
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bis(trimethylsilyl)phosphide chemistry: a half-century of advances across the periodic table
Jack
Baldwin
and
David P.
Mills
 *
*
Department of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.mills@manchester.ac.uk
First published on 12th March 2025
Abstract
Whilst bis(trimethylsilyl)amide has been used extensively as a ligand across the periodic table, the chemistry of its heavier group 15 congeners is relatively underdeveloped. However, bis(trimethylsilyl)phosphide coordination chemistry has provided unique structural motifs and has also shown potential applications in catalysis, materials science, and bioinorganic chemistry. This review, which marks 55 years since the first report of a bis(trimethylsilyl)phosphide complex, provides a comprehensive overview of the synthesis, characterisation and reactivity of structurally authenticated s-, p-, d- and f-block metal complexes of this ligand, focusing on salient single crystal XRD and NMR spectroscopic data. We discuss the factors influencing the diverse coordination modes and reactivity profiles of bis(trimethylsilyl)phosphide complexes, together with an overview of their potential as precursors for novel solid-state materials, aiming to inspire future research endeavours using this ligand. We also review the small number of bis(triisopropylsilyl)phosphide complexes, in order to provide motivation for the future study of other bis(silyl)phosphide ligands.
 Jack Baldwin | Jack Baldwin is from Beverley, East Yorkshire, in the UK. He completed his MChem (2019) at Newcastle University and his PhD (2024) at The University of Manchester, where he also carried out postdoctoral work. His research is centred around the synthesis and characterisation of low coordinate f-block metal complexes with silylphosphide ligands. |
 David P. Mills | David P. Mills is from Caerphilly, South Wales, in the UK. He did his MChem (2004) and PhD (2008) degrees at Cardiff University, and postdoctoral work at the University at Nottingham. He moved to the University of Manchester in 2012 as a lecturer, and was promoted to Professor of Inorganic Chemistry in 2021. His research group focuses on the synthesis of metal complexes with unusual bonding motifs and electronic structures, predominantly those containing f-block metals. |
1. Introduction
Bis(trimethylsilyl)amide, {N(SiMe3)2} (N′′), and derivatives thereof, have been used extensively across the periodic table to provide landmark complexes1–7 since the first s-block examples were reported by Wannagat in 1961.8 The popularity of N′′ in coordination chemistry can be attributed to a combination of: (i) kinetic protection of the metal coordination sphere and inter-ligand dispersion force stabilisation provided by sterically demanding SiMe3 groups; (ii) the absence of potential β-hydride elimination decomposition pathways; (iii) the high lipophilicity of SiMe3 groups increasing complex solubility in non-polar solvents and facilitating crystal growth for structure determination; (iv) negative hyperconjugation by the silyl groups making the ligand charge more diffuse, resulting in softer N-donor atoms and promoting additional stabilising M⋯Si–C and M⋯C–H interactions; (v) NMR spectra that are typically easy to interpret; and, (vi) the ease of preparation and commercial availability of s-block ligand transfer agents.1–7The coordination chemistry of bis(trimethylsilyl)phosphide, {P(SiMe3)2} (P′′), is immature in comparison to that of the lighter congener N′′, despite it having many of the same advantages. This is evident from a survey of the Cambridge Structural Database (CSD), which shows 210 entries for P′′ vs. 4062 entries for N′′ structurally authenticated metal complexes (non-metals and the metalloids B, Si, As, Sb and Te were excluded from this search and are outside the scope of this review); the 210 P′′ metal complexes consist of 94 p-, 64 d-, 30 s- and 22 f-block examples (Fig. 1).9 This can be ascribed to difficulties in handling s-block P′′ salts, which are pyrophoric, malodorous and toxic, and the synthesis of these ligand transfer agents typically using P(SiMe3)3 as a starting material (Scheme 1).10–13 As well as having similar handling concerns to P′′, P(SiMe3)3 is relatively expensive, and thus is typically synthesised from red phosphorus, sodium and chlorotrimethylsilane on large scales in dimethoxyethane (DME) (Scheme 1); this synthetic procedure can discourage investigations as it is inherently hazardous.14 However, an advantage of P′′ over N′′ is that the 100% abundant I = ½ metal-bound 31P nuclei can provide a useful spectroscopic handle, e.g. by using NMR or EPR spectroscopy. P′′ complexes could also potentially deliver unique applications as P′′ is softer than N′′ and has a higher propensity to bridge metal ions as M–P bonds are longer than M–N bonds.15–17 Since the first reported syntheses of s-block P′′ complexes18 they have been used in a plethora of reactions, ranging from nucleophilic additions to radical-mediated processes;19 they have also been shown to be versatile reducing agents,20 and a precursor to phosphorus-centred radicals.19
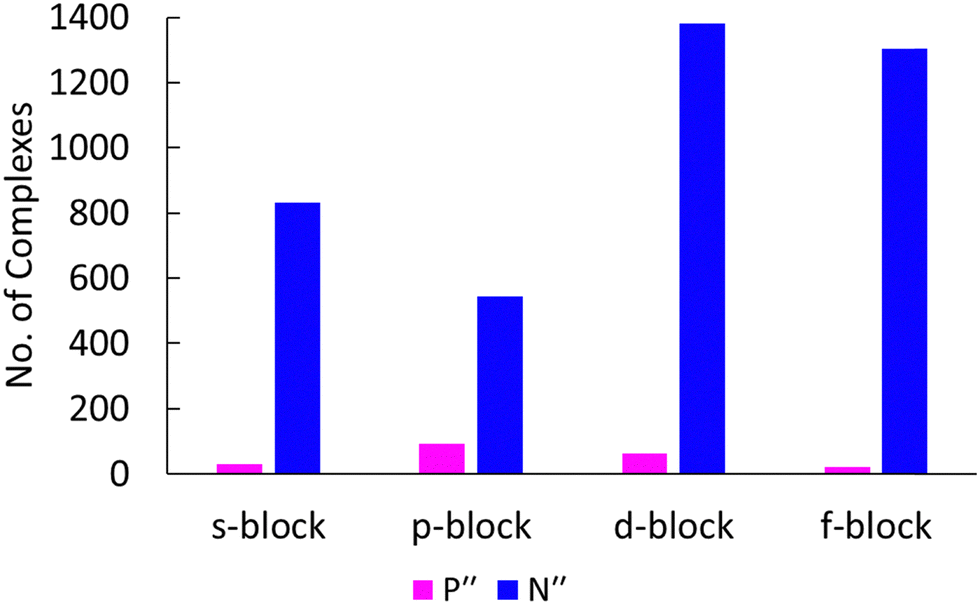 | ||
| Fig. 1 Graph depicting the number of structurally authenticated N′′ vs. P′′ metal complexes containing elements from each block of the periodic table.9 | ||
 | ||
| Scheme 1 Synthesis of KP′′ from red phosphorus, sodium and trimethylsilylchloride.10–13 | ||
Here we provide a review of P′′ coordination chemistry, focusing on structurally authenticated examples of complexes containing direct M–P bonds. When appropriate we make comparisons to complexes of the lighter congener, N′′; previous reviews of metal phosphido chemistry have tended to cover a wider range of ligand substituents and a narrower range of elements.21–23 The review is divided into sections of s-, p-, d-, and f-block metal P′′ complexes; these sections are subdivided by group number, with heterometallic ‘ate’ complexes containing s-block elements included in the p-, d- or f-block sections as appropriate. Where M–P bond lengths from single crystal XRD studies and chemical shifts and coupling constants from 31P and 29Si (4.7% abundant, I = ½) NMR spectroscopic data have been reported they are compiled in Table 1 (calculation of the standard error of the mean (SEM) = SD/√N, where SD = standard deviation and N = sample size). We focus on discussions of these data in this review, which we intend to provide a useful resource to the community and to inspire future research in P′′ coordination chemistry. We also provide coverage of nascent bis(triisopropylsilyl)phosphide chemistry, in order to show that other bis(silyl)phosphide ligands should provide new avenues to explore in future.
| Complex | Mean M–P (Å) | Solvent | 31P Chemical shift (δ) | 31P Coupling constant (Hz) | 29Si Chemical shift (δ) | 29Si Coupling constant (Hz) | Ref. |
|---|---|---|---|---|---|---|---|
| a Refers to stile. b Refers to rung bonds of ladder complexes. c Refers to bridging. d Refers to terminal P′′ ligands. e Refers to syn-isomers. f Refers to anti-isomers. g Refers to cis-isomers. h Refers to trans-isomers. i Refers to the anti-conformation. j Refers to the gauche-conformation. | |||||||
| s-Block | 2.62(2) | d 8-Toluene | −297.6, s | — | — | — | 24 |
| 1 [Li(μ-P′′)(THF)2]2 | |||||||
| 2 [Li(μ-P′′)(DME)]2 | 2.559(4) | — | — | — | — | — | 25 |
| 3 [Li4(μ2-P′′)2(μ3-P′′)2(THF)2] | 2.50(4)a | d 8-Toluene | −297.7, s | — | — | — | 24 |
| 2.56(4)b | |||||||
| 4 [Li6(μ2-P′′)2(μ3-P′′)4] | 2.51(1)a | — | — | — | — | — | 26 |
| 2.517(16)b | |||||||
| 5-K [K4(μ2-P′′)2(μ3-P′′)2]∞ | 3.4168(11)a | THF | −293.4, s | — | 0.70, s | 1 J PSi = 54 | 27 |
| 3.171(6)b | |||||||
| 5-Rb [Rb4(μ2-P′′)2(μ3-P′′)2]∞ | 3.485(2)a | THF | −287.1, s | — | 0.41, s | 1 J PSi = 55 | 27 |
| 3.4157(12)b | |||||||
| 5-Cs [Cs4(μ2-P′′)2(μ3-P′′)2]∞ | 3.656(2)a | THF | −270.0, s | — | 0.50, s | 1 J PSi = 52 | 27 |
| 3.5816(12)b | |||||||
| 6 [Cs(μ-P′′)(μ-1,4-dioxane)3(1,4-dioxane)]∞ | 3.6137(17) | d 8-THF | −276.1, s | — | — | — | 28 |
| 7 [Mg(μ-P′′)(Br)(THF)]2 | 2.5624(16) | d 6-Benzene | −296.9, br | — | 2.9, br | — | 20 |
| 8 [Mg3(μ-P′′)4(P′′)2] | 2.594(7)c | d 8-Toluene | −242.55, tc | 2 J PP = 17.9 | 3.74, s | — | 29 |
| 2.455(4)d | −275.44, td | ||||||
| 9 [Mg{P′′C(Ph)C(C2Ph)}(μ-P′′)]2 | 2.5576(19)c | d 8-Toluene | −262.29, tc | 2 J PP = 5 | 4.56, sc | 1 J PSi = <2c | 30 |
| 2.6998(17) | −98.4, t | 3.63, d | 1 J PSi = 6.3 | ||||
| 10 [Mg(P′′)2(DME)] | 2.487(2) | — | — | — | — | — | 31 |
| 11 [Mg(P′′)2(THF)2] | 2.5023(19) | d 8-Toluene | −294.7, s | — | 1.81, s | 1 J PSi = 33.0 | 32 |
| 12-Ca [Ca(N′′)(μ-P′′)(sol)]2 | 2.9269(18) | d 6-Benzene | −229.96, se | — | 5.42, de | 1 J PSi = 18.7e | 33 |
| −254.82, sf | — | −1.77, df | 1 J PSi = 22.0f | ||||
| 12-Ba [Ba(N′′)(μ-P′′)(sol)]2 | 3.289(2) | d 8-THF | −222.05, s | — | 1.03, s | 1 J PSi = 34.8 | 34 |
| 13-Ca trans-[Ca(P′′)2(THF)4] | 2.917(4) | — | — | — | — | — | 33 |
| 13-Sr trans-[Sr(P′′)2(THF)4] | 3.021(10) | d 8-Toluene | −256.2, s | — | 1.11, s | 1 J PSi = 34.1 | 35 |
| 13-Ba trans-[Ba(P′′)2(THF)4] | 3.175(10) | d 8-THF | −251.0, s | 2 J PP = 23.5 | 1.08, s | 1 J PSi = 44.5 | 36 |
| 3 J PSi = 1.0 | |||||||
| 14 [Sr(P′′)(μ-P′′)3Sr(THF)3] | 3.118(8)c | d 8-Toluene | −276.78, sd | 2 J PP = 22.5d | 1.60, s | 1 J PSi = 38.4d | 35 |
| 3.042(4)d | |||||||
| 15 [Ca(P′′)2(TMTA)2] | 2.995(2) | d 8-THF | −276.8, sd | 2 J PP = 14.5 | 0.87, s | 1 J PSi = 34.4 | 36 |
| 3 J PSi = −2.7 | |||||||
| p-Block | |||||||
| 16 [Al(P′′)(tmp)2] | 2.361(2) | d 6-Benzene | −238.00, s | — | — | — | 37 |
| 17-AlMe [Al(Me)2(P′′)(dmap)] | 2.3768(9) | d 6-Benzene | — | — | — | — | 38 |
| 17-GaMe [Ga(Me)2(P′′)(dmap)] | 2.372(1) | d 8-Toluene | −273.80, s | — | — | — | 39 |
| 17-GatBu [Ga(tBu)2(P′′)(dmap)] | 2.3948(6) | d 6-Benzene | −281.00, s | — | — | — | 40 |
| 18 [Ga(P′′)I(tBu-DAB)] | 2.2991(11) | — | — | — | — | — | 41 |
| 19 [Ga(P′′)2(tBu-DAB)] | 2.3426(18) | — | — | — | — | — | 41 |
| 20-Ga [Ga(DippNacnac)(P′′)(Cl)] | 2.3310(9) | d 6-Benzene | −255.00, s | — | — | — | 42 |
| 20-In [In(DippNacnac)(P′′)(Cl)] | 2.4806(8) | d 6-Benzene | −252.90, s | — | — | — | 42 |
| 21-AlMe [Al(Me)2(μ-P′′)]2 | 2.457(2) | d 8-Toluene | −226.7, s | — | 6.24, Vir. t | 1 J PSi = 9.9 | 43 and 44 |
| 21-AlEt [Al(Et)2(μ-P′′)]2 | 2.458(1) | d 6-Benzene | −246.90, s | — | — | — | 45 |
| 21-AlCH2iPr [Al(CH2iPr)2(μ-P′′)]2 | 2.476(2) | d 6-Benzene | −245.30, s | — | — | — | 43 |
| 21-AlCH2SiMe3 [Al(CH2SiMe3)2(μ-P′′)]2 | 2.483(1) | d 8-Toluene | −231.49, s | — | 6.38, s | 1 J PSi = 6.2 | 46 |
| 21-GaMe [Ga(Me)2(μ-P′′)]2 | 2.450(1) | d 6-Benzene | −219.20, s | — | 6.41, s | — | 40 and 47 |
| 21-GaEt [Ga(Et)2(μ-P′′)]2 | 2.4558(7) | d 8-Toluene | −227.80, s | — | — | — | 48 |
| 21-GanBu [Ga(nBu)2(μ-P′′)]2 | 2.4533(6) | d 6-Benzene | −227.00, s | — | — | — | 49 |
| 21-GaCH2tBu [Ga(CH2tBu)2(μ-P′′)]2 | 2.517(3) | d 6-Benzene | −215.24, s | — | — | — | 50 |
| 21-GaCH2SiMe3 [Ga(CH2SiMe3)2(μ-P′′)]2 | 2.4887(16) | d 6-Benzene | −205.88, s | — | — | — | 51 |
| 21-InMe [In(Me)2(μ-P′′)]2 | 2.630(1) | d 8-Toluene | −239.80, s | — | — | — | 52 |
| 21-InEt [In(Et)2(μ-P′′)]2 | 2.645(1) | d 6-Benzene | −242.90, s | — | — | — | 53 |
| 21-InPh [In(Ph)2(μ-P′′)]2 | 2.612(1) | d 6-Benzene | −221.59, s | — | — | — | 54 |
| 21-InCH2Ph [In(CH2Ph)2(μ-P′′)]2 | 2.6123(6) | d 2-DCM | −220.30, s | — | — | — | 55 |
| 21-InCH2SiMe3 [In(CH2SiMe3)2(μ-P′′)]2 | 2.655(3) | d 6-Benzene | −227.36, s | — | — | — | 56 |
| 21-TlMe [Tl(Me)2(μ-P′′)]2 | 2.692(3) | d 6-Benzene | −234.00, s | 1 J TlP = 2462 | — | — | 57 |
| 22 [Al(Me)2(dmap)(μ-P′′)Ga(Me)3] | 2.416(1) | d 8-Toluene | −262.40, s | — | — | — | 58 |
| 23-CH2SiMe3 [In(Me)(CH2SiMe3)(μ-P′′)]2 | 2.635(2) | d 6-Benzene | −234.32, sg | — | — | — | 59 |
| −234.67, sh | |||||||
| 23-CH2tBu [In(Me)(CH2tBu)(μ-P′′)]2 | 2.637(3) | d 6-Benzene | −239.56, sg | — | — | — | 60 |
| −239.42, sh | |||||||
| 24 [{In(CH2tBu)2}2{μ-P′′}{μ-PH(SiMe3)}] | 2.650(5) | d 6-Benzene | −209.92, s | 1 J PH = 473 | — | — | 60 |
| −209.12, s | |||||||
| 25-AlBrCH2SiMe3 [Al(CH2SiMe3)(Br)(μ-P′′)]2 | 2.436(4) | d 6-Benzene | −215.15, s | — | — | — | 61 |
| 25-GaClMe [Ga(Me)(Cl)(μ-P′′)]2 | 2.4106(10) | — | — | — | — | — | 62 |
| 25-GaClCH2tBu [Ga(Cl)(CH2tBu)(μ-P′′)]2 | 2.422(3) | d 6-Benzene | −233.86, s | — | — | — | 50 |
| 25-GaBrCH2SiMe3 [Ga(CH2SiMe3)(Br)(μ-P′′)]2 | 2.424(3) | d 6-Benzene | −227.61, s | — | — | — | 50 |
| 25-InClCH2SiMe3 [In(CH2SiMe3)(Cl)(μ-P′′)]2 | 2.593(2) | d 6-Benzene | −241.43, s | — | — | — | 59 |
| 25-InClCp* [In(Cp*)(Cl)(μ-P′′)]2 | 2.621(2) | d 6-Benzene | −148.60, s | — | — | — | 63 |
| 26-GaPh [{Ga(Ph)2}2(μ-P′′)(μ-Cl)] | 2.391(4) | d 6-Benzene | −214.74, s | — | — | — | 64 |
| 26-GaCH2tBu [{Ga(CH2tBu)2}2(μ-P′′)(μ-Cl)] | 2.451(3) | d 6-Benzene | −210.22, s | — | — | — | 50 |
| 26-GaCH2SiMe3 [{Ga(CH2SiMe3)2}2(μ-P′′)(μ-Cl)] | 2.416(6) | d 6-Benzene | −213.27, s | — | — | — | 50 |
| 26-InCH2tBu [{In(CH2tBu)2}2(μ-P′′)(μ-Cl)] | 2.622(5) | d 6-Benzene | −227.34, s | — | — | — | 60 |
| 26-InCH2SiMe3 [{In(CH2SiMe3)2}2(μ-P′′)(μ-Cl)] | 2.603(4) | d 6-Benzene | −218.99, s | — | — | — | 56 |
| 27-Al [{Al(Et)2}2(μ-P′′)(μ-As′′)] | 2.497(1) | d 6-Benzene | −240.93, s | — | — | — | 65 |
| −245.83, s | |||||||
| 27-In [{In(CH2SiMe3)2}2(μ-P′′)(μ-As′′)] | 2.691(3) | d 8-Toluene | −229.84, s | — | — | — | 66 |
| −230.72, s | |||||||
| 29 [{Ga(Et)2}2(μ-P′′)(μ-Sb′′)] | 2.574(2) | d 8-Toluene | — | — | — | — | 48 |
| 30-ClP [{Ga(Cl)2}2(μ-P′′)(μ-P′′)] | 2.379(3) | d 8-Toluene | −158.40, s | — | — | — | 67 |
| 30-BrP [{Ga(Br)2}2(μ-P′′)(μ-P′′)] | 2.386(3) | d 8-Toluene | −158.40, s | — | — | — | 68 |
