
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diferrocenyl tosyl hydrazone with an ultrastrong NH⋯Fe hydrogen bond as double click switch†
Christoph
Förster
,
Philipp
Veit
,
Vadim
Ksenofontov
and
Katja
Heinze
*
Institute of Inorganic Chemistry and Analytical Chemistry, Johannes Gutenberg-University of Mainz, Duesbergweg 10-14, 55128 Mainz, Germany. E-mail: katja.heinze@uni-mainz.de; Fax: +49-6131-39-27277; Tel: +49-6131-39-25886
First published on 11th December 2014
Abstract
The ultrastrong and short intramolecular NH⋯Fe hydrogen bond in diferrocenyl hydrazone 2 raises the barrier for intramolecular electron transfer in its mixed-valent cation 2+ and is only disrupted by double oxidation to 22+.
Tosyl hydrazones have found increasing applications in organic synthesis as safe reagents for diazo compound generation.1 The redoxactive diferrocenyl tosylhydrazone 2 had been previously employed for the in situ generation of the highly reactive and elusive diferrocenyl carbene.2 Some spectroscopic data of 2 have been reported before but no further interpretations were given.2aN,N-Dimethyl diferrocenyl hydrazone and diferrocenyl hydrazone have been reported previously by Bildstein, but no unusual properties have been reported either.32 is prepared straightforwardly from the diferrocenyl ketone 1 and p-toluenesulfonyl hydrazide (Scheme 1). Initially, we were mainly interested in the redox properties of 2.
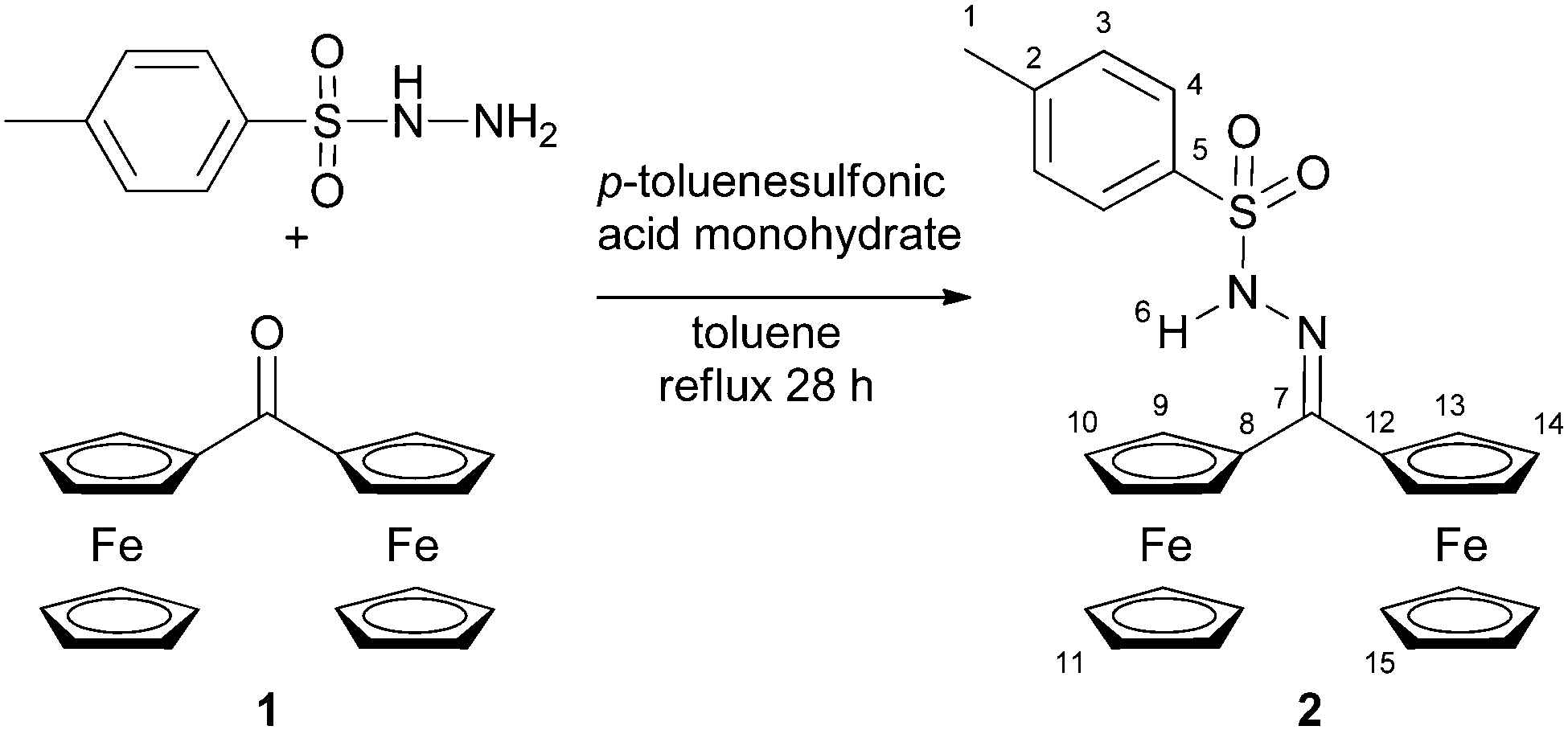 | ||
| Scheme 1 Synthesis of diferrocenyl tosylhydrazone 2 from diferrocenyl ketone 1.4 | ||
