structural modifications remains problematic, despite some progress has achieved in this area. The discovery of appropriate synthesis and optimization routes still presents difficult and often time-intensive challenge task, which computations still only marginally address the key issues.
6. Future research directions
The practical applications of Li-ion batteries can be viewed as being adequate for some portable electronic devices, but improved capacities, asymmetric charge–discharge rates, increase tap and energy densities, safety and cycle life would be welcome, particularly for low cost materials and electrolytes. Significant challenges still exist for the widespread deployment in EV and related applications. Conventional Li-ion batteries operate at relatively high voltages, maintaining the structural integrity and chemical stability of electrodes upon their continuous exposure to extremely low and high electrochemical potentials vs. Li (or other alkali or transition metals). Continuous cycling of almost all tested materials degrades their operational and calendar life. The dominant non-aqueous electrolytes containing corrosive fluorinated salts such as LiPF6 and flammable organic solvents such as ethylene carbonate and dimethyl carbonate, are stable to ∼4.2 V vs. Li, and the development of ionic liquid-based electrolytes and entirely new electrolytes driven by the decomposition and catalytic reactivity seen in aprotic Li–O2 battery chemistries will be required for higher voltage systems. Computational approaches are best placed to predict, examine and filter new electrolytes and their electrochemical and thermodynamic characteristics to guide experimentation and testing.545,546Li-ion batteries are intrinsically unsafe in the charged state with delithiated Li-ion battery cathodes such as Li1−xCoO2 capable of self-discharge under certain conditions. Detailed DFT investigations of lithiation and delithation mechanisms, crystal structure polymorphisms, electronic structure and thermodynamics can screen potential materials for insertion and deinsertion voltages for oxidizable cathodes, and anodes operating at voltages close to metallic lithium (despite the spontaneous formation of a protective solid–electrolyte interphase near the anode) when developed in tandem with electrolyte reactions can postulate options for safer cell chemistries, particularly for emerging alternative rechargeable chemistries. The significant improvements needed for new and existing battery materials can be aided by focused theoretical or computational modelling as powerful methods indispensable to modern materials and energy storage research (Fig. 52).547–549
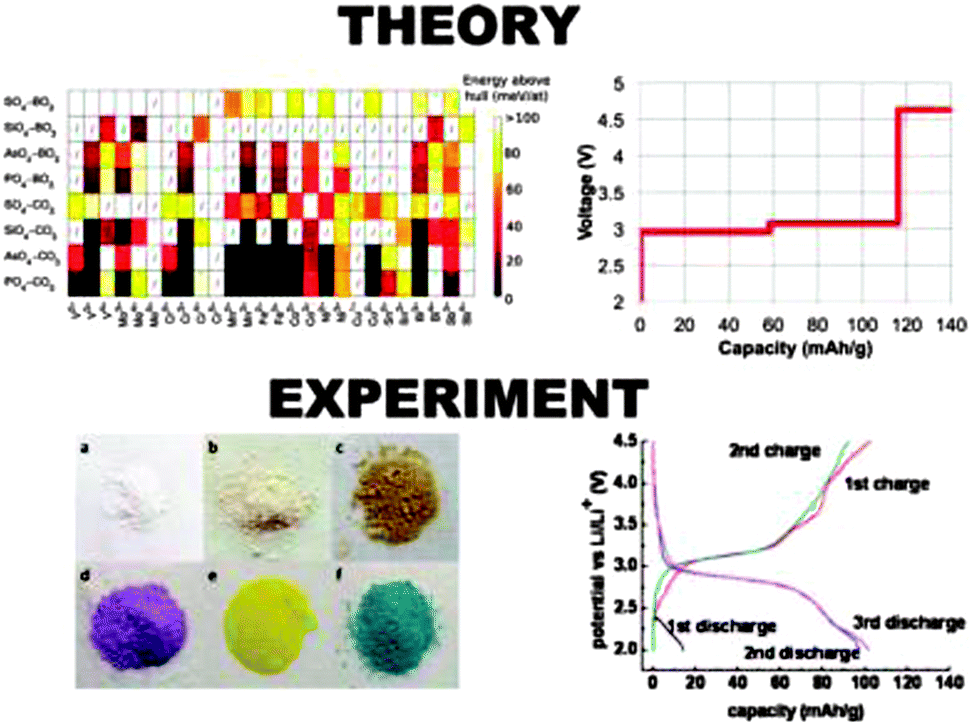 | ||
| Fig. 52 Example of computationally driven design of new Li-ion battery cathodes. (top-left) A family of compounds containing Na, a transition metal, and mixed polyanion group is investigated computationally for stability (the corresponding Li compounds were computed to be too unstable for direct synthesis). Reprinted with permission from ref. 547. Copyright 2011 Royal Society of Chemistry. The ground state “hull” connects the energy of all ground state phases in an energy-composition diagram. The energy above hull is a computed descriptor of the stability of a compound, and in essence describes the thermodynamic decomposition energy of the compound into the most stable phases. Thermodynamically stable compounds exhibit an energy above hull of zero, with greater values indicating decreasing stability. (top-right) Electrochemical properties are calculated for Li versions of compounds predicted to be stable in the Na form, such as the Fe-containing phosphocarbonate. (bottom-left) Hydrothermal synthesis produces a colorful family of sodium metal phosphocarbonate materials as predicted by computation. Reprinted with permission from ref. 548. Copyright 2012 American Chemical Society. (bottom-right) The Na compounds are ion exchanged to form their Li analogues, and predicted battery properties are confirmed. Reprinted with permission from ref. 549. Copyright 2012 Royal Society of Chemistry. | ||
Computational models can support the exploration of new and existing materials as well as developing better materials through so-called data-driven materials design pioneered by several groups and large scale collaborative projects such as the Materials Project (http://www.materialsproject.org) as one pertinent example. Computational methods may also identify the role of functional disorder in the crystal structure of over-lithiated battery materials that allows for improved cycling capabilities. Probably one of the most exciting and useful aspects of theoretical materials chemistry, solid state ionics and computational electrochemistry is the screening and identification of the best materials and electrolytes for very new prototypical battery technologies for higher energy density, safer and low cost production for electric vehicles and power grid applications.163,545,550 In Mg–ion batteries, computational methods may reduce the time necessary to reach a similar state of the art currently seen in Li-ion batteries, by working in parallel with experimental developments for a faster technology development in Li–air batteries,551 Li–sulfur batteries,552 Na–ion batteries,553 Na–sulfur batteries,554 and Mg–ion batteries.555 Moreover, computations have provided insights into the electrochemical processes occurring at different interfaces in Li-ion batteries. Despite the recent progress of Li-ion batteries and their applications to portable electronic devices, modelling conversion reactions involving large structural changes remains difficult. Therefore, the improved techniques based on theoretical studies should be devoted to model the complex properties of electrolytes and to better understand the Li intercalation kinetics in the thousands of systems experimentally investigated over the decades, particularly those with mixed redox couples and also in scenarios where more than one charge is transferred per redox event, which could yield higher energy densities.556 Computational identification of the electrochemical properties and crystal structure evolution of these materials, and their response to large ionic radius cations would be extremely useful.
The discovery of new materials, appropriate synthesis and optimization routes will be a challenging task, but recent investigations that combine experimental and computational investigations have shed light on a class of Li-ion battery materials that are not the more classically investigated ordered and rock-salt structured oxides and ordered spinels.73,74 Ceder et al. examined a class of cathode materials in which Li and transition metals (TMs) share the same sub-lattice in a disordered manner, and benefit from enhanced cation hopping to increase energy density and capacity for rechargeable Li-ion battery oxides.557 In Fig. 53, the experimental the computational calculations indicate that in cation-disordered oxides, excess Li excess opens up percolating networks of 0-TM channels in rocksalt-type lithiated TM oxides.
 | ||
| Fig. 53 Li1.211Mo0.467Cr0.3O2 shows high Li cycling capacity even after substantial cation disordering. (A) XRD patterns of C-coated Li1.211Mo0.467Cr0.3O2 (LMCO/C) electrodes before and after 1, 2, and 10 cycles, 1.5 to 4.3 V, C/10. The inset image shows the C-coating layer. (B) Left: STEM images along the [010] zone axis in a LMCO/C particle before cycling and after 1 and 10 cycles, 1.5 to 4.3 V, C/20. Right: corresponding line profiles of the Z-contrast information with the measured spacing of Li–Mo–Cr layers. (C) Voltage profile of LMCO/C, 1.5 to 4.3 V, C/20. (A) Computed probability of finding a percolating network of 0-TMchannels (color) versus Li content (x in LixTM2–xO2) and cation mixing (TMLi layers/TMTM layers × 100%). (B) Accessible Li content by a percolating 0-TM network (color) versus Li content and cation mixing. In the simulation, cations were randomly distributed at each cation-mixing level. Reprinted with permission from ref. 557. Copyright 2014 AAAS publication. | ||
These new findings are encouraging since disordered Li-excess rocksalts are proposed to have significant benefits over layered materials including (i) small lattice parameter and thus volume changes during charging/discharging, (ii) homogeneous cation density limits changes in Li mobility often associated with changes in the layered of some metal oxides, and potentially independent of Li concentration. While promising, further investigations will work out the voltage profiles, with the aim of maintaining invariant profile over longer times, which may stem from a reduced Li–Li interaction in spite of a less defined local density of Li active sites in the material. Computational investigations in these systems, and many others, will pave a path for significant advancements in rechargeable battery energy storage solutions.
Acknowledgements
This research has received funding from the Seventh Framework Programme FP7/2007-2013 (Project STABLE) under grant agreement no. 314508.References
- J. M. Tarascon and M. Armand, Nature, 2001, 414, 359 CrossRef CAS PubMed.
- H. Takeshita, Portable Li-ion, Worldwide, Proc. Conf. Power 2000, San Diego, 25 September 2000.
- M. S. Whittingham, MRS Bull., 2008, 33, 411 CrossRef CAS.
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- J. Liu, J.-G. Zhang, Z. Yang, J. P. Lemmon, C. Imhoff, G. L. Graff, L. Li, J. Hu, C. Wang, J. Xiao, G. Xia, V. V. Viswanathan, S. Baskaran, V. Sprenkle, X. Li, Y. Shao and B. Schwenzer, Adv. Funct. Mater., 2013, 23, 929 CrossRef CAS.
- M. R. Palacin, Chem. Soc. Rev., 2009, 38, 2565 RSC.
- R. A. Huggins, Advanced Batteries: Materials Science Aspects, Springer, New York, 2009 Search PubMed.
- D. Linden and T. B. Reddy, Handbook of Batteries, McGraw-Hill, New York, 3rd edn, 2002 Search PubMed.
- M.-K. Song, S. Park, F. M. Alamgir, J. Cho and M. Liu, Mater. Sci. Eng., R, 2011, 72, 203 CrossRef PubMed.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- A. Yoshino, Development of Secondary Battery Materials, CMC Publishing Co., Ltd, 2008 Search PubMed.
- C.-M. Park, J.-H. Kim, H. Kim and H.-J. Sohn, Chem. Soc. Rev., 2010, 39, 3115–3141 RSC.
- R. Kanno, Lithium-ion Battery Materials from a Structural Viewpoint, GS Yuasa Technical Report, 2006, vol. 3, no. 1, pp. 1–11.
- T. Abe, Current Status and Future of Lithium-ion Batteries, Secondary Battery Material Seminar (hosted by HORIBA, Ltd.) 2009.
- F. Cheng, J. Liang, Z. Tao and J. Chen, Adv. Mater., 2011, 23, 1695 CrossRef CAS PubMed.
- M. Osiak, H. Geaney, E. Armstrong and C. O'Dwyer, J. Mater. Chem. A, 2014, 2, 9433 CAS.
- J. Cabana, L. Monconduit, D. Larcher and M. R. Palacin, Adv. Mater., 2010, 22, E170 CrossRef CAS PubMed.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 4, 7854 Search PubMed.
