
Highlights from Faraday Discussion 172: Carbon in Electrochemistry, Sheffield, UK, July 2014
Boris
Dyatkin
*a,
Philip A.
Ash
*b and
Surbhi
Sharma
*c
aA.J. Drexel Nanomaterials Institute and the Department of Materials Science and Engineering, Drexel University, Philadelphia, PA, USA
bInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK
cCentre for Hydrogen and Fuel Cell Research, School of Chemical Engineering, University of Birmingham, B15 2TT, UK
First published on 12th January 2015
Traditionally, Faraday Discussions have been very true to their name, focussing genuinely on healthy, in-depth, and fruitful discussions rather than just serving as a platform for one to one researcher interaction and collaboration. So the first experience of a Faraday Discussion is always an eye-opener about how lively conference discussions can become. The format follows that presenters submit full papers a few months in advance which are lightly reviewed before all the papers are circulated to each of the registered participants. All participants arrive at the conference prepared with their queries and arguments after having read the circulated manuscripts. At the conference the authors present their work for five minutes each followed by extensive discussion of 20–25 minutes, which is practically a “live peer-review”. This experience of a discussion-cum-peer review with participants ranging for PhD students to experts in the relevant field is both a daunting and a very enriching experience at the same time. This has been the trend since the first Faraday Discussions held in London in 1907 in London which debated “osmotic pressure”,1 and this format clearly sets it apart from any other conference to date. All discussions form part of the peer review process and are sequentially recorded and published with the accepted manuscripts.The first day began with researchers from UK and the rest of the world forming a major part of the Sheffield train station crowd. The local taxi services were surely the first to know there was something going on at the University of Sheffield! As the academics and researchers arrived at the venue in the early afternoon hours of Monday 28th July, the posters started going up on the boards. The finger lunch already laid out for much awaited academics in the poster area, which created a welcoming ambience for the academics to engage with each other.
The conference began with the welcoming note from the conference chair Katherine Holt (University College London), who also headed the scientific committee constituting Peter Hall (University of Sheffield), Professor John Foord (University of Oxford), Professor Ian Kinloch (University of Manchester) and Professor Julie Macpherson (University of Warwick). Before the beginning of the first session, Professor Richard McCreery (University of Alberta) was invited to deliver the introductory lecture. He delivered a captivating lecture on diazonium reduction carbon molecular electronics with the highlight being the possibility of using carbon junctions to replace diodes in electric guitars in the very near future, which set the mood for rest of the conference. Richard McCreery and his group have been doing some fascinating work on investigating the use of molecules as active components for fabrication of solid-state electronic devices.2–5
Session 1, chaired by Katherine Holt and focussing on “the many faces of carbon in electrochemistry”, began with a presentation by Abruña and co-workers6 (Cornell University), which demonstrated the fabrication of electrochromic electrodes from large area graphene using poly(3,4-ethylenedioxythiophene) (PEDOT) for optical and biosensing device applications. In particular, their work revealed that graphene can easily be modified with polymers using simple electropolymerization processes to help improve its use in various devices while achieving good sensitivity. The next paper, by Bergonzo and co-workers7 (CEA-LIST), focussed on a novel fabrication technique for development of boron-doped diamond microelectrode arrays prepared for neurointerfaces and stimulation which enabled long-term in vivo stability. The 3D surgical implant was also studied in rat sub-retinal region (Fig. 1) for stimulation of the retinal region in blind patients. Guldi and co-workers8 (Friedrich-Alexander-Universität Erlangen-Nürnberg) synthesised the oligomer para-phenylenevinylene with Pd(II)pc and Zn(II)pc as excited-state electron donors which were immobilised onto single-wall carbon nanotubes and peapods and investigated the electronic interactions in ground and excited states using Raman spectroscopy and microscopic techniques to reveal that ground-state charge transfer upon photoexcitation leads to electron injection into the conduction band of SWNTs and peapods. This was followed by a combined but detailed discussion on the three papers where the questions from the audience displayed their keen interest in the topics. The questions and queries regarding the paper by Abruña and co-workers by the actively involved audience of reviewers varied from inquiring the reason for the authors' choice of the graphene-PEDOT system to deliberating the dependence of electronic properties on pH and the possible oxygen functional groups responsible for it. Particular interest was seen in the work on neurointerfaces using BDD, electrode fouling and their self-cleaning properties were discussed extensively, followed by defects and sp2 regions in the nanodiamonds. The contribution of Guldi and co-workers also generated a lot of interest; initial questions regarding the selection criteria for the nanotubes and the degree of filling were followed by those looking for in-depth understanding of the reasons for the gain or loss of the absorption spectra peaks and role of charge transfer in the dispersion of the functionalised nanotubes.
 | ||
| Fig. 1 (A) A view of a non-3D diamond MEA implant for retinal stimulation. The left part consists of 64 diamond electrodes as shown in (B). (C) An in vivo MEA array as implanted in P23H rats at the Paris Vision Institute (ref. 7). | ||
The second half of session 1 continued after the afternoon tea with a contribution from Shaffer and co-workers9 (Imperial College London), who discussed the challenges associated with producing multifunctional composite materials with both good mechanical strength and the ability to store and deliver electrical charge. A supercapacitor system was demonstrated using laminates of carbon-fibre fabric electrodes modified with a carbon aerogel matrix, glass-fibre separators and structural electrolytes. Despite fairly modest performance of the structural capacitors, Shaffer and co-workers pointed out that these multifunctional materials need only offer a better performance relative to the combined weight of individual structural and electrochemical systems. As such, the multifunctional approach they presented could offer greater scope for weight saving than adhesion of a supercapacitor film onto a structural substrate. During the discussions it was also noted that device performance could be greatly improved through modification of carbon surface termination, and enhanced conductivity of the structural electrolytes which is at present rather low. The final paper of the session, by Chen and co-workers,10 considered direct carbon energy storage via the electrodeposition and re-oxidation of solid carbon in CO32−-containing molten salts. Although charge recovery following deposition was found to be limited by side reactions such as formation of CO and alkali metals, it was suggested during the discussions that this could be improved by electrode design and optimisation, and a better understanding of the microstructure of the carbon deposits. It was also noted by several members of the audience that there are difficulties in assessing the true cost of carbon produced using this method, but nonetheless the concept of energy storage and transport in the form of carbon is appealing.