However, we were puzzled by some unexpected spectroscopic data of 2. The absorption band for the NH stretching vibration of 2 is found at 3101 cm−1 in the solid state, significantly lower than that of the analogous diphenyl tosylhydrazone 3 (3219 cm−1; with intermolecular NH⋯OS hydrogen bonds in the solid state;5 ESI†). The NH stretch of 2 is clearly identified by HD exchange and the appearance of the ND stretching absorption at 2317 cm−1 (ESI†). Similarly, in CD2Cl2 solution this band of 2 occurs at 3118 cm−1 while the corresponding band of 3 is detected at significantly higher energies (3267 cm−1 in CD2Cl2). These data clearly advocate intramolecular interactions in 2 but not in 3. Similarly, the resonance of the NH6 proton of 2 (δ = 10.45 ppm) appears at significantly lower field than that of 3 (δ = 7.35 ppm) in CDCl3.
Especially intriguing is the NOE contact NH6⋯Cp(H11) (Scheme 1 and Fig. 1) as the NH group then necessarily points towards a C5H5 ring and hence towards an iron(II) centre. Indeed, DFT calculations suggest that the conformer with an intramolecular NH⋯Fe hydrogen bond (Fe⋯N distance 3.55 Å; Fe⋯H distance 2.67 Å) is more stable by 10 kJ mol−1 with respect to a non-hydrogen bonded conformer. The conceivable zwitterionic species with protonated iron and deprotonated hydrazone is higher in energy by more than 160 kJ mol−1. Hence full proton transfer from the hydrazone to ferrocene is thermodynamically unfeasible. Furthermore, protonation at iron should yield a high-field proton resonance around δ = −2 ppm6 which is not observed for 2.
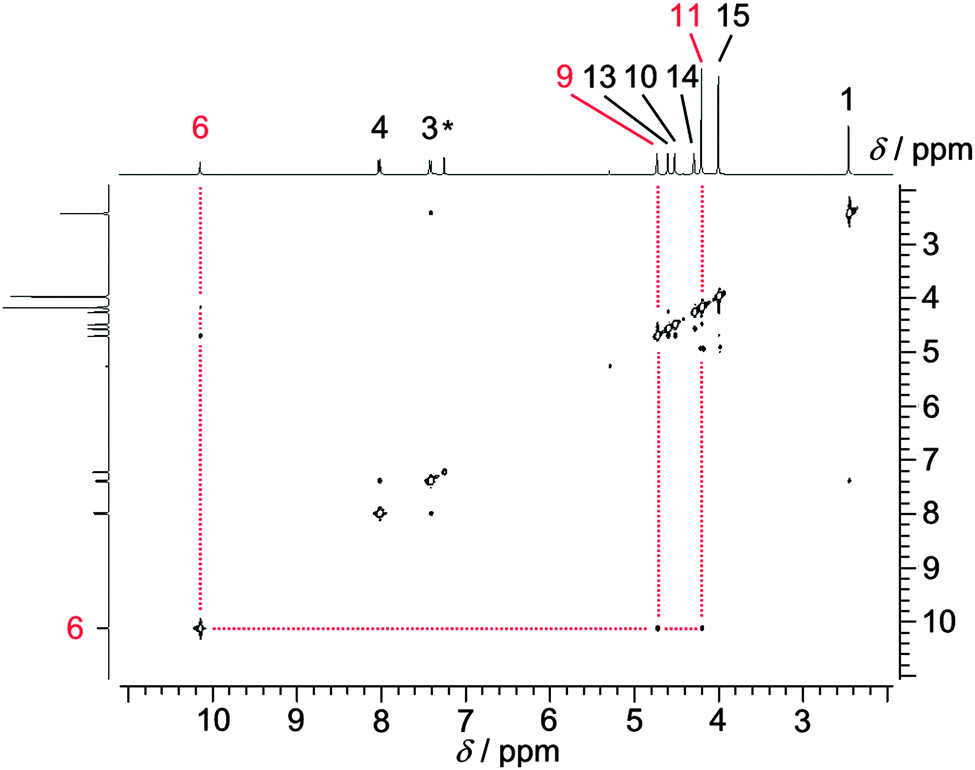 | ||