- C. Masquelier and L. Croguennec, Chem. Rev., 2013, 113, 6552 CrossRef CAS PubMed.
- M. Armand and J.-M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- M. J. Armstrong, C. O'Dwyer, W. J. Macklin and J. D. Holmes, Nano Res., 2014, 7, 1–62 CrossRef CAS.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J.-M. Tarascon, Nat. Mater., 2012, 11, 19 CrossRef CAS PubMed.
- M. Park, X. Zhang, M. Chung, G. B. Less and A. M. Sastry, J. Power Sources, 2010, 195, 7904 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS PubMed.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287 CAS.
- J. B. Goodenough, Chem. Mater., 2010, 22, 587 CrossRef CAS.
- J. B. Goodenough and K.-S. Park, J. Am. Chem. Soc., 2013, 135, 1167 CrossRef CAS PubMed.
- V. Etacheri, R. Marom, R. Elazari, G. Sailtra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 CAS.
- K. Takada, Acta Mater., 2013, 61, 759 CrossRef CAS PubMed.
- Z. Gong and Y. Yang, Energy Environ. Sci., 2011, 4, 3223 CAS.
- J. W. Fergus, J. Power Sources, 2010, 195, 939 CrossRef CAS PubMed.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691 CrossRef CAS.
- B. Xu, D. Qian, Z. Wang and Y. S. Meng, Mater. Sci. Eng., R, 2012, 73, 51 CrossRef CAS PubMed.
- P. He, H. Yu, D. Li and H. Zhou, J. Mater. Chem., 2012, 22, 3680 RSC.
- J. B. Goodenough, J. Power Sources, 2007, 174, 996 CrossRef CAS PubMed.
- K. Zaghib, A. Guerfi, P. Hovington, A. Vijh, M. Trudeau, A. Mauger, J. B. Goodenough and C. M. Julien, J. Power Sources, 2013, 232, 357 CrossRef CAS PubMed.
- B. L. Ellis and L. F. Nazar, Curr. Opin. Solid State Mater. Sci., 2012, 16, 168 CrossRef CAS PubMed.
- S. W. Kim, D.-H. Seo, X. Ma, G. Ceder and K. Kang, Adv. Energy Mater., 2012, 2, 710 CrossRef CAS.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. Carretero-Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- J. W. Fergus, Solid State Ionics, 2012, 227, 102 CrossRef CAS PubMed.
- M. D. Slater, D. Kim, E. Lee and C. S. Johnson, Adv. Funct. Mater., 2013, 23, 947 CrossRef CAS.
- N. Yabuuchi, M. Kajiyama, J. Iwatate, H. Nishikawa, S. Hitomi, R. Okuyama, R. Usui, Y. Yamada and S. Komaba, Nat. Mater., 2012, 11, 512 CrossRef CAS PubMed.
- A. Ponrouch, R. Dedryvere, D. Monti, A. E. Demet, J. M. Ateba Mba, L. Croguennec, C. Masquelier, P. Johansson and M. R. Palacin, Energy Environ. Sci., 2013, 6, 2361 CAS.
- M. Wakihara and O. Yamamoto, Lithium Ion Batteries: Fundamentals and Performance, Wiley-VCH, Weinheim, 1998 Search PubMed.
- W. A. van Schalkwijk and B. Scrosati, Advances in Lithium-Ion Batteries, Kluwer Academic/Plenum, New York, 2002 Search PubMed.
- G. A. Nazri and G. Pistoia, Lithium Batteries: Science and Technology, Springer, New York, 2009 Search PubMed.
- M. Winter, J. O. Besenhard, M. E. Spahr and P. Novák, Adv. Mater., 1998, 10, 725 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- H. Li, Z. X. Wang, L. Q. Chen and X. J. Huang, Adv. Mater., 2009, 21, 4593 CrossRef CAS.
- H. Pan, Y.-S. Hu and L. Chen, Energy Environ. Sci., 2013, 6, 2338 CAS.
- L. Jabbour, R. Bongiovanni, D. Chaussy, C. Gerbaldi and D. Beneventi, Cellulose, 2013, 20, 1523 CrossRef CAS.
- Lithium ion technical handbook, Gold Peak Industries Ltd, November 2003 Search PubMed.
- H. C. Choi, Y. M. Jung, I. Noda and S. B. Kim, J. Phys. Chem. B, 2003, 107, 5806 CrossRef CAS.
- Y. S. Meng and M. E. A. de Dompablo, Energy Environ. Sci., 2009, 2, 589 CAS.
- C. Wolverton and A. Zunger, J. Electrochem. Soc., 1998, 145, 2424 CrossRef CAS PubMed.
- D. Carlier, L. Croguennec, G. Ceder, M. Menetrier, Y. Shao-Horn and C. Delmas, Inorg. Chem., 2004, 43, 914 CrossRef CAS PubMed.
- G. Ceder, Science, 1998, 280, 1099 CrossRef CAS.
- U. Amador, J. M. Gallardo-Amores, G. Heymann, H. Huppertz, E. Moran and M. E. A. de Dompablo, Solid State Sci., 2009, 11, 343 CrossRef CAS PubMed.
- M. E. A. de Dompablo, U. Amador and F. Garcia-Alvarado, J. Electrochem. Soc., 2006, 153, A673 CrossRef PubMed.
- P. Balog, D. Orosel, Z. Cancarevic, C. Schon and M. Jansen, J. Alloys Compd., 2007, 429, 87 CrossRef CAS PubMed.
- J. M. Gallardo-Amores, N. Biskup, U. Amador, K. Persson, G. Ceder, E. Moran and M. E. A. de Dompablo, Chem. Mater., 2007, 19, 5262 CrossRef CAS.
- M. E. A. de Dompablo, J. M. Gallardo-Amores, J. Garcia-Martinez, E. Moran, J. M. Tarascon and M. Armand, Solid State Ionics, 2008, 179, 1758 CrossRef PubMed.
- M. V. Koudriachova and N. M. Harrison, J. Mater. Chem., 2006, 16, 1973 RSC.
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Phys. Rev. Lett., 2001, 86, 1275 CrossRef CAS.
- A. Kuhn, P. Diaz-Carrasco, M. E. A. de Dompablo and F. Garcia-Alvarado, ChemInform, 2007, 38, 3375 CrossRef.
- D. Balachan, D. Morgan, G. Ceder and A. Ven de Walle, J. Solid State Chem., 2003, 173, 462 CrossRef.
- D. Morgan, D. Balachandaran and G. Ceder, Mater. Res. Soc. Symp. Proc., 2003, 755, 43 Search PubMed.
- P. Tang and N. A. W. Holzwarth, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 68, 165107 CrossRef.
- M. E. Arroyo-de Dompablo, J. M. Gallardo-Amores, M. T. Azcondo, F. Garcia-Alvarado and U. Amador, J. Phys. Chem. Solids, 2006, 67, 1243 CrossRef CAS PubMed.
- M. E. Arroyo-de Dompablo, J. M. Gallardo-Amores and U. Amador, Electrochem. Solid-State Lett., 2005, 8, A564 CrossRef CAS PubMed.
- M. S. Whittingham, Chem. Rev., 2004, 104(10), 4271 CrossRef CAS.
- J. Reed and G. Ceder, Chem. Rev., 2004, 104(10), 4513 CrossRef CAS.
- A. J. Cohen, P. M. Sanchez and W. Yang, Chem. Rev., 2002, 112, 289 CrossRef PubMed.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140(4a), A1133 CrossRef.
- T. Nagaura and K. Tozawa, Prog. Batteries Sol. Cells, 1990, 9, 209 CAS.
- L. Heyne, in Fast Ion Transport in Solids, ed. W. Van Gool, North-Holland, Amsterdam, 1973, p. 123 Search PubMed.
- M. S. Whittingham, Prog. Solid State Chem., 1978, 12, 41 CrossRef CAS.
- M. Aydinol, A. F. Kohan, G. Ceder, K. Cho and J. Joannopoulos, Phys. Rev. B: Condens. Matter Mater. Phys., 1997, 56(3), 1354 CrossRef CAS.
- Y. Kim, D. Kim and S. Kang, Chem. Mater., 2011, 23(24), 5388 CrossRef CAS.
- M. E. A. de Dompablo, C. Marianetti, A. Van der Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63(14), 144107 CrossRef.
- L. Wu, K. Nam, X. Wang, Y. Zhou, J.-C. Zheng, X.-Q. Yang and Y. Zhu, Chem. Mater., 2011, 23(17), 3953 CrossRef CAS.
- G. Ceder and A. Van der Ven, Electrochim. Acta, 1999, 45(1–2), 131 CrossRef CAS.
- A. Van der Ven, M. K. Aydinol, G. Ceder, G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58(6), 2975 CrossRef CAS.
- S. Laubach, S. Laubach, P. C. Schmidt, D. Ensling, S. Schmid, W. Jaegermann, A. Thißen, K. Nikolowski and H. Ehrenberg, Phys. Chem. Chem. Phys., 2009, 11(17), 3278 RSC.
- J. M. Wang, J. P. Hu, C. Y. Ouyang, S. Q. Shi and M. S. Lei, Solid State Commun., 2011, 151(3), 234 CrossRef CAS PubMed.
- M. K. Aydinol, A. F. Kohan and G. Ceder, J. Power Sources, 1997, 68(2), 664 CrossRef CAS.
- M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1997, 144(11), 3832 CrossRef CAS PubMed.
- A. Van der Ven, M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1998, 145(6), 2149 CrossRef CAS PubMed.
- D. Kramer and G. Ceder, Chem. Mater., 2009, 21(16), 3799 CrossRef CAS.
- L. Daheron, H. Martinez, R. Dedryvere, I. Baraille, M. Ménétrier, C. Delmas and D. Gonbeau, J. Phys. Chem. C, 2009, 113(14), 5843 CAS.
- K. Kang and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74(9), 094105 CrossRef.
- K. Kang, Y. S. Meng, J. Berger, C. P. Grey and G. Ceder, Science, 2006, 311(5763), 977 CrossRef CAS PubMed.
- A. Van der Ven, G. Ceder, M. Asta and P. D. Tepestch, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64(18), 184307 CrossRef.
- S. J. Hwang, H. S. Park, J. H. Choy and G. Campet, Chem. Mater., 2000, 12(7), 1818 CrossRef CAS.
- R. Prasad, R. Benedek and M. M. Thackeray, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71(13), 134111 CrossRef.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22(3), 691 CrossRef CAS.
- J. Reed and G. Ceder, Electrochem. Solid-State Lett., 2002, 5(7), A145 CrossRef CAS PubMed.
- Y. Hinuma, Y. S. Meng, K. S. Kang and G. Ceder, Chem. Mater., 2007, 19, 1790 CrossRef CAS.
- K. Kang, Y. S. Meng, J. Breger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- A. Van der Ven and G. Ceder, Electrochem. Commun., 2004, 6, 1045 CrossRef CAS PubMed.
- Y. S. Meng and M. E. Arroyo-de Dompablo, Energy Environ. Sci., 2009, 2, 589 CAS.
- J. Breger, Y. S. Meng, Y. Hinuma, S. Kumar, K. Kang, Y. Shao-Horn, G. Ceder and C. P. Grey, Chem. Mater., 2006, 18, 4768 CrossRef CAS.
- B. Xu, C. R. Fell, M. Chi and Y. S. Meng, Energy Environ. Sci., 2011, 4(6), 2223 CAS.
- Y. K. Sun, S. T. Myung, B. C. Park, J. Prakash, I. Belharouak and K. Amine, Nat. Mater., 2009, 8(4), 320 CrossRef CAS PubMed.
- G. Ceder, Y. M. Chiang, D. R. Sadoway, M. K. Aydinol, Y. I. Jang and B. Huang, Nature, 1998, 392, 694 CrossRef CAS PubMed.