While the official poster presentation did not begin until after the conclusion of the first session, 44 posters were displayed prominently even before the introductory lecture of the meeting. The posters, which were conveniently located immediately adjacent to the main conference hall, generated great interest during the entire meeting. To start the session, students presented 30-second elevator pitches of their posters in a summarized form for the conference attendees. The following poster session (Fig. 2) featured posters that covered a very broad spectrum of research related to carbon in electrochemistry. Carbon-based composites were particularly well represented, spanning applications such as chemical sensors (Nolan et al., CRANN, Ireland), polymer composites (Ko et al., University of Ulsan, South Korea), and catalyst supports (Lyth et al., Kyushu University, Japan). Several posters focused on supercapacitors and fundamental carbon properties affecting electrochemical performance, such as work by Dyatkin et al. (Drexel University, USA). Additionally, many researchers explored high-end energy storage devices, including flexible felts (Smith et al., University of Liverpool, UK), CNT-graphene electrodes (Yu, University of Cambridge, UK), battery storage (Dongrag et al., University of Ulsan, South Korea.) and fuel cells (Sharma et al., University of Birmingham, UK). Additional topics included fundamental characterisation of pore shapes (Kurig et al., University of Tartu, Estonia) and capacitive deionization (Porada et al., Leibniz Institute for New Materials, Germany). Many authors who contributed manuscripts for the main discussion showcased separate research efforts in their posters, such as work by Prof. Lawrence Hardwick's group (Wu et al., University of Liverpool, UK). Researchers all over the world showcased their posters at the session, but Prof. Clare Grey's group from the University of Cambridge was best represented with four separate contributions. During the conference dinner, three winners of the Skinner prize were announced by the RSC Committee: Alexander Force from the University of Cambridge (“NMR Studies of the Carbon-Electrolyte Interface in Ionic Liquid Supercapacitors”), Christian van Engers11 from the University of Oxford (“The Electrochemical Graphene Surface Force Balance”) and Robert McSweeny from the University of Nottingham (“Single-walled carbon nanotubes as electrodes for the electrochemical characterisation of guest molecules”).
 | ||
| Fig. 2 Poster session discussion. | ||
The second session of the meeting commenced the following morning and was dedicated entirely to a broad and highly demanded research area of carbon electrodes for energy storage, including Faradaic and non-Faradaic charging mechanisms. The first paper, by Gogotsi12et al. (Drexel University) focused on electrosorption of ions in porous carbide-derived carbon electrodes and the challenges of understanding the significance of functional groups and carbon ordering on capacitance and electrochemical stability (Fig. 3). Theoretical predictions,13 as well as prior research efforts,14 have predicted improved capacitance (a more robust superionic state induced by metallic pore walls) and fewer Faradaic reactions that reduce system lifetimes. Contrary to expectations, defunctionalization and graphitization induced by vacuum annealing of TiC-CDCs and SiC-CDCs at 700–1800 °C decreased capacitance and did not significantly extend the operating voltage window. However, electrode conductivity improved without sacrificing 0.68 nm pore diameter and yielded improved rate handling during cyclic voltammograms at sweep rates above 100 mV s−1. Additionally, annealing reduced diffusion limitations and enhanced “butterfly-shaped” voltage-dependent capacitance behaviors. The exact explanations are still being investigated, but quantum capacitance contributions from graphitic defects (vacancies, Stone–Wales transformations) and functional groups (adatoms) likely significantly affect charge storage and confinement of ions in carbon pores. The subsequent audience discussion of the presented work focused on the fundamental properties and unanswered questions regarding charge storage. In particular, with respect to ion electrosorption in pores of supercapacitor electrodes, there is quite a bit of uncertainty regarding oxygen species concentrations on pore walls. These adatoms may govern both gravimetric capacitance and electrochemical stabilities of the systems. Several of the attendees debated the pros and cons of using oxygen adsorption–desorption to quantify these functional groups and correlating them with electrochemical behaviour.1,21 Additional inquiries focused on the key influences of surface properties on capacitance. Some members of the audience debated the influence of pore structure and whether the narrow pore size distributions could effectively consider ion electrosorption similar to ion intercalation. Professor Gogotsi highlighted the significance of quantum capacitance contributions and the need for additional research to investigate this issue.