| Fig. 1 NOE spectrum of 2 in CDCl3. Relevant correlations involving H6 are indicated by red dotted lines (the asterisk denotes a solvent resonance). | ||
Final proof of the suspected hydrogen bond also in the solid state is provided by an XRD analysis of a single crystal of 2 (Fig. 2). Indeed, the NH vector points to the iron(II) centre Fe2 of one ferrocenyl substituent forming a six-membered ring Fe2–C12–C11–N1–N2–H2N with an Fe2⋯N2 distance of 3.46 Å (Fig. 2). The six-membered ring has already been shown to promote the strongest intramolecular OH⋯Fe bond in ferrocenyl alcohols.7 To the best of our knowledge the N⋯Fe distance in 2 is among the shortest hydrogen bond donor to iron distances in iron complexes observed so far.8,9 Furthermore, 2 features the first NH⋯Fe hydrogen bond with a six-membered ring as the only three previously reported NH⋯Fe hydrogen bonds comprise five-membered rings.8g–i The Cp rings of the hydrogen bonded ferrocenyl substituent are tilted by 6.2° accommodating the NH group. The Cp rings of the other ferrocenyl substituent are essentially coplanar. Both ferrocenyl moieties are syn oriented with respect to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–N hydrazone plane.
N–N hydrazone plane.
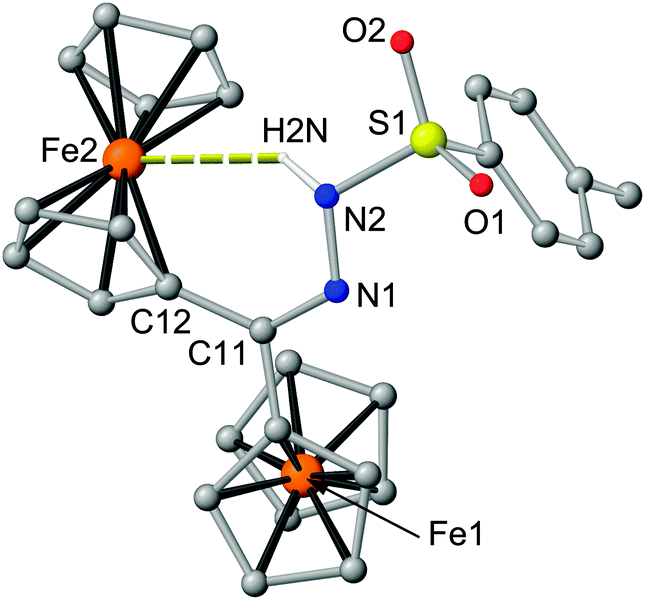 | ||
| Fig. 2 Molecular structure of 2 from single crystal XRD (CH hydrogen atoms omitted). | ||
The different symmetry and coordination of the ferrocene sites is not reflected in the Mößbauer parameters and only a sharp doublet is obtained (δ = 0.4484/0.5273 mm s−1; ΔEQ = 2.300/2.3080 mm s−1 both at 293 K and 90 K, respectively; ESI†) which is rather common in substituted ferrocene derivatives.