- J. S. Braithwaite, C. R. A. Catlow, J. D. Gale, J. H. Harding and P. E. Ngoepe, J. Mater. Chem., 2000, 10, 239 RSC.
- A. Dianat, N. Seriani, M. Bobeth and G. Cuniberti, J. Mater. Chem. A, 2013, 1, 9273 CAS.
- P. Xiao, Z. Q. Deng, A. Manthiram and G. Henkelman, J. Phys. Chem. C, 2012, 116, 23201 CAS.
- Z. Wang, Q. Su and H. Deng, Phys. Chem. Chem. Phys., 2013, 15, 8705 RSC.
- T. Jiang and M. L. Falk, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85(24), 245111 CrossRef.
- M. S. Whittingham, P. Zavalij, K. Ngala and F. Zhang, 25 years of intercalation chemistry for battery materials, 2000.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461 CrossRef CAS.
- J. M. Tarascon, E. Wang, F. K. Shokoohi, W. R. McKinnon and S. Colson, J. Electrochem. Soc., 1991, 138, 2859 CrossRef CAS PubMed.
- J. Bhattacharya and A. Van der Ven, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 104304 CrossRef.
- J. Bhattacharya and C. Wolverton, Phys. Chem. Chem. Phys., 2013, 13, 6486 RSC.
- L. X. Yuan, Z. H. Wang, W. X. Zhang, X. L. Hu, J. T. Chen, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4(2), 269 CAS.
- M. M. Thackeray, L. A. Depicciotto, A. Dekock, P. J. Johnson, V. A. Nicholas and K. T. Adendor, J. Power Sources, 1987, 21, 1 CrossRef CAS.
- M. M. Thackeray, Prog. Solid State Chem., 1997, 25, 1 CrossRef CAS.
- X. Li, Y. Wei, F. Du, C. Wang and G. Chen, China Pat., 200710055272, 2007 Search PubMed.
- G. T. K. Fey, W. Li and J. R. Dahn, J. Electrochem. Soc., 1994, 141, 2279 CrossRef CAS PubMed.
- G. T. K. Fey and W. Perng, Mater. Chem. Phys., 1997, 47, 47279 CrossRef.
- F. Orsini, E. Baudrin, S. Denis, L. Dupont, M. Touboul, D. Guyomard, Y. Piffard and J. M. Tarascon, Solid State Ionics, 1998, 107, 123 CrossRef CAS.
- C. Rossignol, G. Ouvrard and E. Baudrin, J. Electrochem. Soc., 2001, 148, A869 CrossRef CAS PubMed.
- P. Kalyani, N. Kalaiselvi and N. Muniyandi, Mater. Chem. Phys., 2003, 77, 662 CrossRef CAS.
- T. Thongtem, S. Kaowphong and S. Thongtem, in EcoMaterials Processing & Design VII, ed. H. S. Kim, Y. B. Li and S. W. Lee, 2006, vol. 510–511, p. 1142 Search PubMed.
- M. V. Reddy, B. Pecquenard, P. Vinatier and A. Levasseur, J. Power Sources, 2007, 163, 1040–1046 CrossRef CAS PubMed.
- D. W. Murphy, M. Greenblatt, S. M. Zahurak, R. J. Cava, J. V. Waszczak, G. W. Hull and R. S. Hutton, Rev. Chim. Miner., 1982, 19, 441 CAS.
- R. J. Cava, D. W. Murphy, S. Zahurak, A. Santoro and R. S. Roth, J. Solid State Chem., 1984, 53, 64 CrossRef CAS.
- K. M. Colbow, J. R. Dahn and R. R. Haering, J. Power Sources, 1989, 26, 397 CrossRef CAS.
- L. A. Depicciotto, M. M. Thackeray and G. Pistoia, Solid State Ionics, 1988, 28, 1364 CrossRef.
- M. M. Thackeray, C. Wolverton and E. D. Isaacs, Energy Environ. Sci., 2012, 5, 7854 CAS.
- M. K. Y. Chan, C. Wolverton and J. P. Greeley, J. Am. Chem. Soc., 2012, 134, 14362 CrossRef CAS PubMed.
- S. Kirklin, B. Meredig and C. Wolverton, Adv. Energy Mater., 2012, 3, 252 CrossRef.
- D. Shin, C. Wolverton, J. R. Croy, M. Balasubramanian, S. H. Kang, C. M. L. Rivera and M. Hamdeni, J. Electrochem. Soc., 2012, 159, A121 CrossRef CAS PubMed.
- C. Wolverton and A. Zunger, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 2242 CrossRef CAS.
- M. K. Aydinol, A. Van der Ven and G. Ceder, Materials for Electrochemical Energy Storage and Conversion Ii-Batteries Capacitors and Fuel Cells, 1998, vol. 496, p. 65 Search PubMed.
- J. Bhattacharya and A. Van der Ven, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 144302 CrossRef.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30 CrossRef CAS PubMed.
- A. Van der Ven, M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1998, 145, 2149 CrossRef CAS PubMed.
- A. Van der Ven and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 742 CrossRef CAS.
- A. Van der Ven, G. Ceder, M. Asta and P. D. Tepesch, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 184307 CrossRef.
- F. Zhou, M. Cococcioni, C. A. Marianetti, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 235121 CrossRef.
- M. Wagemaker, A. Van Der Ven, D. Morgan, G. Ceder, F. M. Mulder and G. J. Kearley, Chem. Phys., 2005, 317, 130 CrossRef CAS PubMed.
- K. Huang, C. Chen, S. Liu, Q. Luo and Z. Liu, J. Cent. South Univ. Technol., 2007, 14(2), 186 CrossRef CAS PubMed.
- M. A. Kebede, M. J. Phasha, N. Kunjuzwa, L. J. le Roux, D. Mkhonto, K. I. Ozoemena and M. K. Mathe, Sustainable Energy Technologies and Assessments, 2014, 5, 44 CrossRef PubMed.
- C. R. A. Catlow, Computer Modelling in Inorganic Crystallography, Academic Press, San Diego, 1997, vol. 61, p. 1795 Search PubMed.
- S. M. Woodley, C. R. A. Catlow, P. Piszora, K. Stempin and E. Wolska, J. Solid State Chem., 2000, 153, 310 CrossRef CAS.
- T. Ohzuku, M. Kitagawa and T. Hirai, J. Electrochem. Soc., 1990, 137, 769 CrossRef CAS PubMed.
- A. Van der Ven, C. Marianetti, D. Morgan and G. Ceder, Solid State Ionics, 2000, 135, 21 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144(4), 1188 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2011, 4(3), 805 CAS.
- C. Delacourt, P. Poizot, M. Morcrette, J. M. Tarascon and C. Masquelier, Chem. Mater., 2004, 16, 93 CrossRef CAS.
- J. Gaubicher, T. L. Mercier, Y. Chabre, J. Angenault and M. Quarton, J. Electrochem. Soc., 1999, 146, 278 CrossRef PubMed.
- S. Patoux, C. Wurm, M. Morcrette, G. Rousse and C. Masquelier, J. Power Sources, 2003, 119, 278 CrossRef.
- M. Saidi, J. Barker, H. Huang, J. L. Swoyer and G. Adamson, J. Power Sources, 2003, 119, 266 CrossRef.
- G. Ceder, Y. M. Chiang, D. R. Sadoway, M. Aydinol, Y. I. Jang and B. Huang, Nature, 1998, 192, 694 CrossRef PubMed.
- A. Van der Ven, M. K. Aydinol and G. Ceder, J. Electrochem. Soc., 1998, 145, 2149 CrossRef CAS PubMed.
- K. Kang, Y. S. Meng, J. Berger, C. P. Grey and G. Ceder, Science, 2006, 311, 977 CrossRef CAS PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190 CrossRef CAS PubMed.
- Y. S. Meng, Y. W. Wu, B. J. Hwang, Y. Li and G. Ceder, J. Electrochem. Soc., 2004, 151, A1134 CrossRef CAS PubMed.
- M. E. Arroyo-deDompablo, R. Dominko, J. M. Gallando-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik and E. Moran, Chem. Mater., 2008, 20, 5574 CrossRef CAS.
- M. Arroyo, Y. De Dompable, U. Amador and J. M. Tarascon, J. Power Sources, 2007, 174, 1251 CrossRef PubMed.
- G. Ceder, G. Hautier, A. Jain and S. P. Ong, MRS Bull., 2011, 36, 185 CAS.
- Y. S. Meng and M. E. Arroyo-deDompablo, Energy Environ. Sci., 2009, 2, 589 CAS.
- A. R. Armstrong, C. Arrouvet, V. Gentili, S. C. Parker, M. S. Islam and P. G. Bruce, Chem. Mater., 2010, 22, 6426 CrossRef CAS.
- F. Zhou, K. Kang, T. Maxisch, G. Ceder and D. Morgan, Solid State Commun., 2004, 132, 181 CrossRef CAS PubMed.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6, 1144 CrossRef CAS PubMed.
- F. Zhou, C. A. Marianetti, M. Cococcioni, D. Morgan and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 201101 CrossRef.
- F. Zhou, T. Maxisch and G. Ceder, Phys. Rev. Lett., 2006, 97, 155704 CrossRef.
- S. P. Ong, L. Wang, B. Kang and G. Ceder, Chem. Mater., 2008, 20, 1798 CrossRef CAS.
- J. C. Kim, C. J. Moore, B. Kang, G. Hautier, A. Jain and G. Ceder, J. Electrochem. Soc., 2011, 158, A309 CrossRef CAS PubMed.
- G. Hautier, A. Jain, S. P. Ong, B. Kang, C. M. R. Doe and G. Ceder, Chem. Mater., 2011, 23, 3495 CrossRef CAS.
- D. Morgan, G. Ceder, M. Y. Saidi, J. Barker, J. Swoyer, H. Huang and G. Adamson, J. Power Sources, 2003, 119, 755 CrossRef.
- D. Morgan, G. Ceder, M. Y. Saidi, J. Barker, J. Swoyer, H. Huang and G. Adamson, Chem. Mater., 2002, 14, 4684 CrossRef CAS.
- A. Duriff, Crystal Chemistry of Condensed Phosphates, Plenum Press, New York, 1995 Search PubMed.
- S. P. Ong, V. L. Chevrier and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 075112 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef PubMed.
- J. Paier, M. Marsman, K. Hummer, G. Kresse, I. C. Gerber and J. C. Angyan, J. Chem. Phys., 2006, 124, 154709 CrossRef CAS PubMed.
- T. Drezen, N. H. Kwon, P. Bowen, I. Teerlinck, M. Isono and I. Exnar, J. Power Sources, 2007, 174, 949 CrossRef CAS PubMed.
- S. K. Martha, B. Makovsky, J. Grinblat, Y. Gofer, O. Haik, E. Zinigrad, D. Aurbach, T. Drezen, D. Wang, G. Deghenghi and I. Exnar, J. Electrochem. Soc., 2009, 156, A541 CrossRef CAS PubMed.
- B. Kang and G. Ceder, J. Electrochem. Soc., 2010, 157, A808 CrossRef CAS PubMed.
- T. Maxisch, F. Zhou and G. Ceder, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 104301 CrossRef.
- A. Yamada, Y. Kudo and K. Y. Liu, J. Electrochem. Soc., 2001, 148, A1153 CrossRef CAS PubMed.
- M. Yonemura, A. Yamada, Y. Takei, N. Sonoyama and R. Kanno, J. Electrochem. Soc., 2004, 151, A1352 CrossRef CAS PubMed.
- A. K. Varanasi, P. K. Sunagavarapu, A. Bhowmik, M. D. Bhardwaj, B. Narayana, U. V. Waghmara, D. Deodhare and A. Sharma, Bull. Mater. Sci., 2014, 36(7), 1331 CrossRef PubMed.
- X. H. Zhu, N. Chen, F. Lian, Y. P. Song and Y. Li, Chin. Sci. Bull., 2011, 56(30), 3229 CrossRef CAS.