 | ||
| Fig. 3 Analysis of asymmetry of cation and anion electrosorption in porous TiC-CDC microparticles (a) and SiC-CDC nanoparticles (b) that were annealed at different temperatures. As functional groups are removed from the material, selective interactions between polar bonds on [EMIm+] or [TFSI−] and surface species are equalized, leading to improved charge distribution, a more pronounced voltage-dependent capacitance, yet a reduced overall storage density (ref. 12). | ||
Instead of modifying the electrode structure to incorporate metal oxide or quinone structures on carbon surfaces, Frackowiak et al.15 (Poznan University of Technology) took advantage of the high accessible surface areas offered by KOH-activated carbons and introduced redox-active electrolyte species. In particular, the team's efforts included hydroquinones, similar benzene derivatives such as resorcinol and catechol, and I2/I− electrolyte systems. The presented work discussed the influence of stereochemistry of dihydroxybenzene electrolytes: while para-hydroxyl group configurations are redox-active, ortho and meta conformations are not. Hydrogen bonding between hydroxyl and quinone groups is presented as a possible factor that affects electrolyte performance (Fig. 4). Finally, Prof. Frackowiak addressed the importance of selecting an appropriate current collector: while stainless steel corrodes (and adds Faradaic reactions that convolute performance) in H2SO4 electrolyte, Pt and Au noble metals are more reliable and appropriate material choices for an acidic medium. The subsequent discussion focused on the distinction between battery and supercapacitor behaviour of the proposed system; despite the presence of redox-active species, cyclic voltammograms retained rectangular shapes and resembled intrinsic capacitors. Additionally, Prof. Frackowiak and several attendees discussed a unique pH response to long-term cycling of the energy storage system.
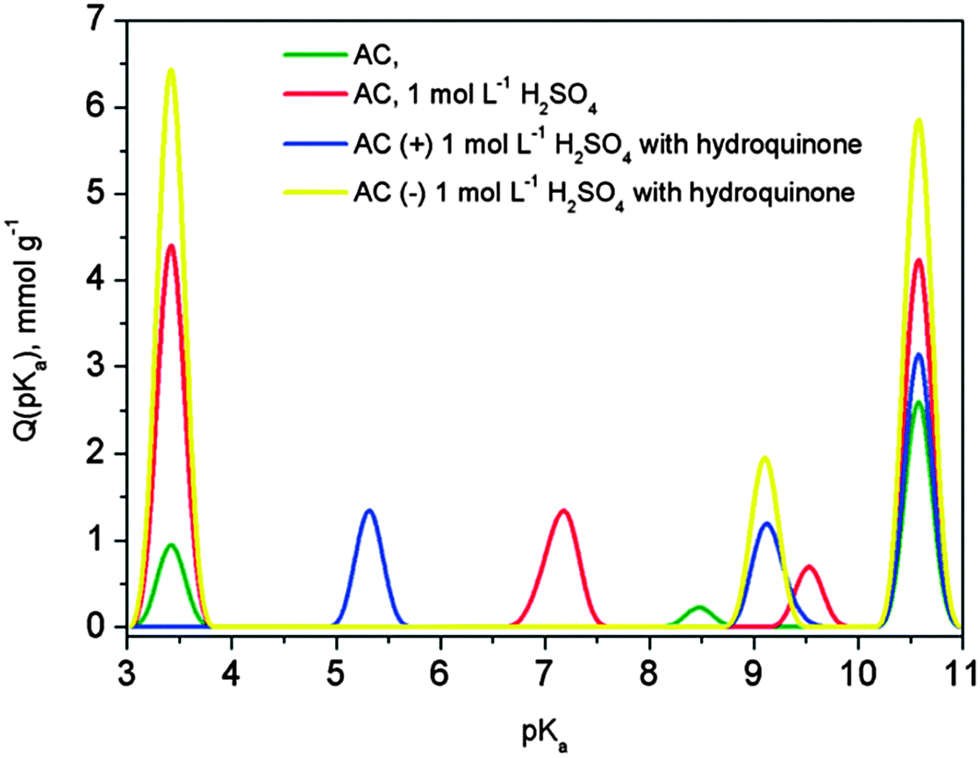 | ||
| Fig. 4 Proton binding curves for activated carbons (ref. 15). | ||
Béguin and co-workers16 (Poznan University of Technology) discussed how the performance of AC/AC electrochemical capacitors can be improved by addition of sodium molybdate. Although the voltage window of traditional aqueous electrolytes is limited by the electrochemical splitting of water at V = 0.846 vs. NHE, addition of small amounts of Na2MoO4 to 1.0 M Li2SO4 electrolyte precluded pitting corrosion of the current collector and maintained good system cyclability up to 1.6 V (Fig. 5). Furthermore, pseudocapacitive (rapid, reversible charge transfer reactions) contributions from the addition of the molybdate ion increased capacitance. System resistance vs. cycle number suggested that MoO42− prevented the formation of an SEI-like layer of breakdown products on the surface of the activated carbon electrodes, which (coupled with prevention of steel current collector corrosion) enhanced the operating potential. The subsequent discussion focused on the specific mechanism that improves electrochemical stability of supercapacitors after the addition of the molybdenum ions to the system.
 | ||
| Fig. 5 Comparison of potential limits of positive (E+) and negative (E−) electrodes vs. rest potential (E0) for (a) pure Li2SO4 electrolyte and (b) Li2SO4 + Na2MoO4 (ref. 16). | ||
Cao et al.17 (Xiamen University) focused on developing a graphene-based composite for Li–O2 batteries. They reported on a solvothermal deposition method, which grafted CoFe2O4 nanoparticles on reduced graphene oxide with a mass loading of 82.7 wt%. The material demonstrated 96.9% coulombic efficiency and good cyclability, owing its performance partially to the reduced state of the graphene substrate and a favorable interface between the rGO and the metal oxide sites. The majority of the subsequent discussion between the audience and the speaker debated whether metal oxides on graphene surfaces for Li–air batteries may be considered catalysts or intercalation sites.