From the 1H NMR and IR data it is obvious that the hydrogen bond persists in chloroform and dichloromethane solution. Moreover, in THF solution the NH vibration of 2 remains essentially unaffected (ESI†). This is in stark contrast to 3 which forms strong NH⋯O hydrogen bonds to THF (Δν ≈ 200 cm−1, ESI†). Similarly, in DMSO the NH proton resonance of 3 is shifted to lower field (Δδ ≈ 3 ppm) while that of 2 in DMSO remains essentially unaffected (ESI†). Hence, the NH⋯Fe hydrogen bond of 2 is even resistant towards THF and DMSO as strong hydrogen bond acceptors. Remarkably, the strongest intramolecular OH⋯Fe bond of ferrocenyl alcohols (namely 2-ferrocenyl ethanol) is fully disrupted already by the weak hydrogen bond acceptor diethylether.7a Enthalpies of XH⋯Y hydrogen bonds have been empirically correlated to Δν of the respective XH vibration.‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10 As the free NH stretch is unavailable for 2 we estimate it from that of 3 in CD2Cl2 (3267 cm−1). Hence, Δν ≈ 149 cm−1 and −ΔH ≈ 13 kJ mol−1, a reasonable value in comparison to the thermodynamic data obtained from DFT calculations (see above). The exceptional resistance of the intramolecular NH⋯Fe hydrogen bond towards splitting by THF or DMSO yielding an intermolecular hydrogen bond is believed to be based both on enthalpic and entropic effects (chelate effect).
10 As the free NH stretch is unavailable for 2 we estimate it from that of 3 in CD2Cl2 (3267 cm−1). Hence, Δν ≈ 149 cm−1 and −ΔH ≈ 13 kJ mol−1, a reasonable value in comparison to the thermodynamic data obtained from DFT calculations (see above). The exceptional resistance of the intramolecular NH⋯Fe hydrogen bond towards splitting by THF or DMSO yielding an intermolecular hydrogen bond is believed to be based both on enthalpic and entropic effects (chelate effect).
Due to the hindered rotation around the CN double bond and the NH⋯Fe hydrogen bond the two sites in the diferrocene 2 are chemically different. No coalescence of ferrocenyl proton resonances and no significant shift of the NH6 resonance are found up to 70 °C in C6D6 (400 MHz) suggesting a high activation barrier for the rotation around the CN double bond and a persistent NH⋯Fe hydrogen bond (ESI†).
Diferrocenes linked by a single atom bridge have found considerable interest due to the intramolecular electron transfer within the corresponding mixed-valent ferrocene–ferrocenium systems. Hence, the electron transfer between the slightly different redox sites with/without an NH⋯Fe hydrogen bond within 2+ was probed. Expectedly, 2 is reversibly oxidized to 2+ and 22+ (at 115 and 595 mV vs. ferrocene, respectively; ESI†). The difference between the potentials for 2+ (ΔE = 480 mV) is larger than the potential difference of the more symmetric ketone 1+ (ΔE = 345 mV; ESI†). First it is mandatory to establish the site of primary oxidation. Paramagnetic 1H NMR spectroscopy was used to identify the localization of the ferrocenium site in 2+.11–15 Upon titration of 2 with substoichiometric amounts of iodine only resonances of protons H13, H14 and H15 (Scheme 1, ESI†) are affected suggesting spin density at the non-hydrogen bonded iron site. This is fully confirmed by DFT calculations on 2+ showing spin density at the non-hydrogen bonded iron site (Fe⋯N distance 3.54 Å; Fe⋯H distance 2.66 Å; Fig. 3). The non-hydrogen bonded conformer of 2+ is calculated 4 kJ mol−1 higher in energy (Fig. 3, ESI†). Upon further oxidation (0.4 eq. iodine) the NH6 proton resonance is broadened and shifted to higher field (ESI†) suggesting the onset of appreciable disproportionation of 2+ into 2 and 22+. DFT calculations of 22+ suggest that the hydrogen bonded conformer is now destabilized by 6 kJ mol−1 (Fig. 3, ESI†). An opening of hydrogen bonds due to accumulation of positive charges has been previously established in oligoferrocenyl peptides featuring conventional NH⋯O hydrogen bonds in the ligand domain.12,13 Similarly, an anionic [1.1]diborataferrocenophane has been shown to bind Li+ between the iron centres, while oxidation of the ferrocenes releases the entrapped Li+ ion.16