| 30-IP [{Ga(I)2}2(μ-P′′)(μ-P′′)] | 2.398(4) | d 8-Toluene | −158.40, s | — | — | — | 68 and 69 |
| 30-IAs [{Ga(I)2}2(μ-P′′)(μ-As′′)] | 2.443(3) | d 6-Benzene | −261.25, s | — | — | — | 69 |
| 31-Al [Al(H)2(μ-P′′)]3 | 2.398(2) | d 8-Toluene | −273.50, s | — | — | — | 70 |
| 31-Ga [Ga(H)2(μ-P′′)]3 | 2.392(3) | d 8-Toluene | −265.80, s | — | — | — | 71 |
| 32-Al [{Al(Me)2}3(μ-P′′){HP(SiMe3)}2] | 2.4287(13) | d 8-Toluene | −253.00, dg | [1JPSi + 2JPP = 85.0]g | — | — | 72 |
| −252.46, dh | [1JPSi + 2JPP = 91.1]h | ||||||
| 32-Ga [{Ga(Me)2}3(μ-P′′){HP(SiMe3)}2] | 2.415(2) | d 8-Toluene | −241.74, tg | 2 J PP = 89.2g | 5.53, dth | 1 J PSi = 8.2h | 72 |
| −241.01, th | 2 J PP = 44.0g | 6.0, dVir.th | 3 J PSi = 6.9h | ||||
| 2 J PP = 84.7h | [1JPSi + 3JPSi = 7.2]h | ||||||
| 2 J PP = 42.3h | |||||||
| 33 [{Ga(Me)2(μ-P′′)}2{μ-P(Me)}]2 | 2.443(9) | d 3-Chloroform | −108.7, t | 2 J PP = 30.5 | — | — | 73 |
| −238.2, t | |||||||
| 34-Al [Al(H)2(μ-P′′)2Li(Et2O)2] | 2.4001(13) | d 8-Toluene | −282.0, br, s | — | — | — | 74 |
| 34-Ga [Ga(H)2(μ-P′′)2Li(Et2O)2] | 2.4122(12) | d 8-Toluene | −277.7, s | — | — | — | 75 |
| 35 [Al(H)2(μ-P′′)2Na(DME)2] | 2.405(1) | d 6-Benzene | −283.0, s | — | 3.7, t | 1 J PSi = 9.8 | 76 |
| 36 [Ge(P′′)(ArMes)] | 2.329(1) | d 6-Benzene | −18.2, s | — | — | — | 77 |
| 37-Ge [Ge(DippNacnac)(P′′)] | 2.3912(8) | d 6-Benzene | −192.7, s | — | 2.0, d | 1 J PSi = 17.1 | 78 and 79 |
| 37-Sn [Sn(DippNacnac)(P′′)] | 2.5526(7) | d 6-Benzene | −183.5, s | 1 J PSi = 17.1 | 4.0, d | 1 J PSi = 17 | 78 |
| 1 J SnP = 1453 | |||||||
| 37-Pb [Pb(DippNacnac)(P′′)] | 2.715(2) | d 6-Benzene | −116.6, s | 1 J PbP = 2852 | 7.4, d | 1 J PSi = 36.0 | 78 and 80 |
| 38 [Ge(Se)(DippNacnac)(P′′)] | 2.2976(7) | d 6-Benzene | −172.6, s | 1 J PSi = 26.0 | — | — | 81 |
| 1 J SeP = 52.0 | |||||||
| 39-Ge [Ge(Ph*)(P′′)] | 2.291(4) | d 6-Benzene | −48.6, s | — | — | — | 82 |
| 39-Sn [Sn(Ph*)(P′′)] | 2.527(1) | d 6-Benzene | −123.1, s | 1 J 117SnP = 1396 | 4.08, d | 1 J PSi = 38.5 | 82 |
| 1 J 119SnP = 1453 | |||||||
| 40 [Pb(P′′)(μ-P′′)]2 | 2.77(1)c | d 6-Benzene | −218.0, sdg | 1 J PbP = 1264dg | — | — | 83 |
| 2.70(1)d | −217.3, sdh | 1 J PbP = 1183dh | |||||
| −281.4, scg | 1 J PbP = 1658cg | ||||||
| −302.4, sch | 1 J PbP = 1598ch | ||||||
| 41 [Sn(P′′)(μ-P′′)2Ca(μ-P′′)2Ca(N′′)] | Sn–P: 2.695(4)c | d 6-Benzene | −220.0, br, s (SnP) | — | 1.75, s (SnPSi2) | — | 84 |
| 2.795(9)d | −232.97, s (CaP) | 5.70, s (CaPSi2) | |||||
| Ca–P: 2.892(7)c | |||||||
| 42 [{Sn(P′′)(μ2-P′′)2}2Ba] | Sn–P: 2.684(7)c | d 8-Toluene | −235.35, s | 1 J SnP = 1068.13 | 4.5, s | — | 34 |
| 2.600(6)d | |||||||
| Ba–P: 3.239(7)c | |||||||
| 43-Ca [Ca(THF)2{Sn(μ2-P′′)(μ3-P′)}2] | Ca–P: 2.903(2) | d 6-Benzene | −223.58, s | 1 J SnP = 710.7 | — | — | 33 |
| Sn–P: 2.642(1) | (SnP) | 3 J SnP = 74.1 | |||||
| −295.15, s | (SnP) | ||||||
| (Sn2P) | 1 J SnP = 578.6 | ||||||
| 2 J PP = 77.3 | |||||||
| (SnP) | |||||||
| 43-Ba [Ba(THF)x{Sn(μ2-P′′)(μ3-P′)}2] | Ba–P: 3.302(12) | d 8-Toluene | −220.30, s | 1 J SnP = 730.0 | — | — | 34 |
| Sn–P: 2.685(9) | |||||||
| d-Block | |||||||
| 37-Cr [Cr(DippNacnac)(P′′)] | 2.3641(3) | — | — | — | — | — | 85 |
| 37-Mn [Mn(DippNacnac)(P′′)(THF)] | 2.461(1) | — | — | — | — | — | 85 |
| 37-Zn [Zn(DippNacnac)(P′′)] | 2.2728(3) | d 6-Benzene | −288.85, s | 1 J PSi = 28.5 | 3.49, d | 1 J PSi = 28.5 | 85 |
| 44 [{Y(P′′)2}2(μ-P′′)2] | 2.849(4)c | d 6-Benzene | −107.80, dpc | 1 J YP = 56.7, 2JPP = 5.0c | — | — | 86 and 87 |
| 2.678(4)d | −104.80, dtd | 1 J YP = 122.4, 2JPP = 5.0d | |||||
| 45 [Sc{C(PPh2S)2}(P′′)(py)2] | 2.618(14) | d 8-Toluene | −176.40, br, s | — | — | — | 88 |
| 46-La [La{P(SiMe3)2}3(THF)2] | 2.886(2) | d 6-Benzene | −113.0, br | — | 2.66, d | 1 J SiP = 22.4 | 89 |
| 47 [Ti(Cp)2(P′′)] | 2.468(1) | — | 251.0, br, s | — | — | — | 90 |
| 48-Hf [Hf(Cp)2(P′′)2] | 2.553(1) | d 6-Benzene | −98.83, s | — | — | — | 91 |
| 48-Zr [Zr(Cp)2(P′′)2] | — | d 6-Benzene | −72.18, s | — | — | — | 91 |
| 49-Hf [Hf(Cp)2(Cl)(P′′)] | — | d 6-Benzene | −153.97, s | — | — | — | 91 |
| 49-Zr [Zr(Cp)2(Cl)(P′′)] | 2.547(6) | d 8-Toluene | −108.90, s | — | — | — | 92 |
| 50-Hf [Hf(Cp)2(Me)(P′′)] | — | d 6-Benzene | −141.92, s | — | — | — | 91 |
| 50-Zr [Zr(Cp)2(Me)(P′′)] | 2.629(3) | — | — | — | — | — | 91 |
| 51 [Zr(C5H4Me)2(P′′)2] | 2.617(3) | d 6-Benzene | −71.20, s | — | — | — | 93 |
| 52 [Zr(Cp)2(Cl){P(SiMe3)2C(H)C(Ph)}] | 2.855(4) | d 6-Benzene | −179.50, s | — | — | — | 94 |
| 53-Me [Zr(Cp)2(Me){P(SiMe3)2C(H)C(Ph)}] | 2.915(3) | d 6-Benzene | −179.30, s | — | — | — | 94 |
| 53-nBu [Zr(Cp)2(nBu){P(SiMe3)2C(H)C(Ph)}] | — | d 6-Benzene | −176.60, s | — | — | — | 94 |
53-C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CPh [Zr(Cp)2(CCPh){P(SiMe3)2C(H)C(Ph)}] CPh [Zr(Cp)2(CCPh){P(SiMe3)2C(H)C(Ph)}] |
2.774(3) | d 6-Benzene | −189.90, s | — | — | — | 95 |
| 54-Cr [Cr(Cp)(μ-P′′)]2 | 2.3839(13) | d 6-Benzene | No resonance observed | — | — | — | 96 |
| 54-Mn [Mn(Cp)([μ-P′′)]2] | 2.5099(7) | — | — | — | — | — | 85 |
| 55 [Li(12-crown-4)2][{Mo(Cp)(CO)2}2(μ-P′′)] | 2.4304(6) | — | — | — | — | — | 97 |
| 56 [1,2-W2(P′′)(NMe2)4] | 2.423(3) | d 6-Benzene | −106.70, si | 1 J WP = 243i | — | — | 98 |
| −88.30, sj | 1 J WP = 250j | ||||||
| 57 [1,2-W2(PCy2)(P′′)(NMe2)4] | 2.425(4) | d 6-Benzene | −106.90, si | 1 J WP = 239i | — | — | 98 |
| −96.30, sj | 1 J WP = 234j | ||||||
| 58 [{Mn(P′′)(μ-P′′)2}{Mn(P′′)(THF)}] | 2.526(5)c | — | — | — | — | — | 82 |
| 2.439(4)d | |||||||
| 59-EtMe4 [Fe(C5EtMe4)(CO)2(P′′)] | 2.359(3) | d 6-Benzene | −219.10, s | — | 6.84, d | 1 J PSi = 46.7 | 99 |
| 59-nBuMe4 [Fe(C5nBuMe4)(CO)2(P′′)] | — | d 6-Benzene | −218.60, s | — | 6.84, d | 1 J PSi = 46.4 | 99 |
| 59-1,3-tBu2H3 [Fe(C5H3nBu2-1,3)(CO)2(P′′)] | — | d 6-Benzene | −266.40, s | — | 7.63, d | 1 J PSi = 53.0 | 99 |
| 60-Et [Ni(κ2-depe)(P′′)] | — | — | — | — | — | — | 100 |
| 60-Cy [Ni(κ2-dcpe)(P′′)] | 2.225(2) | — | — | — | — | — | 100 |
| 60-Ph [Ni(κ2-dppe)(P′′)] | — | — | — | — | — | — | 100 |
| 61-NinBu [Ni(Br)(nBu2-bimy)2(P′′)] | — | d 6-Benzene | −197.30, s | — | — | — | 101 |
| 61-NiiPr [Ni(I)(iPr2-bimy)2(P′′)] | — | d 6-Benzene | −178.60, s | — | — | — | 101 |
| 61-PdnBu [Pd(I)(nBu2-bimy)2(P′′)] | 2.3648(17) | d 6-Benzene | −192.80, s | — | — | — | 101 |
| 61-PdiPr [Pd(I)(iPr2-bimy)2(P′′)] | 2.3442(12) | d 6-Benzene | −181.70, s | — | — | — | 101 |
| 62 [Ni(PMe3)(μ-P′′)]2 | 2.381(1)c | — | — | — | — | — | 102 |
| 2.129(2)d | |||||||
| 63-Pd [{Pd(PPh3)}2(P′′){Si[N(tBu)]2CPh}] | — | d 8-THF | −173.00, t | 2 J PP = 5.2 | 0.01, d | 1 J PSi = 6.0 | 103 |
| 63-Pt [{Pt(PPh3)}2(P′′){Si[N(tBu)]2CPh}] | 2.364(3) | d 6-Benzene | −94.60, t | 2 J PP = 33.3 | 8.3, d | 1 J PSi = 7.4 | 103 |
| 1 J PSi = 219.2 | |||||||
| 1 J PtP = 1633 | |||||||
| 64 [Cu(iPr2-bimy)2(P′′)] | 2.291(7) | d 6-Benzene | −261.00, s | — | — | — | 104 |
| 65 [Au(iPr2-bimy)(P′′)] | — | d 6-Benzene | −235.00, s | — | — | — | 105 |
| 66-Cu [Cu(iPr)(P′′)] | 2.1913(15) | d 6-Benzene | −268.00, s | — | — | — | 104 |
| 66-Au [Au(iPr)(P′′)] | 2.3174(10) | d 6-Benzene | −235.70, s | — | — | — | 105 |
| 67 [Au(CAACMeEt)(P′′)] | 2.3253(5) | d 6-Benzene | −233.10, s | — | — | — | 106 |
| 68-Cu [Cu(μ-P′′)]6 | 2.210(4) | d 6-Benzene | −149.00, s | — | — | — | 104 |
| 68-Ag [Ag(μ-P′′)]6 | 2.404(4) | d 6-Benzene | −236.00, s | — | — | — | 104 |
| 69 [Hg(P′′)2] | 2.406(1) | d 6-Benzene | −162.00, s | — | — | — | 83 |
| 70-Zn [Zn(P′′)(μ-P′′)]2 | 2.420(1)c | d 6-Benzene | −183.00, br, sc | ||||
| 2.295(1)d | −237.30, br, sd | — | — | — | 83 | ||
| 70-Cd [Cd(P′′)(μ-P′′)]2 | 2.594(1)c | d 6-Benzene | −180.10, br, sc | — | — | — | 83 |
| 2.459(1)d | −229.50, br, sd | ||||||
| 71-tBu [Zn(tBu)(μ-P′′)]2 | — | d 6-Benzene | −216.70, sc | — | 4.17, s | — | 107 |
| 71-iPr [Zn(iPr)(μ-P′′)]2 | 2.411(4) | d 6-Benzene | −216.30, s | — | 4.24, s | — | 107 |
| 71-CH2SiMe3 [Zn(CH2SiMe3)(μ-P′′)]2 | 2.415(1) | d 6-Benzene | −205.80, s | — | 4.38, s | — | 107 |
| 72-Zn [Zn(PnPr3)Cl(μ-P′′)]2 | 2.437(1)c | — | — | — | — | — | 108 |
| 2.419(1)d | |||||||
| 72-Cd [Cd(PnPr3)Br(μ-P′′)]2 | 2.590(1)c | — | — | — | — | — | 108 |
| 2.578(1)d | |||||||
| 73 [{Zn(Cl)(MeCN)(μ-P′′)2}2{Zn(μ-Cl)}2] | 2.374(3) | — | — | — | — | — | 108 |
| 74 [NnBu4]2[Cd4(I)8(P′′)2] | 2.507(7) | — | — | — | — | — | 108 |
| 75-Me [Zn(Me)(P′′)]3 | 2.390(3) | d 6-Benzene | −246.80, s | — | 4.62, s | — | 107 |
| 75-Et [Zn(Et)(P′′)]3 | — | d 6-Benzene | −246.90, s | — | 4.61, s | — | 107 |
| 75-iPr [Zn(iPr)(P′′)]3 | 2.408(6) | d 6-Benzene | −243.60, s | — | — | — | 107 |
| 75-nBu [Zn(nBu)(P′′)]3 | 2.388(4) | d 6-Benzene | −246.00, s | — | 4.67, s | — | 107 |
| 76-Cr [Cr(CO)5(μ-P′′){Al(dmap)(Me2)}] | 2.528(1) | d 2-DCM | −278.10, s | — | — | — | 39 |
| 76-Fe [Fe(CO)4(μ-P′′){Al(dmap)(Me2)}] | 2.377(1) | d 8-Toluene | −258.40, s | — | — | — | 39 |
| 76-Ni [Ni(CO)3(μ-P′′){Al(dmap)(Me2)}] | 2.315(2) | d 6-Benzene | −275.20, s | — | — | — | 39 and 109 |
| 77-Cr [Zr(C5H4Me)2(μ-P′′)2Cr(CO)n] | 2.656(6) | d 6-Benzene | −38.40, s | — | — | — | 110 |
| 77-Mo [Zr(Cp)2(μ-P′′)2Mo(CO)4] | 2.6648(11) | d 6-Benzene | −57.90, s | — | — | — | 110 |
| 77-Ni [Zr(Cp)2(μ-P′′)2Ni(CO)2] | 2.654(1) | d 6-Benzene | −42.10, s | — | — | — | 111 |
| f-Block | |||||||
| 46-Ce [Ce(P′′)3(THF)2] | 2.849(3) | d 6-Benzene | 616.7, br | — | 5.30, s | — | 89 |
| 46-Pr [Pr(P′′)3(THF)2] | 2.837(3) | d 6-Benzene | 1894.2, br | — | 15.65, s | — | 89 |
| 46-Nd [Nd(P′′)3(THF)2] | 2.818(2) | d 6-Benzene | 2570.3, br | — | 42.94, s | — | 89 and 112 |
| 46-Sm [Sm(P′′)3(THF)2] | 2.789(3) | d 6-Benzene | −259.2, br | — | 0.52, s | — | 89 |
| 46-Tm [Tm(P′′)3(THF)2] | 2.707(16) | d 6-Benzene | — | — | — | — | 113 |
| 78 [Sm(P′′)(μ-P′′)3Sm(THF)3] | 3.039(5)c | d 8-THF | — | — | — | — | 114 |
| 3.027(3)d | |||||||
| 79-Sm [{Sm(P′′)3(THF)}2(μ-I)K3(THF)] | 3.033(5) | d 6-Benzene | — | — | — | — | 115 |
| 79-Eu [{Eu(P′′)3(THF)}2(μ-I)K3(THF)] | 3.0316(4) | d 6-Benzene | — | — | — | — | 115 |
| 80 [KYb(P′′)3{μ-K(P′′)}2]∞ | 2.952(10) | d 6-Benzene | — | — | — | — | 115 |
| 81-Sm trans-[Sm(P′′)2(py)4] | 3.0342(9) | d 6-Benzene | — | — | — | — | 115 |
| 81-Eu trans-[Eu(P′′)2(py)4] | 3.0364(7) | d 6-Benzene | — | — | — | — | 115 |
| 81-Yb trans-[Yb(P′′)2(py)4] | 2.928(13) | d 6-Benzene | −253.93, s | 1 J YbP = 925 | 1.58, Vir. t | 1 J PSi = 16 | 115 |
| 82-Sm [Sm(P′′)2(18-crown-6)] | 3.089(3) | d 6-Benzene | — | — | — | — | 115 |
| 82-Eu [Eu(P′′)2(18-crown-6)] | 3.096(4) | d 6-Benzene | — | — | — | — | 115 |
| 82-Yb [Yb(P′′)2(18-crown-6)] | 2.9662(10) | d 6-Benzene | −265.58, s | 1 J YbP = 977 | 1.94, Vir. t | 1 J PSi = 17 | 115 |
| 83-Th [Th(P′′)(Cp*)2(Cl)] | — | d 6-Benzene | 109.0, s | — | — | — | 116 |
| 83-U [U(P′′)(Cp*)2(Cl)] | 2.788(4) | d 6-Benzene | — | — | — | — | 116 |
| 84-Th [Th(P′′)(Cp*)2(Me)] | 2.888(4) | d 6-Benzene | 115.22, s | — | — | — | 116 |
| 84-U [U(P′′)(Cp*)2(Me)] | 2.893(4) | d 6-Benzene | — | — | — | — | 116 |
| 85-Th [Th{P(SiMe3)(SiMe2CH2)}(Cp*)2] | — | d 6-Benzene | 95.88, s | — | — | — | 116 |
| 85-U [U{P(SiMe3)(SiMe2CH2)}(Cp*)2] | 2.655(6) | d 6-Benzene | — | — | — | — | 116 |
| 86-Th [Th(TrenDMBS)(P′′)] | 2.9406(11) | d 6-Benzene | −100.09, s | — | — | — | 117 |
| 86-U [U(TrenDMBS)(P′′)] | 2.8646(14) | d 6-Benzene | 2055.21, s | — | — | — | 117 |
| 87-Th [Th(TrenTIPS)(P′′)] | 2.9020(13) | d 6-Benzene | −66.45, s | — | 8.49, s | — | 117 |
| 87-U [U(TrenTIPS)(P′′)] | 2.8391(9) | d 6-Benzene | — | — | −97.09, s | — | 117 |
2.1 s-Block P′′ complexes
Alkali metal and alkaline earth metal P′′ complexes are used extensively as ligand transfer agents, and therefore are important starting materials to p-, d- and f-block P′′ complexes. Surprisingly, to date there are no structurally characterised examples of Na P′′ complexes on the CSD,9 though various synthetic routes to group 1 P′′ salts including Na derivatives have been reported extensively.11,12 A Be P′′ complex has yet to be structurally authenticated to the best of our knowledge,9 thus there are obvious gaps in s-block P′′ chemistry that can be relatively easily addressed.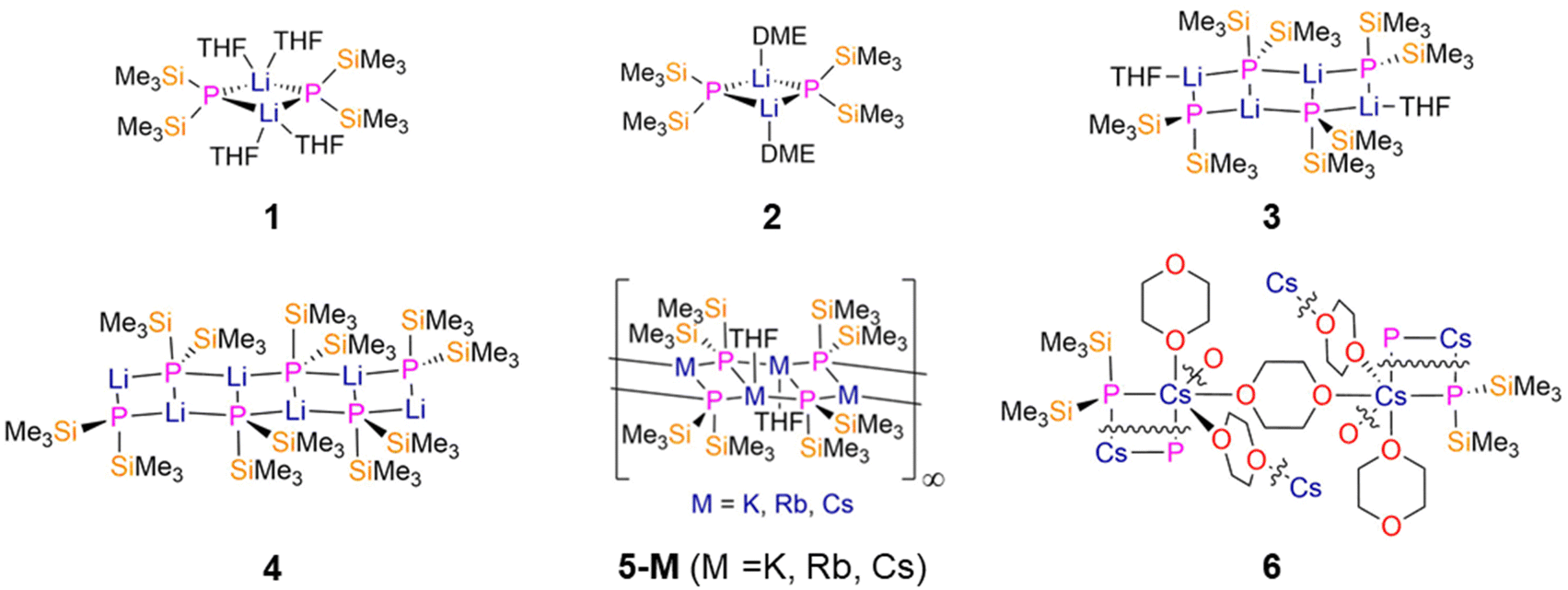 | ||
| Fig. 2 Group 1 M P′′ complexes [Li(μ-P′′)(THF)2]2 (1), [Li4(μ2-P′′)2(μ3-P′′)2(THF)2] (2), [Li(μ-P′′)(DME)]2 (3), [Li6(μ2-P′′)2(μ3-P′′)4] (4), [M4(μ2-P′′)2(μ3-P′′)2]∞ (5-M; M = K, Rb, Cs), [Cs(μ-P′′)(μ-1,4-dioxane)3(1,4-dioxane)]∞ (6). | ||
Similar ladder-like structures were obtained for P′′ complexes of the larger group 1 metals K, Rb and Cs by Ruhlandt-Senge and Uhlig in 1998.27 The polymeric heavy alkali metal P′′ complexes [M4(μ2-P′′)2(μ3-P′′)2]∞ (5-M; M = K, Rb, Cs, Fig. 2) were synthesised by the treatment of P(SiMe3)3 with the parent metal alkoxide MOtBu (M = K, Rb, Cs) in THF, and were found to contain four-coordinate metal centres and five-coordinate phosphorus atoms in the solid state.27 Whilst complexes 5-K and 5-Rb were found to react rapidly with air and moisture, 5-Cs spontaneously ignites upon contact with air, owing to the increase in reactivity with the size of the alkali metal ion.27 A higher degree of oligomerisation was observed for the heavy group 1 P′′ complexes in the solid state in comparison to their lighter Li analogues;26,27 this is limited to growth of the Stiles of the ladder, signifying that the steric bulk of P′′ is sufficient to prevent oligomer growth in other directions. The M–P bond lengths of 5-M increase as the size of the metal increases from K-Cs; however, the K–P bond lengths of 5-K (3.3169(7), 3.4063(8), and 3.4273(8) Å) are slightly longer than expected from ionic radii trends. This was attributed to the steric demand of P′′ having a greater effect on the smaller K ion than for Rb or Cs, with 5-Rb and 5-Cs having mean M–P bond lengths of 3.486(2) Å and 3.656(2) Å, respectively.27,123