The final paper of the session was presented by Prof. Hardwick18 (University of Liverpool). His work focused on Faradaic charge storage and the process of lithium staging between graphite layers. While prior research efforts have correlated changes in the G band (1580 cm−1) of Raman spectra with Li-ion intercalation,19 the presented work focused on both first-order and second-order (2D band at 2681 cm−1) bands. The research group designed a battery coin cell modified with a window for spectroscopic analysis and correlated electrochemical results with Raman modes. In particular, as voltage-dependent lithiation approaches stage 1, the 2D resonance mode blue-shifts to 2611 cm−1 and eventually disappears (Fig. 6). This corresponds with similar shifts, splits, and eventual disappearance of the G-band as lithium intercalation transforms the anode to a graphite-intercalated compound. As with many spectroelectrochemical approaches,20 this in situ technique is very versatile and can be applied for fundamental analysis of many different charge storage mechanisms. A number of follow-up questions debated the nature of the Raman shift, as well as its correspondence with either the Daumas–Herold or Rudorff models that explain staging of Li+ cation intercalation into graphite anodes. There was also considerable discussion of the influence of mechanical strain on detected Raman shifts and the required efforts that are needed to deconvolute it from intrinsic structural changes.
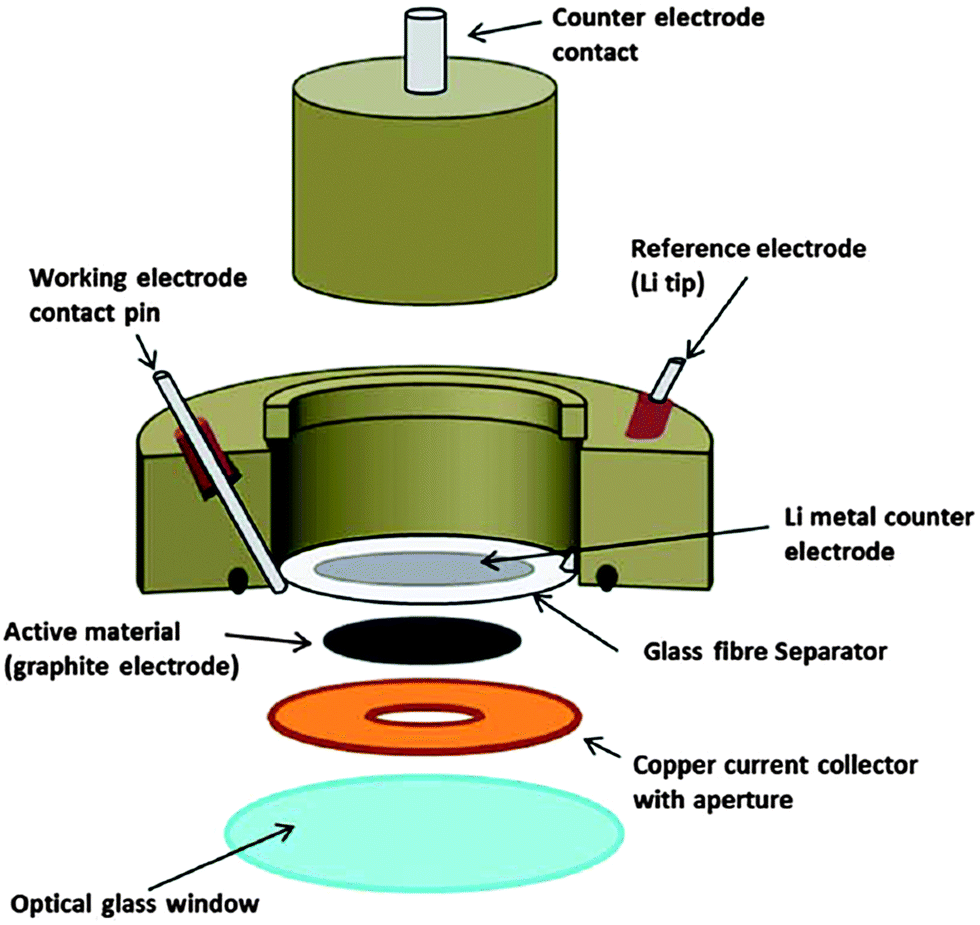 | ||
| Fig. 6 Schematic of in situ coin cell used for Raman spectroscopy in conjunction with electrochemistry (ref. 18). | ||
Session 3 focussing on the “Role of surface contaminants, functionalities, defects and electronic structure” began after lunch, and was jointly chaired by Professor Julie Macpherson and Professor John Foord. The first paper by Dryfe and co-workers22 (University of Manchester) investigated the effect of polymer and solvent residues on the surface transfer kinetics of mechanically exfoliated graphene layers. Using Raman spectroscopy and atomic force microscopy studies, the authors observed that despite the various procedures such as solvent washing, vacuum annealing and exposure to controlled gaseous environment used for cleaning polymer residues, the most common residues in the CVD and mechanically exfoliated graphene was poly(methyl methacrylate) or PMMA which is commonly used for transfer processes. The next paper, from Stevenson et al.23 (The University of Texas at Austin) looked into electrochemically assisted grafting of bis(4-nitrophenyl)iodonium tetrafluoroborate for doping single layer epitaxial graphene. Electrochemical investigations combined with scanning tunnelling microscopy and simulation studies revealed that reproducible and controlled grafts with three-fold symmetry could be achieved. Holt and co-workers24 (University College London) focussed on electrochemical investigations of graphene nanoflakes unzipped from single-walled carbon nanotubes and edge functionalised with carboxylic and amide functionalities. They found the carboxyl-terminated graphene response in the presence of ferricyanide and hydroquinone was highly pH dependent. The authors report that the high density of the carboxylic or amide functionalities did not affect the fast electron transfer kinetics reported and observed for graphene electrodes.