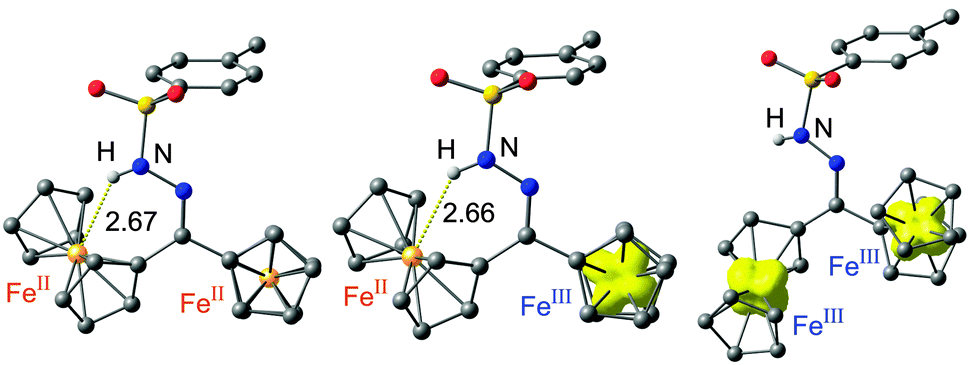 | ||
| Fig. 3 DFT [B3LYP/LANL2DZ (Fe), 6-31G* (C, H, N, O, S)] calculated structures and spin densities of 2, 2+ and 22+ (CH hydrogen atoms omitted, Fe⋯H distances in Å, isosurface contour value 0.01 a.u.). | ||
Indeed, oxidation of 2 with one equivalent AgSbF6 in CD2Cl2 (E1/2 = 650 mV17) to 2+ leaves the energy of the NH vibration essentially unperturbed (ESI†). Oxidation of 2 to 22+ with two equivalents AgSbF6 in CD2Cl2 yields a new band at 3274 cm−1 (ESI†). This energy is very similar to that of 3 in CD2Cl2 without a hydrogen bond (vide supra). This experimental observation strongly confirms the opening of the NH⋯Fe hydrogen bond in the dication 22+ but not in the monocation 2+ as suggested by the DFT calculations.
According to time-dependent DFT calculations the intervalence charge transfer band (IVCT)18 of 2+ is calculated at 718 nm originating from the charge transfer between the δ orbitals of the different ferrocene and ferrocenium sites (ESI†). For the similar mixed-valent ketone 1+ lacking the hydrogen bond this band is calculated at significantly lower energy (1147 nm, ESI†). The latter is a typical value for symmetric mixed-valent ferrocene–ferrocenium complexes with short bridges (Robin-Day class II)18 while the former is quite high in energy. The NH⋯Fe hydrogen bond lowers the ferrocene δ orbitals in 2+ and hence significantly increases the energy required for Fc(⋯HN) → Fc+ intervalence charge transfer. Possibly, electron transfer within 2+ approaches the Robin-Day class I regime.18
Experimentally, a weak near-infrared band is observed for 1+ at ≈1244 nm (IVCT) while no near-infrared band is discernible for 2+ (ESI†). However, the UV/Vis spectrum of 2+ is not exactly the average spectrum of 2 and 22+ in the visible region and an additional band can be suspected from the difference spectrum at around ≈665 nm which might be assignable to a high energy IVCT (ESI†). With this interpretation of the absorption bands the activation barriers for the thermal electron transfer18b within the mixed-valent cations 1+ and 2+ are estimated from ΔG≠ET = λ/4 + ΔG0/2 + (ΔG0)2/(4(λ − 2HAB)) − HAB + HAB2/(λ + ΔG0) with λ = Eop − ΔG0 as ΔG≠ET = 22 ± 2 and 46 ± 2 kJ mol−1, respectively (assuming HAB ≤ 300 cm−1 (ref. 13, 14 and 19)).§ Hence, the intramolecular electron transfer barrier in 2+ is substantially higher than that in 1+.
In summary, the diferrocenyl hydrazone 2 features an ultrastrong intramolecular NH⋯Fe hydrogen bond. To the best of our knowledge, this NH⋯Fe hydrogen bond is one of the strongest reported so far for ferrocenyl hydrogen bond acceptors. This hydrogen bond confines charge and spin in the ground state of the mixed-valent cation 2+ to the non-hydrogen bonded site. Electron transfer between ferrocene and ferrocenium in 2+ requires quite high energies due to the electronic dissymmetry. Upon double oxidation of 2 to 22+ the NH⋯Fe hydrogen bond is fully disrupted. Hence, the redox sequence 2 → 2+ → 22+ constitutes a switch with a structural response at the NH⋯Fe hydrogen bond after two redox events. The exceptional hydrogen bond strength allows accessing and exploiting the NH⋯Fe hydrogen bond in terms of modulating (intramolecular) electron transfer (2+) and vice versa in terms of modulating NH⋯Fe hydrogen bonding by (intermolecular) electron transfer (2/22+) resulting in a redox-stimulated switch.