A 1,4-dioxane-bridged polymeric Cs P′′ complex [Cs(μ-P′′)(μ-1,4-dioxane)3(1,4-dioxane)]∞ (6, Fig. 2) was synthesised in 2008 by Ionkin by the reaction of P(SiMe3)3 with CsF in 1-4-dioxane at 90 °C for 3 hours.28 This study focussed on using CsF, P(SiMe3)3 and 2,4,6-tri-tert-butylphenylchloride (Mes*Cl) in order to form Mes*P′′, as this reaction does not occur at room temperature without reactive alkali metal precursors present.28 The reaction progress was monitored by 31P{1H} NMR spectroscopy as 6 shows a distinctive chemical shift of −276.10 ppm.27,28 As seen previously for the ladder-like oligomers 5, larger alkali metals like Cs give higher coordination numbers and higher order aggregates.28 The mean Cs–P bond length of 6 (3.6137(17) Å) is slightly longer than those seen for 5-Cs.27,28
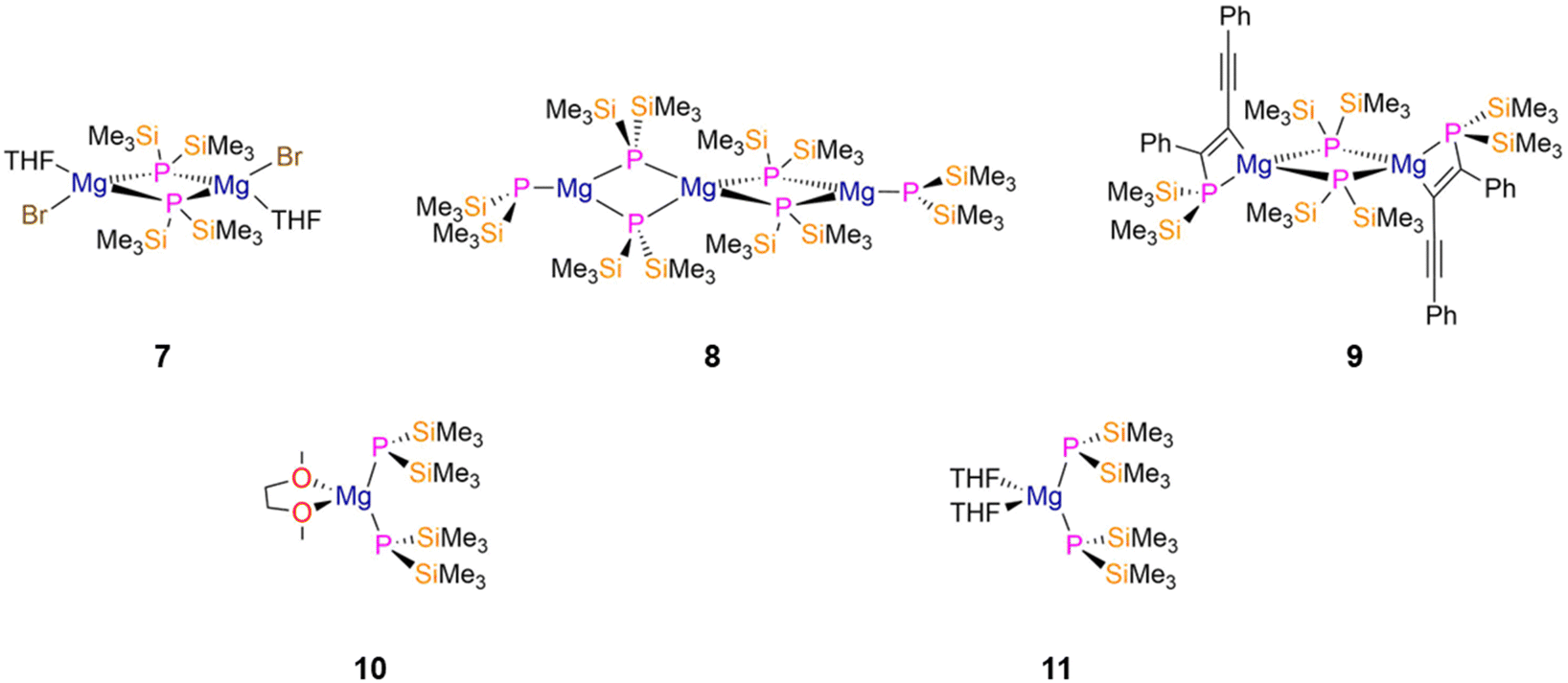 | ||
| Fig. 3 Mg P′′ complexes [Mg(μ-P′′)(Br)(THF)]2 (7), [Mg3(μ-P′′)4(P′′)2] (8), [Mg{P′′C(Ph)C(C2Ph)}(μ-P′′)]2 (9), [Mg(P′′)2(DME)] (10), [Mg(P′′)2(THF)2] (11). | ||
In 1998, Westerhausen reported the reaction of [Mg(P′′)2]n with diphenylbutadiyne to give the dimeric [2 + 2]-cycloaddition product [Mg{P′′C(Ph)C(C2Ph)}(μ-P′′)]2 (9, Fig. 3), which contains MgPC2 four-membered rings with mean Mg–Pbridging bond lengths of 2.5576(19) Å.30 The formation of 9 was confirmed by 31P{1H} NMR spectroscopy, with two triplets at −98.4 and −262.3 ppm for the phosphine and bridging phosphide groups, respectively (2JPP = 5 Hz).30 Evidence of weak conjugation of the triple and double bonds within 9 was provided by their respective stretching bands in the IR spectrum at 2131 and 1596 cm−1.30
The monomeric solvated Mg P′′ complexes [Mg(P′′)2(DME)] (10, Fig. 3) and [Mg(P′′)2(THF)2] (11, Fig. 3) were synthesised by Westerhausen in 1994 and 1995, respectively, by the separate protonolysis reactions of two equivalents of HP′′ with MgR2 (R = nBu, secBu, N′′) in the respective donor solvent.31,32 Both complexes exhibit distorted tetrahedral geometries about the Mg ion, with the phosphorus atoms showing pyramidal geometries in the solid state with Mg–P bond lengths of 2.487(2) Å (10) and 2.5023(19) Å (11).31,32 Complex 11 exhibits a single resonance in the 31P{1H} NMR spectrum at −294.7 ppm; the respective NMR data for 10 were not reported but a similar chemical shift would be expected.31,32
The dimeric heavy group 2 metal P′′ complexes, [M(N′′)(μ-P′′)(sol)]2 (12-Ca: M = Ca, sol = THF; 12-Ba: M = Ba, sol = DME; Fig. 4) were synthesised by Westerhausen in 1995 and 1996, respectively, by the separate reactions of [Ca(N′′)2(THF)2] or [Ba(N′′)2] with HP′′ in toluene or DME.33,34 These bimetallic complexes, which represented the first mixed bis(trimethylsilyl)pnictide complexes at the time, consist of two four-coordinate (12-Ca) or five-coordinate (12-Ba) metal centres, each with a bridging P′′, a terminal N′′, and monodentate THF for 12-Ca or bidentate DME for 12-Ba.33,34 The 31P{1H} NMR spectrum of 12-Ca indicates that a mixture of geometrical isomers is present in solution, with the syn-conformer resonance at (−229.96 ppm) and the anti-conformer at −254.82 ppm, whereas only one conformer is present in solution for 12-Ba (−222.05 ppm).33,34 For 12-Ca, it was noted that the central Ca2P2 ring is highly distorted, with Ca–P bond lengths of 2.927(2) and 3.005(2) Å, whereas the Ba2P2 ring in 12-Ba is essentially planar with Ba–P bond lengths of 3.289(2) and 3.340(2) Å; this is likely a consequence of the larger Ba ion better accommodating the bulky P′′ ligands.33,34
 | ||
| Fig. 4 Heavy group 2 P′′ complexes [M(N′′)(μ-P′′)(sol)]2 (12-Ca: M = Ca, sol = THF; 12-Ba: M = Ba, sol = DME), trans-[M(P′′)2(THF)4] (13-M; M = Ca, Sr, Ba), [Sr(P′′)(μ-P′′)3Sr(THF)3] (14), [Ca(P′′)2(TMTA)2] (15, TMTA = 1,3,5-trimethyl-1,3,5-triazinane). | ||
The protonolysis reaction of 12-Ca with HP′′ in THF gave the monomeric solvated P′′ complex trans-[Ca(P′′)2(THF)4] (13-Ca, Fig. 4).33 Similarly, the separate reactions of [M(N′′)2(THF)n] (M = Sr, n = 0; M = Ba, n = 2) with two equivalents of HP′′ in THF gave the heavier homologues, trans-[M(P′′)2(THF)4] (13-Ba,3613-Sr, Fig. 4).35 All three complexes exhibit distorted octahedral geometries with two axial P′′ and four equatorial THF, with M–P distances of 2.911(2) and 2.924(2) Å (13-Ca),33 3.035(6) and 3.006(6) Å (13-Sr),35 and 3.158(6) and 3.190(6) Å (13-Ba),36 and near-linear P–M–P angles of 175.16(7)° (13-Ca),33 174.2(2)° (13-Sr)35 and 174.9(1)° (13-Ba).36 When 13-Sr is heated under vacuum, the bimetallic complex, [Sr(P′′)(μ-P′′)3Sr(THF)3] (14, Fig. 4) forms, which has one four-coordinate Sr ion with three bridging and one terminal P′′, and one six-coordinate Sr ion with three bridging P′′ and three bound THF.35 Complex 14 is structurally analogous to that of the bimetallic Sm species discussed in Section 2.4.1.125
Finally, the reaction of [Ca(N′′)(μ-N′′)]2 with four equivalents each of HP′′ and 1,3,5-trimethyl-1,3,5-triazinane (TMTA) gave the eight-coordinate C2-symmetric Ca complex, [Ca(P′′)2(TMTA)2] (15, Fig. 4).36 This complex contains two P′′ and two tridentate TMTA ligands in chair conformations, with two identical Ca–P bond lengths of 2.994(2) Å, and a wide range of Ca–N bond lengths between 2.575(4)–2.704(5) Å, which was attributed to steric buttressing.36
2.2 p-Block complexes
As stated previously p-block P′′ compounds containing direct E–P bonds with E = non-metals and the metalloids B and Si are not within the scope of this review. Surprisingly no structurally authenticated examples of As, Sb, Bi or Te P′′ complexes were found in the CSD;9 we note that a synthetic route to a trimeric Bi(I) complex [Bi(μ-P′′)]3 has been reported and it has been claimed that the solid-state structure has been determined, but this dataset has not been deposited on the CSD.12e The absence of group 15 and 16 metal(loid) P′′ complexes to date is noteworthy given that this chemistry has been developed for the lighter congener N′′.7 This section is therefore divided into group 13 and group 14 metal P′′ complexes, and subdivided into homo- and hetero-metallic examples, with the heavier metalloid Ge included for comparisons with heavier homologous Sn and Pb complexes.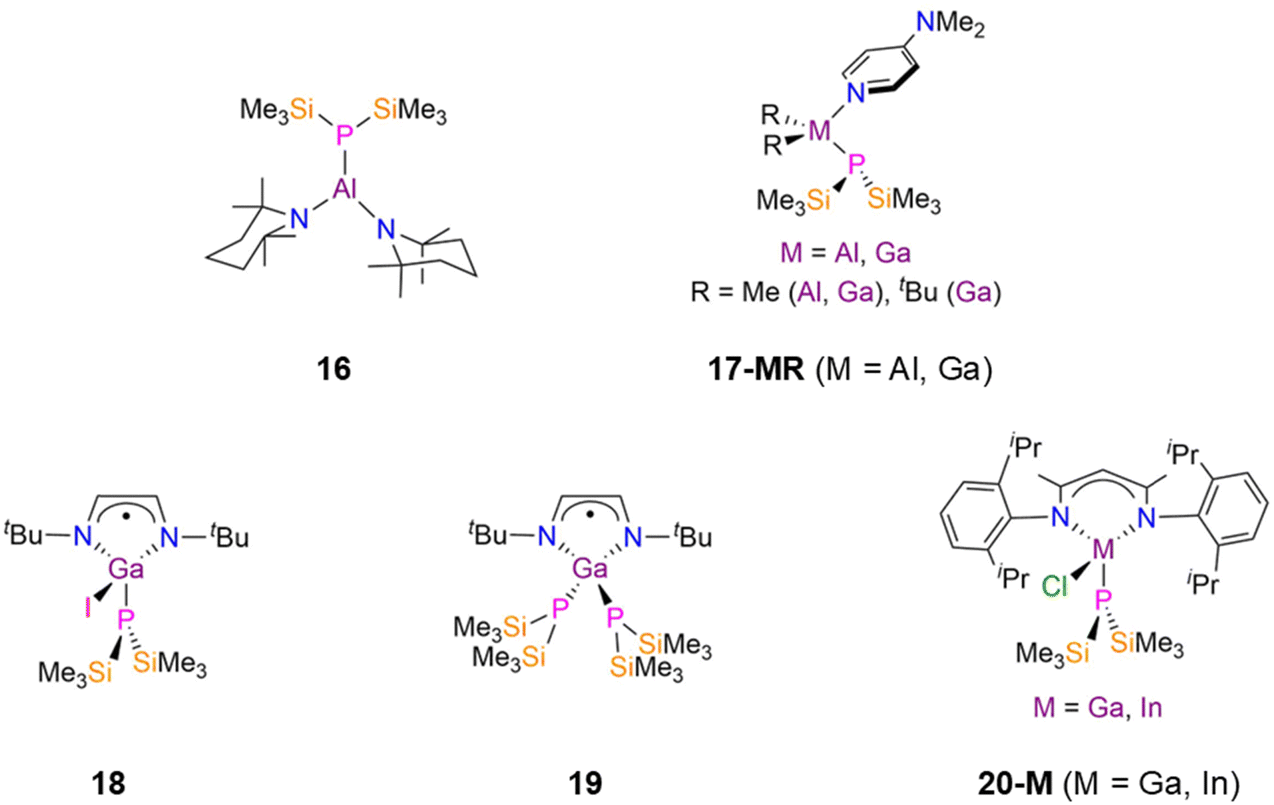 | ||
| Fig. 5 Monomeric group 13 P′′ complexes [Al(P′′)(tmp)2] (tmp = 2,2,6,6-tetramethyl-piperidino) (16), [M(R)2(P′′)(dmap)] (17-MR. 17-AlMe: M = Al, R = Me; 17-GaMe: M = Ga, R = Me; 17-GatBu: M = Ga, R = tBu; dmap = 4-(dimethylamino)pyridine), [Ga(P′′)(I)(tBu-DAB)], (18, tBu-DAB = {N(tBu)C(H)}2), [Ga(P′′)2(tBu-DAB)] (19), [M(DippNacnac)(P′′)(Cl)] (DippNacnac = {HC[C(Me)N(Dipp)]2}, Dipp = C6H3iPr2-2,6) (20-M; M = Ga, In). | ||
In 2005, Jones and Murphy reported the synthesis of the Ga(III) P′′ complexes, [Ga(P′′)(I)(tBu-DAB)] (18, Fig. 5) and [Ga(P′′)2(tBu-DAB)] (19, Fig. 5) (tBu-DAB = {N(tBu)C(H)}2), by the reaction of the Ga(II) precursors [Ga(tBu-DAB)(I)]2 with either two or four equivalents of MP′′ (M = Li, Na) in Et2O at −78 °C.41 The mechanism of formation of 18 is unclear, but involves both salt elimination and Ga–Ga bond cleavage, with oxidation of Ga(II) to Ga(III). When only one equivalent of MP′′ was used the unreacted Ga(II) starting material was isolated, whilst the addition of a further equivalent of MP′′ to 18 gave 19 in moderate yield.41 Both of these Ga(III) P′′ complexes have distorted tetrahedral geometries, with the Ga–P bond lengths being longer for 19 (2.343(2) Å mean) than for 18 (2.300(2) Å) due to the increased steric bulk about the metal centre.41 Complexes 18 and 19 did not give NMR data that could be interpreted due to their paramagnetism, but EPR spectroscopy showed that there was a small amount of unpaired spin density at the P atoms due to the splitting of the signal from coupling to 31P nuclei. A smaller line width was seen for 19 due to increased spin delocalisation at the P′′ ligands; despite this giving increased spin density at gallium, the unpaired electrons are mostly localised on the diazabutadiene ligands.41 In 2019, Scheer investigated a series of group 13 phosphido complexes for their potential applications in nanoparticles, semiconductors and optoelectronic layers.42 The authors postulated that bulky co-ligands would be needed in order to isolate mononuclear parent phosphido complexes, as there is a tendency towards oligomerisation due to the Lewis acidity of the group 13 metal centres and Lewis basicity of PH2.42 The reaction of [M(DippNacnac)(Cl)2] (M = Ga, In; DippNacnac = {HC[C(Me)N(Dipp)]2}, Dipp = C6H3iPr2-2,6) with excess LiP′′ in toluene gave the monosubstituted complexes [M(DippNacnac)(P′′)(Cl)] (20-M; M = Ga, In, Fig. 5); the disubstituted P′′ complexes could not be prepared due to the steric bulk of the β-diketiminate ligand.4231P{1H} NMR spectroscopy revealed singlet resonances at −255.0 ppm (20-Ga) and −252.9 ppm (20-In), whilst single crystal XRD showed that these complexes have heavily distorted tetrahedral geometries, with N–M–N angles of 96.03(13)° (20-Ga) and 90.33(10)° (20-In); the M–P bond length of 2.3310(9) Å (20-Ga) is shorter than that found for 20-In (2.4806(8) Å).42
There have been numerous published examples of symmetric dimeric group 13 P′′ complexes, [M(R)2(μ-P′′)]2 (21-MR; M = Al, Ga, In, Tl; R = Me, Et, Ph, nBu, CH2Ph, CH2iPr, CH2tBu, CH2SiMe3, Fig. 6), as these have found interest as precursors for III–V semiconductor materials.43–57 Each of these complexes consists of two four-coordinate distorted tetrahedral metal centres bridged by two P′′ ligands to give M2P2 cores. In 21-AlR the mean Al–P bond lengths increase as the steric influence of the R group increases: 21-AlMe, 2.457(2) Å; 21-AlEt, 2.458(1) Å; 21-AlCH2iPr, 2.476(2) Å; 21-AlCH2SiMe3, 2.483(1) Å.43–46 This trend is also seen for the 21-GaR series: 21-GaMe, 2.450(1) Å; 21-GaEt, 2.4558(7) Å; 21-GanBu, 2.4533(6) Å; 21-GaCH2tBu, 2.517(3) Å; 21-GaCH2SiMe3, 2.4887(16) Å.40,47–51 Finally, the In–P bond lengths in 21-InR vary similarly: 21-InMe, 2.630(1) Å; 21-InEt, 2.645(1) Å; 21-InPh, 2.612(1) Å; 21-InCH2Ph, 2.6123(6) Å; 21-CH2SiMe3, 2.655(3) Å.52–55 The variation of R groups in these complexes changes their functionalities; for example, [In(tBu)2(μ-P′′)]2 (21-IntBu) was employed in the fabrication of nanowires of approximately 10–100 nm diameter.55 In 2003 Schulz disclosed that the combination of 17-Al with GaMe3 in the presence of dmap at −30 °C yielded the adduct [Al(Me)2(dmap)(μ-P′′)Ga(Me)3] (22, Fig. 6), as well as [Ga(Me)2(μ-P′′)]2 (21-GaMe) [AlMe3(dmap)]; the latter products formed upon decomposition when warming to room temperature as a result of a methyl group transfer from Ga to Al with concomitant Al–P bond cleavage.58 This rearrangement was monitored by VT 31P{1H} NMR spectroscopy; from −50 to −10 °C there was only a single resonance corresponding to 22 (−262 ppm), but above −10 °C an additional signal appears at −219 ppm due to the formation of 21-GaMe.58
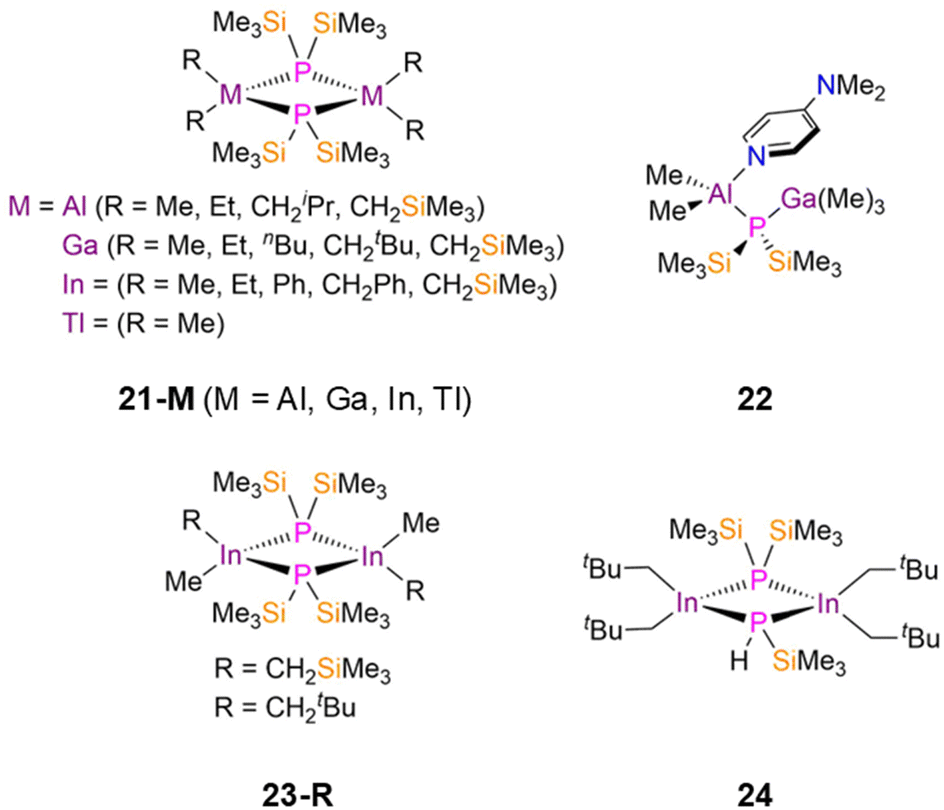 | ||
| Fig. 6 Dinuclear group 13 P′′ complexes [M(R)2(μ-P′′)]2 (21-MR; M = Al, Ga, In, Tl; R = Me, Et, Ph, nBu, CH2Ph, CH2iPr, CH2tBu, CH2SiMe3), [Al(Me)2(dmap)(μ-P′′)Ga(Me)3] (22), [In(Me)(R)(μ-P′′)]2 (23-R; R = CH2SiMe3, CH2tBu), [{In(CH2tBu)2}2(μ-P′′){μ-PH(SiMe3)}] (24). | ||
In both 1992 and 1993, Wells reported the synthesis of three dimeric In(III) P′′ complexes, [In(Me)(R)(μ-P′′)]2 (23-R; R = CH2SiMe3, CH2tBu, Fig. 6) and [{In(CH2tBu)2}2(μ-P′′){μ-PH(SiMe3)}] (24, Fig. 6).59,60 Complexes 23-R exhibit puckered In2P2 cores, with each In atom bound to two different alkyl groups, and mean In–P bond lengths of 2.635(2) Å (23-CH2SiMe3) and 2.637(3) Å (23-CH2tBu).59,60 Complex 24 was isolated as a minor product from the reaction of [In(CH2tBu)2(Cl)] and P(SiMe3)3, and represented the first example of a dinuclear In phosphide complex in which the phosphorus centres were substituted asymmetrically; complex 24 exhibits a mean In–P bond length of 2.650(3) Å and hydrocarbon solutions of this complex were found to be unstable at room temperature.60
There are numerous examples of dinuclear group 13 bridged P′′ complexes that contain either terminal or bridging halides as these are often used as precursors to functionalised complexes via salt metathesis reactions. For example, [M(R)(X)(μ-P′′)]2 (25-MXR; M = Al, Ga, In; X = Cl, Br; R = Me, CH2tBu, CH2SiMe3, η1-Cp*; Cp* = C5Me5, Fig. 7) were synthesised from parent [M(R)(X)2] with either P(SiMe3)3 or LiP′′ between 1991–2004 by Schulz, Wells, and Theopold.50,59,61–63,126 Each of these complexes has a M2P2 core, with the four-coordinate group 13 metals having distorted tetrahedral geometries.50,59,61–63,126 Complex 25-AlBrCH2SiMe3 exhibits a mean Al–P bond length of 2.436(4) Å, whilst the Ga–P bond lengths of 25-GaXR range from 2.4106(10)–2.424(3) Å, and the In–P distances of 25-InXR vary from 2.593(2)–2.621(2) Å.50,59,61,63 Wells reported several examples of group 13 metal P′′ complexes where a chloride has been incorporated into the central four membered ring, [{M(R)2}2(μ-P′′)(μ-Cl)] (26-MR; M = Ga, In; R = Ph, CH2tBu, CH2SiMe3, Fig. 7).50,56,60,64 For 26-GaCH2tBu, the central Ga2PX core is planar, but 26-GaCH2SiMe3 deviates from planarity by 23° and 26-GaCH2Ph deviates by 6.4° due to steric effects.50,64 In contrast, the In P′′ complexes 26-InCH2tBu and 26-InCH2SiMe3 were found to be planar, due to the increased size of In vs. Ga allowing for the large steric bulk of both alkyl and silylphosphide ligands.56,60 This increase in size from Ga to In is reflected in the respective mean M–P bond lengths; 26-GaR (2.391(4)–2.451(3) Å) and 26-InR (2.603(4)–2.622(5) Å).50,56,60,64