The following discussions focussed on the chloroform and acetone intercalation in graphene and the consequent effects on the Fermi level shift along with the depression of outer-sphere electron transfer rate observed in hexamine ruthenium chloride, on graphene, possibly due to the proximity of its redox potential to the Dirac point. Work on covalent modification of epitaxial single layer graphene also generated interesting discussion and debate on the effect of the size of single-layer graphene on the double-layer capacitance as well as the relative reactivity of graphene vs. HOPG, reactivity of edge planes in HOPG, and control over the reaction conditions during functionalization of graphene in order to not disturb the hexagonal symmetry. Work on edge functionalisation of graphene by Daren et al. led to further interesting conversations around a possible systematic study of the effect of edge termination on the rate of electron transfer, which could provide insight into desirable linker characteristics for covalent attachment of quantum dots to electron acceptors. Elaborate discussions were also focused on porosity of the graphene films, presence of other possible oxygen defects on basal and edge planes and the use of Raman or IR spectroscopy and spectroelectrochemistry to identify ‘blocking’ species responsible for irreversibility of the ferri/ferrocyanide redox couple with COOH terminated flakes.
Following the refreshing afternoon tea in the poster room, the next paper, presented by Hodge et al.25 (Imperial College London), investigated the reduction potentials of highly charged nanocarbon (single-walled nanotubes) polyelectrolyte ions by studying their chemical reactivity towards metal salts and complexes. While the authors found the results for potassium graphenide obtained from charge utilisation yields, combined with thermogravimetric analysis and inductively coupled plasma- atomic emission spectroscopy, matched well with the previously reported results and theoretical predictions, the exact electrochemical potentials of the charged nanocarbons was difficult to observe. Juarez et al.26 (Ulm University) delved into the decoration of the step-edges of highly ordered pyrolytic graphite using Ag, Au and Pt. The results found that metallic nanowires could be formed on bare or functionalised step-edges and the interaction between the metal wires and carbon followed the sequence Pt > Au > Ag. The final paper of the session, by Varley et al.27 (University College London), studied the effect of nanodiamond immobilised on boron-doped diamond electrodes for reversible oxidation of ferrocene methanol. Significant current enhancement was observed and was found to have an inverse relation with the nanodiamond diameter (Fig. 7). Enthusiastic discussions followed with questions enquiring the possible use of metal deposition to prevent re-aggregation of graphene and the degree of reduction of potassium. The ab initio and theoretical study presented by Juarez et al. generated interest and queries regarding the distribution of carboxylic acid groups on the zigzag edges of graphene. Leading to the discussion on the validity of the study since with HOPG, the deposition environment involves step edges – several layers in height – on a terrace, whilst the modelling considers only the edge of a single graphene layer and how the modelling can be improved to reflect this. The contribution of Varley et al. was also received with great interest and debates continued on the furnace treatment of nanodiamond surfaces to remove any graphitic sp2 carbon from the surface and to maximise the number of oxygen terminating groups and what the surfaces of the two different nanodiamonds (detonation and HPHT) looked like after the experimental treatment process along with the variation in density of the sp2 or “defect” sites between detonation diamond and mechanically ground HPHT. Further discussion built on the possible nature on interaction between the nanodiamond surface and the electrode and the effect of nanodiamond size on the current response and evaluation of the microelectrode effect by changing the mass loading of nanodiamonds.
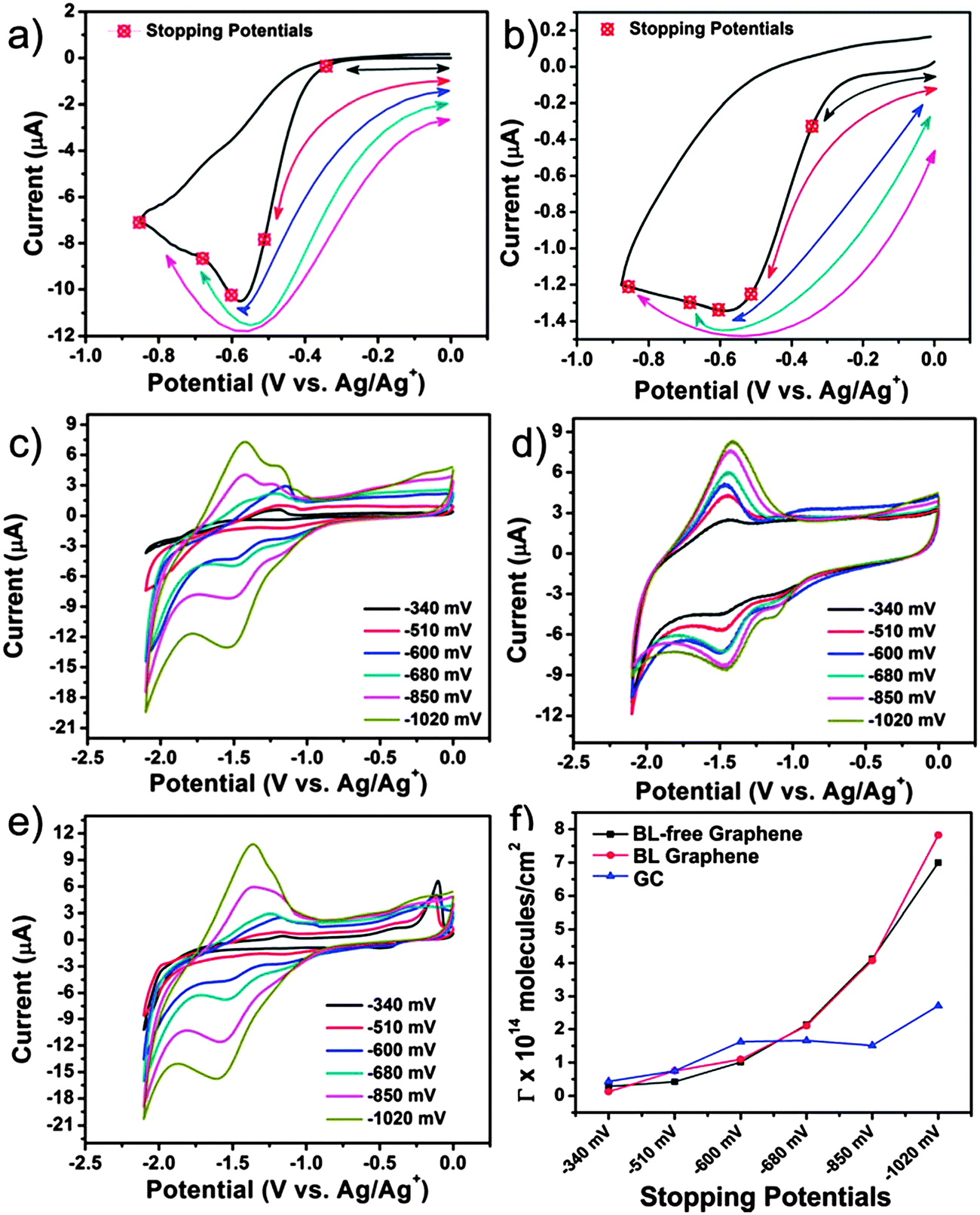 | ||
| Fig. 7 Stopping potentials overlaid on the voltammogram for covalent functionalization with BNPI on BL-free graphene on insulating SiC (a) and glassy carbon (b). A final stopping potential at −1.02 V was also performed for each carbon substrate (point not shown in graphs (a) and (b)). The reduction of the p-nitrophenyl moiety after grafting to BL graphene on insulating SiC (c), GC (d) and BL-free graphene on insulating SiC (e). (f) The grafting coverage estimated for each of the stopping potentials (ref. 23). | ||
After the closing of the sessions for the day, pre-drinks were followed by a generous conference dinner. The highlight of the conference dinner was the “Loving Cup Ceremony”, using the traditional silver cup, ably demonstrated by the Faraday Committee chair Professor Graham Hutchings and Dr Katherine Holt (Fig. 8).