- H. J. Feng and F. M. Liu, Chin. Phys. B, 2009, 18, 2481 CrossRef CAS.
- A. Nyten, A. Abouimrane, A. Armand, T. Gustaffson and J. O. Thomas, Electrochem. Commun., 2005, 7(2), 156 CrossRef CAS PubMed.
- R. Dominko, M. Bele, M. Gaberseck, A. Meden, M. Remskar and J. Jamnik, Electrochem. Commun., 2006, 8, 217 CrossRef CAS PubMed.
- A. R. West and F. P. Glasser, J. Solid State Chem., 1972, 4, 20 CrossRef CAS.
- V. V. Politaev, A. A. Petrenko, V. B. Nalbandyan, B. S. Medvedev and E. S. Shvetsova, J. Solid State Chem., 2007, 180(3), 1045 CrossRef CAS PubMed.
- M. E. A. de Dompablo, J. M. Gallardo-Amores, L. Dupont, G. Mali, H. Ehrenberg, J. Jamnik and E. Moran, Chem. Mater., 2008, 20, 5574 CrossRef.
- S. I. Nishimura, S. Hayase, P. Kanno, M. Yashima, N. Nakayama and A. Yamada, J. Am. Chem. Soc., 2008, 130(40), 13212 CrossRef CAS PubMed.
- A. Boulineau, C. Sirisopanaporn, R. DoDominko, A. R. Armstrong, P. G. Bruce and C. Masquelier, Dalton Trans., 2010, 39(27), 6310 RSC.
- G. Mali, A. Meden and R. Dominko, Chem. Commun., 2010, 46(19), 6310 RSC.
- G. Zhong, Y. Li, P. Yan, Z. Liu, M. Xie and H. Lin, J. Phys. Chem. C, 2010, 114, 3693 CAS.
- F. Zhou, M. Cococcioni, K. Kang and G. Ceder, Electrochem. Commun., 2004, 6(11), 1144 CrossRef CAS PubMed.
- N. Kuganathan and M. S. Islam, Chem. Mater., 2009, 21(21), 5196 CrossRef CAS.
- P. Larsson, R. Ahuja, A. Nyten and J. O. Thomas, Electrochem. Commun., 2006, 8(5), 797 CrossRef CAS PubMed.
- M. E. A. de Dompablo, M. Armand, J. M. Tarascon and U. Amador, Electrochem. Commun., 2006, 8(8), 1292 CrossRef PubMed.
- A. R. Armstrong, N. Kuganathan, M. S. Islam and P. G. Bruce, J. Am. Chem. Soc., 2011, 133(33), 13031 CrossRef CAS PubMed.
- A. Kokalj, R. Dominko, G. Mali, A. Meden, M. Gaberscek and J. Jamnik, Chem. Mater., 2007, 19(15), 3633 CrossRef CAS.
- P. Larsson, R. Ahuja, A. Liivat and J. O. Thomas, Comput. Mater. Sci., 2010, 47(3), 678 CrossRef CAS PubMed.
- M. Armand and M. E. A. de Dompablo, J. Mater. Chem., 2011, 21(27), 10026 RSC.
- M. E. A. de Dompablo, U. Amador and F. Garcia-Alvarado, J. Electrochem. Soc., 2006, 153(4), A673 CrossRef PubMed.
- D. H. Seo, Y. U. Park, S. W. Kim, I. Park, R. A. Shakoor and K. Kang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83(20), 205127 CrossRef.
- A. Liivat and J. O. Thomas, Comput. Mater. Sci., 2010, 50(1), 191 CrossRef CAS PubMed.
- Y. Li, X. Cheng and Y. Zhang, ECS Trans., 2012, 41(41), 87 CAS.
- J. Barker, M. Y. Saidi and J. L. Swoyer, J. Electrochem. Soc., 2003, 150(10), A1394 CrossRef CAS PubMed.
- N. Recham, J. N. Chotard, L. Dupont, C. Delacourt, W. Walker, M. Armand and J. M. Tarascon, Nat. Mater., 2010, 9(1), 68 CrossRef CAS PubMed.
- B. L. Ellis, W. R. M. Makahnouk, Y. Makimura, K. Toghill and L. F. Nazar, Nat. Mater., 2007, 6(10), 749 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 62, 6671 CrossRef.
- A. I. Liechtenstein, V. I. Anisimov and J. Zaanen, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, R5467 CrossRef CAS.
- G. Mills, H. Jonsson and G. K. Schenter, Surf. Sci., 1995, 324, 305 CrossRef CAS.
- M. Ramzan, S. Lebegue, T. W. Kang and R. Ahuja, J. Phys. Chem. C, 2011, 115, 2600 CAS.
- R. Tripathi, G. R. Gardiner, M. S. Islam and L. F. Nazar, Chem. Mater., 2011, 23(8), 2278 CrossRef CAS.
- T. Mueller, G. Hautier, A. Jain and G. Ceder, Chem. Mater., 2011, 23(17), 3854 CrossRef CAS.
- Z. Liu and X. Huang, Solid State Ionics, 2010, 181(25–26), 1209 CrossRef CAS PubMed.
- S. C. Chung, P. Barpanda, S. Nishimura, Y. Yamada and A. Yamada, Phys. Chem. Chem. Phys., 2012, 14, 8678 RSC.
- M. B. Yahia, F. Lemoigno, G. Rousse, F. Boucher, J. M. Tarascon and M. L. Doublet, Energy Environ. Sci., 2012, 5, 9584 Search PubMed.
- V. Legagneur, Y. An, A. Mosbah, R. Portal, A. Le Gal La Salle, A. Verbaere, D. Guyomard and Y. Piffard, Solid State Ionics, 2001, 139, 37 CrossRef CAS.
- D. H. Seo, Y. U. Park, S. W. Kim, I. Park, R. A. Shakoor and K. Kang, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 205127 CrossRef.
- A. J. Cohen, P. Mori-Sanchez and W. Yang, Chem. Rev., 2011, 112, 289 CrossRef PubMed.
- M. E. Arroyo de Dompablo and J. A. Morales, J. Electrochem. Soc., 2006, 153, A2098 CrossRef PubMed.
- T. F. Yi, Y. R. Zhu and R. S. Zhu, Solid State Ionics, 2008, 179, 2132 CrossRef CAS PubMed.
- J. Shu, T. F. Yi, M. Shui, Y. Wang, R. S. Zhu, X. F. Chu, F. Huang, D. Xu and L. Hou, Comput. Mater. Sci., 2010, 50, 776 CrossRef CAS PubMed.
- M. C. Yang, B. Xu, J. H. Cheng, C. J. Pan, B. J. Hwang and Y. S. Meng, Chem. Mater., 2011, 23, 2832 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CrossRef CAS PubMed.
- S. Yang, Y. Gong, Z. Liu, L. Zhan, D. P. Hashim, L. Ma, R. Vajtai and P. M. Ajayan, Nano Lett., 2013, 13, 1596 CAS.
- Z. S. Hu, W. Ren, L. Xu, F. Li and H. M. Cheng, ACS Nano, 2011, 5, 5463 CrossRef PubMed.
- D. Zhou, Y. Cui and B. Han, Chin. Sci. Bull., 2012, 57, 2983 CrossRef CAS.
- E. Yoo, J. Kim, E. Hosono, H. S. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS PubMed.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS PubMed.
- A. H. C. Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109 CrossRef.
- O. V. Yazvez, Acc. Chem. Res., 2013, 46, 2319 CrossRef PubMed.
- C. Lee, X. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- A. Politano, A. R. Marino, D. Campi, D. Farias, R. Miranda and G. Chiarello, Carbon, 2012, 50, 4903 CrossRef CAS PubMed.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491 CrossRef CAS PubMed.
- Y. Hernandez, V. Nicolosi, M. Lotya, F. M. Blighe, Z. Sun, S. De, I. T. McGovern, B. Holland, M. Byrne, Y. K. Gunko, J. J. Boland, P. Niraj, G. Duesberg, S. Krishnamurthy, R. Goodhue, J. Hutchison, V. Scardaci, A. C. Ferrari and J. N. Coleman, Nat. Nanotechnol., 2008, 3, 563 CrossRef CAS PubMed.
- R. M. Tromp and J. B. Hannon, Phys. Rev. Lett., 2009, 102, 106104 CrossRef CAS.
- B. Borca, S. Barja, M. Garnica, M. Minniti, A. Politano, J. M. Rodriguez-Garcia, J. J. Hinarejos, D. Farias, A. L. V. de Parga and R. Miranda, New J. Phys., 2010, 12, 093018 CrossRef.
- E. Cazzanelli, T. Caruso, M. Castriota, A. R. Marino, A. Politano, G. Chiarello, M. Giarola and G. Moriotto, J. Raman Spectrosc., 2013, 44, 1393 CrossRef CAS.
- M. Kantha, N. A. Cordero, L. M. Molina, J. A. Alonso and L. A. Girifalco, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 125422 CrossRef.
- H. Tachikawa, Y. Nagoya and T. Fukuzumi, J. Power Sources, 2010, 195, 6148 CrossRef CAS PubMed.
- T. Bhardwaj, A. Antic, B. Pavan, V. Barone and B. D. Fahlman, J. Am. Chem. Soc., 2010, 132, 12556 CrossRef CAS PubMed.
- D. H. Wu, Y. F. Li and Z. Zhou, Theor. Chem. Acc., 2011, 130, 209 CrossRef CAS.
- A. L. M. Reddy, A. Srivastava, S. R. Gowda, H. Gullapalli, M. Dubey and P. M. Ajayan, ACS Nano, 2010, 4, 6337 CrossRef CAS PubMed.
- D. Das, S. Kim, K. R. Lee and A. K. Singh, Phys. Chem. Chem. Phys., 2013, 15, 15128 RSC.
- C. C. Ma, X. H. Shao and D. P. Cao, J. Mater. Chem., 2012, 22, 8911 RSC.
- X. L. Wang, Z. Zeng, H. Ahn and G. X. Wang, Appl. Phys. Lett., 2009, 95(18), 183103 CrossRef PubMed.
- C. K. Yang, Appl. Phys. Lett., 2009, 94(16), 163115 CrossRef PubMed.
- C. Uthaisar and V. Barone, Nano Lett., 2010, 10(8), 2838 CrossRef CAS PubMed.
- J. Wolfenstine, J. Mater. Sci., 2010, 45, 3954 CrossRef CAS.
- G. Y. Aleshin, D. A. Semenenko, A. I. Belova, T. K. Zakharchenko, D. M. Itkis, E. A. Goodilin and Y. D. Tretyakov, Solid State Ionics, 2011, 184, 62 CrossRef CAS PubMed.
- T. Kuboki, T. Okuyama, T. Ohsaki and N. Takami, J. Power Sources, 2005, 146, 766 CrossRef CAS PubMed.
- T. T. Truong, Y. Qin, Y. Ren, Z. Chen, M. K. Chan, J. P. Greeley, K. Amine and Y. Sun, Adv. Mater., 2011, 23, 4947 CrossRef CAS PubMed.
- S. E. Burkhardt, J. Bois, J. M. Tarascon, R. G. Henning and H. D. Abruna, Chem. Mater., 2013, 25, 132 CrossRef CAS.
- J. P. Coleman, Electrochemistry of Carboxylic Acids and Derivatives: Cathodic Reductions, in Acid Derivatives, ed. S. Patai, John Wiley & Sons, Ltd., Chichester, U.K., 1979, vol. 2 Search PubMed.
- W. McSweeney, O. Lotty, C. Glynn, H. Geaney, J. D. Holmes and C. O'Dwyer, Electrochim. Acta, 2014, 135, 356–367 CrossRef CAS PubMed.
- V. V. Viswanathan, K. S. Thygesen, J. S. Hummelshoj, J. K. Norskov, G. Girishkumar, B. D. McCloskey and A. C. Luntz, J. Chem. Phys., 2011, 135, 3 CrossRef PubMed.