Notes and references
-
(a) J. R. Fulton, V. K. Aggarwal and J. de Vicente, Eur. J. Org. Chem., 2005, 1479–1492 CrossRef CAS
; (b) Q. Xiao, Y. Zhang and J. Wang, Acc. Chem. Res., 2013, 46, 236–247 CrossRef CAS PubMed
-
(a) K.-Y. Kay, L. H. Kim and I. C. Oh, Tetrahedron Lett., 2000, 41, 1397–1400 CrossRef CAS
- B. Bildstein and P. Denifl, Synthesis, 1994, 158–159 CrossRef CAS
-
(a) M. D. Rausch, E. O. Fischer and H. Grubert, J. Am. Chem. Soc., 1960, 82, 76–82 CrossRef CAS
- S. Roy and A. Nangia, Cryst. Growth Des., 2007, 7, 2047–2058 CAS
-
(a) T. J. Curphey, J. O. Santer, M. Rosenblum and J. H. Richards, J. Am. Chem. Soc., 1960, 82, 5249–5250 CrossRef CAS
-
(a) D. S. Trifan and R. Bacskai, J. Am. Chem. Soc., 1960, 82, 5010–5011 CrossRef CAS
- The CSD was searched (ConQuest) to extract data reported in this paper (http://www.ccdc.cam.ac.uk) on October 29th 2014. References reporting iron complexes with short X⋯Fe distances (2–4 Å; X = element of groups 15–17) in XH⋯Fe hydrogen bonds (O⋯Fe 3.353–3.480 Å, seven examples; N⋯Fe 3.433–3.585 Å, three examples):
(a) H. Schottenberger, M. Buchmeiser, C. Rieker, P. Jaitner and K. Wurst, J. Organomet. Chem., 1997, 541, 249–260 CrossRef CAS
-
(a) L. Brammer, Dalton Trans., 2003, 3145–3157 RSC
-
(a) Y. S. Shubina and L. M. Epstein, J. Mol. Struct., 1992, 265, 367–384 CrossRef
- T.-Y. Dong, M.-Y. Hwang, T.-L. Hsu, C.-C. Schei and S.-K. Yeh, Inorg. Chem., 1990, 29, 80–84 CrossRef CAS
- D. Siebler, M. Linseis, T. Gasi, L. M. Carrella, R. F. Winter, C. Förster and K. Heinze, Chem. – Eur. J., 2011, 17, 4540–4551 CrossRef CAS PubMed
- D. Siebler, C. Förster and K. Heinze, Dalton Trans., 2011, 40, 3558–3575 RSC
- D. Siebler, C. Förster, T. Gasi and K. Heinze, Organometallics, 2011, 30, 313–327 CrossRef CAS
- K. Hüttinger, C. Förster and K. Heinze, Chem. Commun., 2014, 50, 4285–4288 RSC
- M. Scheibitz, R. F. Winter, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., 2003, 115, 954–957 (
Angew. Chem., Int. Ed.
, 2003
, 42
, 924–927
) CrossRef
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed
-
(a) M. B. Robin and P. Day, Adv. Inorg. Chem. Radiochem., 1967, 10, 247–422 CrossRef CAS
- T. Kienz, C. Förster and K. Heinze, Organometallics, 2014, 33, 4803–4812 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, spectral and X-ray diffraction details, DFT calculations. CCDC 1031559. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4cc08868a |
| ‡ The empirical correlation of hydrogen bond enthalpies and XH wavenumber shifts is as follows: −ΔH = 18 Δν/(Δν + 720); ΔH in kcal mol−1; Δν in cm−1.10 |
| § For the symmetric mixed-valence isomers of 1+ ΔG0 = 0. For the mixed-valence isomers of 2+ ΔG0 has been estimated from the higher relative FeII/III redox potential of the 2+/22+ couple (potential difference ΔE(2+) = 480 mV) featuring the hydrogen bond with respect to the 1+/12+ couple (potential difference ΔE(1+) = 345 mV) lacking the hydrogen bond. Hence, the hydrogen bonded FeII centre is more difficult to oxidise to FeIII than a non-hydrogen bonded FeII centre by ΔE(2+–1+) = ΔE(2+) − ΔE(1+) = 135 mV. Everything else being equal this value approximately accounts for the difference in ΔG0 for 1+ (ΔG0 = 0) and 2+ (ΔG0 = 0.135 eV). |
| This journal is © The Royal Society of Chemistry 2015 |