 | ||
| Fig. 8 Loving Cup Ceremony at the Conference Dinner on Tuesday, 29th July, 2014. | ||
The final session of discussions began on Wednesday morning and focussed on the use of carbon interfaces in synthesis, sensing and electrocatalysis. Contributions covered a diverse range of topics, from the use of diamond interfaces for light-induced reduction reactions and electrochemical phenol–arene cross-coupling to fundamental aspects of electron transfer processes at carbon surfaces, with discussion of electrochemistry at individual single-walled carbon nanotubes (SWCNTs) and the effects of temperature on outer-sphere processes at boron-doped diamond (BDD) electrodes. There were also papers concerning carbon-based platforms allowing immobilisation of enzymes for a range of electrocatalytic and biosensing applications.
The session began with a paper by Hamers et al.28 (University of Wisconsin-Madison, USA) investigating the dynamics and reactivity of electrons emitted into water as a result of above-bandgap illumination of diamond thin films grown on niobium. The lifetime of the solvated electrons was monitored using transient absorption spectroscopy; it was found that the lifetime of electrons emitted from diamond films was greater than the lifetime of electrons emitted from the neat niobium surface, due to the absence of electronic states in the bandgap of diamond that are degenerate with the solvated electrons (as compared to the case of a metallic surface which has a continuum of empty states leading to facile recombination). An interesting consequence of the essential irreversibility of electron emission from diamond thin films is the possibility for new routes to high-energy reactions such as the reduction of N2 (Fig. 9), even if (as stated by Prof. Hamers) this approach is not likely to challenge the Haber–Bosch process! These results stimulated a discussion about the nature of the conduction band in water and how the position of this band affects the distance that an emitted electron can travel before it becomes solvated. The lifetime of photo-excited electrons injected into the conduction band of the diamond film was said to account for the dependence of solvated electron yield on film thickness, as the diffusion length is limited to a few hundred nanometres within the diamond film. Following on the theme of novel electrochemical reactivity at diamond, Waldvogel and co-workers29 (Johannes Gutenberg-Universität, Germany) described a green synthetic route to dibenzofurans via a direct oxidative phenol–arene cross-coupling reaction that avoided the need for organometallic catalysts or harsh reaction environments and using electrons as the sole ‘reagent’. Despite the low overall yields, the simplicity of this approach could make it an interesting synthetic alternative with further development, and consideration of effects such as surface termination (which is dependent upon electrode preparation and pre-treatment) and the heterogeneity of polycrystalline BDD as an electrode material.
 | ||
| Fig. 9 Photoemission of electrons may allow the use of diamond to induce new chemistry. Energy level diagram for a H-terminated diamond surface in contact with water, and reduction potentials of relevant reactions that may be initiated at the diamond surface (ref. 28). | ||
Boron-doped diamond has a very high thermal conductivity, greater than that of copper, and as such was the electrode material of choice for Newton and co-workers30 (University of Warwick, UK) in their study considering the effect of temperature on outer-sphere electron transfer processes. Temperature pulse voltammetric experiments were described, using a pulsed near-infrared diode laser to heat the back face of a freestanding BDD disc electrode. Temperature pulses were found to perturb the double layer, leading to the superposition of capacitive decay-like features onto voltammograms recorded in the presence of fast, outer-sphere redox mediators. The temperature profile induced in the vicinity of the electrode surface was calculated using a finite element model. Several interesting applications for the laser-heated electrodes were suggested during the discussion. Through use of a laser capable of accessing shorter timescales it should be possible to obtain information about the dynamics of reorganisation of the double layer at BDD, extending work at gold disc electrodes reported by Brennan et al.,31 or even to perform an electrochemical ‘freeze-quench’ experiment where the laser is used to melt a layer of electrolyte close to the electrode surface which could then become trapped in an unusual state upon re-freezing. The effect of different surface functionalities on surface-sensitive redox processes could also be examined.