- M. Osiak, E. Armstrong, T. Kennedy, C. M. S. Torres, K. M. Ryan and C. O'Dwyer, ACS Appl. Mater. Interfaces, 2013, 5, 8195–8202 CAS.
- C. K. Chan, H. Peng, G. Liu, K. McIlwrath, X. F. Zhang, R. A. Huggins and Y. Cui, Nat. Nanotechnol., 2008, 3, 31 CrossRef CAS PubMed.
- S.-H. Ng, J. Wang, D. Wexler, K. Konstantinov, Z.-P. Guo and H.-K. Liu, Angew. Chem., Int. Ed., 2006, 45, 6896 CrossRef CAS PubMed.
- Y. Liu, Z. Y. Wen, X. Y. Wang, X. L. Yang, A. Hirano, N. Imanishi and Y. Takeda, J. Power Sources, 2009, 189, 480 CrossRef CAS PubMed.
- R. Ruffo, S. S. Hong, C. K. Chan, R. A. Huggins and Y. Cui, J. Phys. Chem. C, 2009, 113, 11390 CAS.
- L.-F. Cui, R. Ruffo, C. K. Chan, H. Peng and Y. Cui, Nano Lett., 2009, 9, 491 CrossRef CAS PubMed.
- T. Song, J. Xia, J.-H. Lee, D. H. Lee, M.-S. Kwon, J.-M. Choi, J. Wu, S. K. Doo, H. Chang, W. I. Park, D. S. Zang, H. Kim, Y. Huang, K.-C. Hwang, J. A. Rogers and U. Paik, Nano Lett., 2010, 10, 1710 CrossRef CAS PubMed.
- B. Hertzberg, A. Alexeev and G. Yushin, J. Am. Chem. Soc., 2010, 132, 8548 CrossRef CAS PubMed.
- H. Wu, G. Zheng, N. Liu, T. J. Carney, Y. Yang and Y. Cui, Nano Lett., 2012, 12, 904 CrossRef CAS PubMed.
- L.-F. Cui, L. Hu, J. W. Choi and Y. Cui, ACS Nano, 2010, 4, 3671 CrossRef CAS PubMed.
- S. W. Lee, M. T. McDowell, J. W. Choi and Y. Cui, Nano Lett., 2011, 11, 3034 CrossRef CAS PubMed.
- J. Graetz, C. C. Ahn, R. Yazami and B. Fultz, J. Electrochem. Soc., 2004, 151, A698 CrossRef CAS PubMed.
- C. K. Chan, X. F. Zhang and Y. Cui, Nano Lett., 2008, 8, 307 CrossRef CAS PubMed.
- M.-H. Park, K. Kim, J. Kim and J. Cho, Adv. Mater., 2010, 22, 415 CrossRef CAS PubMed.
- X. H. Liu, S. Huan, S. T. Picraux, J. Li, T. Zhu and J. Y. Huang, Nano Lett., 2011, 11, 3991 CrossRef CAS PubMed.
- M.-H. Park, Y. Cho, K. Kim, J. Kim, M. Liu and J. Cho, Angew. Chem., Int. Ed., 2011, 123, 9821 CrossRef.
- L. P. Tan, Z. Lu, H. T. Tan, J. Zhu, X. Rui, Q. Yan and H. H. Hng, J. Power Sources, 2012, 206, 253 CrossRef CAS PubMed.
- F.-W. Yuan, H.-J. Yang and H.-Y. Tuan, ACS Nano, 2012, 6, 9932 CrossRef CAS PubMed.
- K. T. Lee, Y. S. Jung and S. M. Oh, J. Am. Chem. Soc., 2003, 125, 5652 CrossRef CAS PubMed.
- M. Valvo, U. Lafont, D. Munao and E. M. Kelder, J. Power Sources, 2009, 189, 297 CrossRef CAS PubMed.
- L. Bazin, S. Mitra, P. L. Taberna, P. Poizot, M. Gressier, M. J. Menu, A. Barnabe, P. Simon and J.-M. Tarascon, J. Power Sources, 2009, 188, 578 CrossRef CAS PubMed.
- Y. Yu, L. Gu, X. Lang, C. Zhu, T. Fujita, M. Chen and J. Maier, Adv. Mater., 2011, 23, 2443 CrossRef CAS PubMed.
- B. Peng, F. Cheng, Z. Tao and J. Chen, J. Chem. Phys., 2010, 133, 034701 CrossRef PubMed.
- J. Wang, I. D. Raistrick and R. A. Huggins, J. Electrochem. Soc., 1986, 133, 457 CrossRef CAS PubMed.
- V. B. Shenoy, P. Johari and Y. Qi, J. Power Sources, 2010, 195, 6825 CrossRef CAS PubMed.
- Y. Kubata, M. C. S. Escano, H. Nakanishi and H. Kasai, J. Appl. Phys., 2007, 102, 053704 CrossRef PubMed.
- Y. H. Xu, G. P. Yin and P. J. Zuo, Electrochim. Acta, 2008, 54, 341 CrossRef CAS PubMed.
- Y. M. Kang, S. B. Suh and Y. S. Kim, Inorg. Chem., 2009, 48, 11631 CrossRef CAS PubMed.
- V. L. Chevier and J. R. Dahn, J. Electrochem. Soc., 2009, 156, A454 CrossRef PubMed.
- H. Kim, K. E. Kweon, C. Y. Chou, J. G. Ekerdt and G. S. Hwang, J. Phys. Chem. C, 2010, 114, 17942 CAS.
- K. Zhao, W. L. Wang, J. Gregoire, M. Pharr, Z. Suo, J. J. Vlassak and E. Kaxiras, Nano Lett., 2011, 11, 2962 CrossRef CAS PubMed.
- V. B. Shenoy, P. Johari and Y. Qi, J. Power Sources, 2010, 195, 6825 CrossRef CAS PubMed.
- C. Y. Chou, H. Kim and G. S. Hwang, J. Phys. Chem. C, 2011, 115, 20018 CAS.
- K. Li, H. Xie, J. Liu, Z. Ma, Y. Zhou and D. Xue, Phys. Chem. Chem. Phys., 2013, 15, 17658 RSC.
- J. Moon, K. Cho and M. Cho, Int. J. Precis. Eng. Manuf., 2012, 13, 1191 CrossRef PubMed.
- Q. Zhang, W. Zhang, W. Wan, Y. Cui and E. Wang, Nano Lett., 2010, 10(9), 3243 CrossRef CAS PubMed.
- T. L. Chan and J. R. Chelikowsky, Nano Lett., 2010, 10, 821 CrossRef CAS PubMed.
- J. Sangster and C. W. Bale, J. Phase Equilib., 1998, 19, 70 CAS.
- M. E. Stournara, P. R. Guduru and V. B. Shenoy, J. Power Sources, 2012, 208, 165 CrossRef CAS PubMed.
- S. C. Jung and Y. K. Han, Phys. Chem. Chem. Phys., 2013, 15, 13586 RSC.
- Q. Zhang, Y. Cui and E. Wang, J. Phys. Chem. C, 2011, 115, 9376 CAS.
- E. Bekaert, F. Robert, P. E. Lippens and M. Menetries, J. Phys. Chem. C, 2010, 114, 6749 CAS.
- T. P. Kumar, R. Ramesh, Y. Y. Lin and G. T. K. Fey, Electrochem. Commun., 2004, 6, 520 CrossRef CAS PubMed.
- M. F. Ng, J. W. Zheng and P. Wu, J. Phys. Chem. C, 2010, 114, 8542 CAS.
- J. W. Zheng, S. M. L. Nai, M. F. Ng, P. Wu, J. Wei and M. Gupta, J. Phys. Chem. C, 2009, 113, 14015 CAS.
- N. Liu, N. Lu, Y. X. Yao, Y. R. Li, C. Z. Wang and K. M. Ho, J. Phys. Chem. C, 2011, 115, 15739 CAS.
- G. Ceder, Y. M. Cheng, D. R. Sadoway, M. Aydinol, Y. I. Jang and B. Huang, Nature, 1998, 192, 694 CrossRef PubMed.
- Y. S. Meng and M. E. Arroyo-deDompablo, Energy Environ. Sci., 2009, 2, 589 CAS.
- M. Armand and J. M. Tarascon, Nature, 2008, 451, 652 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS PubMed.
- X. P. Gao, J. L. Bao, G. L. Pan, H. Y. Zhu, P. X. Huang, F. Wu and D. Y. Song, J. Phys. Chem. B, 2004, 108, 5547 CrossRef CAS.
- S. M. Yuan, J. X. Li, L. T. Yang, L. W. Su, L. Liu and Z. Zhou, ACS Appl. Mater. Interfaces, 2011, 3, 705 CAS.
- L. Liu, Y. Li, S. M. Yuan, M. Ge, M. M. Ren, C. S. Sun and Z. Zhou, J. Phys. Chem. C, 2010, 114, 251 CAS.
- K. J. Nakahara, R. Nakajima, T. Matsushima and H. Majima, J. Power Sources, 2003, 117, 131 CrossRef CAS.
- G. X. Wang, D. H. Bradhurst, S. X. Dou and H. K. Liu, J. Power Sources, 1999, 83, 156 CrossRef CAS.
- K. N. Jung, S. I. Pyun and S. W. Kim, J. Power Sources, 2003, 119, 637 CrossRef.
- C. Y. Ouyang, Z. Y. Zhong and M. S. Lei, Electrochem. Commun., 2007, 9, 1107 CrossRef CAS PubMed.
- Z. Zhong, C. Ouyang, S. Shi and M. Lei, ChemPhysChem, 2008, 9, 2104 CrossRef CAS PubMed.
- K. Saravanan, K. Ananthanarayan and P. Balaya, Energy Environ. Sci., 2010, 3, 939 CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- A. R. Armstrong, G. Armstrong, J. Canales, R. Garcia and P. G. Bruce, Adv. Mater., 2005, 17, 862 CrossRef CAS.
- M. Wagemaker, A. P. M. Kentgens and F. M. Mulder, Nature, 2002, 418, 397 CrossRef CAS PubMed.
- H. Zhang, G. R. Li, L. P. An, T. Y. Yan, X. P. Gao and H. Y. Zhu, J. Phys. Chem. C, 2007, 111, 6143 CAS.
- L. Kavan, M. Kalbac, M. Zukalova, I. Exnar, V. Lorenzen, R. Nesper and M. Graetzel, Chem. Mater., 2004, 16, 477 CrossRef CAS.
- Y. S. Hu, L. Kienle, Y. G. Gao and J. Maier, Adv. Mater., 2006, 18, 1421 CrossRef CAS.
- G. Armstrong, A. Armstrong, P. G. Bruce, P. Reale and B. Scrosati, Adv. Mater., 2006, 18, 2597 CrossRef CAS.
- M. Wagemaker, W. J. H. Borghols and F. M. Mulder, J. Am. Chem. Soc., 2007, 129, 4323 CrossRef CAS PubMed.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588 CrossRef CAS PubMed.
- A. S. Dalton, A. A. Belak and A. Vander Ven, Chem. Mater., 2010, 24, 1568 CrossRef.
- Y. S. Hu, L. Kienle, Y. G. Gao and J. Maier, Adv. Mater., 2006, 18, 1421 CrossRef CAS.
- J. Y. Sin, J. H. Joo, D. Samuelis and J. Mier, Chem. Mater., 2012, 24, 543 CrossRef.
- R. Vande Krol, A. Goossens and E. A. Meulenkamp, J. Electrochem. Soc., 1999, 146, 3150 CrossRef CAS PubMed.
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 69, 54106 CrossRef.
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Phys. Rev. Lett., 2001, 86, 1275 CrossRef CAS.
- S. Kerisit, K. M. Rosso and Z. Yang, J. Phys. Chem. C, 2010, 114(18), 8542 Search PubMed.
- M. L. Sushko, K. M. Rosso and J. Liu, J. Phys. Chem. C, 2010, 114, 20277 CAS.
- H. Yildrim, J. Greeley and S. K. R. S. Sankarnarayan, J. Phys. Chem. C, 2009, 115, 15661 Search PubMed.
- A. Stashans, S. Lunell, R. Bergstrom, A. Hegfeldt and E. S. Lindquist, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 53, 159 CrossRef CAS.
- M. V. Koudriachova, N. M. Harrison and S. W. de Leeuw, Comput. Mater. Sci., 2002, 24, 235 CrossRef CAS.
- M. Vijaykumar, S. Kerisit, C. Wang, Z. Nie, K. M. Rosso, Z. Yang, G. Graff, J. Liu and J. Hu, J. Phys. Chem. C, 2009, 113, 14567 Search PubMed.
- W. J. H. Borghols, M. Wagemaker, U. Lafont, E. M. Kelder and F. M. Mulder, Chem. Mater., 2008, 20, 2949 CrossRef CAS.
- R. J. Cava, A. Santoro, D. W. Murphy, S. M. Zahurak and R. S. Roth, J. Solid State Chem., 1984, 83, 64 CrossRef.
- S. Lunell, A. Stashans, L. Ojamae, H. Lindstrom and A. Hagfeldt, J. Am. Chem. Soc., 1997, 119, 7374 CrossRef CAS.
- H. Lindstrom, S. Sodergren, A. Solbrand, H. Rensmo, J. Hjelm, A. Hagfeldt and S. Lindquist, J. Phys. Chem. B, 1997, 101, 7717 CrossRef.
- T. Okumura, T. Fukutsuka, A. Yanagihara, Y. Orikasa, H. Arai, Z. Ogumi and Y. Uchimoto, J. Mater. Chem., 2011, 21, 15369 RSC.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930 CrossRef CAS PubMed.
- C. Arrouvel, S. C. Parker and M. S. Islam, Chem. Mater., 2009, 21(20), 4778 CrossRef CAS.
- Y. Yang and J. M. F. Ferreira, Mater. Res. Bull., 1998, 33, 389 CrossRef.
- M. Hirano, C. Nakahara, K. Ota and M. Inagaki, J. Am. Chem. Soc., 2002, 85, 1333 CAS.
- M. Hirano, C. Nakahara, K. Ota, O. Tanaike and M. Inagaki, J. Solid State Chem., 2003, 170, 39 CrossRef CAS.
- N. A. Kaskhedikar and J. Maier, Adv. Mater., 2009, 21, 2664 CrossRef CAS.
- Z. G. Zhang, J. L. Zhang, M. C. W. Kintner-Meyer, X. C. Lu, D. W. Choi, J. P. Lemmon and J. Liu, Chem. Rev., 2011, 111, 3577 CrossRef PubMed.
- H. Shimoda, B. Gao, X. P. Tang, A. Kleinhammes, H. Fleming, Y. Wu and O. Zhou, Phys. Rev. Lett., 2002, 88, 015502 CrossRef CAS.
- D. Qian, G. J. Wagner, W. K. Liu, M. F. Yu and R. S. Ruoff, Appl. Mech. Rev., 2002, 55, 495 CrossRef.
- H. Dai, Surf. Sci., 2002, 500, 218 CrossRef CAS.
- M. S. Dresselhaus, G. Dresselhaus and P. C. Eklund, Science of Fullerenes and Carbon Nanotubes, Academic Press, San Diego, CA, USA, 1996 Search PubMed.
- L. C. Qin, Phys. Chem. Chem. Phys., 2007, 9, 31 RSC.
- S. A. Hodge, M. K. Bayazit, K. S. Coleman and M. S. P. Shaffer, Chem. Soc. Rev., 2012, 41, 4409 RSC.
- Z. H. Yang and H. Q. Wu, Mater. Chem. Phys., 2001, 71, 7 CrossRef CAS.
- J. Zhao, A. Buldum, J. Han and J. P. Lu, Phys. Rev. Lett., 2000, 85, 1706 CrossRef CAS.
- M. Senami, Y. Lkeda, A. Fukushima and A. Tachibana, AIP Adv., 2011, 1, 042106 CrossRef PubMed.
- B. Song, J. Yang, J. Zhao and H. Fang, Energy Environ. Sci., 2011, 4, 1379 CAS.
- M. Khantha, N. A. Cordero, J. A. Alonso, M. Cawkwell and L. A. Girifalco, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 115430 CrossRef.
- M. Zhao, Y. Xia, X. Liu, Z. Tan, B. Huang, F. Li, Y. Ji and C. Song, Phys. Lett. A, 2005, 340, 434 CrossRef CAS PubMed.
- Y. Liu, H. Yukawa and M. Morinaga, Comput. Mater. Sci., 2004, 30, 50 CrossRef CAS PubMed.
- J. Yang, H. J. Liu and C. T. Chan, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 64, 085420 CrossRef.
- V. Meunier, J. Kephart, C. Roland and J. Bernholc, Phys. Rev. Lett., 2002, 88, 075506 CrossRef.
- P. Dubot and P. Cenedese, Phys. Rev. B: Condens. Matter Mater. Phys., 2001, 63, 241402 CrossRef.
- C. Garau, A. Frontera, D. Quinonero, A. Costa, P. Ballester and P. M. Deya, Chem. Phys., 2004, 297, 85 CrossRef CAS PubMed.
- C. Garau, A. Frontera, D. Quinonero, A. Costa, P. Ballester and P. M. Deya, Chem. Phys. Lett., 2003, 374, 548 CrossRef CAS.
- K. N. Ishidate and M. Hasegawa, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 245418 CrossRef.
- K. N. Ishidate, K. Sasaki, Y. Oikawa, M. Baba and M. Hasegawa, e-J. Surf. Sci. Nanotechnol., 2005, 3, 358 CrossRef.
- S. B. Fagan, S. Guerini, J. Mendes Filho and V. Lemos, Microelectron. J., 2005, 36, 499 CrossRef CAS PubMed.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. H. Lee, S. G. Kim and A. G. Rinzler, Science, 1996, 273, 483 CAS.
- Y. Ando, X. Zhao, H. Shimoyama, G. Sakai and K. Kaneto, Int. J. Inorg. Mater., 1999, 1, 77 CrossRef CAS.
- M. F. Yu, O. Lourie, M. J. Dyer, K. Moloni, T. F. Kelley and R. S. Ruoff, Science, 2000, 287, 637 CrossRef CAS.
- A. Udomvech and T. Kerdcharoen, J. Korean Phys. Soc., 2008, 52, 1350 CrossRef CAS.
- V. Meunier, J. Kephart, C. Ronald and J. Bernholc, Phys. Rev. Lett., 2002, 88, 075506 CrossRef.
- I. Mukhopadhyay, N. Hoshino, S. Kawasaki, F. Okino, W. K. Hsu and H. Toudhara, J. Electrochem. Soc., 2002, 149, A39 CrossRef CAS PubMed.
- Y. F. Li, Z. Zhou and L. B. Wang, J. Chem. Phys., 2008, 129, 104703 CrossRef CAS PubMed.
- J. Zhao, B. Wen, Z. Zhou, Z. Chen and P. V. R. Schleyer, Chem. Phys. Lett., 2005, 415, 323 CrossRef CAS PubMed.
- H. L. Wang, L. F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Y. Liang, Y. Cui and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS PubMed.
- Y. K. Sun, Z. H. Chen and H. J. Noh, Nat. Mater., 2012, 11, 942 CrossRef CAS PubMed.
- B. Dunn, H. Kamath and J. M. Tarascon, Science, 2011, 334, 928 CrossRef CAS PubMed.
- J. M. Clark, S. I. Nishimura, A. Yamada and M. S. Islam, Angew. Chem., Int. Ed., 2012, 51, 1 CrossRef.
- D. W. Shin and A. Manthiram, Electrochem. Commun., 2011, 13, 1213 CrossRef CAS PubMed.
- J. Liu and A. Manthiram, Chem. Mater., 2009, 21, 1695 CrossRef CAS.
- B. D. McCloskey, D. S. Bethune, R. M. Shelby, G. Girishkumar and A. C. Luntz, J. Phys. Chem. Lett., 2011, 2, 1161 CrossRef CAS.
- J. Read, J. Electrochem. Soc., 2006, 153, A96 CrossRef CAS PubMed.
- C. O. Laoire, S. Mukerjee, E. J. Plichta, M. A. Handrickson and K. M. Abraham, J. Electrochem. Soc., 2011, 158, A302 CrossRef CAS PubMed.
- W. Xu, J. Xiao, D. Y. Wang, J. Zhang and J. G. Zhang, Electrochem. Solid-State Lett., 2010, 13, A48 CrossRef CAS PubMed.
- W. Xu, J. Xiao, D. Y. Wang, J. Zhang and J. G. Zhang, J. Electrochem. Soc., 2010, 157, A219 CrossRef CAS PubMed.
- Y. Watanabe, S. I. Kanoshita, S. Wada, K. Hoshino, H. Morimoto and S. I. Tobishima, J. Power Sources, 2002, 179(2), 770 CrossRef PubMed.
- X. G. Sun and C. A. Angell, Electrochem. Commun., 2009, 11(7), 1418 CrossRef CAS PubMed.
- A. Abouimrane, I. Belharouak and K. Amine, Electrochem. Commun., 2009, 11(5), 1388 CrossRef PubMed.
- M. Nagahama, N. Hasegawa and S. Okada, J. Electrochem. Soc., 2010, 157(6), A748 CrossRef CAS PubMed.
- M. Galinsky, A. Lewandowski and I. Stepniak, Electrochim. Acta, 2006, 51, 5567 CrossRef PubMed.
- A. Chagnes, H. Allouchi, B. Carre and D. Lemordant, Solid State Ionics, 2005, 176, 1419 CrossRef CAS PubMed.
- A. Chagnes, M. Diaw, B. Carre, P. Willmann and D. Lemordant, J. Power Sources, 2005, 145, 82 CrossRef CAS PubMed.
- S. Zugmann, D. Moosbauer, M. Amereller, C. Schreiner, F. Wudy, R. Schmitz, P. Isken, C. Dippel, R. Muller, M. Kunze, A. Lex-Balducci, M. Winter and H. J. Gores, J. Power Sources, 2011, 196, 1417 CrossRef CAS PubMed.
- G. C. Farrington, in Proceedings of the NATO Advanced Study Institute on Solid State Batteries, Alcabideche, Portugal, ed. C. A. C. Sequeria and A. Hooper, September 19–26, 1984 Search PubMed.
- A. M. Christie, S. J. Lilley, E. Staunton, Y. G. Andrew and P. G. Bruce, Nature, 2005, 433, 50 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS.
- D. M. Tigellar, M. A. B. Meador and W. R. Bennett, Macromolecules, 2007, 40, 4159 CrossRef.
- R. L. Kerr, S. A. Miller, R. K. Shoemaker, B. J. Elliott and D. L. Gin, J. Am. Chem. Soc., 2009, 131, 15972 CrossRef CAS PubMed.
- S. W. Choi, J. R. Kim, Y. R. Ahn, S. M. Jo and E. J. Cairns, Chem. Mater., 2007, 19, 104 CrossRef CAS.
- K. U. Jeong, H. D. Chae, C. I. Lim, H. K. Lee, J. H. Ahn and C. Nah, Polym. Int., 2010, 59, 249 CrossRef CAS.
- F. Croce, G. B. Appetecchi, L. Persi and B. Scrosati, Nature, 1998, 394, 456 CrossRef CAS PubMed.
- D. R. MacFarlane, J. H. Huang and M. Forsyth, Nature, 1999, 402, 792 CrossRef CAS PubMed.