 | ||
| Fig. 7 Dimeric group 13 P′′ complexes [M(R)(X)(μ-P′′)]2 (25-MXR: M = Al, Ga, In; X = Cl, Br; R = Me, CH2tBu, CH2SiMe3, η1-Cp*; Cp* = C5Me5), [{M(R)2}2(μ-P′′)(μ-Cl)] (26-MR: M = Ga, In; R = Ph, CH2tBu, CH2SiMe3), [{M(R)2}2(μ-P′′)(μ-As′′)] (27-M: M = Al, R = Et, M = In, R = CH2SiMe3), [M(R)2(μ-As′′)]2 (28-M: M = Al, R = Et, M = In, R = CH2SiMe3), [{Ga(Et)2}2(μ-P′′)(μ-Sb′′)] (29), [{Ga(X)2}2(μ-P′′){μ-E(SiMe3)2}] (30-ClP: X = Cl, Pn′′ = P′′; 30-BrP: X = Br, Pn′′ = P′′; 30-IP: X = I, Pn′′ = P′′; 30-IAs: X = I, Pn′′ = As′′). | ||
There are also examples of dinuclear group 13 P′′ complexes that contain bridging {Pn(SiMe3)2} ligands; Pn = As (As′′) or Sb (Sb′′). In 1994 and 1995, Wells synthesised the respective Al and In P′′ complexes [{M(R)2}2(μ-P′′)(μ-As′′)] (27-Al: M = Al, R = Et; 27-In: M = In, R = CH2SiMe3; Fig. 7) by the separate reactions of [M(R)2(P′′)]2 with [M(R)2(As′′)]2 (28-Al: M = Al, R = Et; 28-In: M = In, R = CH2SiMe3, Fig. 7),65,66 whilst an analogous Ga complex, [{Ga(Et)2}2(μ-P′′)(μ-Sb′′)] (29, Fig. 7), was synthesised by Wells in 2000 via the combination of [Ga(Et)2(Cl)] with P(SiMe3)3 and Sb(SiMe3)3; the As analogue of 29 was not structurally authenticated.48 Complexes 27-M and 29 represent rare examples of two different group 15 elements bridging between Al, Ga or In; in the solid state planar central M2PE cores (M = Al, In, E = As; M = Ga, E = Sb) were observed in each case, with the metal centres displaying distorted tetrahedral geometries.48,65,66 The mean M–P bond lengths increase with the size of the metal (27-Al: 2.497(1) Å; 29: 2.574(2) Å; 27-In: 2.691(3) Å).48,65,66 Between 1993–1995, Wells reported several other examples of dimeric Ga(III) P′′ halide complexes, namely [{Ga(X)2}2(μ-P′′)(μ-Pn′′)] (30-ClP: X = Cl, Pn′′ = P′′; 30-BrP: X = Br, Pn′′ = P′′; 30-IP: X = I, Pn′′ = P′′; 30-IAs: X = I, Pn′′ = As′′; Fig. 7).67–69 These complexes were isolated from the separate reactions of GaX3 (X = Cl, Br, I) with either two equivalents of P(SiMe3)3 (30-ClP, 30-BrP, 30-IP) or one equivalent each of P(SiMe3)3 and As(SiMe3)3 (30-IAs).67–69 These complexes are the first examples of Ga pnictide dimers that contain only exocyclic halogen ligands, and in common with the previously discussed bridging pnictide complexes they all display a planar central Ga2PE core with the Ga atoms exhibiting distorted tetrahedral geometries. The mean Ga–P bond lengths range from 2.379(3)–2.398(4) Å, with the increase in size of the terminal halogen leading to longer bridging Ga–P bond distances.67–69 The incorporation of the bridging As′′ ligand in 30-IAs gives a longer Ga–P bond length (2.443(3) Å) than the mean Ga–P distance in 30-IP (2.398(4) Å).69 The authors anticipated that these complexes would be viable starting materials for introducing further substitution at the Ga centres through dehalosilylation and salt elimination reactions.67–69
Although there are numerous reports of dimeric group 13 P′′ complexes, there are just a handful of structurally characterised tri- or tetra-nuclear examples. The trinuclear complexes [M(H)2(μ-P′′)]3 (M = Al, 31-Al; M = Ga, 31-Ga, Fig. 8), were reported by Wells in 1997 and 1998, and [{M(Me)2}3(μ-P′′){HP(SiMe3)}2] (M = Al, 32-Al; M = Ga, 32-Ga, Fig. 8), were reported by Robinson and Weidlein in 1994 and 1999, respectively.70–72 These complexes were synthesised by the reaction of parent H3M·NMe3 (M = Al, Ga) with P(SiMe3)3 for 31-M, or from parent [M(Me)2(OMe)]3 (M = Al, Ga) and HP′′ for 32-M. Each complex features six-membered M3P3 rings with three four-coordinate distorted tetrahedral Al/Ga atoms, with 31-M exhibiting planar rings and 32-M having twisted conformations in order to relieve steric congestion.70–72 The mean M–P bond lengths increase from 31-M to 32-M: 2.398(4) Å (31-Al), 2.4287(13) Å (32-Al), 2.392(3) Å (31-Ga), and 2.415(2) Å (32-Ga).70–72 The tetranuclear Ga(III) P′′ complex [{Ga(Me)2(μ-P′′)}2{μ-P(Me)}]2 (33, Fig. 8), reported by Robinson in 1994, was prepared from the reaction of the Lewis acid–base adduct [GaMe3-PMe3] with P(SiMe3)3.73 Complex 33 is bicyclic and features two fused Ga2P3 five-membered rings, with distorted tetrahedral geometries of both Ga and P atoms, mean Ga–P bond lengths of 2.443(9) Å and a P–P single bond distance of 2.25(3) Å.73 Remarkably, unlike the vast majority of P′′ complexes, 31-M, 32-M and 33 are all stable in air for a limited amount of time.70–73
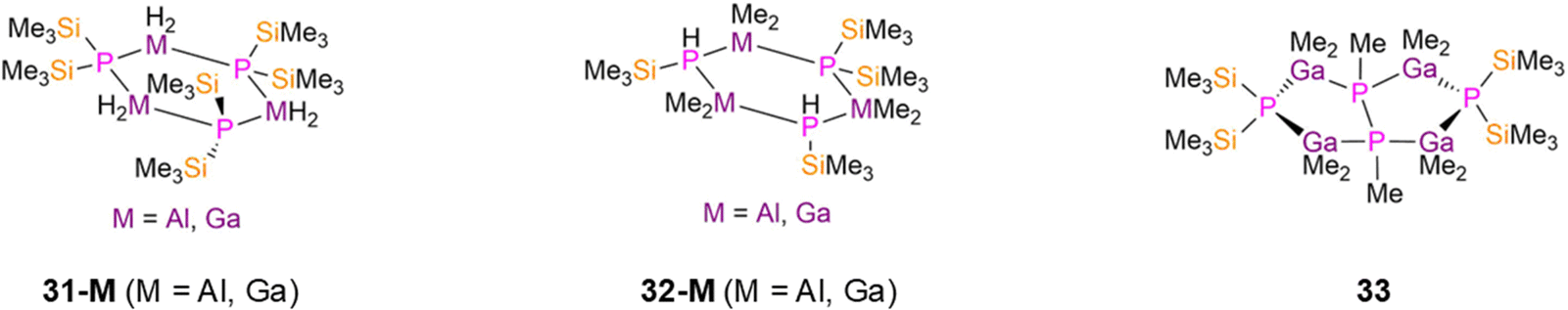 | ||
| Fig. 8 Polynuclear 13 P′′ complexes [M(H)2(μ-P′′)]3 (31-M; M = Al, Ga), [{M(Me)2}3(μ-P′′){HP(SiMe3)}2] (32-M; M = Al, Ga), [{Ga(Me)2(μ-P′′)}2{μ-P(Me)}]2 (33). | ||
2.2.1.1 Heterometallic group 13 complexes. There are a handful of structurally characterised examples of heterometallic mixed s- and p-block P′′ complexes, which are often used as precursors for complexes that possess bonds between group 13–15 elements.74 The Li group 13 ‘ate’ P′′ complexes, [M(H)2(μ-P′′)2Li(Et2O)2] (34-M; M = Al, Ga; Fig. 9) were synthesised by Wells in 1997 and 1998, respectively, via the reaction of LiMH4 with two equivalents of P(SiMe3)3 (M = Al, Ga) in diethyl ether.74,75 The LiP2M (M = Al, Ga) cores of these complexes were confirmed to be planar by both NMR spectroscopy and single crystal XRD, and show mean M–P bond lengths of 2.4001(13) Å (34-Al) and 2.4122(12) Å (34-Ga).74,75 Both the hydrogen and carbon atoms of the SiMe3 groups gave triplet resonances in the 1H and 13C{1H} NMR spectra from second order coupling to two identical 31P nuclei, and signals at −282.0 and −277.7 ppm were seen in the 31P{1H} NMR spectra of 34-Al and 34-Ga, respectively.74,75 The M–H bonds were evidenced by stretching absorptions in IR spectra at 1752 cm−1 (34-Al) and 1838 cm−1 (34-Ga), together with broad resonances in 1H NMR spectra at 4.3 ppm (34-Al) and 4.7 ppm (34-Ga).74,75 A structurally analogous Al P′′ complex, [Al(H)2(μ-P′′)2Na(DME)2] (35, Fig. 9), was synthesised by Hänisch in 2004 via the reaction of NaAlH4 with four equivalents of the primary phosphine, H2PSiMe3 in DME.76 Similarly to 34-M, complex 35 has a planar four-membered NaP2Al ring, mean Al–P bond lengths of 2.405(1) Å, triplet resonances in both its 1H and 13C{1H} NMR spectra for the SiMe3 groups, and a single resonance at −283.0 ppm in its 31P{1H} NMR spectrum. Both symmetric and asymmetric stretches of the Al–H bonds were observed in the IR spectrum of 35 at 1762 and 1728 cm−1.76
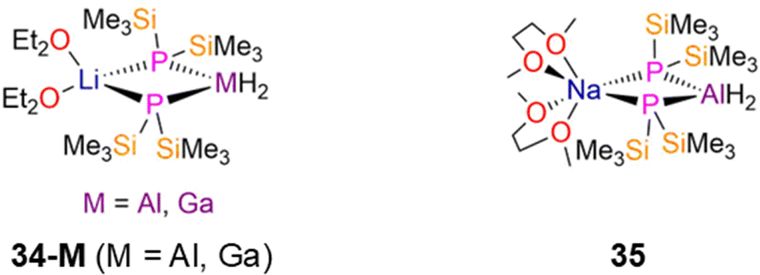 | ||
| Fig. 9 Heterometallic group 13 P′′ complexes [M(H)2(μ-P′′)2Li(Et2O)2] (34-M; M = Al, Ga), [Al(H)2(μ-P′′)2Na(DME)2] (35). | ||
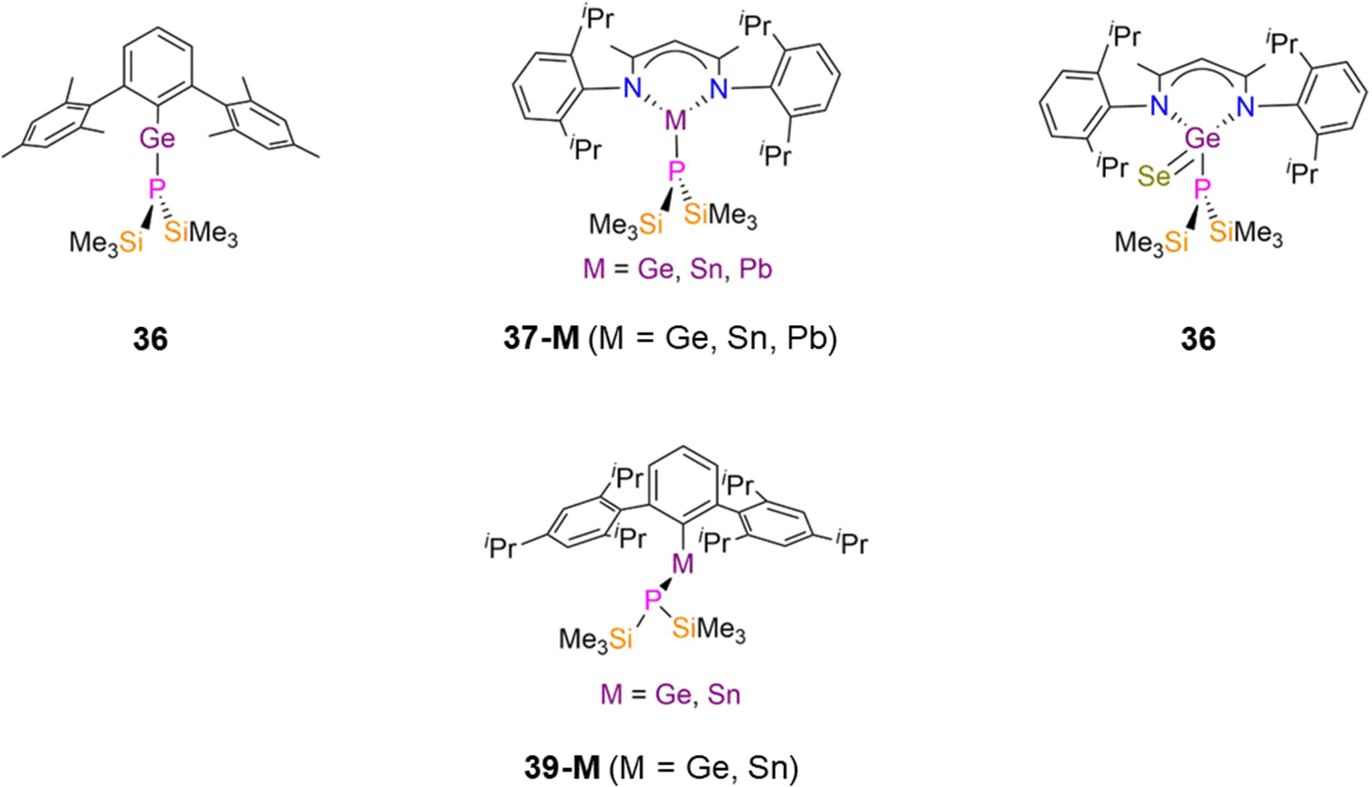 | ||
| Fig. 10 Mononuclear group 14 P′′ complexes [Ge(P′′)(ArMes)] (ArMes = C6H3Mes2-2,6) (36), [M(DippNacnac)(P′′)] (37-M; M = Ge, Sn, Pb, DippNacnac = CH{(CH3)2CN(Dipp)}2; Dipp = C6H3iPr2-2,6), [Ge(Se)(DippNacnac)(P′′)] (38), [M(Ph*)(P′′)] (39-M; M = Ge, Sn, Ph* = C6H3Trip-2,6, Trip = C6H2iPr3-2,4,6). | ||
In 1993, Buhro reported the dimeric Pb(II) P′′ complex, [Pb(P′′)(μ-P′′)]2 (40, Fig. 11) which was synthesised by the protonolysis reaction of [Pb(N′′)2] with two equivalents of HP′′; the Sn(II) analogue of 40 was synthesised by the same methods but was not structurally authenticated.83 Complex 40 was reported to decompose to Pb and the diphosphine (Me3Si)2P–P(SiMe3)2 in refluxing benzene. Complex 40 exhibits a puckered central M2P2 core and crystallises with the trigonal pyramidal terminal P′′ ligands mutually syn-, with mean Pb–P bond lengths of 2.77(1) Å (terminal) and 2.70(1) Å (bridging).83 An equilibrium between the syn- and anti-conformers of 40 is seen in solution, with the 31P{1H} NMR spectrum containing two sets of resonances in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 1:1 ratio (−217.3 ppm, 1JPbP = 1264 Hz, anti-terminal; −218.0 ppm, 1JPbP = 1183 Hz, syn-terminal; −281.4 ppm, 1JPbP = 1598 Hz, syn-bridging; −302.4 ppm, 1JPbP = 1658 Hz, anti-bridging).83 Further assignment of these conformers was enabled by 1H NMR spectroscopy as the SiMe3 groups on the bridging P′′ are inequivalent for the syn- and equivalent for the anti-conformer, thus three resonances are associated with the former and two for the latter conformer.83 It was deduced from the intensities of the resonances at room temperature that solutions of 40 contained nearly equal amounts of syn- and anti-conformers.83
:1 and 1:1 ratio (−217.3 ppm, 1JPbP = 1264 Hz, anti-terminal; −218.0 ppm, 1JPbP = 1183 Hz, syn-terminal; −281.4 ppm, 1JPbP = 1598 Hz, syn-bridging; −302.4 ppm, 1JPbP = 1658 Hz, anti-bridging).83 Further assignment of these conformers was enabled by 1H NMR spectroscopy as the SiMe3 groups on the bridging P′′ are inequivalent for the syn- and equivalent for the anti-conformer, thus three resonances are associated with the former and two for the latter conformer.83 It was deduced from the intensities of the resonances at room temperature that solutions of 40 contained nearly equal amounts of syn- and anti-conformers.83
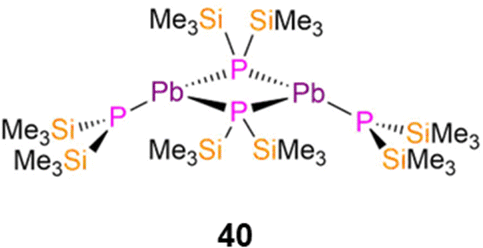 | ||
| Fig. 11 Complex [Pb(P′′)(μ-P′′)]2 (40). | ||
2.2.2.1 Heterometallic group 14 complexes. In 1994 Westerhausen reported the synthesis of two Sn(II) P′′ complexes, [Sn(P′′)(μ-P′′)2Ca(μ-P′′)2Ca(N′′)] (41, Fig. 12) and [{Sn(P′′)(μ2-P′′)2}2Ba] (42, Fig. 12), by the separate reactions of the respective M(II) group 2 bis-amide complexes, [M(N′′)2] (M = Ca, Ba) with [Sn(N′′)2] and five or six equivalents of HP′′ in toluene.34,84 Both complexes exhibit central {M(μ-P′′)4} (M = Ca, Ba) cores, with 41 featuring one terminal CaN′′ and one terminal SnP′′ fragment, and 42 having two terminal SnP′′ moieties.34,84 The central s-block atom of both complexes have distorted tetrahedral geometries, the terminal Ca atom in 41 is planar, and the terminal Sn atoms of both 41 and 42 are pyramidal. The bridging Sn–P bond distances in 41 are longer than that of the terminal Sn–P bond by approximately 0.09 Å, whereas the corresponding difference in 42 is ca. 0.11 Å.34,84 The cluster complexes, [M(THF)x{Sn(μ2-P′′)(μ3-P′)}2] (M = Ca, x = 2, 43-Ca; M = Ba, x = 3, 4, 43-Ba, P′ = {P(SiMe3)}; Fig. 12), were synthesized by Westerhausen in 1994 and 1995 using analogous methods but with THF as the reaction solvent. Both 43-M feature two three-coordinate Sn atoms, each bound to one bridging P′′ and two dianionic bridging silylphosphadiides, {P(SiMe3)}, to the respective s-block metal; 43-Ca and 43-Ba have mean Sn–P bond lengths of 2.642(1) Å and 2.885(9) Å, respectively.33,34
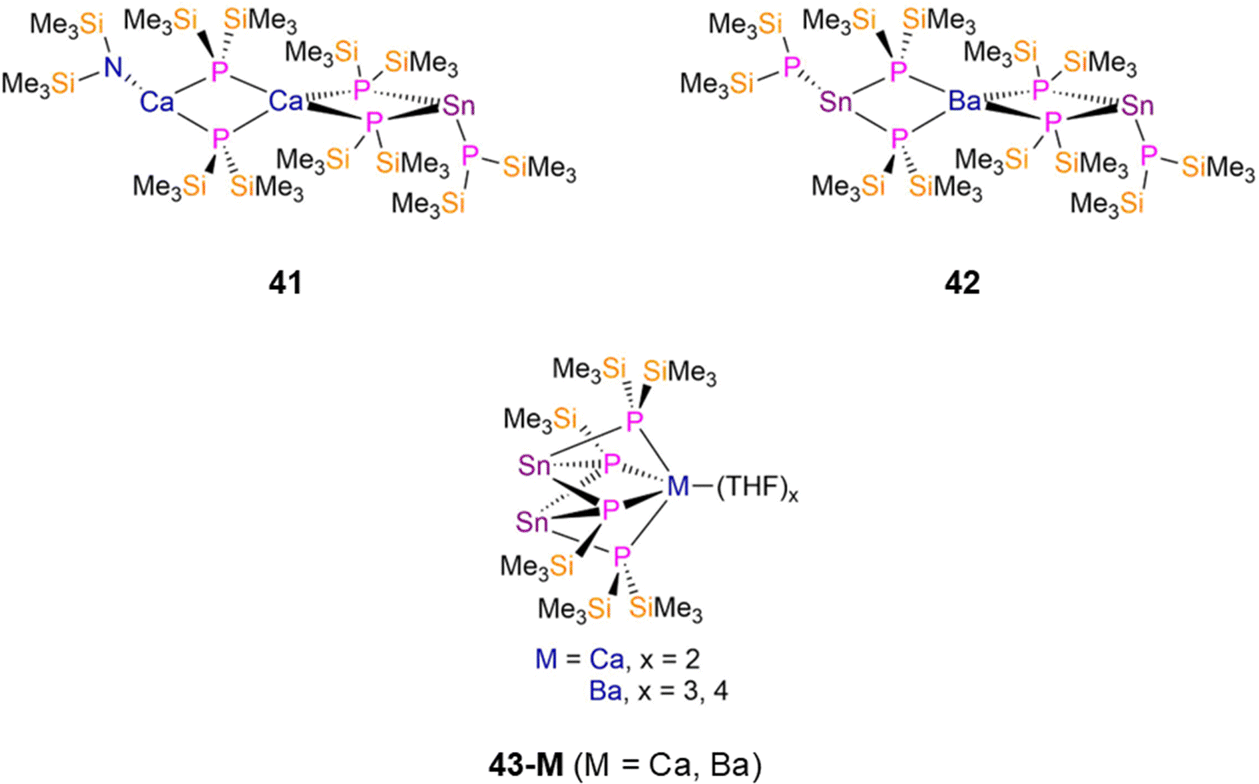 | ||
| Fig. 12 Heterometallic group 14 P′′ complexes [Sn(P′′)(μ-P′′)2Ca(μ-P′′)2Ca(N′′)] (41), [{Sn(P′′)(μ-P′′)2}2Ba] (42), [M(THF)x{Sn(μ2-P′′)(μ3-P′)}2] (43-M; M = Ca, x = 2, M = Ba, x = 3, 4, P′ = {P(SiMe3)}). | ||
2.3 d-Block P′′ complexes
d-Transition metal P′′ complexes are known to be versatile reagents for the formation of alkylidene-phosphides,134,135 diphosphenyls136 and cyclophosphines;137 bulky supporting ligands are required in order to prevent oligomerisation to give monomeric complexes, and for homoleptic complexes oligomers tend to form. As expected and in common with the vast majority of other ligands,22,23 first row d-transition metal P′′ chemistry is more developed than for the heavier second and third row metals. It is noteworthy that no group 5 or 9 P′′ complexes were found in the CSD search described above, and there are also no structurally authenticated Tc, Re, Ru or Os P′′ complexes,9 thus there are multiple opportunities here to expand d-block P′′ chemistry rapidly.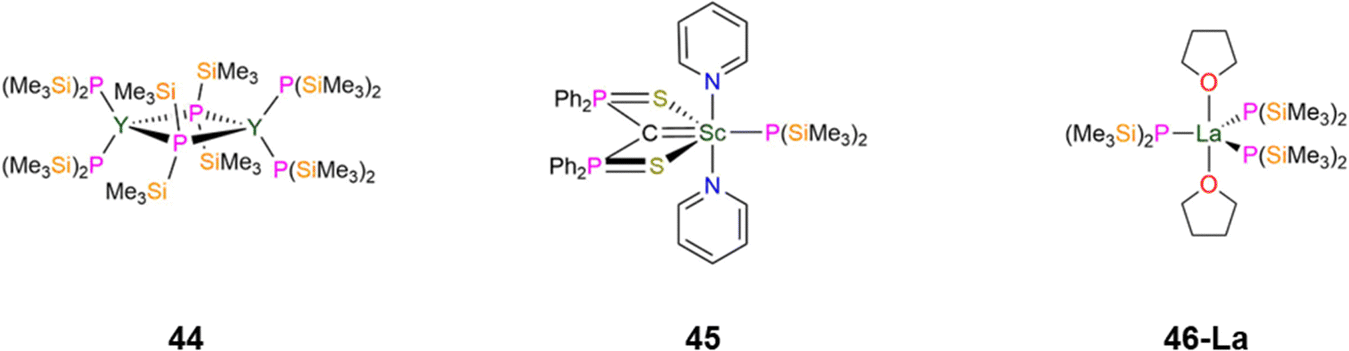 | ||
| Fig. 13 Group 3 P′′ complexes [{Y(P′′)2}2(μ-P′′)2] (44), [Sc{C(PPh2S)2}(P′′)(py)2] (45), [La{P(SiMe3)2}3(THF)2] (46-La). | ||
In 2015, as part of a study by Mézailles into how the anionic ligand trans- to a bound carbon atom in Sc(III) methanediide complexes influences complex geometry, the P′′ complex, [Sc{C(PPh2S)2}(P′′)(py)2] (45, Fig. 13) was synthesised.88 Complex 45 has a Sc–P bond length of 2.618(14) Å; it was found that the methanediide carbon atom is planar for all Sc(III) complexes explored in this study save the N′′ analogue, [Sc{C(PPh2S)2}(N′′)(THF)], which has one THF bound to Sc rather than two pyridines. Density Functional Theory (DFT) calculations indicated that the coordinating solvent made little impact on the planarity of the methanediide. Natural Bond Orbital (NBO) analysis revealed that in 45 the donation from both the C and P atoms are stronger than the corresponding C and N atoms in [Sc{C(PPh2S)2}(N′′)(THF)], which leads to the difference in methanediide geometry.88 In 2024, Mills and co-workers reported the synthesis of [La{P(SiMe3)2}3(THF)2] (46-La, Fig. 13) by a salt metathesis reaction of the respective [La(I)3(THF)4] starting material with three equivalents of KP′′.89 Single crystal XRD showed that 46-La to exhibit a distorted trigonal bipyramidal geometry, with three equatorial P′′ and two axial THF molecules, with the O–La–O angle showing a small deviation from linearity: 175.14(8)° and a mean La–P bond length of 2.886(2) Å. Two of the equatorial P′′ in 46-La show pyramidal geometries about the phosphorus atom, whereas the third P′′ exhibits a planar geometry. Solid state 31P MAS NMR spectroscopy of 46-La revealed two components in their spectra in a 2:1 ratio, with the major component assigned to pyramidal and the minor component to planar P environments. This is in contrast to the solution 31P{1H} NMR spectrum of 46-La, which exhibits one broad resonance due to dynamic processes and quadrupolar broadening due to the 99.9% abundant 77La I = 7/2 nuclei.
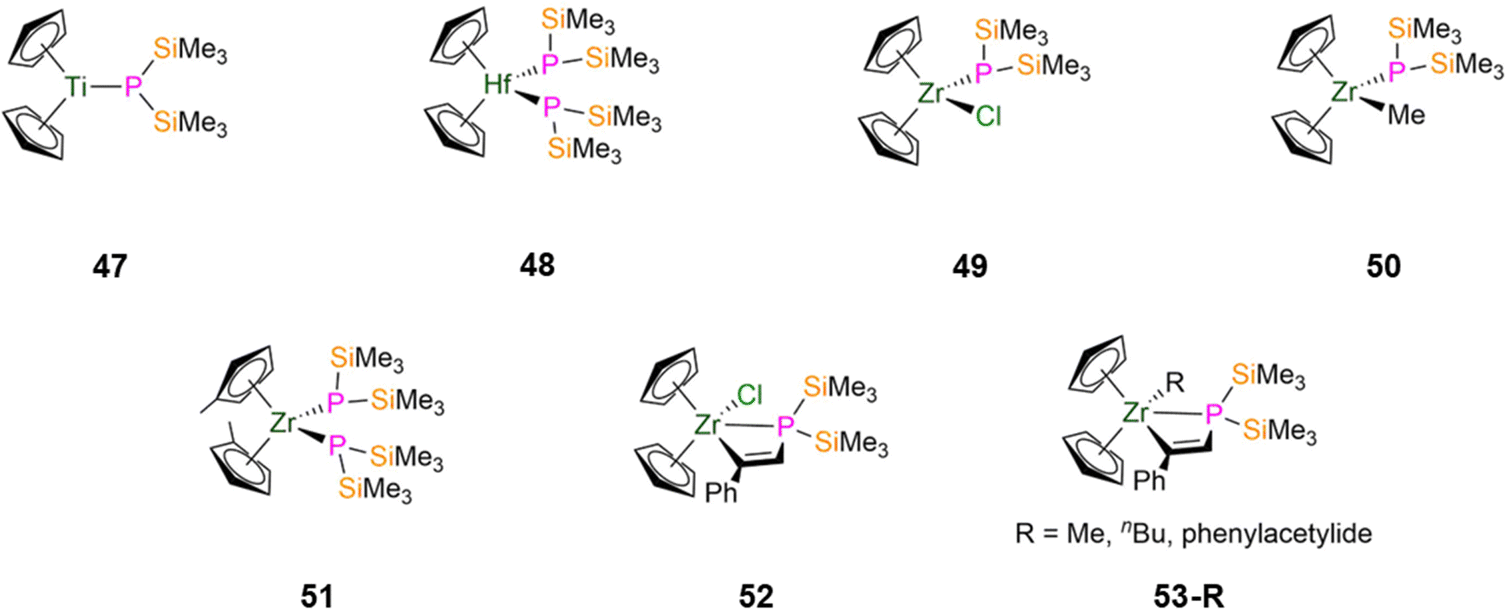 | ||
| Fig. 14 Group 4 P′′ complexes [Ti(Cp)2(P′′)] (47), [Hf(Cp)2(P′′)2] (48), [Zr(Cp)2(Cl)(P′′)] (49), [Zr(Cp)2(Me)(P′′)] (50), [Zr(C5H4Me)2(P′′)2] (51), [Zr(Cp)2(Cl){P(SiMe3)2C(H)C(Ph)}] (52), [Zr(Cp)2(R){P(SiMe3)2C(H)C(Ph)}] (53-R; R = Me, nBu, CCPh). | ||
Between 1992–1995, Hey-Hawkins synthesised zirconocene alkenyl complexes that feature P′′ incorporated into metallacycles.94,95 The insertion reaction of [Zr(Cp)2(P′′)(Cl)] with phenylacetylene in toluene at reflux gave [Zr(Cp)2(Cl){P(SiMe3)2C(H)C(Ph)}] (52, Fig. 14), which can undergo subsequent salt metathesis reactions with RLi (R = Me, nBu, CCPh) to yield [Zr(Cp)2(R){P(SiMe3)2C(H)C(Ph)}] (R = Me, nBu, CCPh, 53-R; Fig. 14); 53-nBu was not structurally characterised.94,9531P{1H} NMR spectroscopy showed modest changes in chemical shifts when Cl is substituted for an R group (R = Cl, −179.5 ppm; Me, −179.3 ppm; nBu, −176.6 ppm; CCPh, −189.9 ppm).94,95 For all of these complexes a doublet is observed in the 1H NMR spectra for the alkenyl proton, with 1JPH coupling constants ranging from 8–12 Hz.94,95 The alkenyl moieties exhibit the Z-configuration in the solid state, and the Zr–P distances (52: 2.855(4) Å; 53-Me: 2.915(3) Å; 53-CCPh: 2.774(3) Å) indicate that Zr–P interactions persist in solution for these complexes.94,95 The phosphorus atoms exhibit pyramidal geometries in 52 and 53-R, due to steric effects causing wide Si–P–Si angles.94,95
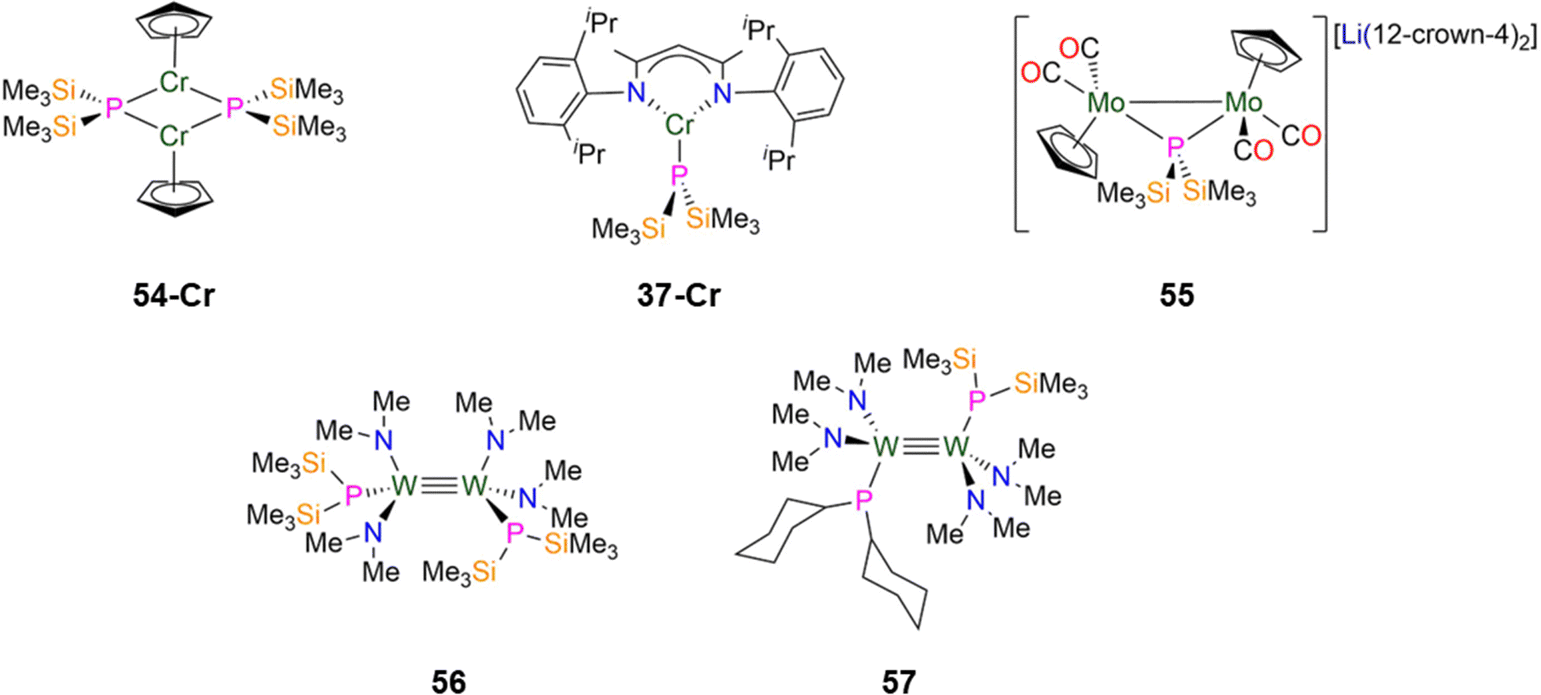 | ||
| Fig. 15 Group 6 P′′ complexes [Cr(Cp)(μ-P′′)]2 (54-Cr), [Cr(DippNacnac)(P′′)] (37-Cr; DippNacnac = CH{(CH3)2CN(Dipp)}2; Dipp = C6H3iPr2-2,6), [Li(12-crown-4)][{Mo(Cp)(CO)2}2(μ-P′′)] (55), [1,2-W2(P′′)(NMe2)4] (56), [1,2-W2(PCy2)(P′′)(NMe2)4] (57). | ||
In 2024, Weigend and Hänisch synthesised a Cr(II) P′′ complex, [Cr(DippNacnac)(P′′)] (37-Cr, Fig. 15) by the salt metathesis reaction of the Cr(II) precursor [Cr(DippNacnac)(Cl)] with one equivalent of LiP′′ in Et2O at −78 °C.140 Complex 37-Cr features a nearly planar P atom (Σ angles = 356°), which allows its lone electron pair to act as a π-donor into a vacant Cr 3d orbital, leading to a short Cr–P bond length of 2.3641(3) Å.140
In 2021, Scheer synthesised the dinuclear Mo(I) P′′ complex, [Li(12-crown-4)2][{Mo(Cp)(CO)2}2(μ-P′′)] (55, Fig. 15) by the addition of LiP′′ to [Mo(Cp)(CO)2]2 in THF; the addition of one equivalent of either AsCl3 or SbCl3 to 55 gives the mixed-dipnictogen complexes [{Mo(Cp)(CO)2}2(μ-PE)] (E = As, Sb), releasing two equivalents of SiMe3Cl in the process.97 The Mo–P distances in 55 are 2.4304(6) Å, which is between that expected for a single or double bond, and the phosphorus atom exhibits a distorted tetrahedral geometry. A long Mo–Mo bond length in 55 (3.1890 Å) is observed compared to the starting material [Mo(Cp)(CO)2]2 (2.4477(12) Å), due to the degradation of the MoMo triple bond to a Mo–Mo single bond.97
In 1992, Chisholm synthesised two examples of dinuclear W(III) complexes that exhibit tungsten-tungsten triple bonds and incorporate both P′′ and amide ligands.98 The salt metathesis reaction of [1,2-W2(Cl)2(NMe2)4] with two equivalents of LiP′′ gave [1,2-W2(P′′)2(NMe2)4] (56), whilst combination of the mono-substituted precursor [1,2-W2(Cl)(P′′)(NMe2)4] with LiPCy2 gave [1,2-W2(PCy2)(P′′)(NMe2)4] (57, Fig. 15).98 Complex 56 exhibits an anti-conformation in the solid-state across the triple bond, with trigonal planar nitrogen atoms and pyramidal phosphorus atoms.98 The WW triple bond lengths of 2.2989(9) Å (56) and 2.3016(10) Å (57) are similar to the mean values for WW triple bonds deposited on the CCDC (2.32(5) Å)9 and the W–P distances are statistically equivalent at 2.423(3) Å (56) and 2.425(4) Å (57).98 These complexes were characterised by both 1H and 31P{1H} NMR spectroscopy, with the latter showing a dynamic equilibrium in solution between the anti- and gauche-conformations; the 31P chemical shifts are −106.7 ppm (56) and −106.9 ppm (57) for the former and −88.3 ppm (56) and −96.3 ppm (57) for the latter.98 It was noted that upon lowering the temperature from 295 to 193 K, the 31P chemical shifts move to a higher field and a change of line shape is observed, which was attributed to inversion of the phosphorus centres.98
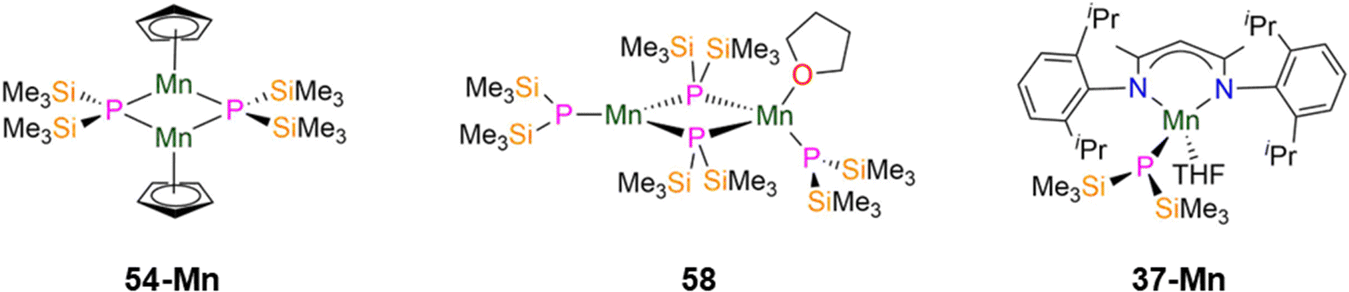 | ||
| Fig. 16 Group 7 P′′ complexes [Mn(Cp)(μ-P′′)]2 (54-Mn), [{Mn(P′′)(μ-P′′)2}{Mn(P′′)(THF)}] (58), [Mn(DippNacnac)(P′′)(THF)] (37-Mn; DippNacnac = CH{(CH3)2CN(Dipp)}2; Dipp = C6H3iPr2-2,6). | ||
In 1993, Buhro and co-workers reported the synthesis of the dinuclear Mn(II) P′′ complex [{Mn(P′′)(μ-P′′)2}{Mn(P′′)(THF)}] (58, Fig. 16) from the protonolysis reaction of [Mn(N′′)2(THF)] with two equivalents of HP′′ in hexane.83 In the solid state 58 consists of one trigonal planar three-coordinate Mn centre with two bridging and one terminal P′′, and one four-coordinate distorted tetrahedral Mn centre with two bridging and one terminal P′′, together with a coordinated THF. It was noted that the bound THF in 58 can be removed when this complex is exposed to a dynamic vacuum.83 The central Mn2P2 core in 58 is nearly planar and the phosphorus atoms of the terminal P′′ adopt pyramidal geometries (mean Σ angles 334.2(3)°).83 The bridging Mn–P distances range from 2.493(2)–2.565(2) Å, whereas the terminal Mn–P bond lengths are 2.417(3) Å for the three-coordinate Mn and 2.461(2) Å for the four-coordinate Mn centre.83 Due to the paramagnetic nature of complex 58, NMR spectroscopic data were intractable, but Evans method magnetic susceptibility measurements gave a μeff value of 3.33μB per Mn atom; this is lower than the spin-only value for a high-spin system with S = 5/2 (5.92μB) and was ascribed to the Mn centres being antiferromagnetically coupled.83
In 2024 Weigend and Hänisch synthesised the Mn(II) P′′ complex [Mn(DippNacnac)(P′′)(THF)] (37-Mn, Fig. 16), in an analogous manner to 37-Cr discussed in the previous section.140 The structure of 37-Mn is similar to 37-Cr but THF is additionally bound to Mn in the solid state; the Mn–P bond length of 37-Mn (2.461(1) Å) is longer than the corresponding Cr–P distance seen in 37-Cr and is similar to the mean Mn–P bond lengths found in 54-M (2.5075(5) Å) and 58 (2.461(2) Å).83,85
In 1992, Weber reported the synthesis of a series of Cp-substituted Fe(II) P′′ complexes [Fe(C5R5)(CO)2(P′′)] (59-R; C5R5 = C5EtMe4, 59-EtMe4; C5nBuMe4, 59-nBuMe4; C5H3tBu2-1,3, 59-1,3-tBu2H3, Fig. 17) by the separate salt metathesis reactions of the parent monobromide complexes, [Fe(C5R5)(CO)2(Br)] (R5 = EtMe4, nBuMe4, 1,3-tBu2H3) with LiP′′.99f The syntheses of three analogous Ru(II) P′′ complexes [Ru(C5R5)(CO)2(P′′)] (R5 = 1,2,4-iPr3H2, 1,3-tBu2H3, 1,3-(SiMe3)2H3) were reported by Weber in 1994 but again these complexes were not structurally authenticated.99g We note that only 59-EtMe4 was structurally characterised by single crystal XRD in the former paper,99f thus comparative discussions of 59-R focus on NMR and IR spectroscopy. The Fe–P distance in 59-EtMe4 is 2.359(3) Å and in the solid state the phosphorus atom has a pyramidal geometry (329.2(2)°), with the trimethylsilyl groups eclipsed with respect to the CO ligands. The 31P{1H} NMR spectra of 59-R exhibit singlets at −219.1 ppm (59-EtMe4), −218.6 ppm (59-nBuMe4) and −266.4 ppm (59-1,3-tBu2H3), whilst the IR spectra contain CO stretching modes at 1986 and 1939 cm−1 (59-EtMe4), 1988 and 1935 cm−1 (59-nBuMe4), and 1995 and 1944 cm−1 (59-1,3-tBu2H3). It was deduced that a reduced donor strength for the 1,3-bis-tert-butyl-substituted C5R5 ligand gives rise to weaker M–C π-backbonding and therefore stronger CO bonding in 59-1,3-tBu2H3. Finally, Weber reported that the separate reactions of 59-R or related Fe(II) and Ru(II) P′′ complexes with Mes*PCl2 gave M(II) diphosphene complexes,99f,g analogously to the reactions described above.99e
 | ||
| Fig. 17 Group 8 P′′ complexes [Fe(C5R5)(CO)2(P′′)] (59-R; C5R5 = C5EtMe4, 59-EtMe4; C5nBuMe4, 59-nBuMe4; C5H3tBu2-1,3, 59-1,3-tBu2H3). | ||
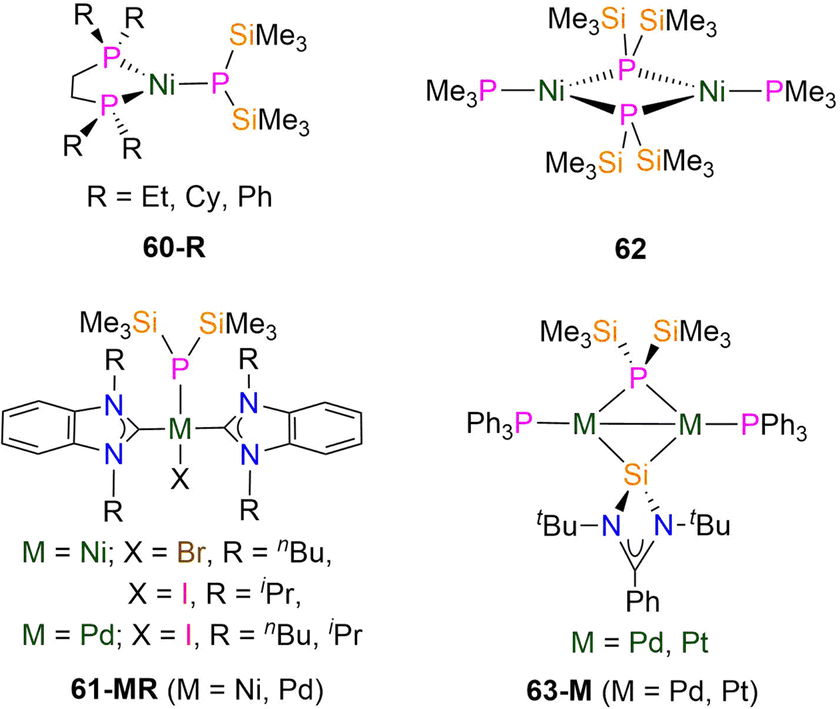 | ||
| Fig. 18 Group 10 P′′ complexes [Ni(κ2-drpe)(P′′)] (60-R; drpe = R2PCH2CH2PR2, R = Et, Cy, Ph), [M(X)(R2-bimy)2(P′′)] (61-MR. M = Ni, X = Br, R = nBu, 61-NinBu; M = Ni, X = I, R = iPr, 61-NiiPr; M = Pd, X = I, R = nBu, 61-PdnBu; iPr, 61-PdiPr; bimy = benzimidazole-2-ylidene), [Ni(PMe3)(μ-P′′)]2 (62), [{M(PPh3)}2(P′′){Si[N(tBu)]2CPh}] (63-M; M = Pd, Pt). | ||
In 1982, Schäfer reported the synthesis of the dimeric Ni(I) P′′ complex [Ni(PMe3)(μ-P′′)]2 (62, Fig. 18) by the decomposition of [Ni(PMe3)2(P′′)2] at room temperature to form both [Ni(PMe3)2{μ-P(SiMe3)}2] and [Ni(PMe3)(μ-P′′)]2.102 In the solid state 62 exhibits a planar central Ni2P2 ring in which each Ni atom is three-coordinate and bound to two bridging P′′ and one terminal trimethylphosphine.102 The Ni–P bond lengths within the central core are 2.186(1) Å and the Ni–P distances for the terminal phosphines are 2.129(2) Å; the Ni⋯Ni separation is 2.381(1) Å.102 In 2013 Inoue reported the synthesis of the dinuclear M(I) P′′ complexes [{M(PPh3)}2(P′′){Si[N(tBu)]2CPh}] (63-M; M = Pd, Pt; Fig. 18); 63-Pt was characterised by both NMR spectroscopy and single crystal XRD, whereas the formation of 63-Pd was determined by NMR spectroscopy and high resolution mass (HRMS) spectrometry only.103 The 1H NMR spectra of 63-M exhibit singlets (0.92 ppm, 63-Pt; 0.86 ppm, 63-Pd) for the tBu groups, as well as a doublet (0.46 ppm, 3JPH = 5.2 Hz, 63-Pt; 0.54 ppm, 3JPH = 5.2 Hz, 63-Pd) for the trimethylsilyl groups. In the 31P{1H} NMR spectrum of 63-Pt a doublet at 56.0 ppm (1JPtP = 5137 Hz and 2JPtP = 60.5 Hz, 33.8% abundant 195Pt I = ½) and a triplet at −94.6 ppm (1JPtP = 1633 Hz) are observed that respectively correspond to PPh3 and P′′, and these are seen at 41.2 and −163.4 ppm, respectively, for 63-Pd.103 The Pt–Pt bond length of 63-Pt (2.6466(5) Å) falls within the reported range of Pt–Pt bonds (2.4059(6)-2.7183(10) Å),9 whilst the P′′ Pt–P bond lengths (2.360(2) and 2.367(2) Å) are longer than the corresponding distances for the coordinated phosphines of 2.222(2) and 2.213(2) Å.103
:1 ratio of iPr2-bimy: P′′, together with one resonance in the 31P{1H} NMR spectrum at −261 ppm; the analogous signal for 65 is at δP = −235 ppm.104,105 The 1H NMR spectrum of 66-Cu showed that the solid-state structure was retained in solution due to the 1:1 ratio of IPr: P′′ ligands. The 31P{1H} NMR spectra of 66-M each show one resonance, at −268 ppm for the Cu analogue and −235.7 ppm for the Au congener.104,105 In 2023 Liu reported the synthesis of [Au(CAACMeEt)(P′′)] (67; CAACMeEt = dimethyl-diethyl-cyclic(alkyl)(amino)carbene, Fig. 19) by the reaction of [Au(CAACMeEt){P(Ph)2}] with KP′′.106 This synthetic route is remarkable as it involves Au–P bond cleavage occurring, despite the strong covalency between Au and P due to relativistic effects which result in Au having the highest electronegativity of any d-transition metal (Pauling χ = 2.54). As with 66-M, 67 exhibits a near-linear geometry (C–Au–P: 174.45(5)°) with a distorted trigonal pyramidal geometry about the phosphorus atom and an Au–P bond length of 2.3253(5) Å. Complex 67 has one resonance in its 31P{1H} NMR spectrum at −233.1 ppm.106
 | ||
| Fig. 19 Group 11 P′′ complexes [Cu(iPr2-bimy)2(P′′)] 64 (iPr2-bimy = 1,3-di-isopropylbenzimidazole-2-ylidene), [Au(iPr2-bimy)(P′′)] (65),; [M(IPr)(P′′)] (66-M; M = Cu, Au), IPr = (1,3-bis(2,6-diisopropylphenyl)-2-ylidene), [Au(CAACMeEt)(P′′)] (67, CAACMeEt = dimethyl-diethyl-cyclic(alkyl)(amino)carbene), [M(μ-P′′)]6 (68-M; M = Cu, Ag). | ||
Group 11 metal P′′ cluster complexes have been synthesised in order to explore their potential as precursors for semiconductor materials. The group 11 hexameric cyclic complexes [M(μ-P′′)]6 (68-M; M = Cu, Ag; Fig. 19) were synthesised in 2015 by Corrigan by the separate reactions of parent polymeric [M(StBu)]n with equimolar P(SiMe3)3via the cleavage of P–Si and M–S bonds and the formation of tBuSSiMe3 and 68-M.104 In the solid state the P atoms of 68-M form the corners of a planar hexagonal structure, and the two-coordinate metal centres form the sides.104 The M–P bond lengths range from 2.2043(16)–2.2094(15) Å for 68-Cu and 2.393(1)–2.411(2) Å for 68-Ag, and the P–M–P angles deviate slightly from linearity in both complexes (∼178°, 68-Cu; ∼177°, 68-Ag).104 The phosphorus atoms in 68-M exhibit distorted tetrahedral geometries such that the trimethylsilyl groups are equally arranged above and below the plane of the ring. It was noted that the voids above and below the centre of the ring are occupied by THF lattice solvent molecules, which is believed to be important for the formation of the cyclic structures, and this sensitivity is in line with the loss of solvent leading to discolouration of solutions of 68-M.104 NMR spectroscopy showed that the symmetrical structures are retained in solution for both complexes, with only one resonance in each 1H NMR spectrum at 0.50 ppm (68-Cu) and 0.48 ppm (68-Ag), and each 31P{1H} NMR spectrum at −149 ppm (68-Cu) and −236 ppm (68-Ag).104
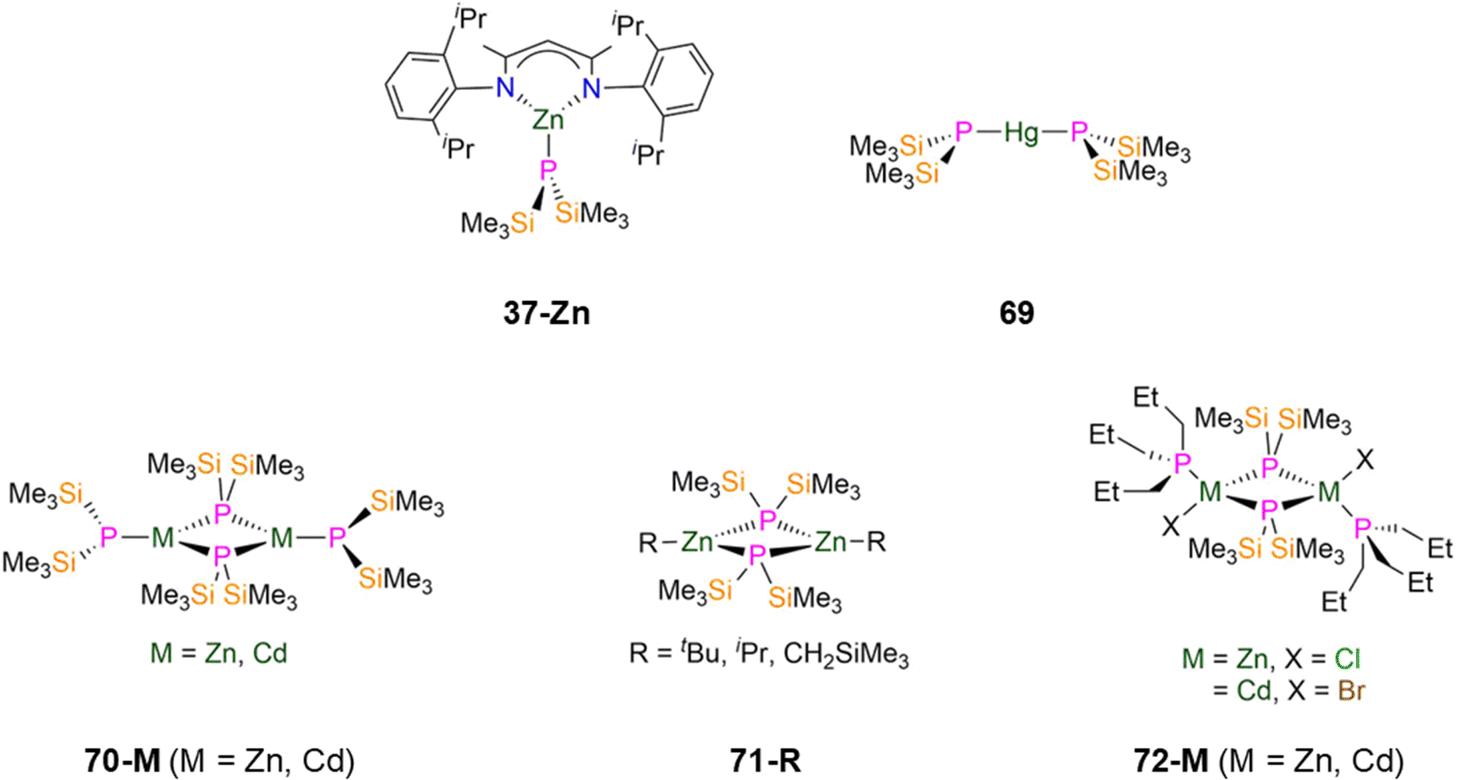 | ||
| Fig. 20 Monomeric and dimeric group 12 P′′ complexes [Zn(DippNacnac)(P′′)] (37-Zn; DippNacnac = CH{(CH3)2CN(Dipp)}2; Dipp = C6H3iPr2-2,6), [Hg(P′′)2] (69), [M(P′′)(μ-P′′)]2 (70-M, M = Zn, Cd), [Zn(R)(μ-P′′)]2 (71-R, R = tBu, iPr, CH2SiMe3), [M(PnPr3)(X)(μ-P′′)]2 (72-Zn: M = Zn, X = Cl; 72-Cd: M = Cd, X = Br). | ||
Many dimeric group 12 complexes have been reported with bridging P′′, with varying terminal ligands such as alkyls, aryls, halides, phosphines and phosphides. In 1993 Buhro synthesised the homoleptic M(II) P′′ complexes [M(P′′)(μ-P′′)]2 (70-M, M = Zn, Cd, Fig. 20) by the protonolysis reaction of [M(N′′)2] (M = Zn, Cd) with HP′′.83 Single crystal XRD studies of 70-M showed that each metal in the dimer has a distorted trigonal planar geometry and is bound by two bridging and one terminal P′′, with the terminal phosphorus atom being pyramidal; the distorted rectangular M2P2 cores in 70-M have bridging M–P–M angles which are closer to 90° rather than the usual 120°. The M–Pbridging (2.419(1) and 2.421(1) Å for 70-Zn; 2.575(1) and 2.612(1) Å for 70-Cd) and M–Pterminal (2.295(1) Å, 70-Zn; 2.459(1) Å, 70-Cd) bond lengths also lie within the expected range for bridging and terminal Zn–P and Cd–P bonds, respectively (CSD: Zn–P (2.212(3)–2.9159(7) Å), Cd–P (2.2379(8)–2.800(6) Å)).9,83 Both complexes exhibit fluxional properties in solution, with low temperature NMR data (230 K) consistent with the presence of the dimer, as evidenced by two resonances in the 31P{1H} NMR spectra for bridging and terminal phosphides (−183.0 ppm, Pbridging, −237.3 ppm Pterminal, 70-Zn; −180.1 ppm, Pbridging, −229.5 ppm, Pterminal, 70-Cd).83 Upon conducting high temperature NMR experiments, line broadening and coalescence of these two resonances is seen for both complexes, which was assigned to bridging-to-terminal site exchange with a calculated barrier of ΔG‡360 = 14.3(2) kcal mol−1 (70-Zn) and ΔG‡321 = 12.7(6) kcal mol−1 (70-Cd).83 The mechanism of this exchange was proposed to be dissociative where the dimers dissociate into the respective monomers and reassociate, or that one of the M–Pbridging bonds is broken, the remaining M–Pbridging bond rotates, and then the cleaved M–Pbridging bond reforms to give the exchanged dimer.83