In an interesting contribution from Güell et al.32 (University of Warwick, UK) the possibilities for studying individual SWNTs using scanning electrochemical cell microscopy in parallel with a range of electrochemical, morphological and spectroscopic techniques were discussed, using the example of serotonin oxidation. Serotonin oxidation is a multi-step process, and the authors showed that electrochemical activity was relatively uniform across the whole surface of the SWNTs. This implies that the electrochemical activity was not localised to defect sites, and provides further evidence that electron-transfer reactions do occur at the walls of pristine SWNTs. The spatial resolution of the scanning electrochemical cell microscopy technique was discussed, and Güell et al. pointed to their earlier work that demonstrated enhanced catalytic oxygen reduction at point defects located at kink sites on individual nanotubes.33
The final two papers of Faraday Discussion 172 focused on the use of enzymes adsorbed onto electrode surfaces. The contribution of Hu et al.34 (University of Oxford, UK) demonstrated the preparation of functioning glutamate biosensors utilising Pt nanoparticles and glutamate oxidase co-immobilised on high surface area BDD or reduced graphene oxide (rGO). During the discussion it was noted that the precise oxygen content of the rGO needed for the device to function was unclear; the electrochemical response of rGO is significantly impaired at oxygen contents greater than about 18%. Oxidation of glutamate by glutamate oxidase in the presence of dioxygen produces stoichiometric peroxide, and subsequent electro-oxidation of this peroxide at 0.5 V vs. Ag/AgCl was used to quantify the original glutamate concentration. The authors stated that the requirement for such a high potential impaired the selectivity of the sensor, but demonstrated that selectivity could be improved by capping the electrode surface with a protective layer of poly(phenylenediamine) in order to suppress diffusion of larger molecules to the electrode surface. The BDD and rGO-based composite biosensors were shown to display far greater long-term stability than the equivalent biosensor constructed on glassy carbon, and the limit of detection for glutamate was found to be comparable to that of similar devices in the literature (ca. 220–350 nM). The paper by Quinson et al.35 (University of Oxford, UK) also demonstrated the application of high surface area electrodes, in this case to determine the effect of carbon material properties on the electrocatalytic H2 oxidation activity of the electrode-immobilised enzyme hydrogenase 1 from E. coli. The authors noted that no significant change in structure of the enzyme was observed upon adsorption; Hu et al. also stated that the measured Michaelis constants of their glutamate oxidase were similar, regardless of whether the enzyme was in solution or adsorbed. The electrocatalytic behaviour of hydrogenase 1 was found to be very similar on the range of carbon materials studied, and high current densities were achieved without pre-treatment of the carbon surface. The fact that hydrogenase 1 performed well on a range of commercially available carbon materials that are already widely used in fuel cells is encouraging for the prospects of future ‘green’ devices based around existing technologies, especially given the recent report of a highly active and oxygen-tolerant hydrogenase by Matsumoto and co-workers, mentioned by Dr Lyth (Kyushu University) during the discussion.36
The concluding remarks were delivered in inimitable style by Patrick Unwin, University of Warwick, who drew an interesting analogy between the Faraday Discussion topic of ‘carbon in electrochemistry’ and Yorkshire, not least due to the highly varied nature of the ‘surface’ of the countryside surrounding Sheffield which mirrors the diversity of carbon interfaces available to electrochemists. The concluding remarks were followed by well-deserved acknowledgements to the hosts and organisers, before one final lunch brought a very successful and enjoyable Faraday Discussion to a close. The delegates went their separate ways with plenty to think about following an intense but stimulating three days of discussion.
Acknowledgements
P.A.A. would like to acknowledge financial support from the European Research Council (ERC, EnergyBioCatalysis-ERC-2010-StG-258600). B.D. was partially supported by the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center funded by the US Department of Energy, Office of Science, Office of Basic Energy Sciences.References
- http://www.rsc.org/ConferencesAndEvents/RSCConferences/FD/history.asp .
- B. Das, R. G. Pillai, Y. Wu and R. L. McCreery, Redox-Gated Three-Terminal Organic Memory Devices: Effect of Composition and Environment on Performance, ACS Appl. Mater. Interfaces, 2013, 5, 11052–11058 CAS.
- L. C. T. Shoute, Y. Wu and R. L. McCreery, Direct Spectroscopic Monitoring of Conductance Switching in Polythiophene Memory Devices, Electrochim. Acta, 2013, 110, 437–445 CrossRef CAS PubMed.
- S. Y. Sayed, A. Bayat, M. Kondratenko, Y. Leroux, P. Hapiot and R. L. McCreery, Bilayer Molecular Electronics: All-Carbon Electronic Junctions Containing Molecular Bilayers Made with “Click” Chemistry, J. Am. Chem. Soc., 2013, 135, 12972–12975 CrossRef CAS PubMed.
- A. J. Bergren, R. L. McCreery, S. R. Stoyanov, S. Gusarov and A. Kovalenko, J. Phys. Chem. C, 2010, 114, 15806–15815 CAS.
- N. L. Ritzert, W. Li, C. Tan, G. G. Rodríguez-Calero, J. Rodríguez-Lopez, K. Hernández-Burgos, S. Conte, J. J. Parks and H. D. Abruña, Faraday Discuss., 2014, 172, 27–45 CAS.