- J. W. Zhang, X. B. Huang, H. Wei, J. W. Fu, Y. W. Huang and X. Z. Tang, J. Appl. Electrochem., 2010, 40(8), 1475 CrossRef CAS PubMed.
- U. V. Alpen, M. F. Bell and W. Wichelhaus, Electrochim. Acta, 1978, 23, 1395 CrossRef.
- H. Y. P. Hong, Mater. Res. Bull., 1978, 13, 117 CrossRef CAS.
- M. Murayama, N. Sonoyama, A. Yamada and R. Kanno, Solid State Ionics, 2004, 170(3–4), 173 CrossRef CAS PubMed.
- R. Kanno, T. Hata, Y. Kawamoto and M. Irie, Solid State Ionics, 2000, 130(1–2), 97 CrossRef CAS.
- A. Hayashi, H. Kitaura, T. Ohtomo, S. Hama and M. Tatsumisago, 15th International Meeting on Lithium Batteries, July 2010, Montreal, Quebec, Canada, 2010.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379 CrossRef CAS PubMed.
- D. Aurbach, K. Gamolsky, B. Markovsky, Y. Gofer, M. Schmidt and U. Heider, Electrochim. Acta, 2002, 47, 1423 CrossRef CAS.
- H. Ota, Y. Sakata, Y. Otake, K. Shima, M. Ue and J. Yamaki, J. Electrochem. Soc., 2004, 151, A1778 CrossRef CAS PubMed.
- Y. Wang, S. Nakamura, K. Tasaki and P. B. Balbuena, J. Am. Chem. Soc., 2002, 124, 4408 CrossRef CAS PubMed.
- S. A. Campbell, C. Bowes and R. S. McMillan, J. Electroanal. Chem., 1990, 284, 195 CrossRef CAS.
- Y. Wang, L. Xing, W. Li and D. Bedrov, J. Phys. Chem. Lett., 2013, 4, 3992 CrossRef CAS.
- Y. X. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123, 11708 CrossRef CAS PubMed.
- K. Kanamura, S. Toriyama, S. Shiraishi and S. Takehara, J. Electrochem. Soc., 1995, 142, 1383 CrossRef CAS PubMed.
- Z. Hong, M. Wei, T. Lan and G. Cao, Nano Energy, 2012, 1, 466 CrossRef CAS PubMed.
- L. Ji, H. Zheng, A. Ismach, Z. Tan, S. Xun, E. Lin, V. Battaglia, V. Srinivasan and Y. Zhang, Nano Energy, 2012, 1, 164 CrossRef CAS PubMed.
- D. Aurbach, M. Daroux, P. Faguy and E. Yeager, J. Electroanal. Chem., 1991, 297, 225 CrossRef CAS.
- T. Ogasawara, A. Debart, M. Holzapfel, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390 CrossRef CAS PubMed.
- A. Debart, A. J. Peterson, J. Bao and P. G. Bruce, Angew. Chem., Int. Ed., 2008, 47, 4521 CrossRef CAS PubMed.
- K. M. Abraham and Z. A. Jiang, J. Electrochem. Soc., 1996, 143, 1 CrossRef CAS PubMed.
- I. Kowaluk, J. Read and M. Solomon, Pure Appl. Chem., 2007, 79, 851 Search PubMed.
- F. Mizuno, S. Nakanishi, Y. Kotani, S. Yokoishi and H. Iba, Electrochemistry, 2010, 78, 403 CrossRef CAS PubMed.
- S. A. Freunberger, Z. Peng, L. J. Hardwick, Y. Chen, F. Barde and P. Bruce, ECS Meeting Abstr., 2010, 340, 1002 Search PubMed.
- J. Y. Song, Y. Y. Wang and C. C. Wan, J. Power Sources, 1999, 77, 183 CrossRef CAS.
- S. S. Sekhon, M. Deepa and S. A. Agnihotry, Solid State Ionics, 2000, 136–137, 1189 CrossRef CAS.
- J. O. Besenhard, M. Winter, J. Yang and W. Biberacher, J. Power Sources, 1995, 54(2), 228 CrossRef CAS.
- A. R. Armstrong, D. W. Tee, F. La Mantia, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2008, 130(11), 3554 CrossRef CAS PubMed.
- P. Arora, R. E. White and M. Doyle, J. Electrochem. Soc., 1998, 145(10), A3647 CrossRef PubMed.
- D. Aurbach, M. D. Levi, E. Levi and A. Schechter, J. Phys. Chem. B, 1997, 101(12), 2195 CrossRef CAS.
- A. Schechter, D. Aurbach and H. Cohen, Langmuir, 1999, 15(9), 3334 CrossRef CAS.
- G. C. Chung, S. H. Jun, K. Y. Lee and M. H. Kim, J. Electrochem. Soc., 1999, 146, A1664 CrossRef PubMed.
- M. D. Levi and D. Aurbach, J. Electroanal. Chem., 1997, 421, 79 CrossRef CAS.
- M. Nishizawa, R. Hashitani, T. Itoh, T. Matsue and I. Uchida, Electrochem. Solid-State Lett., 1998, 1, 10 CrossRef CAS PubMed.
- P. Yu, B. N. Popov, J. A. Ritter and R. E. White, J. Electrochem. Soc., 1999, 146, 8 CrossRef CAS PubMed.
- M. D. levi and D. Aurbach, J. Phys. Chem. B, 1997, 101, 4641 CrossRef CAS.
- G. Botte, B. A. Johnson and R. E. White, J. Electrochem. Soc., 1999, 146, 914 CrossRef CAS PubMed.
- T. Li and P. B. Balbuena, Chem. Phys. Lett., 2000, 317, 422 CrossRef.
- K. A. Hirasawa, T. Sato, S. Yamaguchi and S. Mori, J. Electrochem. Soc., 1997, 144, L81 CrossRef CAS PubMed.
- K. Kanamura, T. Umegaki, M. Ohashi, S. Toriyama, S. Shiraishi and Z. I. Takehara, Electrochim. Acta, 2001, 47, 433 CrossRef CAS.
- K. Kanamura, S. Toriyama, S. Shiraishi and Z. I. Takehara, Electrochim. Acta, 1995, 142, 1383 CAS.
- M. Arakawa and J. I. Yamaki, J. Power Sources, 1995, 54, 250 CrossRef CAS.
- L. Xing, C. Wang, W. Li, M. Xu, X. Meng and S. Zhao, J. Phys. Chem. B, 2009, 113, 5181 CrossRef CAS PubMed.
- L. Xing, C. Wang, O. Borodin, G. D. Smith and W. Li, J. Phys. Chem. A, 2011, 115, 13896 CrossRef CAS PubMed.
- L. Xing, W. Li, C. Wang, F. Gu, M. Xu, C. Tan and J. Yi, J. Phys. Chem. B, 2009, 113, 16596 CrossRef CAS PubMed.
- P. Verma, P. Maire and P. Novak, Electrochim. Acta, 2010, 55(22), 6332 CrossRef CAS PubMed.
- M. A. McArthur, S. Trussler and J. R. Dahn, J. Electrochem. Soc., 2012, 159(3), A198 CrossRef CAS PubMed.
- N. S. Choi, K. H. Yew, K. Y. Lee, M. Sung, H. Kim and S. S. Kim, J. Power Sources, 2006, 161(2), 1254 CrossRef CAS PubMed.
- D. Aurbach, Y. Gofer, M. Benzion and P. Aped, J. Electroanal. Chem., 1992, 339(1–2), 451 CrossRef CAS.
- V. Etacheri, O. Haik, Y. Gofer, G. A. Roberts, I. C. Stefan, R. Fasching and D. Aurbach, Langmuir, 2012, 28(1), 965 CrossRef CAS PubMed.
- S. Dalavi, P. Guduru and B. L. Lucht, J. Electrochem. Soc., 2012, 159(5), A642 CrossRef CAS PubMed.
- I. A. Profatilova, C. Stock, A. Schmitz, S. Passerini and M. Winter, J. Power Sources, 2013, 222, 140 CrossRef CAS PubMed.
- K. Leung, S. B. Rempe, M. E. Foster, Y. Ma, J. M. Martinez del la Hoz, N. Sai and P. B. Balbuena, J. Electrochem. Soc., 2014, 161(3), A213 CrossRef CAS PubMed.
- M. D. Bhatt, M. Cho and K. Cho, Appl. Surf. Sci., 2010, 257, 1463 CrossRef CAS PubMed.
- M. D. Bhatt, M. Cho and K. Cho, J. Solid State Electrochem., 2012, 16, 435 CrossRef CAS PubMed.
- B. Kalssen, R. Aroca, M. Nazri and G. A. Nazri, J. Phys. Chem. B, 1998, 102, 4795 CrossRef.
- T. Li and P. B. Balbuena, J. Electrochem. Soc., 1999, 146(10), 3613 CrossRef CAS PubMed.
- M. D. Bhatt, M. Cho and K. Cho, Can. J. Chem., 2011, 89, 1525 CrossRef CAS.
- M. D. Bhatt, M. Cho and K. Cho, Modell. Simul. Mater. Sci. Eng., 2012, 20, 065004 CrossRef.
- E. G. Leggesse, R. T. Lin, T. F. Teng, C. L. Chen and J. C. Jiang, J. Phys. Chem. A, 2013, 117, 7959 CrossRef CAS PubMed.
- K. Tasaki, J. Phys. Chem. B, 2005, 109, 2920 CrossRef CAS PubMed.
- Y. X. Wang and P. B. Balbuena, J. Phys. Chem. B, 2003, 107(23), 5503 CrossRef CAS.
- Y. X. Wang, S. Nakamura, K. Tasaki and P. B. Balbuena, J. Am. Chem. Soc., 2002, 124(16), 4408 CrossRef CAS PubMed.
- Y. X. Wang, S. Nakamura, M. Ue and P. B. Balbuena, J. Am. Chem. Soc., 2001, 123(47), 11708 CrossRef CAS PubMed.
- Y. K. Han and S. U. Lee, Theor. Chem. Acc., 2004, 112(2), 106 CrossRef CAS.
- J. M. Vollmer, L. A. Curtiss, D. R. Vissers and K. Amine, J. Electrochem. Soc., 2004, 151(1), A178 CrossRef CAS PubMed.
- P. Johansson, J. Phys. Chem. A, 2006, 110(44), 12077 CrossRef CAS PubMed.
- K. Leung and J. L. Budzien, Phys. Chem. Chem. Phys., 2010, 12(25), 6583 RSC.
- K. Leung, Y. Qi, K. R. Zavadil, Y. S. Jung, A. C. Dillon, A. S. Cavanagh, S. H. Lee and S. M. George, J. Am. Chem. Soc., 2011, 133(37), 14741 CrossRef CAS PubMed.
- L. D. Xing, O. Borodin, G. D. Smith and W. S. Li, J. Phys. Chem. A, 2011, 115(47), 13896 CrossRef CAS PubMed.
- O. Borodin and G. D. Smith, J. Phys. Chem. B, 2009, 113(6), 1763 CrossRef CAS PubMed.
- D. Bcdrov, G. D. Smith and A. C. T. van Duin, J. Phys. Chem. A, 2012, 116(11), 2978 CrossRef PubMed.
- Y. Ma and P. B. Balbuena, J. Electrochem. Soc., 2014, 161(8), E3097 CrossRef CAS PubMed.
- Y. K. Han and S. U. lee, Bull. Korean Chem. Soc., 2005, 26(1), 43 CrossRef CAS.
- Y. Wang and P. B. Balbuena, J. Phys. Chem. B, 2002, 106, 4486 CrossRef CAS.
- J. Yu, P. B. Balbuena, J. Budizen and K. Leung, J. Electrochem. Soc., 2011, 158(4), A400 CrossRef CAS PubMed.
- Y. Wang and P. B. Balbuena, J. Phys. Chem. A, 2002, 106, 9582 CrossRef CAS.
- C. O. Laoire, S. Mukerjee, K. M. Abraham, E. J. Plichta and M. A. Hendrickson, J. Phys. Chem. C, 2010, 114, 9178 CAS.
- S. A. Freunberger, Y. Chen, Z. Peng, J. M. Graffin, L. J. Hardwick, F. Barde, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2011, 133, 8040 CrossRef CAS PubMed.
- W. Xu, K. Xu, V. V. Viswanathan, S. A. Towne, J. S. Hardy, J. Xiao, Z. Nie, D. Hu, D. Wang and J. G. Zhang, J. Power Sources, 2011, 196, 9631 CrossRef CAS PubMed.
- J. S. Wilkes and M. J. Jaworotko, J. Chem. Soc., Chem. Commun., 1992, 13, 965 RSC.
- C. Nanjundiah, S. F. McDevitt and V. R. Koch, J. Electrochem. Soc., 1997, 144, 3392 CrossRef CAS PubMed.
- J. Fuller, R. T. Karlin and R. A. Osteryoung, J. Electrochem. Soc., 1997, 144, 3881 CrossRef CAS PubMed.
- C. J. Allen, S. Mukerjee, E. J. Plichta, M. A. Hendrickson and K. M. Abraham, J. Phys. Chem. Lett., 2011, 2, 2420 CrossRef CAS.
- D. Zhang, R. S. Li, T. Huang and A. S. Yu, J. Power Sources, 2010, 195, 1202 CrossRef CAS PubMed.
- F. Mizuno, S. Nakanishi, A. Shirasawa, K. Takechi, T. Shiga, H. Nishikoori and H. Iba, Electrochemistry, 2011, 79, 876 CrossRef CAS PubMed.
- S. P. Ong, O. Andreussi, Y. Wu, N. Marzari and G. Ceder, Chem. Mater., 2011, 23, 2979 CrossRef CAS.
- K. Angenendt and P. Johansson, J. Phys. Chem. C, 2010, 114, 20577 CAS.
- K. Angenendt and P. Johansson, J. Phys. Chem. B, 2011, 115, 7808 CrossRef CAS PubMed.
- K. M. Abraham and Z. Jiang, J. Electrochem. Soc., 1996, 143, 1 CrossRef CAS PubMed.
- B. Kumar, J. Kumar, R. Leese, J. P. Fellner, S. J. Rodrigues and K. M. Abraham, J. Electrochem. Soc., 2010, 157, A50 CrossRef CAS PubMed.
- S. Ogata, N. Ohba and T. Kouno, J. Phys. Chem. C, 2013, 117, 17960 CAS.
- Z. Q. Wang, M. S. Wu, G. Liu, X. L. Lei, B. Xu and C. Y. Ouyang, Int. J. Electrochem. Sci., 2014, 9, 562 CAS.
- P. B. Balbuena, E. J. Lamas and Y. Wang, Electrochim. Acta, 2005, 50, 3788 CrossRef CAS PubMed.
- P. Johansson, J. Tegenfeldt and J. Lindgren, Polymer, 1999, 40, 4399 CrossRef CAS.
- M. C. Lopez, G. F. Ortiz, E. M. A. de Dompablo and J. L. Tirado, Inorg. Chem., 2014, 53, 2310 CrossRef CAS PubMed.
- C. Tran, X. Q. Yang and D. Y. Qu, J. Power Sources, 2010, 195, 2057 CrossRef CAS PubMed.
- H. S. Lee, X. Q. Yang, C. L. Xiang, J. McBreen and L. S. Choi, J. Electrochem. Soc., 1998, 145, 2813 CrossRef CAS PubMed.
- L. F. Li, H. S. Lee, H. Li, X. Q. Yang, K. W. Nam, W. S. Yoon, J. McBreen and X. J. Huang, J. Power Sources, 2008, 184, 517 CrossRef CAS PubMed.
- B. Xie, H. S. Lee, H. Li, X. Q. Yang, J. McBreen and L. Q. Chen, Electrochem. Commun., 2008, 10, 1195 CrossRef CAS PubMed.
- L. F. Li, H. S. Lee, H. Li, X. Q. Yang and X. J. Huang, Electrochem. Commun., 2009, 11, 2296 CrossRef CAS PubMed.
- L. F. Li, B. Xia, H. S. Lee, H. Li, X. Q. Yang, J. McBreen and X. J. Huang, J. Power Sources, 2009, 189, 539 CrossRef CAS PubMed.
- J. Besenhard and H. Fritz, J. Electroanal. Chem. Interfacial Electrochem., 1974, 53, 329 CrossRef CAS.
- M. Chhowalla, H. S. Shin, G. Eda, L.-J. Li, K. P. Loh and H. Zhang, Nat. Chem., 2013, 5, 263 CrossRef PubMed.
- V. Nicolosi, M. Chhowalla, M. G. Kanatzidis, M. S. Strano and J. N. Coleman, Science, 2013, 340, 1226419 CrossRef.
- D. Aurbach, B. Markovsky, I. Weissman, E. Levi and Y. Ein-Eli, Electrochim. Acta, 1999, 45, 67 CrossRef CAS.
- M. Fujimoto, Y. Shoji, Y. Kida, R. Ohshita, T. Nohma and K. Nishio, J. Power Sources, 1998, 72, 226 CrossRef CAS.
- H. Kaneko, K. Sekine and T. Takamura, J. Power Sources, 2005, 146, 142 CrossRef CAS PubMed.
- H.-L. Zhang, C.-H. Sun, F. Li, C. Liu, J. Tan and H.-M. Cheng, J. Phys. Chem. C, 2007, 111, 4740 CAS.
- Y.-K. Han, S. U. Lee, J.-H. Ok, J.-J. Cho and H.-J. Kim, Chem. Phys. Lett., 2002, 360, 359 CrossRef CAS.
- T. D. Bogart, A. M. Chockla and B. A. Korgel, Curr. Opin. Chem. Eng., 2013, 2, 286 CrossRef PubMed.
- T. Kennedy, E. Mullane, H. Geaney, M. Osiak, C. O'Dwyer and K. M. Ryan, Nano Lett., 2014, 14(2), 716 CrossRef CAS PubMed.
- M. Bhatt and C. O'Dwyer, J. Electrochem. Soc., 2014, 161(9), A1415 CrossRef CAS PubMed.
- K. Ushirogata, K. Sodeyama, Y. Okuno and Y. Tateyama, J. Am. Chem. Soc., 2013, 135, 11967 CrossRef CAS PubMed.
- S. Tobishima and A. Yamaki, Electrochim. Acta, 1983, 28, 1067 CrossRef CAS.
- B. Klassen, R. Aroca, M. Nazri and G. A. Nazri, J. Phys. Chem. B, 1998, 102, 4795 CrossRef CAS.
- S. Tobishima and A. Yamaki, Electrochim. Acta, 1984, 29, 267 CrossRef CAS.
- C. R. Yang, Y. Y. Wang and C. C. Wan, J. Power Sources, 1998, 72, 66 CrossRef CAS.
- Y. Wang and P. B. Balbuena, Int. J. Quantum Chem., 2005, 102, 724 CrossRef CAS.
- R. J. Blint, J. Electrochem. Soc., 1995, 142, 696 CrossRef CAS PubMed.
- R. J. Blint, J. Electrochem. Soc., 1997, 144, 787 CrossRef CAS PubMed.
- X. Xuan, H. Zhang, J. Wang and H. Wang, J. Phys. Chem. A, 2004, 108, 7513 CrossRef CAS.
- M. Masia and R. Rey, J. Phys. Chem. B, 2004, 108, 17992 CrossRef CAS.
- M. D. Bhatt and C. O'Dwyer, Curr. Appl. Phys., 2014, 14, 349 CrossRef PubMed.
- G. M. Veith, J. Nanda, I. H. Delmau and N. J. Dudney, J. Phys. Chem. Lett., 2012, 3, 1242 CrossRef CAS.
- G. M. Veith, N. J. Dudney, J. Howe and J. Nanda, J. Phys. Chem. C, 2011, 115, 14325 CAS.
- S. Oswald, D. Mikhailova, F. Scheiba, P. Reichel, A. Fieldler and E. Ehrenberg, Anal. Bioanal. Chem., 2011, 400, 691 CrossRef CAS PubMed.
- S. H. Oh, T. Yim, E. Pomeranstseva and L. F. Nazar, Electrochem. Solid-State Lett., 2011, 14, A185 CrossRef CAS PubMed.
- S. E. Sloop, J. K. Pugh, S. Wang, J. B. Kerr and K. Kinoshita, Electrochem. Solid-State Lett., 2001, 4, A42 CrossRef CAS PubMed.
- C. L. Campion, B. L. Lucht, R. Boris, J. DiCario, R. Gitzendanner and K. M. Abraham, Abstract 58, The 224th American Chemical Society Meeting, Boston, MA, August 18–22, 2002.
- M. Takehara, M. Ue, N. Sato and Y. Sakata, The 41st Battery Symposium in Japan, Nagoya, November 20–22, 2000, p. 278.
- A. F. Janzen, X. Ou and M. G. Sowa, J. Fluorine Chem., 1997, 83, 27 CrossRef CAS.
- T. Sonoda, H. Kamizori, S. Ikeda, H. Nagashima, K. Momota, T. Hashimoto, A. Kimiura, J. Yamaki, R. Hagiwara and F. Kita, The 40th Battery Symposium in Japan, Kyoto, November 14–16, 1999, p. 447.
- K. Tasaki, K. Kanda, S. Nakamura and M. Ue, J. Electrochem. Soc., 2003, 150(12), A1628 CrossRef CAS PubMed.
- S. D. Han, J. L. Allen, E. Jonsson, P. Johansson, D. W. McOwen, P. D. Boyle and W. A. Henderson, J. Phys. Chem. C, 2013, 117, 5521 CAS.
- G. Ceder, MRS Bull., 2010, 35, 693 CrossRef CAS.
- A. Jain, S. P. Ong, G. Hautier, W. Chen, W. D. Richards, S. Dacek, S. Cholia, D. Gunter, D. Skinner, G. Ceder and K. A. Persson, APL Mater., 2013, 1, 011002 CrossRef PubMed.
- G. Hautier, A. Jain, H. Chen, C. Moore, S. P. Ong and G. Ceder, J. Mater. Chem., 2011, 21, 17147 RSC.
- H. Chen, G. Hautier and G. Ceder, J. Am. Chem. Soc., 2012, 134, 19619 CrossRef CAS PubMed.
- H. Chen, G. Hautier, A. Jain, C. J. Moore, B. Kang, R. Doe, L. Wu, Y. Zhu and G. Ceder, Chem. Mater., 2012, 24, 2009 CrossRef CAS.
- J. Wen. Y. Yu and C. Chen, Mater. Express, 2012, 2(3), 197 CrossRef PubMed.
- M. D. Bhatt, H. Geaney, M. Nolan and C. O'Dwyer, Phys. Chem. Chem. Phys., 2014, 16, 12093 RSC.
- Y. X. Yin, S. Xin, Y. G. Guo and L. J. Wan, Angew. Chem., Int. Ed., 2013, 52, 13186 CrossRef CAS PubMed.
- V. Palomares, P. Serras, I. Villaluenga, K. B. Hueso, J. C. Gonzalez and T. Rojo, Energy Environ. Sci., 2012, 5, 5884 CAS.
- K. B. Hueso, M. Armand and T. Rojo, Energy Environ. Sci., 2013, 6, 734 CAS.
- H. D. Yoo, I. Shterenberg, Y. Gofer, G. Gershinsky, N. Pour and D. Aurbach, Energy Environ. Sci., 2013, 6, 2265 CAS.
- G. Hautier, A. Jain, T. Mueller, C. Moore, S. P. Ong and G. Ceder, Chem. Mater., 2013, 25, 2064 CrossRef CAS.
- J. Lee, A. Urban, X. Li, D. Su, G. Hautier and G. Ceder, Science, 2014, 343, 519 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2015 |