The synthesis of the dimeric Zn(II) P′′ complexes [Zn(R)(μ-P′′)]2 (71-R; R = tBu, iPr, CH2SiMe3; Fig. 20) was reported in 1995 by Westerhausen by the reaction of parent ZnR2 (R = tBu, iPr, CH2SiMe3) with HP′′; however 71-tBu was not structurally characterised.107 Complexes 71-iPr and 71-CH2SiMe3 are dimers that consist of two trigonal planar Zn centres with bridging P′′ and terminal alkyl groups, with nearly planar Zn2P2 cores. The Zn–P bond lengths are 2.405(3) and 2.416(3) Å for 71-iPr and 2.394(1) and 2.436(1) Å for 71-CH2SiMe3.10731P{1H} NMR spectroscopy showed one resonance for each complex, at −216.3 ppm (71-iPr), −205.8 ppm (71-CH2SiMe3) and −216.7 ppm (71-tBu).107 Dimeric group 12 metal P′′ complexes [M(PnPr3)(X)(μ-P′′)]2 (72-Zn: M = Zn, X = Cl; 72-Cd: M = Cd, X = Br; Fig. 20) featuring both a terminal phosphine and halide were synthesised in 1996 by Fenske via the separate reactions of MX2 (M = Zn, X = Cl; M = Cd, X = Br) with P(SiMe3)3 in the presence of PnPr3.108 Both complexes exhibit distorted tetrahedral metal centres in the solid state, with the central M2P2 rings showing planar geometries with typical M–Pbridging (2.426(1) and 2.447(1) Å for 72-Zn; 2.571(1) and 2.609(1) Å for 72-Cd) and M–Pterminal (2.408(1) and 2.430(1) Å for 72-Zn; 2.584(1) and 2.571(1) Å for 72-Cd) bond distances.108
In 1996, Fenske reported the synthesis of a Zn(II) P′′ cluster complex, [{Zn(Cl)(MeCN)(μ-P′′)2}2{Zn(μ-Cl)}2] (73, Fig. 21), by the reaction of ZnCl2 with equimolar P(SiMe3)3 in MeCN.108 Single crystal XRD revealed that the four Zn and four P atoms form a Zn4P4 eight membered ring, with two chlorides bridging two of the Zn centres to give a Zn2Cl2 core. The two Zn atoms that are not involved in the central four-membered ring are distorted tetrahedral and coordinated by two bridging P′′ (mean Zn–P: 2.405(2) Å), a terminal chloride and MeCN, whereas the coordination spheres of the other two Zn atoms are completed by bridging P′′, with a shorter mean Zn–P bond length (2.343(2) Å) giving much more strongly distorted tetrahedra (P–Zn–P: 153.88(4)°). Solutions of 73 decompose upon warming to room temperature, precluding the collection of NMR spectroscopic data that could be confidently assigned.108 In the same report a Cd(II) cluster complex, [NnBu4]2[Cd4(I)8(P′′)2] (74, Fig. 21), was formed from the reaction of [Cd(I)2{P(SiMe3)3}]2 with NnBu4I, with ISiMe3 eliminated as a byproduct.108 Complex 74 consists of four C atoms with four bridging iodides and two bridging P′′ to yield an adamantane-like skeleton; each distorted tetrahedral Cd atom also bears one terminal iodide.108 The mean Cd–P bond distance is 2.507(7) Å, which is shorter than those found for other Cd–Pbridging bonds in 70-Cd (2.612(1) Å) and 72-Cd (2.609(1) Å).83,108 The trimeric Zn(II) P′′ complexes [Zn(R)(P′′)]3 (75-R; R = Me, Et, iPr, nBu, Fig. 21) were reported in 1994 by Westerhausen, via the addition of ZnR2 to HP′′. Single crystal XRD showed that these complexes contain six-membered Zn3P3 rings in twisted boat conformations, with the three Zn atoms having near trigonal planar geometries with terminal alkyl groups, and the phosphorus atoms showing distorted tetrahedral geometries; the solid-state structure of 75-Et was not reported.107 In solution this series of complexes were found to exhibit equilibria with the dimeric complexes discussed earlier in this section (71-R), with the larger alkyl groups favouring the formation of dimers.107 For complex 75-Me, two of the phosphorus atoms are situated 0.525 Å and 0.857 Å above and below the plane, respectively, whereas in 75-nBu these two phosphorus atoms are located more symmetrically at ±0.640 Å.107 The mean Zn–P bond lengths of 2.390(3) Å (75-Me), 2.408(6) Å (75-iPr) and 2.388(4) Å (75-nBu) fall within the expected range for bridging Zn–P bonds (2.245(4)–2.8049(7) Å).9,107 Chemical shifts in the 31P{1H} NMR spectra for the trimeric complexes of −246.8 ppm (75-Me), −246.9 ppm (75-Et), −243.6 ppm (75-iPr), −246.0 ppm (75-nBu) are shifted to lower chemical shifts than the related dimers, which are seen at–216.3 ppm (71-iPr) and −216.7 ppm (71-tBu).107
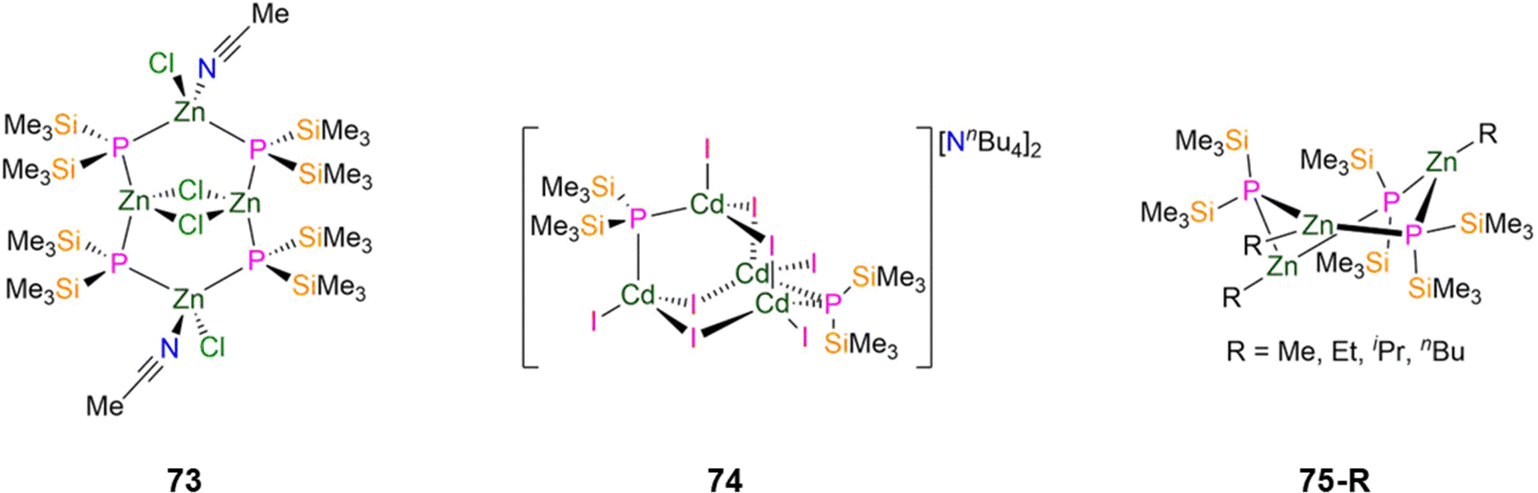 | ||
| Fig. 21 Group 12 P′′ cluster complexes [{Zn(Cl)(MeCN)(μ-P′′)2}2{Zn(μ-Cl)}2] (73), [NnBu4]2[Cd4(I)8(P′′)2] (74), [Zn(R)(P′′)]3 (75-R; R = Me, Et, iPr, nBu). | ||
 | ||
| Fig. 22 Heterometallic P′′ complexes [M(CO)n(μ-P′′){Al(dmap)(Me2)}] (76-M; M = Cr, n = 5, 76-Cr; M = Fe, n = 4, 76-Fe; M = Ni, n = 3, 76-Ni), [Zr(Cp)2(μ-P′′)2M(CO)n] (77-M; M = Mo, n = 4, 77-Mo; M = Ni, n = 2, 77-Ni). | ||
2.4 f-Block P′′ complexes
:1 ratio, with the major component assigned to pyramidal and the minor component to planar P environments.89 This is in contrast to the one resonance observed in the solution 31P{1H} NMR spectra of 46-Ln due to dynamic processes and broad resonances.89
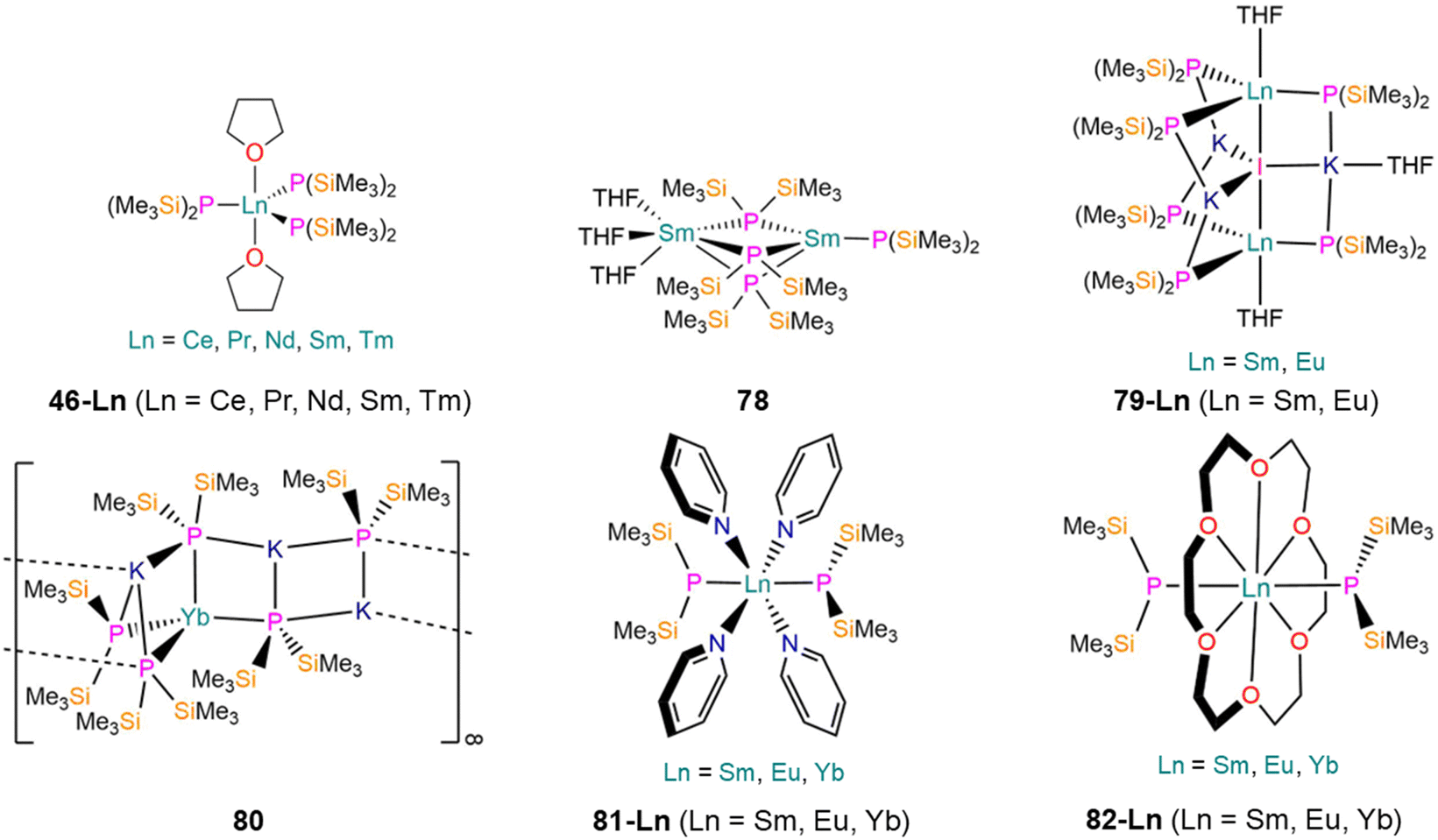 | ||
| Fig. 23 Ln P′′ complexes [Ln(P′′)3(THF)2] (46-Ln; Ln = Ce, Pr, Nd, Sm, Tm), [Sm(P′′)(μ-P′′)3Sm(THF)3] (78), [{Ln(P′′)3(THF)}2(μ-I)K3(THF)] (79-Ln; Ln = Sm, Eu), [KYb(P′′)3{μ-K(P′′)}2]∞ (80), trans-[Ln(P′′)2(py)4] (81-Ln; Ln = Sm, Eu, Yb), [Ln(P′′)2(18-crown-6)] (82-Ln; Ln = Sm, Eu, Yb). | ||
In 1996, Rabe reported the synthesis of the bimetallic Sm(II) complex [Sm(P′′)(μ-P′′)3Sm(THF)3] (78, Fig. 23) by the salt metathesis reaction of two equivalents of KP′′ with SmI2(THF)2 in THF at room temperature, analogously to the preparation of 14 discussed in Section 2.1.1.125 Single crystal XRD showed that 78 features one four-coordinate Sm ion with one terminal and three bridging P′′, and one six-coordinate Sm ion with three bridging P′′ and three bound THF. The N′′ analogue of 78, [Sm(N′′)2(THF)2],145 is monometallic, following a general trend that higher nuclearities tend to be observed for P′′ complexes compared to their N′′ counterparts.146 The solid state structure of 78 differs from those of the six-coordinate monometallic Ln(II) bis-(diphenyl)phosphide complexes, [Ln(PPh2)2(THF)4] (Ln = Sm, Yb) and [Sm(PPh2)2(N-methylimidazole)4],147 and bears more resemblance to the solvent-free Eu(II) N′′ ‘ate‘ complex, [Eu(N′′)(μ-N′′)2Na].148 The terminal Sm–P bond length of 78 (3.027(3) Å) is similar to the mean bridging Sm–P distances (3.039(5) Å), and all are shorter than those reported for six-coordinate [Sm(η1-dibenzophospholyl)2(THF)4] (3.1908(6) Å),148 [Sm(PPh2)2(N-methylimidazole)4] (3.139(3) Å),147 and [Sm(η5-C8H4PMe2-2,3)2(THF)2] (3.0775(1) Å).149 VT 1H NMR spectra of 78 were collected in both d8-toluene and d8-THF in order to determine its structure in solution, but these studies were inconclusive. In d8-THF at 293 K there is a singlet at 0.42 ppm (ν1/2 = 10 Hz), and at 173 K this becomes two singlets at 3.94 ppm (ν1/2 = 1 6 Hz) and −0.01 ppm (ν1/2 = 8 Hz). In d8-toluene at 293 K there is one singlet at 0.99 ppm (ν1/2 = 20 Hz), but at 193 K seven broad resonances were observed at 13.60, 8.50, 6.10, 3.80, 3.10, −0.54 and −2.93 ppm.125
In 2024, Mills and co-workers showed that the addition of two or three equivalents of KP′′ to [LnI2(THF)2] (Ln = Sm, Eu, Yb) in Et2O at −78 °C gave the dimeric Ln(II) P′′ complexes [{Ln(P′′)3(THF)}2(μ-I)K3(THF)] (79-Ln; Ln = Sm, Eu) and polymeric [KYb(P′′)3{μ-K(P′′)}2]∞ (80), Fig. 23.115 The addition of pyridine to either 79-Ln or 80 at room temperature gave the monomeric pyridine-solvated Ln(II) P′′ complexes trans-[Ln(P′′)2(py)4] (81-Ln; Ln = Sm, Eu, Yb), whereas the addition of 18-crown-6 to 79-Ln and 80 at room temperature afforded [Ln(P′′)2(18-crown-6)] (82-Ln; Ln = Sm, Eu, Yb), Fig. 23.115 The dimeric Ln(II) ‘ate’ complexes 79-Ln consist of two {KLn(P′′)3(THF)} fragments with KI encapsulated between them. The Ln centres exhibit distorted trigonal bipyramidal geometries with three equatorial P′′, one axial THF and one axial iodide, which bridges the two fragments.115 The mean Ln–P bond lengths in 79-Ln (Ln = Sm, 3.033(7) Å; Eu, 3.038(7) Å) are shorter than the bridging Sm–P′′ bonds in 78 but are statistically equivalent to the terminal Sm–P′′ bond length of 78.115,125 Due to the paramagnetic nature of Sm(II) and Eu(II) ions, no resonances were observed in the 31P{1H} NMR spectra of 79-Ln.115 In the solid state 80 is a 1D coordination polymer with P′′ bridging Yb and K cations.115 The Yb centres are each coordinated by four P′′ and exhibit highly distorted tetrahedral geometries (P–Yb–P angles: 93.79(13)°, 98.4(2)°, 98.5(2)°, 105.94(14)°, 126.55(14)° and 125.8(2)°).115 The Yb–P bond lengths in 80 range between 2.837(7)–3.043(3) Å, with the mean Yb–P bond length (2.952(10) Å) being shorter than the Ln–P distances in 79-Ln due to the smaller ionic radius of Yb(II) compared with Sm(II) and Eu(II).115 Despite Yb(II) being a diamagnetic ion, it was noted that no resonances could be observed in the 171Yb{1H} NMR spectrum of 80. However, multiple resonances were observed in the 1H, 13C, 29Si DEPT90 and 31P{1H} NMR spectra. VT 1H and 31P{1H} NMR experiments between 213–323 K did not provide signals that could be confidently assigned to 80, which was attributed to rapid aggregation processes.115 Upon cooling, the major signal at δP = −219.0 ppm broadens further and two new signals form at ca. −211 and −243 ppm, but no 171Yb (14.3% abundant, I = ½) satellites could be assigned. It was therefore concluded that 80 converts into a complex mixture of aggregates in solution, which are in constant dynamic equilibria.115
In the solid state 81-Ln exhibit distorted octahedral geometries, with trans-configurations consisting of two axial P′′ and four equatorial pyridines.115 Complexes 81-Sm and 81-Eu are more distorted from the ideal octahedral geometry than 81-Yb, with the former exhibiting three N–Ln–N angles between 74.87(12)–77.75(14)° and a fourth of 133.3(2)° for 81-Sm and 131.81(12)° for 81-Eu, whereas for 81-Yb the range of N–Ln–N angles is much closer to ideal 90° (86.16(8)–92.09(7)°).115 This was attributed to the larger Sm(II) and Eu(II) ions allowing for two Me groups from P′′ ligands to form additional electrostatic interactions, with Ln⋯C distances of 3.809(5) Å (81-Sm) and 3.840(4) Å (81-Eu).115 Unlike for 80, all multinuclear NMR spectra could be assigned for the diamagnetic complex 81-Yb, though it was noted that drops of pyridine needed to be added to the sample in order to provide solution stability.115 The 13C{1H} NMR spectrum of 81-Yb exhibits silyl group resonances that are virtual triplets due to splitting by strongly coupled 31P nuclei (δC = 7.85 ppm, 2JPC = 5.4 Hz).115 These higher order effects are also seen in the 29Si DEPT90 NMR spectrum (δSi = 1.58 ppm, 1JPSi = 16 Hz).115 The 31P{1H} NMR spectrum of 81-Yb exhibits a resonance at −253.93 ppm, with satellites due to 171Yb nuclei with a 1JYbP coupling constant of 925 Hz. 147 The 171Yb{1H} NMR spectrum of 81-Yb shows a triplet resonance at 1075.50 ppm, with the same 1JYbP coupling constant as seen in the 31P{1H} NMR experiment.115
For complexes 82-Ln, distorted hexagonal bipyramidal geometries are observed, with mutually trans-P′′, and 18-crown-6 coordinated about the equatorial plane. Longer Ln–P bond lengths are observed for 82-Ln (82-Sm: 3.089(3) Å; 82-Eu: 3.086(6) Å; 82-Yb: 2.9662(11) Å) compared to those seen in 79-Ln, which is attributed to the presence of the puckered 18-crown-6 ligand.115 The P–Ln–P angles in 82-Ln deviate from linearity, with angles of 161.65(7)° (82-Sm), 154.80(11)° (82-Eu) and 173.98(4)° (82-Yb), again due to the puckered macrocyclic ligand.115 As for 81-Yb, the 13C{1H} NMR spectrum of 82-Yb exhibits virtual triplets for the silyl groups due to coupling with the 31P nuclei (δC = 8.43 ppm, 2JPC = 5.6 Hz),115 and higher order effects were also seen in the 29Si DEPT90 NMR spectra of 82-Yb (δSi =1.98 ppm, 1JPSi = 17 Hz), manifesting as virtual triplets.115 The 31P{1H} NMR spectra of 82-Yb exhibits a resonance at −265.58 ppm, with satellites to 171Yb nuclei with a 1JYbP coupling constant of 977 Hz. Similarly to 81-Yb, the 171Yb{1H} NMR spectra of 82-Yb exhibits the expected triplet resonance at 176.88 ppm.115
 | ||
| Fig. 24 An P′′ complexes [An(P′′)(Cp*)2(Cl)] (83-An; An = Th, U), [An(P′′)(Cp*)2(Me)] (84-An; An = Th, U), [An{P(SiMe3)(SiMe2CH2)}(Cp*)2] (85-An; An = Th, U), [An(TrenDMBS)(P′′)] (86-An; An = Th, U), [An(TrenTIPS)(P′′)] (87-An; An = Th, U). TrenDMBS = {N(CH2CH2NSiMe2tBu)3}, TrenTIPS = {N(CH2CH2NSiiPr3)3}. | ||
In 2018, Liddle, Scheer and co-workers reported the synthesis of a series of An(IV) pnictogen complexes containing bulky triamidoamine ancillary ligands, including the P′′ complexes [An(TrenDMBS)(P′′)] (86-An) and [An(TrenTIPS)(P′′)] (87-An) (An = Th, U) (TrenDMBS = {N(CH2CH2NSiMe2tBu)3}, TrenTIPS = {N(CH2CH2NSiiPr3)3}, Fig. 24) by the reaction of the respective triamidoamine precursor complexes [An(TrenR)(L)][BPh4] (An = Th, L = DME; U, L = THF) with one equivalent of KP′′.117 It was noted that for 86-Th the Th–P bond length (2.9406(11) Å) was 0.08 Å longer than the U–P bond in 86-U (2.8646(14) Å) despite the single bond covalent radius of Th only being 0.05 Å larger than U.90 Conversely, the U–P bond distance in 87-U (2.8391(9) Å) is shorter than that seen for 87-U, and the geometry about the P atom is more planar in the former complex (359.94(5)°) than the latter (354.06(8)°); this was determined by the sum of the angles about the phosphorus atom and the deviation of this value from 360°, which can be attributed to the more sterically demanding TrenTIPS ligand.117 As part of a study to probe the propensity of An–E (An = Th, U; E = P, As, Sb, Bi) bonds to undergo either homolytic cleavage or acid–base/dehydrocoupling to occur, 86-U and 87-U were heated to 80 °C and exposed to a 125 W UV lamp for 2 h. It was noted that both complexes are remarkably robust, and undergo <5% thermal and photolytic decomposition under these conditions, likely by homolytic cleavage, whereas acid–base/dehydrocoupling is the most likely decomposition route for more redox-robust Th(IV)–E bonds.
3. Bis(triisopropylsilyl)phosphide (P††) complexes
In accord with the paucity of structurally authenticated P′′ complexes compared to N′′ across the periodic table, there are even fewer examples of complexes containing bulkier bis(trialkylsilyl)phosphides.146,152–160 As bulkier bis(trialkylsilyl)amides such as bis(triisopropylsilyl)amide ({N(SiiPr3)2}, N††) are now starting to grow in popularity as ligands in low-coordinate s-block and f-block chemistry,5,161 we also cover bis(triisopropylsilyl)phosphide ({P(SiiPr3)2}, P††) chemistry here; only seven structurally authenticated P†† complexes have been reported to date.In 1996, Driess reported the synthesis of [{Li(μ-P††)}3{Li[μ-P(SiiPr3)H]}] (88) by the lithiation of HP(SiiPr3)2 with nBuLi in toluene.155 Complex 88 is an eight-membered ring consisting of four Li centres, three bridging P†† and one bridging HP(SiiPr3) ligand.155 Each Li atom in 88 is two-coordinate and the mean Li–P bond length is 2.44(5) Å which is shorter than those seen for the ladder-like Li–P′′ complexes 3 and 4; bulky P†† supresses the rearrangement to a ladder-like conformation. The 31P NMR spectrum of a solution of 88 in d8-toluene at −70 °C exhibited two singlets at −338 and −370 ppm and a broad doublet at −351 ppm (1JPH = 170 Hz). This provided evidence that in solution 88 is a mixed aggregate made up of various [Li{P(SiiPr3)2}(H)] and [Li(P††)] building blocks.155 In 2004, von Haenisch published the solid-state structure of [Li(μ-P††)(THF)]2 (89), Fig. 25, via a CSD communication.162 Later, in 2005, Westerhausen also reported the synthesis of 89 by analogous methods to that of 88, but with THF used as the solvent.163 Complex 89 is a dimeric complex consisting of two three-coordinate Li centres bridged by two P††, with each Li bound by one THF.163 The Li–P bond length in 89 is 2.533(6) Å, which falls within the range observed for Li–P′′ complexes discussed in this review. It is noted however that although this bond length is neither longer nor shorter than for Li–P′′ complexes, the coordination number of Li in 89 is only three, compared to four for 1.163 The 29Si{1H} NMR spectrum of 89 shows a single resonance at 21.6 ppm for the SiiPr3 groups, which exhibits coupling to the 31P nuclei (1JPSi = 49.7 Hz), and one resonance was observed in the 31P{1H} NMR spectrum at −374.7 ppm.163 In 2024, Kays and Mills separately reported the synthesis of NaP†† by the addition of two or three equivalents of SiiPr3Cl to a refluxing DME solution of red phosphorus, Na metal and naphthalene to yield crystals of either [Na(P††)(DME)2] (90), or [Na(μ-P††)(THF)]2 (91) following treatment with THF, Fig. 25.164,165 In contrast to P′′ chemistry, where Na salts are yet to be structurally authenticated (see above), the solid-state structures of both 90 and 91 were determined by single crystal XRD. Complex 90 consists of a five-coordinate Na centre with one P†† and two bound DME solvent molecules,164 whilst 91 is dimeric with a Na2P2 core and bridging P††, with the Na coordination spheres completed by a single THF each.165 The mean Na–P bond length in 90 is 2.824(3) Å,164 compared to 2.805(2) Å for 91,165 which fall within the mean bond lengths discussed for both Li–P′′ and K–P′′ complexes. The geometry of the phosphorus centre in 90 is trigonal pyramidal, with the sum of angles about the phosphorus centre adding to 332.53(10)°.164 The 29Si DEPT90 NMR spectrum of 90 or 91 in d8-THF contains a doublet at 20.65 ppm with 1JPSi = 60.2 Hz, whilst a singlet is observed in the 31P{1H} NMR spectrum at −384.26 ppm with satellites observed with 1JPSi = 59.8 Hz.164,165
 | ||
| Fig. 25 P†† complexes [{Li(μ-P††)}3{Li[μ-P(SiiPr3)H]}] (88), [Li(μ-P††)(THF)]2 (89), [Na(P††)(DME)2] (90), [Na(μ-P††)(THF)]2 (91), [W(P††)(CO)5][Li(THF)4] (92), [M(P††)2] (93-M; M = Zn, Cd, Hg), [Ln(P††)2(THF)3] (94-Ln; Ln = Sm, Eu), [Yb(P††)2(THF)2] (94-Yb). | ||
In 2005, Westerhausen reported the synthesis of a W(0) P†† complex by addition of a THF solution of [W(CO)5(THF)] to 89 to yield [W(P††)(CO)5][Li(THF)4] (92), Fig. 25.163 Complex 92 exhibits a distorted octahedral geometry with five coordinated CO and one P††.163 The W–P bond length of 92 is 2.6665(7) Å and the geometry about the phosphorus centre is trigonal pyramidal (350.38(6)°), which was noted to reflect the shift in the trans-CO stretching vibration to smaller wavenumbers (1904 cm−1).163 As phosphides are strong σ-donors but weak π-acceptors, the π-backbonding from the W to the trans-CO ligand is increased, which weakens this W–C bond.163 The 29Si{1H} NMR spectrum of 92 exhibits a triplet at 21.1 ppm with coupling to 31P of 1JPSi = 40.8 Hz, and a singlet resonance at −409.2 ppm is observed in its 31P{1H} NMR spectrum.163 In 2024, Kays reported the synthesis of the group 12 M(II) complexes [M(P††)2] (93-M; M = Zn, Cd, Hg) by the salt metathesis reactions of parent ZnCl2, CdI2 or HgBr2 with two equivalents of 90.165 The average M–P bond lengths of 2.2263(5) (93-Zn), 2.4215(10) (93-Cd) and 2.3938(7) (93-Hg) Å are consistent with those seen for the terminal P′′ ligands in 68 and 69-M.83,165 In addition, complexes 93-M exhibit an increasing P–M–P angle from Zn to Hg (93-Zn; 168.747(12)°, 93-Cd; 169.215(19)°, 93-Hg; 170.086(16)°).165 The 31P{1H} NMR spectra for 93-M exhibit singlets at −287.8, −284.0 and −209.2 for the Zn, Cd and Hg complexes respectively.165 For 93-Cd, the 113Cd NMR spectrum exhibited a triplet resonance at 137.68 ppm (1JCdP = 350 Hz, 12.2% abundant 113Cd I = ½) whereas the 199Hg NMR spectrum of 93-Hg contains a triplet at 13.2 ppm (1JHgP = 407.7 Hz, 16.9% abundant 199Hg I = ½).165 Interestingly, the 29Si NMR spectra for 93-Zn and 93-Cd display apparent doublets, whereas for 93-Hg the 29Si NMR spectrum is consistent with an AA′XX′ spin system with virtual coupling. This spectrum was accurately simulated using 1JPSi = 50.6 Hz, 3JP′Si = 0.0 Hz and 2JPP′ = 19.0 Hz.165
In 2024, Mills and co-workers reported the synthesis of a series of three Ln(II) P†† complexes by the addition of two equivalents of 90 to [LnI2(THF)2] (Ln = Sm, Eu, Yb) to yield [Ln(P††)2(THF)x] (Ln = Sm, x = 3, 94-Sm; Eu, x = 3, 94-Eu; Yb, x = 2, 94-Yb), Fig. 25.164 Complexes 94-Sm and 94-Eu exhibit five-coordinate Ln centres which are bound by two P†† and three THF, whereas 94-Yb features four-coordinate Yb bound by two P†† and two THF.164 The mean Ln–P bond lengths of 3.0336(13) (94-Sm), 3.0237(18) (94-Eu) and 2.8065(13) (94-Yb) Å are shorter than those seen for the Ln(II) P′′ complexes 79-M, 81-M and 82-M despite the increased bulk of the P†† ligand, though the coordination numbers of 94-Ln are lower.164 Complexes 94-M exhibit bent geometries, as evidenced by the P–Ln–P angles (156.11(3)°, 94-Sm; 156.10(3)°, 94-Eu; 133.48(3)°, 94-Yb).164 For the diamagnetic complex 94-Yb, a virtual triplet is observed in the 29Si DEPT90 NMR spectrum at 24.30 ppm, with coupling to 31P nuclei 1JPSi = 15.8 Hz. The 31P{1H} NMR spectrum contains a singlet at −301.10 ppm which shows satellites from coupling to 171Yb, with 1JYbP = 1382.1 Hz, as well as to 29Si, 1JPSi = 18.1 Hz.164 The 171Yb{1H} NMR spectrum of 94-Yb contains a triplet resonance at 682 ppm due to coupling to two equivalent 31P nuclei (1JYbP = 1382.9 Hz).
4. Conclusions
We have shown that metal P′′ coordination chemistry lags far behind that of its lighter congener N′′, and that there are therefore a huge number of avenues to be explored and exploited. Although the CSD search performed above gave 210 P′′ vs. 4062 N′′ metal complexes, we note that an analogous search for the other heavy group 15 congeners As′′ (101), Sb′′ (58) and Bi′′ (22) returns progressively fewer hits upon descent of the group.9 Perhaps the most surprising omissions in s-block metal P′′ chemistry are that there are no structurally characterised examples of Na or Be P′′ complexes to date.9 The former of these absences is quite remarkable considering that synthetic routes to Na P′′ salts are well-known,12 and that solid-state structures of solvated Na P†† complexes are already known despite the chemistry of this bulkier bis(silyl)phosphide ligand being in its relative infancy.164,165 We have found there to be relatively few p-block metal P′′ complexes, including no structurally authenticated group 15 and 16 metal(loid) P′′ complexes to date.9 This is quite astonishing considering the potential of such molecular precursors in the synthesis of solid-state III–V semiconductors;42–57 we noted that a Bi P′′ complex has been reported without a solid-state structure,12e and this is also the case for Sb.166 We have identified multiple gaps in d-block metal P′′ chemistry that can be exploited, mainly for the second and third row d-transition metals. There are ten d-transition metal M–P bonds that have yet to be structurally authenticated for this ligand, surprisingly including the entirety of groups 5 and 9.9 We have discussed that numerous 18 e− Fe(II), Ru(II) and Os(II) P′′ complexes have been synthesised but not yet structurally characterised;99 similarly, there are numerous other 18 e− d-transition metal P′′ complexes that are known but lack solid-state structural data, including Mn(I)167 and Re(I)168 examples. The synthesis of group 7 M(I) and group 8 M(II) P′′ complexes are particularly promising avenues to pursue as these are diamagnetic and follow the 18 e− rule, allowing reactions to be easily monitored by 31P NMR spectroscopy and in principle providing relatively stable products.22,23 Finally, there is also a notable paucity of f-block metal P′′ complexes, considering how crucial the N′′ ligand has been to the development of molecular f-block chemistry,5 and this has only started to be addressed relatively recently.115,116,164The structural data discussed herein has showcased how the flexibility of coordination modes and geometries of P′′ provides rich coordination chemistry that juxtaposes its lighter congener N′′ and other bulky bis(silyl)phosphide ligands such as P†† and {P(SiPh3)2}.12e,146,169 The softer nature of P′′ vs. N′′ could be exploited in the stabilisation of low oxidation state metal complexes and unusual structural motifs, and the greater tendency of P′′ to bridge metals and form oligomers can be harnessed to provide multimetallic complexes that can be used as precursors for catalysis, superconductors and magnetic materials. With the exception of s-block P′′ complexes, which have been widely applied as ligand transfer and reducing agents,12,18–20 we note that relatively few reactivity studies have been reported for other structurally characterised p-, d- and f-block metal P′′ complexes to date. This is an unusual observation given the rich chemistry of metal N′′ complexes across the periodic table,1–7 and that both insertion of unsaturated substrates into M–P bonds and cleavage of P–Si bonds are relatively facile processes for P′′ complexes.15–23,99 We envisage that future investigations of P′′ coordination chemistry, as well as its heavier As, Sb and Bi congeners and bulkier derivatives such as P†† and {P(SiPh3)2} in tandem, will provide examples that complement and contrast to those of better-understood N′′ complexes.
Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the European Research Council for a Consolidator Grant (CoG-816268) for D. P. M. and for a PhD studentship for J. B., as well as the University of Manchester for support.References
- in Metal Amide Chemistry, ed. M. Lappert, A. Protchenko, P. Power and A. Seeber, John Wiley and Sons Ltd., Chichester, 2008 Search PubMed.
- H. Bürger, Angew. Chem., Int. Ed. Engl., 1980, 19, 656–657 CrossRef.
- D. H. Harris and M. F. Lappert, J. Organomet. Chem., 1976, 2, 13–122 Search PubMed.
- R. D. Dicken, A. Motta and T. J. Marks, ACS Catal., 2021, 11, 2715–2734 Search PubMed.
- C. A. P. Goodwin and D. P. Mills, Silylamides: towards a half-century of stabilising remarkable f-element chemistry, in Specialist Periodical Reports: Organometallic Chemistry, ed. I. J. S. Fairlamb, J. Lynam, N. J. Patmore and P. Elliott, RSC Publishing, Cambridge, vol. 41, 2017, pp. 123–156 Search PubMed.
- M. P. Coles, Coord. Chem. Rev., 2015, 297–298, 2–23 CrossRef.
- M. P. Coles, Coord. Chem. Rev., 2015, 297–298, 24–39 CrossRef.
- U. Wannagat and H. Niederprüm, Chem. Ber., 1961, 94, 1540–1547 CrossRef.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B:Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef PubMed (search date: 13/11/24).
- S. A. Kosarev and S. J. Collier, Tris(trimethylsilyl)phosphine, in Encyclopedia of Reagents for Organic Synthesis, ed. A. Charette, J. Bode, T. Rovis and R. Shenvi, John Wiley & Sons, Chichester, 2011 Search PubMed.
- G. Fritz, H. Schäfer and W. Hölderich, Z. Anorg. Allg. Chem., 1974, 407, 266–286 Search PubMed.
- (a) F. Uhlig, S. Gremler, M. Dargatz, M. Scheer and E. Herrmann, Z. Anorg. Allg. Chem., 1991, 606, 105–108 Search PubMed; (b) M. Scheer, S. Gremler, E. Herrmann, U. Grünhagen, M. Dargatz and E. Kleinpeter, Z. Anorg. Allg. Chem., 1991, 606, 203–210 CrossRef; (c) F. Uhlig and R. Hummeltenberg, J. Organomet. Chem., 1993, 452, C9–C10 Search PubMed; (d) M. Scheer, S. Gremler, E. Herrman, M. Dargatz and H.-D. Schädler, Z. Anorg. Allg. Chem., 1993, 619, 1047–1052 CrossRef; (e) S. C. Goel, M. A. Matchett, D. Cha, M. Y. Chiang and W. E. Buhro, Phosphorus, Sulfur, Silicon Relat. Elem., 1993, 76, 289–292 CrossRef; (f) H. H. Karsch, F. Bienlein, T. Rupprich, F. Uhlig, E. Herrmann and M. Scheer, in Synthetic Methods of Organometallic and Inorganic Chemistry: Phosphorus, Arsenic, Antimony and Bismuth, ed. W. A. Herrmann, Stuttgart, 1996, vol. 3, pp. 58–65 Search PubMed.
- C. A. Russell and N. S. Townsend, Catalysis: Design and Synthesis, in Phosphorus(III) Ligands in Homogeneous Catalysis, ed. P. W. N. M. van Leeuwen and P. C. J. Kamer, John Wiley & Sons, Chichester, 2012 Search PubMed.
- G. Becker, G. Ditten, K. Hübler, U. Hübler, K. Merz, M. Niemeyer, N. Seidler, M. Westerhausen and Z. Zheng, The Function of the Trimethylsilyl Substituent in the Syntheses of Low-Valent Phosphorus and Arsenic Containing Compounds, in Organosilicon Chemistry Set: From Molecules to Materials, ed. N. Auner and J. Weis, Wiley-VCH, Weinheim, 2005, pp. 161–186 Search PubMed.
- G. Becker, G. Gresser and W. Uhl, Z. Naturforsch., B, 1981, 36, 16–19 CrossRef.
- R. Appel, D. Gudat, E. Niecke, M. Nieger, C. Porz and H. Westermann, Z. Naturforsch., B, 1991, 46, 865–883 CrossRef CAS.
- M. D. Healy, P. E. Laibinis, P. D. Stupik and A. R. Barron, J. Chem. Soc., Chem. Commun., 1989, 6, 359 RSC.
- H. Bürger and H. J. Neese, Inorg. Nucl. Chem. Lett., 1970, 6, 299–304 CrossRef.
- B. Das, A. Makol and S. Kundu, Dalton Trans., 2022, 51, 12404–12426 RSC.
- B. M. Day and M. P. Coles, Eur. J. Inorg. Chem., 2010, 5471–5477 CrossRef CAS.
- J. Du, P. J. Cobb, J. Ding, D. P. Mills and S. T. Liddle, Chem. Sci., 2024, 15, 13–45 RSC.
- Comprehensive Organometallic Chemistry IV, ed. G. Parkin, K. Meyer and D. O’Hare, Elsevier B. V., Amsterdam, 2022 Search PubMed.
- Comprehensive Coordination Chemistry III, ed. E. C. Constable and L. Que Jr., Elsevier B. V., Amsterdam, 2007 Search PubMed.
- E. Hey, P. B. Hitchcock, M. F. Lappert and A. K. Rai, J. Organomet. Chem., 1987, 325, 1–12 CrossRef CAS.
- G. Becker, H.-M. Hartmann and W. Schwarz, Z. Anorg. Allg. Chem., 1989, 577, 9–22 CrossRef CAS.
- E. Hey-Hawkins and E. Sattler, J. Chem. Soc., Chem. Commun., 1992, 24, 775–776 RSC.
- U. Englich, K. Hassler, K. Ruhlandt-Senge and F. Uhlig, Inorg. Chem., 1998, 37, 3532–3537 CrossRef CAS PubMed.
- A. S. Ionkin, W. J. Marshall, B. M. Fish, A. A. Marchione, L. A. Howe, F. Davidson and C. N. McEwen, Eur. J. Inorg. Chem., 2008, 2386–2390 CrossRef CAS.
- M. Westerhausen, M. H. Digeser, B. Wieneke, H. Nöth and J. Knizek, Eur. J. Inorg. Chem., 1998, 517–521 CrossRef CAS.
- M. Westerhausen, M. H. Digeser, H. Nöth, T. Seifert and A. Pfitzner, J. Am. Chem. Soc., 1998, 120, 6722–6725 CrossRef CAS.
- M. Westerhausen and W. Schwarz, Z. Anorg. Allg. Chem., 1994, 620, 304–308 CrossRef CAS.
- M. Westerhausen and A. Pfitzner, J. Organomet. Chem., 1995, 487, 187–195 CrossRef CAS.
- M. Westerhausen and W. Schwarz, Z. Anorg. Allg. Chem., 1996, 622, 903–913 CrossRef CAS.
- M. Westerhausen, H.-D. Hausen and W. Schwarz, Z. Anorg. Allg. Chem., 1995, 621, 877–888 CrossRef CAS.
- M. Westerhausen, J. Organomet. Chem., 1994, 479, 141–151 CrossRef CAS.
- M. Westerhausen and W. Schwarz, J. Organomet. Chem., 1993, 463, 51–63 CrossRef CAS.
- T. Habereder, H. Nöth and R. T. Paine, Eur. J. Inorg. Chem., 2007, 4298–4305 CrossRef.
- F. Thomas, S. Schulz and M. Nieger, Eur. J. Inorg. Chem., 2001, 161–166 CrossRef.
- F. Thomas, S. Schulz, M. Nieger and K. Nättinen, Chem. – Eur. J., 2002, 8, 1915 CrossRef PubMed.
- M. Matar, S. Schulz and U. Flörke, Z. Anorg. Allg. Chem., 2007, 633, 162–165 CrossRef.
- K. L. Antcliff, R. J. Baker, C. Jones, D. M. Murphy and R. P. Rose, Inorg. Chem., 2005, 44, 2098–2105 CrossRef PubMed.
- B. Li, S. Bauer, M. Seidl, A. Y. Timoshkin and M. Scheer, Chem. – Eur. J., 2019, 25, 13714–13718 CrossRef PubMed.
- L. K. Krannich, C. L. Watkins, S. J. Schauer and C. H. Lake, Organometallics, 1996, 15, 3980–3984 CrossRef.
- E. Hey-Hawkins, M. F. Lappert, J. L. Atwood and S. G. Bott, J. Chem. Soc., Dalton Trans., 1991, 939–948 RSC.
- R. L. Wells, A. T. McPhail, M. F. Self and J. A. Laske, Organometallics, 1993, 12, 3333–3339 CrossRef.
- M. Westerhausen, C. Birg, H. Nöth, J. Knizek and T. Seifert, Eur. J. Inorg. Chem., 1999, 2209–2214 CrossRef.
- D. Wiedmann, H.-D. Hausen and J. Weidlein, Z. Anorg. Allg. Chem., 1995, 621, 1351–1357 CrossRef.
- R. J. Jouet, R. L. Wells, A. L. Rheingold and C. D. Incarvito, J. Organomet. Chem., 2000, 601, 191–198 CrossRef.
- S. T. Barry, S. Belhumeur and D. S. Richeson, Organometallics, 1997, 16, 3588–3592 CrossRef.
- R. L. Wells, R. A. Baldwin, P. S. White, W. T. Pennington, A. L. Rheingold and G. P. A. Yap, Organometallics, 1996, 15, 91–97 CrossRef.
- R. L. Wells, E. E. Foos, R. A. Baldwin, A. L. Rheingold and G. P. A. Yap, Heteroat. Chem., 1998, 9, 147–150 CrossRef.
- F. Thomas, S. Schulz and M. Nieger, Z. Anorg. Allg. Chem., 2002, 628, 235–242 CrossRef.
- C. von Hänisch, Z. Anorg. Allg. Chem., 2001, 627, 68–72 CrossRef.
- R. L. Wells, A. T. McPhail, L. J. Jones and M. F. Self, Polyhedron, 1993, 12, 141–147 CrossRef.
- C. Banerjee, D. L. Hughes, M. Bochmann and T. Nann, Dalton Trans., 2012, 41, 7244 RSC.
- R. L. Wells, A. T. McPhail and M. F. Self, Organometallics, 1992, 11, 221–225 CrossRef.
- F. Thomas, S. Schulz, H. Mansikkamäki and M. Nieger, Angew. Chem., Int. Ed., 2003, 42, 5641–5644 CrossRef PubMed.
- F. Thomas, S. Schulz, H. Mansikkamäki and M. Nieger, Organometallics, 2003, 22, 3471–3477 CrossRef.
- R. L. Wells, A. T. McPhail, L. J. Jones III and M. F. Self, J. Organomet. Chem., 1993, 449, 85–94 CrossRef.
- R. L. Wells, A. T. McPhail and M. F. Self, Organometallics, 1993, 12, 3363–3367 CrossRef.
- R. L. Wells, E. E. Foos, A. L. Rheingold, G. P. A. Yap, L. M. Liable-Sands and P. S. White, Organometallics, 1998, 17, 2869–2875 CrossRef.
- M. Nieger, F. Thomas and S. Schulz, CCDC 244687: Experimental Crystal Structure Determination, 2004.
- T. Douglas and K. H. Theopold, Inorg. Chem., 1991, 30, 594–596 CrossRef.
- R. L. Wells, S. R. Aubuchon, M. F. Self, J. P. Jasinski, R. C. Woudenberg and R. J. Butcher, Organometallics, 1992, 11, 3370–3375 CrossRef.
- J. A. L. Cooke, R. L. Wells and P. S. White, Organometallics, 1995, 14, 3562–3565 CrossRef.
- L. J. I. Jones, A. T. McPhail and R. L. Wells, Organometallics, 1994, 13, 2504–2507 CrossRef.
- R. L. Wells, M. F. Self, A. T. McPhail, S. R. Aubuchon, R. C. Woudenberg and J. P. Jasinski, Organometallics, 1993, 12, 2832–2834 CrossRef.
- S. R. Aubuchon, A. T. McPhail, R. L. Wells, J. A. Giambra and J. R. Bowser, Chem. Mater., 1994, 6, 82–86 CrossRef.
- R. L. Wells, S. R. Aubuchon, M. S. Lube and P. S. White, Main Group Chem., 1995, 1, 81–88 CrossRef.
- J. F. Janik, R. L. Wells and P. S. White, Inorg. Chem., 1998, 37, 3561–3566 CrossRef CAS PubMed.
- J. F. Janik, R. L. Wells, V. G. Young, A. L. Rheingold and I. A. Guzei, J. Am. Chem. Soc., 1998, 120, 532–537 CrossRef CAS.
- A. Schaller, H.-D. Hausen, J. Weidlein and P. Fischer, Z. Anorg. Allg. Chem., 2000, 626, 616–618 CrossRef CAS.
- J. A. Burns, W. T. Pennington and G. H. Robinson, Organometallics, 1995, 14, 1533–1535 CrossRef CAS.
- J. F. Janik, R. L. Wells and P. S. White, Organometallics, 1998, 17, 2361–2365 CrossRef CAS.
- J. F. Janik, R. L. Wells, V. G. Young and J. A. Halfen, Organometallics, 1997, 16, 3022–3026 CrossRef CAS.
- C. von Hänisch and B. Rolli, Z. Anorg. Allg. Chem., 2004, 630, 1987–1990 CrossRef.
- M. Usher, A. V. Protchenko, A. Rit, J. Campos, E. L. Kolychev, R. Tirfoin and S. Aldridge, Chem. – Eur. J., 2016, 22, 11685–11698 CrossRef CAS PubMed.
- E. C. Y. Tam, N. A. Maynard, D. C. Apperley, J. D. Smith, M. P. Coles and J. R. Fulton, Inorg. Chem., 2012, 51, 9403–9415 CrossRef CAS PubMed.
- S. Yao, M. Brym, K. Merz and M. Driess, Organometallics, 2008, 27, 3601–3607 CrossRef CAS.
- S. Yao, S. Block, M. Brym and M. Driess, Chem. Commun., 2007, 3844–3846 RSC.
- L. M. Harris, E. C. Y. Tam, S. J. W. Cummins, M. P. Coles and J. R. Fulton, Inorg. Chem., 2017, 56, 3087–3094 CrossRef CAS PubMed.
- B. P. Johnson, S. Almstätter, F. Dielmann, M. Bodensteiner and M. Scheer, Z. Anorg. Allg. Chem., 2010, 636, 1275–1285 CrossRef CAS.
- S. C. Goel, M. Y. Chiang, D. J. Rauscher and W. E. Buhro, J. Am. Chem. Soc., 1993, 115, 160–169 CrossRef CAS.
- M. Westerhausen, M. M. Enzelberger and W. Schwarz, J. Organomet. Chem., 1995, 491, 83–90 CrossRef CAS.
- S. Scheuermayer, F. Tuna, M. Bodensteiner, M. Scheer and R. A. Layfield, Chem. Commun., 2012, 48, 8087–8089 RSC.
- M. Westerhausen, S. Schneiderbauer, M. Hartmann, M. Warchhold and H. Nöth, Z. Anorg. Allg. Chem., 2002, 628, 330–332 CrossRef CAS.
- M. Westerhausen, M. Hartmann and W. Schwarz, Inorg. Chim. Acta, 1998, 269, 91–100 CrossRef CAS.
- M. Fustier, X.-F. Le Goff, M. Lutz, J. C. Slootweg and N. Mézailles, Organometallics, 2015, 34, 63–72 CrossRef CAS.
- J. Baldwin, K. L. Bonham, T. R. C. Thompson, G. K. Gransbury, G. F. S. Whitehead, I. J. Vitorica-Yrezabal, D. Lee, N. F. Chilton and D. P. Mills, JACS Au, 2025 DOI:10.1021/jacsau.4c01018.
- D. Fenske, A. Grissinger, E. M. Hey-Hawkins and J. Magull, Z. Anorg. Allg. Chem., 1991, 595, 57–66 CrossRef CAS.
- L. Weber, G. Meine, R. Boese and N. Augart, Organometallics, 1987, 6, 2484–2488 CrossRef CAS.
- E. Hey, M. F. Lappert, J. L. Atwood and S. G. Bott, Polyhedron, 1988, 7, 2083–2086 CrossRef CAS.
- F. Lindenberg and E. Hey-Hawkins, J. Organomet. Chem., 1992, 435, 291–297 CrossRef CAS.
- E. Hey-Hawkins and F. Lindenberg, Chem. Ber., 1992, 125, 1815–1819 CrossRef CAS.
- F. Lindenberg and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 1995, 621, 1531–1534 CrossRef CAS.
- S. Scheuermayer, F. Tuna, E. M. Pineda, M. Bodensteiner, M. Scheer and R. A. Layfield, Inorg. Chem., 2013, 52, 3878–3883 CrossRef CAS PubMed.
- L. Dütsch, C. Riesinger, G. Balázs and M. Scheer, Chem. – Eur. J., 2021, 27, 8804–8810 CrossRef PubMed.
- W. E. Buhro, M. H. Chisholm, K. Folting, J. C. Huffman, J. D. Martin and W. E. Streib, J. Am. Chem. Soc., 1992, 114, 557–570 CrossRef CAS.
- (a) H. Schäfer, Z. Anorg. Allg. Chem., 1980, 467, 105–122 CrossRef; (b) L. Weber, K. Reizig and R. Boese, Chem. Ber., 1985, 118, 1193–1203 CrossRef CAS; (c) L. Weber, K. Reizig and R. Boese, Organometallics, 1985, 4, 2097–2101 CrossRef CAS; (d) L. Weber and D. Bungardt, J. Organomet. Chem., 1986, 311, 269–280 CrossRef CAS; (e) L. Weber, K. Reizig, D. Bungardt and R. Boese, Organometallics, 1987, 6, 110–114 CrossRef CAS; (f) L. Weber, I. Schumann, H.-G. Stammler and B. Neumann, Z. Naturforsch., 1992, B47, 1134–1140 CrossRef; (g) L. Weber, H. Misiak, S. Buchwald, H.-G. Stammler and B. Neumann, Organometallics, 1994, 13, 2194–2204 CrossRef CAS.
- H. Schäfer, D. Binder, B. Deppisch and G. Mattern, Z. Anorg. Allg. Chem., 1987, 546, 79–98 CrossRef.
- M. Madadi, B. Khalili Najafabadi, M. A. Fard and J. F. Corrigan, Eur. J. Inorg. Chem., 2015, 3094–3101 CrossRef CAS.
- B. Deppisch and H. Schäfer, Z. Anorg. Allg. Chem., 1982, 490, 129–135 CrossRef CAS.
- N. C. Breit, T. Szilvási, T. Suzuki, D. Gallego and S. Inoue, J. Am. Chem. Soc., 2013, 135, 17958–17968 Search PubMed.
- B. Khalili Najafabadi and J. F. Corrigan, Dalton Trans., 2015, 44, 14235–14241 RSC.
- B. Khalili Najafabadi and J. F. Corrigan, Can. J. Chem., 2016, 94, 593–598 CrossRef CAS.
- R. Wei, X. Wang and L. Leo Liu, Z. Anorg. Allg. Chem., 2023, 649, e202300005 CrossRef CAS.
- B. Rademacher, W. Schwarz and M. Westerhausen, Z. Anorg. Allg. Chem., 1995, 621, 287–300 CrossRef CAS.
- O. Fuhr and D. Fenske, Z. Anorg. Allg. Chem., 1999, 625, 1229–1236 CrossRef CAS.
- F. Thomas, S. Schulz and M. Nieger, Organometallics, 2001, 20, 2405–2408 CrossRef CAS.
- F. Lindenberg, T. Gelbrich and E. Hey-Hawkins, Z. Anorg. Allg. Chem., 1995, 621, 771–778 CrossRef.
- F. Lindenberg, T. Shribman, J. Sieler, E. Hey-Hawkins and M. S. Eisen, J. Organomet. Chem., 1996, 515, 19–25 CrossRef CAS.
- G. W. Rabe and J. W. Ziller, Inorg. Chem., 1995, 34, 5378–5379 CrossRef CAS.
- G. W. Rabe, J. Riede and A. Schier, J. Chem. Soc., Chem. Commun., 1995, 5, 577–578 RSC.
- W. A. Herrmann, R. Anwander, F. C. Munck, W. Scherer, V. Dufaud, N. W. Huber and G. R. J. Artus, Z. Naturforsch., B, 1994, 49, 1789–1797 CrossRef CAS.
- J. Baldwin, A. Brookfield, G. F. S. Whitehead, L. S. Natrajan, E. J. L. McInnes, M. S. Oakley and D. P. Mills, Inorg. Chem., 2024, 63, 18120–18136 CrossRef CAS PubMed.
- S. W. Hall, J. C. Huffman, M. M. Miller, L. R. Avens, C. J. Burns, A. P. Sattelberger, D. S. J. Arney and A. F. England, Organometallics, 1993, 12, 752–758 CrossRef CAS.
- T. M. Rookes, E. P. Wildman, G. Balázs, B. M. Gardner, A. J. Wooles, M. Gregson, F. Tuna, M. Scheer and S. T. Liddle, Angew. Chem., Int. Ed., 2018, 57, 1332–1336 CrossRef CAS PubMed.
- G. Fritz and W. Hölderich, Z. Anorg. Allg. Chem., 1976, 422, 104–114 CrossRef CAS.
- W. N. Setzer and P. von R. Schleyer, Adv. Organomet. Chem., 1985, 24, 353–451 CrossRef CAS.
- R. D. Rogers, J. L. Atwood and R. Grüning, J. Organomet. Chem., 1978, 157, 229–237 CrossRef CAS.
- D. Mootz, A. Zinnius and B. Böttcher, Angew. Chem., Int. Ed. Engl., 1969, 8, 378–379 CrossRef CAS.
- L. M. Engelhardt, A. S. May, C. L. Raston and A. H. White, J. Chem. Soc., Dalton Trans., 1983, 8, 1671–1673 RSC.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, A32, 751–767 CrossRef CAS.
- R. E. Mulvey and S. D. Robertson, Modern Developments in Magnesium Reagent Chemistry for Synthesis, in Alkaline-Earth Metal Compounds: Oddities and Applications, ed. S. Harder, Springer Berlin, Heidelberg, 2013 Search PubMed.
- G. W. Rabe, J. Riede and A. Schier, Organometallics, 1996, 15, 439–441 CrossRef CAS.
- A. Kuczkowski, S. Schulz and M. Nieger, Appl. Organomet. Chem., 2004, 18, 244–251 CrossRef CAS.
- D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC.
- Y. Mizuhata, T. Sasamori and N. Tokitoh, Chem. Rev., 2009, 109, 3479–3511 CrossRef CAS PubMed.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400–6441 CrossRef CAS PubMed.
- P. P. Power, Acc. Chem. Res., 2011, 44, 627–637 CrossRef CAS PubMed.
- R. C. Fischer and P. P. Power, Chem. Rev., 2010, 110, 3877–3923 CrossRef CAS PubMed.
- P. P. Power, Nature, 2010, 463, 171–177 CrossRef PubMed.
- L. Weber, K. Reizig and M. Frebel, Chem. Ber., 1986, 119, 1857–1867 CrossRef.
- L. Weber and D. Bungardt, J. Organomet. Chem., 1986, 311, 269–280 CrossRef.
- L. Weber, K. Reizig, D. Bungardt and R. Boese, Organometallics, 1987, 6, 110–114 Search PubMed.
- L. Weber and G. Meine, Chem. Ber., 1987, 120, 457–459 CrossRef.
- H. M. Dietrich, C. Meermann, K. W. Törnroos and R. Anwander, Organometallics, 2006, 25, 4316–4321 CrossRef.
- P. Pyykkö, J. Phys. Chem. A, 2015, 119, 2326–2337 CrossRef PubMed.
- C. von Hänisch, K. Dollberg, J. Moritz, K. Mészáros, C. Jakobi, R.-M. Richter and F. Weigend, Z. Anorg. Allg. Chem., 2024, e202400023 Search PubMed.
- R. A. Andersen, D. H. Templeton and A. Zalkin, Inorg. Chem., 1978, 17, 2317–2319 CrossRef.
- M. Niemeyer, Z. Anorg. Allg. Chem., 2002, 628, 647–657 CrossRef.
- D. C. Bradley, J. S. Ghotra and F. A. Hart, J. Chem. Soc., Dalton Trans., 1973, 10, 1021–1023 RSC.
- G. Occhipinti, C. Meermann, H. M. Dietrich, R. Litlabø, F. Auras, K. W. Törnroos, C. Maichle-Mössmer, V. R. Jensen and R. Anwander, J. Am. Chem. Soc., 2011, 133, 6323–6337 CrossRef PubMed.
- W. J. Evans, D. K. Drummond, H. Zhang and J. L. Atwood, Inorg. Chem., 1988, 27, 575–579 CrossRef.
- M. A. Matchett, M. Y. Chiang and W. E. Buhro, Inorg. Chem., 1994, 33, 1109–1114 CrossRef.
- G. W. Rabe, G. P. A. Yap and A. L. Rheingold, Inorg. Chem., 1995, 34, 4521–4522 CrossRef.
- T. D. Tilley, R. A. Andersen and A. Zalkin, Inorg. Chem., 1984, 23, 2271–2276 CrossRef.
- F. Nief and L. Ricard, J. Organomet. Chem., 1994, 464, 149–154 CrossRef.
- J. M. Manriquez, P. J. Fagan, T. J. Marks, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1978, 100, 7112–7114 CrossRef.
- S. J. Simpson, H. W. Turner and R. A. Andersen, Inorg. Chem., 1981, 20, 2991–2995 CrossRef.
- M. Westerhausen, M. H. Digeser, H. Nöth and J. Knizek, Z. Anorg. Allg. Chem., 1999, 2, 215–220 Search PubMed.
- M. Driess, U. Winkler, W. Imhof, L. Zsolnai and G. Huttner, Chem. Ber., 1994, 6, 1031–1035 CrossRef.
- M. Westerhausen, G. Lang and W. Schwarz, Chem. Ber., 1996, 9, 1035–1040 CrossRef.
- M. Driess and H. Pritzkow, Z. Anorg. Allg. Chem., 1996, 9, 1524–1530 CrossRef.
- A. S. Ionkin and W. J. Marshall, Organometallics, 2003, 22, 4136–4144 CrossRef.
- A. Lorbach, S. Breitung, I. Sänger, F. Schödel, M. Bolte, M. Wagner and H.-W. Lerner, Inorg. Chim. Acta, 2011, 1, 1–9 CrossRef.
- M. Petrie and P. P. Power, J. Chem. Soc., Dalton Trans., 1993, 11, 1737–1745 RSC.
- C. von Hänisch and D. Nikolova, Eur. J. Inorg. Chem., 2006, 4770–4773 CrossRef.
- M. Driess, R. Janoschek, H. Pritzkow, S. Rell and U. Winkler, Angew. Chem., Int. Ed. Engl., 1995, 15, 1614–1616 CrossRef.
- F. Ortu and D. P. Mills, Low Coordinate Rare Earth and Actinide Complexes, in Handbook on the Physics and Chemistry of Rare Earths, ed. J.-C. G. Bünzli and V. K. Pecharsky, Elsevier B. V., Amsterdam, 2019, ch. 306, 55, pp. 1–87 Search PubMed.
- C. von Hänisch, CCDC 154239: Experimental Crystal Structure Determination, 2004.
- M. Westerhausen, T. Rotter, H. Gorls, C. Birg, M. Warchhold and H. Noth, Z. Naturforsch., B, 2005, 60, 766–770 CrossRef.
- J. Baldwin, A. Brookfield, G. F. S. Whitehead, L. S. Natrajan, E. J. L. McInnes, M. S. Oakley and D. P. Mills, Inorg. Chem., 2024, 63, 20295–20306 CrossRef PubMed.
- O. P. Churchill, A. Dase, L. J. Taylor, S. P. Argent, N. T. Coles, G. S. Walker and D. L. Kays, Inorg. Chem., 2024, 63, 20286–20294 CrossRef PubMed.
- B. Ross, J. Belz and M. Nieger, Chem. Ber., 1990, 123, 975–978 CrossRef.
- L. Weber and G. Meine, Chem. Ber., 1987, 120, 457–459 CrossRef.
- L. Weber, G. Meine, R. Boese and D. Bläser, Chem. Ber., 1988, 122, 853–857 CrossRef.
- G. Lacorre and H. Grützmacher, Dalton Trans., 2022, 51, 3497–3501 RSC.
| This journal is © The Royal Society of Chemistry 2025 |