- C. Hérbert, E. Scorsone, A. Bendali, R. Kiran, M. Cottance, H. A. Girard, J. Degardin, E. Dubus, G. Lissorgues, L. Rousseau, S. Picaud and P. Bergonzo, Faraday Discuss., 2014, 172, 47–59 Search PubMed.
- V. Straub, A. Gallego, G. de la Torre, T. W. Chamberlain, A. N Khlobytsov, T. Torres and D. M. Guldi, Faraday Discuss., 2014, 172, 61–79 Search PubMed.
- N. Shirshova, H. Qian, M. Houllé, J. H. G. Steinke, A. R. J. Kucernak, Q. P. V. Fontana, E. S. Greenhalgh, A. Bismarcka and M. S. P. Shaffer, Faraday Discuss., 2014, 172, 81–103 CAS.
- H. V. Ijije, R. C. Lawrence, N. J. Siambun, S. M. Jeong, D. A. Jewell, D. Hu and G. Z. Chen, Faraday Discuss., 2014, 172, 105–116 CAS.
- J. Britto, N. E. A. Cousens, S. W. Coles, C. D. van Engers, V. Babenko, A. T. Murdock, A. Koós, S. Perkin and N. Grobert, Langmuir, 2014, 30(38), 11485–11492 CrossRef PubMed.
- B. Dyatkin and Y. Gogotsi, Faraday Discuss., 2014, 172, 139–162 CAS.
- S. Kondrat, N. Georgi, M. V. Fedorov and A. A. Kornyshev, Phys. Chem. Chem. Phys., 2011, 13, 11359–11366 RSC.
- J. Chmiola, S. Osswald and Y. Gogotsi, Carbon, 2012, 50, 4880–4886 CrossRef PubMed.
- E. Frackowiak, M. Meller, J. Menzel, D. Gastol and K. Fic, Faraday Discuss., 2014, 172, 179–198 CAS.
- Q. Abbas, P. Ratajczak and F. Béguin, Faraday Discuss., 2014, 172, 199–214 CAS.
- Y. Cao, S.-R. Cai, S.-C. Fan, W.-Q. Hu, M.-S. Zheng and Q.-F. Dong, Faraday Discuss., 2014, 172, 215–221 CAS.
- C. Sole, N. E. Drewett and L. J. Hardwick, Faraday Discuss., 2014, 172, 223–237 CAS.
- M. Inaba, H. Yoshida, Z. Ogumi, T. Abe, Y. Mizutani and M. Asano, J. Electrochem. Soc., 1995, 142, 20–26 CrossRef CAS PubMed.
- F. W. Richey, B. Dyatkin, Y. Gogotsi and Y. A. Elabd, J. Am. Chem. Soc., 2013, 135, 12818–12826 CrossRef CAS PubMed.
- K. J. Hüttinger, Carbon, 1990, 28, 453–456 CrossRef.
- H. V. Paten, M. Velicky, N. Clark, C. A. Muryn, I. A. Kinloch and R. A. W. Dryfe, Faraday Discuss., 2014, 172, 261–272 Search PubMed.
- K. J. Stevenson, P. A. Veneman, R. I. Gearba, K. M. Mueller, B. J. Holliday, T. Ohta and C. K. Chan, Faraday Discuss., 2014, 172, 273–291 CAS.
- M. M. Lounasvouri, M. Rosillo-Lopez, C. G. Salzmann, D. J. Caruana and K. B. Holt, Faraday Discuss., 2014, 172, 293–310 RSC.
- S. A. Hodge, H. H. Tay, D. B. Anthony, R. Menzel, D. J. Buckley, P. L. Cullen, N. T. Skipper, C. A. Howard and M. S. P. Shaffer, Faraday Discuss., 2014, 172, 311–325 CAS.
- M. F. Juarez, S. Fuentes, G. J. Soldano, L. Avalle and E. Santos, Faraday Discuss., 2014, 172, 327–347 CAS.
- T. S. Varley, M. Hirani, G. Harrison and K. B. Holt, Faraday Discuss., 2014, 172, 349–364 RSC.
- R. J. Hamers, J. A. Brandy, D. Zhu and L. Zhang, Faraday Discuss., 2014, 172, 397–411 CAS.
- B. Elsler, D. Schollmeyer and S. R. Waldvogel, Faraday Discuss., 2014, 172, 413–420 CAS.
- L. Meng, J. G. Iacobini, M. B. Joseph, J. V. Macpherson and M. E. Newton, Faraday Discuss., 2014, 172, 421–438 CAS.
- J. L. Brennan and R. J. Forster, J. Phys. Chem. B, 2003, 107, 9344–9350 CrossRef CAS.
- A. G. Güell, K. E. Meadows, P. V. Dudin, N. Ebejer, J. C. Beyers, J. V. Macpherson and P. R. Unwin, Faraday Discuss., 2014, 172, 439–455 Search PubMed.
- J. C. Beyers, A. G. Güell and P. R. Unwin, J. Am. Chem. Soc., 2014, 136, 11252–11255 CrossRef PubMed.
- J. Hu, S. Wisetsuwannaphum and J. S. Foord, Faraday Discuss., 2014, 172, 457–472 CAS.
- J. Quinson, R. Hidalgo, P. A. Ash, F. Dillon, N. Grobert and K. A. Vincent, Faraday Discuss., 2014, 172, 473–496 CAS.
- T. Matsumoto, S. Eguchi, H. Nakai, T. Hibino, K.-S. Yoon and S. Ogo, Angew. Chem., Int. Ed., 2014, 53, 8895–8898 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |