DNA damage by oxo- and peroxo-chromium(V) complexes: insight into the mutation and carcinogenesis mechanisms
Lamis
Judah†
a,
Roberto
Marin
a,
Diane
Stroup
b,
Chrys
Wesdemiotis
c and
Rathindra N.
Bose
*a
aDepartment of Chemistry, University of Houston, Houston, TX 77204, USA. E-mail: rnbose@uh.edu
bDepartment of Chemistry, Kent State University, Kent, OH 44242, USA
cDepartment of Chemistry, University of Akron, Akron, Ohio 44325, USA
First published on 23rd September 2013
Abstract
The carcinogenic and mutagenic properties of chromium(VI) compounds are attributed to hypervalent chromium species, especially metastable oxo- and peroxo-chromium(V)/(IV) complexes. To understand the nature of DNA damage induced by various hypervalent chromium complexes, reactions of sodium bis(2-ethyl-2-hydroxybutanato)oxochromate(V) (I) with single- and double-stranded DNA were investigated in the presence of hydrogen peroxide and a radical scavenger. In these reactions, three DNA damaging agents, oxo- and peroxo-chromium(V) complexes and hydroxyl radicals, were generated. Both double and single-stranded DNA cleavages were largely observed after piperidine treatment, indicating base-labile scissions. Although these cleavages appeared to be random in most cases, a modest selectivity towards the G base was observed for the double stranded oligonucleotides. A variety of sugar and base oxidation products were detected after enzymatic digestion of the reaction mixtures with exo- and endo-nucleases. These products include furfural, 5-methylene furanone, 8-oxo-7,8-dihydro-2′-guanosine (8-oxodGuo), thymine propenal, deoxyribonucleoside derivative of 5-guanidinohydantoin (dGh), cytosine glycol and 5-hydroxy cytosine. The first three mentioned products were observed for reactions of compound I with DNA in the absence of peroxide. Since compound I generated a persistent peroxo-chromium(V) complex in situ and oxidation of DNA by the oxo-chromium(V) was relatively slow, it appears that the peroxo-species was largely responsible for the formation of dGh through a four-electron oxidation of the guanine residue following the mechanism proposed by Meunier and coworkers. Likewise, the formation of propenals follows a similar pathway observed for bleomycine-mediated DNA oxidation that follows Hecht's mechanism. On the other hand, 5,6-dihydroxy-5,6-dihydro-2′-deoxycytidine (dCydGl) and 5-hydroxy-2′-deoxycytidine (5-OHdCyd) were most likely formed from the reactions of DNA with the hydroxyl radical. We hypothesize that base oxidations and the formation of abasic sites are most likely responsible for transverse DNA mutation.
Introduction
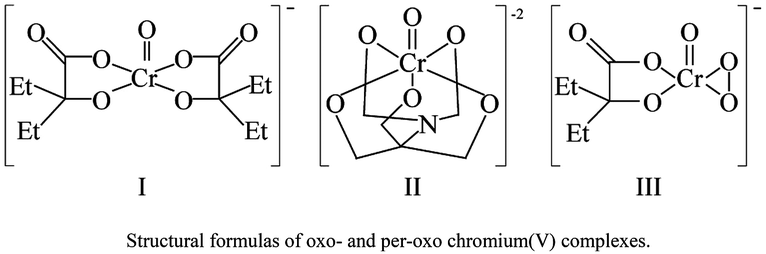
Chromium(VI) compounds, such as chromate and dichromate, are well known carcinogenic and mutagenic agents. Workers exposed to chromium dust show a high percentage of lung and other bronchial cancers.1–4 Since reactions between Cr(VI) or Cr(III) and DNA have shown insignificant damage to DNA,5 it is believed that metastable hypervalent chromium species generated in the cellular milieu cause DNA damage and mutations. In fact, reductants such as glutathione,5–8 ascorbic acid9,10 and cytochrome P-45011 indeed reduce Cr(VI) to Cr(III) through detectable Cr(V) and Cr(IV) intermediates. In addition to cancer, prolonged exposures to chromium and other metals such as nickel and arsenic are linked to cardiovascular disease, neurological disorders, and hypertension.12 It has been suggested that post-translational histone modifications might be linked to a variety of toxic effects.12Since oxo- and peroxo-chromium(V/IV) complexes are believed to be putative DNA damaging agents, systematic studies evaluating the ability of each of the reactive species might provide insight into the nature and extent of DNA damage caused by these species. Our earlier work reported specific pathways of DNA damage by two oxo-chromium(V) complexes (I and II).13,14 Furthermore, the DNA damaging ability of peroxo-chromium(IV) complexes15 has been demonstrated recently by using an authentic diperoxo-complex, although the mechanistic details of DNA damage by the peroxo-chromium(IV) complex are yet to be unveiled. The present study focuses on the DNA damaging ability of compound I in the presence of peroxide in which a peroxo-chromium(V) complex (III) was generated in situ.16 We envisioned that by comparing the products observed in this study with those known from earlier studies, insight into the DNA damage by the peroxo-complex can be gained.
The impetus for the present study is based on the increasing evidence indicating the involvement of molecular oxygen in chromium-induced carcinogenesis. For example, Cr(VI)-induced mutations on certain strains of Salmonella were observed only in the presence of O2.17 Also, it has been shown that the absence of O2 or the presence of catalase significantly reduced the in vitro DNA damage induced by Cr(VI) in the presence of ascorbic acid and glutathione.17,18 These studies initially suggested that the hydroxyl radical formed in O2-dependent, Fenton-like reactions was the active species in the abovementioned systems. However, subsequent studies have demonstrated that the nature of DNA damage observed is inconsistent with the action of the hydroxyl radical.19 The extent of DNA damage was related to both Cr(V) intermediates and O2, which led to the suggestion that the possible DNA-damaging species are Cr(V/IV) peroxo or superoxo complexes.20–22 In fact, Cr(V)-peroxo and peroxo-ascorbate complexes were detected by electron paramagnetic resonance spectroscopy in aerated aqueous media.23 In another study, Pourahmad and O'Brien24 showed that adding Cr(VI) to isolated rat hepatocytes results in rapid glutathione oxidation, reactive oxygen species formation, lipid peroxidation, decreased mitochondrial membrane potential, and lysosomal membrane rupture before hepatocyte lysis occurred. When reactive oxygen species scavengers, antioxidants, and glutamine were used, cytotoxicity was prevented. Furthermore, glutathione-depleted hepatocytes were resistant to Cr(VI) toxicity. This evidence led to the conclusion that Cr(VI) cytotoxicity is associated with mitochondrial/lysosomal toxicity by the biological reactive intermediates Cr(V/IV) and reactive oxygen species.
Experimental section
Reagents: Unless otherwise specified, all reagents were of highest purity and used without further purification. Disodium ethylenediaminetetraacetate (EDTA), sodium dichromate, sodium bicarbonate, 2-ethyl-2-hydroxybutyric acid (HEBA), and 2-furaldehyde were purchased from Aldrich Chemical Company. Bis(2-hydroxyethyl)iminotris(hydroxymethyl)methane (BT), all bases (A, C, G, and T) and their 2′-deoxyribonucleoside counterparts (dAdo, dCyd, and dGuo), thymidine (dThd), calf thymus double-stranded DNA (dsDNA), hydrogen peroxide (30%), 5,5-dimethyl-1-pyrroline-N-oxide (DMPO), alkaline phosphatase (calf intestines), nuclease P1 (snake venom), ammonium formate, N,N′-methylene-bis-acrylamide(bis-acrylamide), N,N,N′,N′-tetramethylethylenediamine (TEMED), ammonium persulfate, formamide, piperidine, tris-base, and boric acid were purchased from Sigma Chemical Company. Acetone, hexane, phosphoric acid (85%), perchloric acid, HPLC grade acetonitrile, chloroform, and methanol were purchased from Fisher Scientific Company. Xylene cyanol and bromophenol blue were obtained from Bio-Rad Laboratories and 8-hydroxy-2′-deoxyguanosine was purchased from ESA Incorporated (Chelmsford, MA). Acrylamide and urea were purchased from Mallinckrodt and Baker Inc. T4 polynucleotide kinase was purchased from Promega (Madison, WI). Oligonucleotides were synthesized by GibcoBRL and [γ-33P]ATP, used for radioactive labeling of the oligonucleotides, was purchased from Amersham (Piscataway, NJ). 5-Methylfuranone (5-MF) was synthesized by following the method of Grundmann and Kober.25Sodium bis(2-ethyl-2-hydroxybutanato)oxochromate(V) was synthesized following the method of Krumpolc and Rocek.26 Solutions of calf thymus ds-DNA were prepared in 20.0 mM BT buffer at pH 7.4. Initial DNA concentrations were estimated spectrophotometrically based on the absorbance at 260 nm. The exact concentrations of ds-DNA, however, were calculated from HPLC data after enzymatic digestion of DNA sample by using standard calibration curves of known concentrations of dA, dC, dG and T.
Physical measurements
High performance liquid chromatographic separations
The HPLC instrument was equipped with a pump (Waters, model 515), a photodiode array detector (Waters, model 996), and a gradient programmer (ISCO, model 2360). The instrument was controlled and operated by the Millennium 3.2 software. A gradient separation using a mobile phase consisting of a mixture of 50.0 mM ammonium formate (pH 4.0, Solvent A) and 100% methanol (Solvent B) with a flow rate of 1.0 mL min−1 was carried out on a symmetry C18 column (Waters, 100 Å pore diameter, 5 μm particle size, and 3.9 × 150 mm). A linear gradient from 100% solvent A to 85% A–15% B was run for the first 15 min followed by a linear increase of solvent B to 100% for the next 25 min. For the products extracted with chloroform, the mobile phase consisted of a 60.0 mM ammonium formate (pH 4.0) and 10% acetonitrile mixture. Products were characterized by comparing retention times and UV-visible spectra of the eluates with those of authentic samples.Unless otherwise specified, for HPLC separations, the reaction of ds-DNA with chromium(V) complexes initiated by incubating calf thymus ds-DNA (1.0 mM) with sodium bis(2-ethyl-2-hydroxybutanato)oxochromate(V) (2.0 mM) was carried out either in excess HEBA ligand (20 mM) or in bis-tris buffer (20 mM) at 7.4. When the reactions ended, EDTA (22 μL, 50 mM, pH 8.0) was added to replace the bound ligand onto chromium(III) by chelating with EDTA. The reaction was then enzymatically digested by first adding nuclease P1 (12 units) for each 200 μL of reaction and incubating for 30 min at 37 °C. This process was repeated a second time to ensure complete digestion. Then, alkaline phosphatase (20 units) for each 200 μL of reaction was added and the reaction was incubated at 37 °C for additional 30 min. This treatment was also repeated a second time to ensure complete DNA digestion. These double enzymatic digestions cleaved both 3′ and 5′ phosphate groups from the 2-deoxyribosyl moiety of DNA and released free nucleosides.
Fast atom bombardment – mass spectrometric (FAB-MS) measurements
FAB-MS measurements were performed using a modified VG AutoSpec tandem mass spectrometer of E1BE2 (electrostatic analyzer 1–magnet–electrostatic analyzer 2) geometry at the University of Akron. This instrument houses one collision cell (C1s-1) in the field free region preceding E1 (FFR-1) followed by two more (C1s-2 and C1s-3). The ions were produced by FAB+ with a Cs+ gun operated at 20 kV. These ions were accelerated at a voltage of 8 keV. The source was at ambient temperature, and the vacuum was operated at 10−7–10−8 mbar. The spectrum produced was the result of multi-scan summations. Collected fractions from HPLC separations were lyophilized and dissolved in 10–15 μL of 3-nitrobenzyl alcohol. Aliquots of 3 μL of the resulting matrix were then applied onto the probe tip and introduced into the ion source.Nuclear magnetic resonance measurements
NMR experiments were performed using a Bruker 500 MHz (DRX 500) instrument, proton decoupled 31P resonances were recorded at 202.45 MHz, and the chemical shifts are reported with respect to 85% phosphoric acid at 0.0 ppm. For recording 31P spectra, a pulse of 8.20 μs with a repetition time of 0.2 s was used. Typically, 64 k data points were collected within a frequency domain of 8090 Hz. A line-broadening factor of 1.0 Hz was introduced before the Fourier transformation. The spectrum of the oligonucleotide, 5′ CTA GAT CAT C 3′ (1.5 mM) in bis-tris (50 mM) in H2O2 (200 mM) was recorded in D2O. The reaction of the oligonucleotide with I (5 mM) was carried out for 12 h. The reaction mixture was treated with EDTA (30 mM) for 45 min to bind Cr(III) before recording the spectra.Reactions of chromium(V) complexes (1.0 mM) with DNA in the presence of H2O2 were carried out in two buffers, HEBA and BT, at pH 7.4. Usually, two sets of duplicate reactions (with or without 10% piperidine) were performed. Control reactions consisted of 2 μL of the labeled oligonucleotide in nuclease free water, 20.0 mM BT or 20 mM HEBA buffer at pH 7.4, and 5.0 mM H2O2. Reaction mixtures were incubated for two hours at 37 °C, and then terminated by adding 10 μL of 0.50 M EDTA (pH 8.0). For the piperidine treated reactions, piperidine was removed by vacuum evaporation. All samples were then re-suspended in 5 μL of formamide loading buffer and heated for 3 min at 90 °C to denature the DNA before loading onto the 6% denaturing polyacrylamide gel using TBE buffer.
The separation was accomplished on an Owl Separation Systems aluminum-backed sequencer (35 × 45 cm) equipped with a power supply (Pac 3000, BioRad). Samples of 1 or 2 μL of the reactions (including the Maxam–Gilbert reactions) were loaded onto the gel and electrophoresed for 90 to 120 min at 60 V. The gel was soaked in a solution of a mixture of 10% acetic acid and 15% methanol to eliminate urea. The gel was then dried and exposed to a phosphor storage screen for 48 to 72 h in a cassette. The screen was scanned with a Molecular Dynamics Typhoon 8600 phosphorimager. Scanned images were analyzed using the software Image Quant Tools (version 2.2) and Image Quant (version 5.2) from Molecular Dynamics.
Results
Nature of chromium(V) complexes in solution
In the presence of hydrogen peroxide, compound I generated a peroxo complex (compound III), and a hydroxyl radical, as reported earlier.4 No other oxo- or peroxo-chromium(V) complexes were detected by the epr spectroscopy, including those reported by Zhang and Lay.21 Chromium(V)-peroxo complex (III) can be formed through several different pathways including via direct substitution of HEBA ligand with a peroxide ion, and through the oxidation of either chromium(III) or reduction of Cr(VI) which are generated by the disproportionation reaction of compound I. If either reduction of Cr(VI) or oxidation of Cr(III) was responsible for its formation, compound III generation would have exhibited an induction period to allow the disproportionation reaction to occur. Following that induction period, Cr(V)-concentrations would either be increased over time or maintained at a steady state status. Since the highest concentration of the Cr(V)-peroxo complex was generated immediately after reacting compound I with peroxide (2 min) and the peroxo-complex decayed exponentially within 90 min, the direct substitution of HEBA ligand by the peroxide ion is the most likely pathway to generate compound III. Although a BT ligand was added to scavenge hydroxyl radicals from the reaction mixtures, a complete scavenging of the radical was not achieved as the spectra exhibited the presence of hydroxyl radicals. The amount of the radical, however, was much less in the presence of BT as compared to its absence. Also, the BT ligand is known to slowly replace both HEBA molecules to form a stable Cr(V)-BT complex.8 The amount of Cr(V)-BT complex (II) was negligible (<10%) compared to the compound I through the duration of the DNA reactions as determined from the intensity of the epr signal for the compound II which appeared at g = 1.965.8 Since the peroxo complex persists just over an hour in solution, reactions with DNA were limited to two hours. Note that DNA oxidations by compounds I and II require 12–24 h. High concentrations of peroxide were used to rapidly generate appreciable amounts of peroxo-chromium(V) complex so that the oxidation of DNA by chromium(V)-peroxo complexes can be studied by minimizing the interference from compounds I and II.Characterization of DNA oxidation products by HPLC, ESI-MS, and FAB-MS
Fig. 1 shows a typical HPLC chromatogram of the products formed in the reaction between compound I and ds-DNA in the presence of peroxide, which were observed after enzymatic digestions of the DNA. The peaks identified as dA, dC, dG, and T in the chromatogram are primarily due to the hydrolysis of unreacted DNA. Naturally, the remaining peaks mainly represent products generated in the reaction. As shown in Fig. 1, the reaction yielded major and minor products, and major products were observed with retention times between 1–2 min and 18.2 min. The designation of major and minor products was based on the intensity of chromatographic peaks of the products. It appears that more than 40% dG was oxidized based on a comparison of the intensities of dG of control experiments with those of DNA reactions under the highest peroxide concentration. However, no attempts were made to quantify the percent yield of any specific products. The characterization of the major and minor products is described below.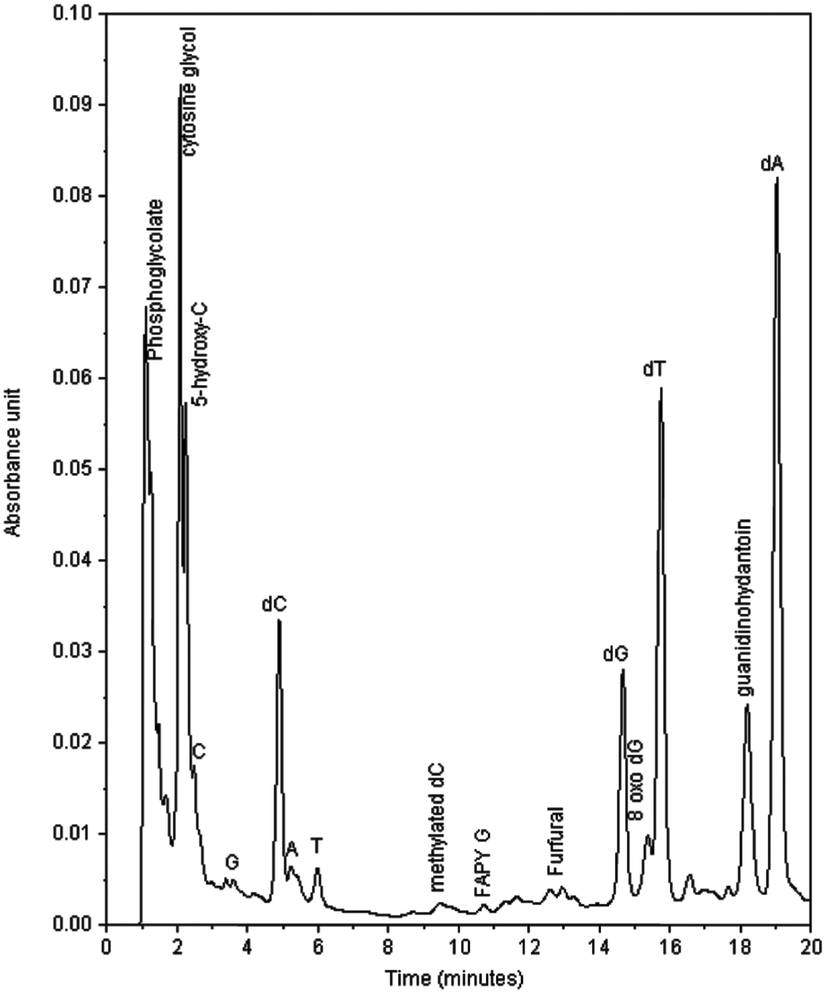 | ||
| Fig. 1 A representative chromatogram of the products of the reaction of ds-DNA (1.0 mM) with I (2.0 mM) in the presence of H2O2 (200.0 mM) and HEBA (20 mM) after enzymatic digestion. Note that methylated-dC is not the product of the reaction. The control sample contained small amount of the methylated base. | ||
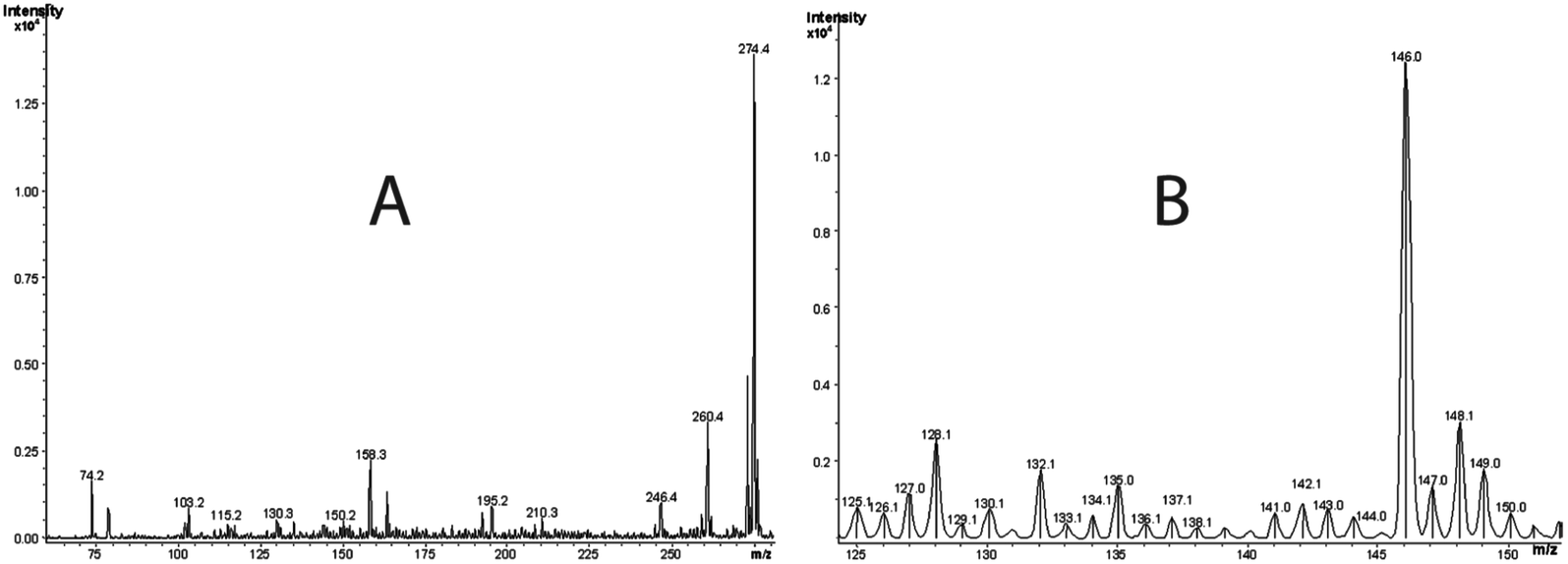 | ||
| Fig. 2 The ESI-MS in the positive ion mode of the 2′-deoxyribonucleoside derivative of 5-guanidinohydantoin (dGh) (A), and 5-hydroxy-C and cytosine glycol (B). Molecular masses are shown at m/z = 274.4 for the 2′-deoxyribonucleoside derivative of 5-guanidinohydantoin (dGh), 128.1 for 5-hydroxy-C, and 146.0 for cytosine glycol. | ||
Fig. 2B represents the mass spectrum of the products collected from 2.2 min retention time in the chromatogram. Note that the peak centered at 2.2 min represents overlapping components of two products. The positive ion mode spectrum showed protonated molecular ion peaks at m/z of 128.1 and 146.0. These molecular masses and their retention times are indicative of 5-hydroxy-2′-deoxycytidine (5-OHdCyd) and cytosine glycol.
The third major fraction corresponding to peaks at 1.5 min in the HPLC chromatogram largely contained a Cr(III) product that exhibited an electronic spectrum identical to that of Cr(III)-EDTA complex.28 To explore whether highly polar and non-UV absorbing molecules such as glycolic acid and phosphoglycolate were present, we adopted a strategy reported by Mazzer et al.29 Essentially, fractions were collected at 0.5 min intervals, and mass spectra of all fractions were recorded. To avoid major fragmentations of the buffer, a softer ionization technique, FAB-MS, was used rather than ESI-MS. Figs. 3A and 3B show spectra of two such fractions. Peaks at m/z 188.0 and 75.0 in the FAB-MS in the negative ion mode represent glycolic acid (m/z = 75.0), which is the fragment resulting from phosphoglycolate (Fig. 3B at m/z = 188.0). Phosphoglycolate in the mass spectrum appeared as an ammonium ion adduct.
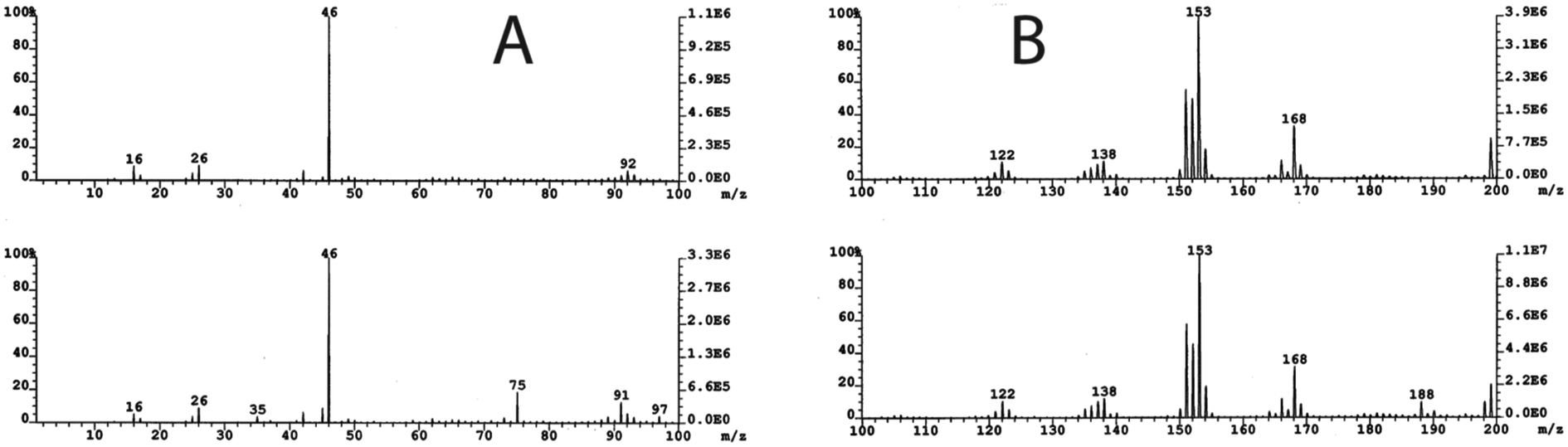 | ||
| Fig. 3 FAB-MS in the negative ion mode of glycolic acid at m/z = 75 (A) and phosphoglycolate at m/z = 188 (B). The top spectrum for A is the matrix, 3-nitrobenzyl alcohol and ammonium formate; and for B is the matrix, 3-nitrobenzyl alcohol plus ammonium formate. | ||
The fourth major product was the modified dG, 8-oxo-dGuo. This product was observed by oxo-chromium(V) compounds as well as by a hydroxyl radical as reported earlier.13,14
To confirm the formation of 5-MF and furfural, aliquots of the reactions of Cr(V) with ss-DNA and ds-DNA in the presence of H2O2 were extracted separately with chloroform in order to detect any products that are more soluble in chloroform such as 5-MF and furfural. Only furfural was observed in the reaction of I with ds-DNA in the presence of H2O2. On the other hand, 5-MF was produced in more abundance than furfural in the reaction with ss-DNA (Fig. 4). It should be noted that furfural and 5-MF might have been underestimated since the precursor 2-deoxyribolactone, which produces furfural and 5-MF through its decomposition,31 has much higher half-lives than the reaction time (2–3 h) reported here.
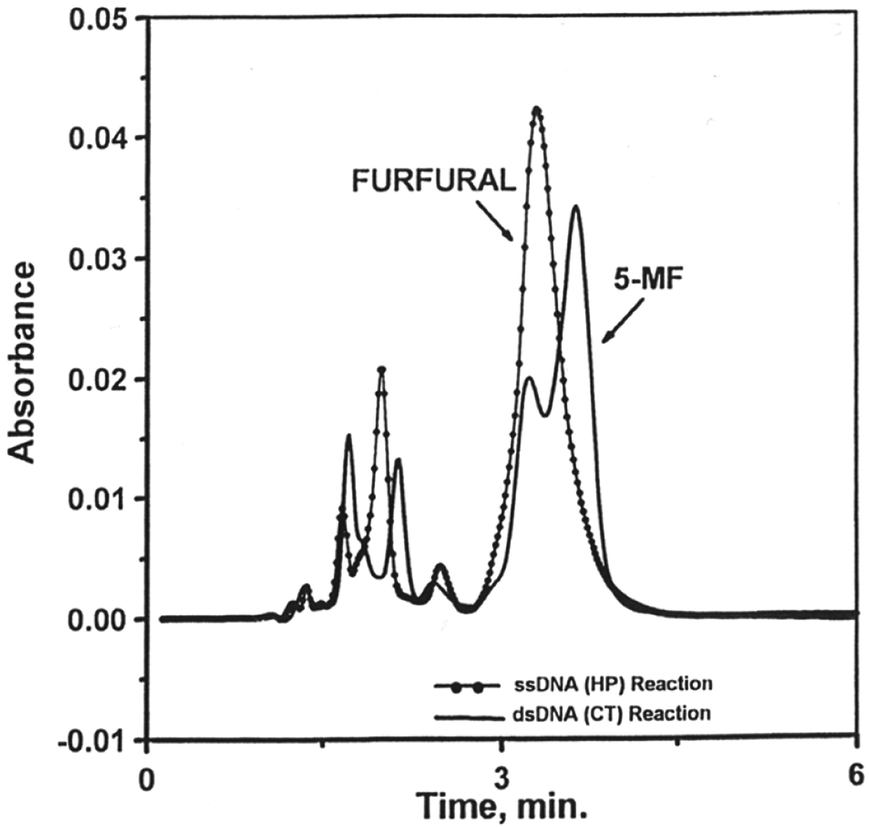 | ||
| Fig. 4 HPLC chromatogram of chloroform extract for the reaction of Cr(V) (1.0 mM), DNA (10.0 mM), BT (15.0 mM), and H2O2 (146.0 mM) at pH 7.0. Note that the product abbreviated as 5-methyl-G was present in the control DNA experiment. | ||
Chromium(V) mediated cleavage of single- and double-stranded oligonucleotides by PAGE experiments
Fig. 5 shows the electrophoretogram for the cleavage of the double stranded oligonucleotide, H1–H2. Very small cleavages were observed in the absence of piperidine, while extensive cleavage was observed in its presence. Similar observations were made for both single-stranded oligos, H1 and H2. These cleavages were significantly increased by increasing peroxide concentrations. The cleavage of oligonucleotides at specific bases was represented as a percentage of total cleavage, which was assumed to be 100%. Table 1 lists the percentage of cleavages at the four bases and compares them with those percentages in natural abundance.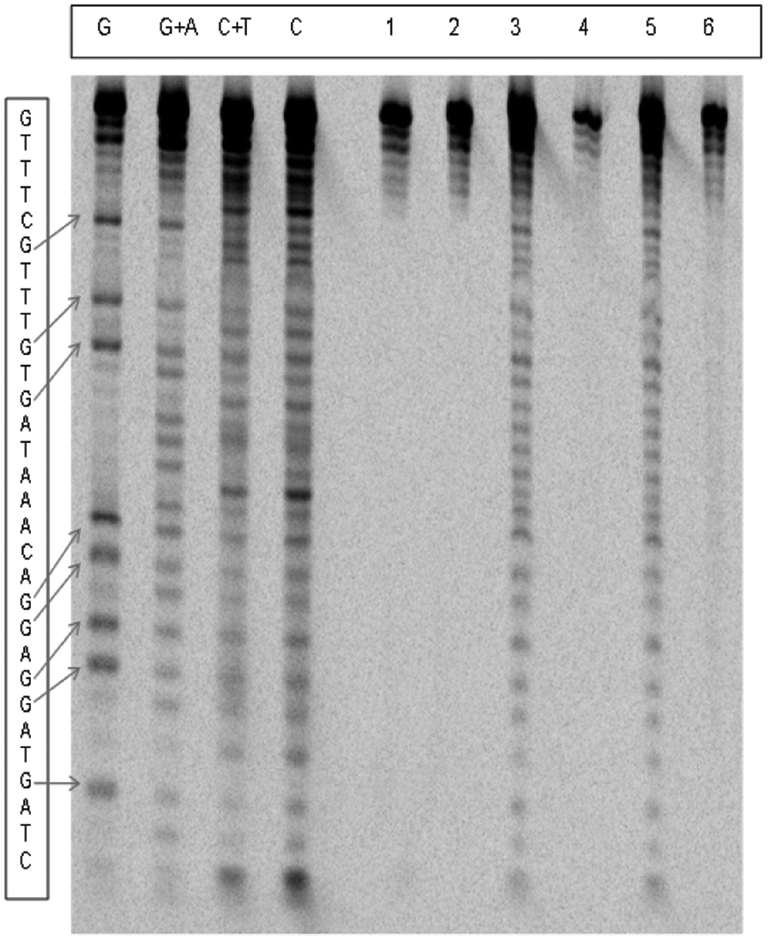 | ||
| Fig. 5 Electrophoretograms of Cr(V)-induced DNA damage of ds-H1–H2 in BT buffer. Oligomer H1 was P-33 labeled on the 5′-end with T4 kinase. Lane 1: control (ds-DNA + 5.0 mM H2O2 + piperidine); lane 2: same as lane 1 without piperidine; lane 3: ds-DNA + 1.0 mM Cr(V) + 5.0 mM H2O2 + piperidine; lane 4: same as lane 3 without piperidine; lane 5: ds-DNA + 1.0 mM Cr(V) + 20.0 mM H2O2 + piperidine; lane 6: same as lane 5 without piperidine. Lanes G, G + A, T + C, and C: Maxam–Gilbert sequencing ladder. | ||
| Oligo | [H2O2] (mM) | Buffer | %G | %C | %T | %A |
|---|---|---|---|---|---|---|
| H1 | 5.0 | BT | 34 (30.3) | 7 (9.1) | 51 (33.3) | 8 (27.3) |
| 20.0 | 31 | 10 | 51 | 8 | ||
| H2 | 5.0 | HEBA | 11 (9.1) | 26 (30.3) | 33 (27.3) | 29 (33.3) |
| 20.0 | 12 | 33 | 33 | 22 | ||
| 5.0 | BT | 4 | 27 | 32 | 37 | |
| 20.0 | 7 | 29 | 33 | 31 | ||
| H1–H2 | 20.0 | HEBA | 36 (30.3) | 7 (9.1) | 38 (33.3) | 19 (22.3) |
| 5.0 | BT | 44 | 6 | 41 | 9 | |
| 20.0 | 45 | 6 | 43 | 7 |
The cleavages appeared to be quite random, except that modest preferences towards G (ds-oligo) and T (H1) were observed, especially in BT buffer.
Strand breaks detected by phosphorus-31 NMR spectroscopy
The reaction of compound I with a small oligonucleotide, 5′-CTAGATCAT C-3′, in the presence of peroxide was monitored by phosphorus-31 NMR spectroscopy. The spectrum of the oligonucleotide showed several resonances in the chemical shift between −0.5 and −1.0 ppm. These resonances are due to the magnetic heterogeneity of the P-31 nuclei present in the molecule. In the presence of peroxide, the oligonucleotide exhibited several new peaks due to its reaction of compound I in BT buffer. The new resonances appeared in the downfield region, 0 to 2 ppm, indicating cleavages of the oligonucleotide (Fig. 6). Although the PAGE experiments did not reveal SSB for 33-mer, such a breakage was observed for a small oligonucleotide by phosphorus-31 NMR. These breakages might be due to the selection of a small oligo-nucleotide whose oxidation chemistry might be different from those observed for the larger oligos, consistent with the fact that the DNA oxidation depends on the length, sequence specificity, and structures of oligonucleotides.32,33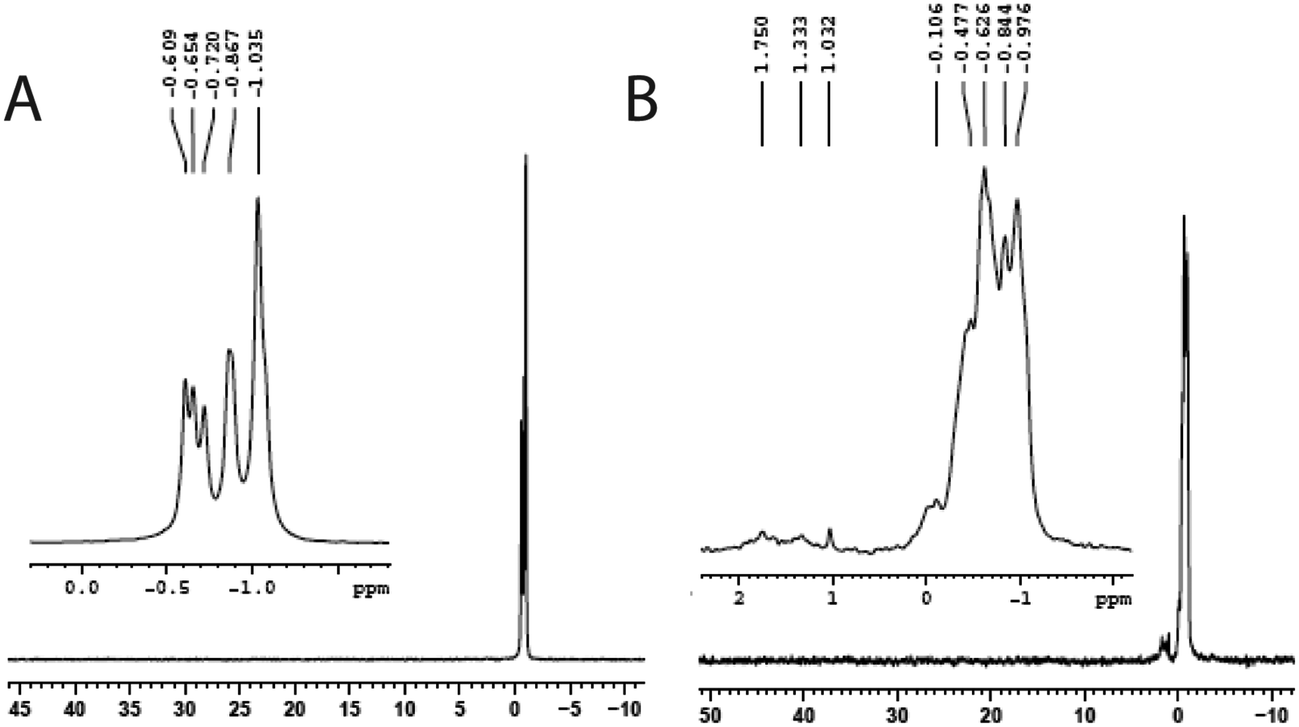 | ||
| Fig. 6 (A) 31P NMR of an oligo 5′-CTA GAT CAT C-3′ (1.5 mM) in bis-tris (50 mM) and H2O2 (200 mM). (B) 31P NMR of the same mixture described in (A) after 12 h of incubation with I (5.0 mM) followed by the treatment with EDTA (30 mM) before recording NMR spectra. | ||
Discussion
At first glance, the reactions reported here appeared complex due to the presence of three DNA damaging agents, oxo-chromium(V), peroxo-chromium(V), and the hydroxyl radical. However, the DNA oxidation products by the two known Cr(V) complexes, Cr(V)-HEBA (I) and Cr(V)-BT (II), are independently documented.13,14 Products from DNA damage by the hydroxyl radical are also reported.34–36 Furthermore, reactions in the present study were conducted for only two hours to minimize DNA oxidations by compounds I and II as they require a much longer time period, usually 12–24 h, to produce significant DNA oxidation products. Therefore, we address the reactions pathways for oxo- and peroxo-chromium(V) by comparing the products observed here with those reported in earlier work.13,14 DNA oxidations by I in the absence13,14 and presence of low hydrogen peroxide concentrations37,38 yielded 5-MF and furfural. These authors37 did not observe any peroxo-chromium(V) formation under low peroxide concentrations and hence the DNA oxidation most likely resulted by complex I as we have observed and by the hydroxyl radical. Finally, Sugden38 has reported Frank scission and alkali labile strand breaks of DNA by compound I in the presence of peroxide. However, no oxidation products were reported or characterized by this author.As indicated above, in the absence of peroxide, compounds I and II largely generated furfural, 5-MF, and 8-hydroxy-dG,13,14 which were observed here as minor products, consistent with the expectation based on the short reaction time allowed for the reactions. The major products, 2′-deoxyribonucleoside derivative of 5-guanidinohydantoin (dGh), phosphoglycolic acid, 5-hydroxy-C (5-OHdCyd), and cytosine glycol, most likely resulted from the reactions of peroxo-chromium(V) species and the hydroxyl radical. The latter two products most likely originated from radical-initiated damage as observed by others. It is highly likely that dGh was formed by the peroxo-species. This is based on the argument that in the absence of hydrogen peroxide, oxo-Cr(V) complexes did not produce dGh. Secondly, Shi et al.39 observed 8-oxo-G as a product, not an intermediate, produced by the reaction of the 2-deoxy-guanosine with the hydroxyl radical. Recently, Madugundu et al.40 have also reported the formation of 8-oxo-G by the hydroxyl radical in abundance over the other products. Note the modified dG base is reportedly further oxidized by 1e inorganic reductants to produce a variety of products including (dGh).41 Therefore, we rule out the possibility that oxo-Cr(V) complexes or the hydroxyl radical were responsible for the formation of dGh. Although two mechanisms for dGh formation have been reported,41,42 we favor the mechanism postulated by Vialas et al.42 They proposed the formation of a guanine cation radical at the C5 site via two-electron oxidation by Mn(V), followed by hydroxylation at the C5 and C8 sites and subsequent two-electron oxidation by a second manganese unit.42 Since the peroxochromium(V) is a four-electron oxidant, one molecule might be engaged in this 4e oxidation process. Alternatively, two peroxo-species might be involved in this process, each unit serving as 2e electron donor producing the oxo-chromium(V) species as shown in Scheme 1. This mechanism is different from that proposed by Muller et al.,41 who proposed the formation of 8-oxo-G as an intermediate followed by further 2e oxidation of the same. Our preference towards the mechanism proposed by Meunier and co-workers42 is based on the argument that we have detected 8-oxo-dGuo in our product, which would not have existed if it were an intermediate.
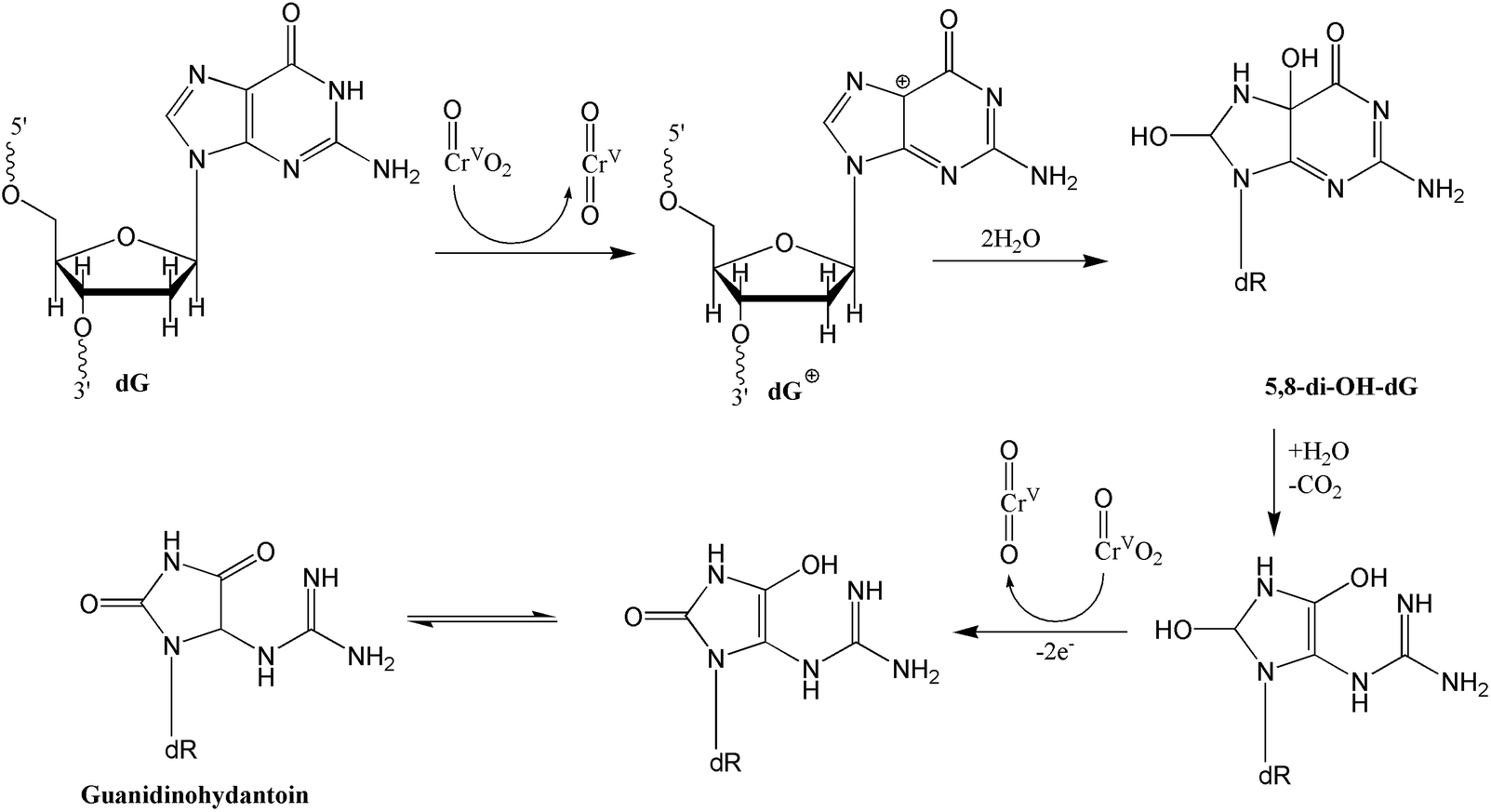 | ||
| Scheme 1 Proposed oxidation mechanism for the formation of the 2′-deoxyribonucleoside derivative of 5-guanidinohydantoin (dGh) by peroxo-chromium(V) complex following the mechanism proposed by Vialas et al.42 | ||
In addition to dGh, oxidations of the guanine base have yielded a variety of products that include imidazolone, oxazolone, 2-imino-5,5′-spirodihydantoin (sp), 4-hydroxy-2,5-dioxo-imidazolidine-4-carboxy acid, dehydroguanidinohydantoin (Gh), and many others. Many of these products are thought to form via further oxidation of 8-oxo-G.43 Since we did not observe these products as major oxidation products, it further supports our hypothesis that peroxo-chromium(V) species oxidizes dGuo to dGh via a different mechanism, not through 8-oxo-dGuo intermediate, as shown above in Scheme 1. However, we cannot eliminate the possibility that the peroxo-species slowly oxidizes 8-oxo-dGuo as we have observed this product in our reactions. As indicated earlier, the propensity of DNA oxidation not only depends on the sequence and structure of the DNA but also on the nature of the oxidizing agents. For example, Sugden and coworkers44 have reported the oxidation of 8-oxoG base in single- and double-stranded DNA containing 8-oxoG by Cr(V)-salen complex to sp while Cr(V)-HEBA yielded Gh. Likewise, Burrows and coworkers41 reported the formation of Gh by iridium(IV) complexes.
The formation of 3′-phosphoglycolate ends and base propenals was also observed in our reactions. This indicates the H-abstraction from the 4′ position as a pathway for oxidatively generated DNA damage. One of the best-characterized pathways of DNA cleavage by H4′ abstraction is that mediated by the antitumor antibiotic bleomycin-Fe(II).45,46 It appears that our results were similar to those observed for bleomycin-Fe(II) activated DNA damage under aerobic conditions, wherein DNA strand scission and the subsequent formation of both base propenal and 3′-phosphoglycolate were observed by activated bleomycin-Fe(II). On the other hand, DNA strand scission with the production of free bases, 3′-phosphate, and 5′-phosphate end groups is due to the activated bleomycin-Fe(II) under anaerobic conditions.35,36 Products of this pathway were not observed in our reactions. Under our conditions, the peroxo-chromium(V) species most likely behaves as the ferric peroxide complex47 shown in Scheme 2. It is interesting to note that the reaction of chromium(VI) in the presence of glutathione and hydrogen peroxide also generated phosphoglycolate and base propenals, implying that peroxo-chromium(V) species might be involved in this reaction for DNA oxidation.29 However, we cannot exclude the possibility of hydroxyl radical-initiated oxidation process at the H4′ site.
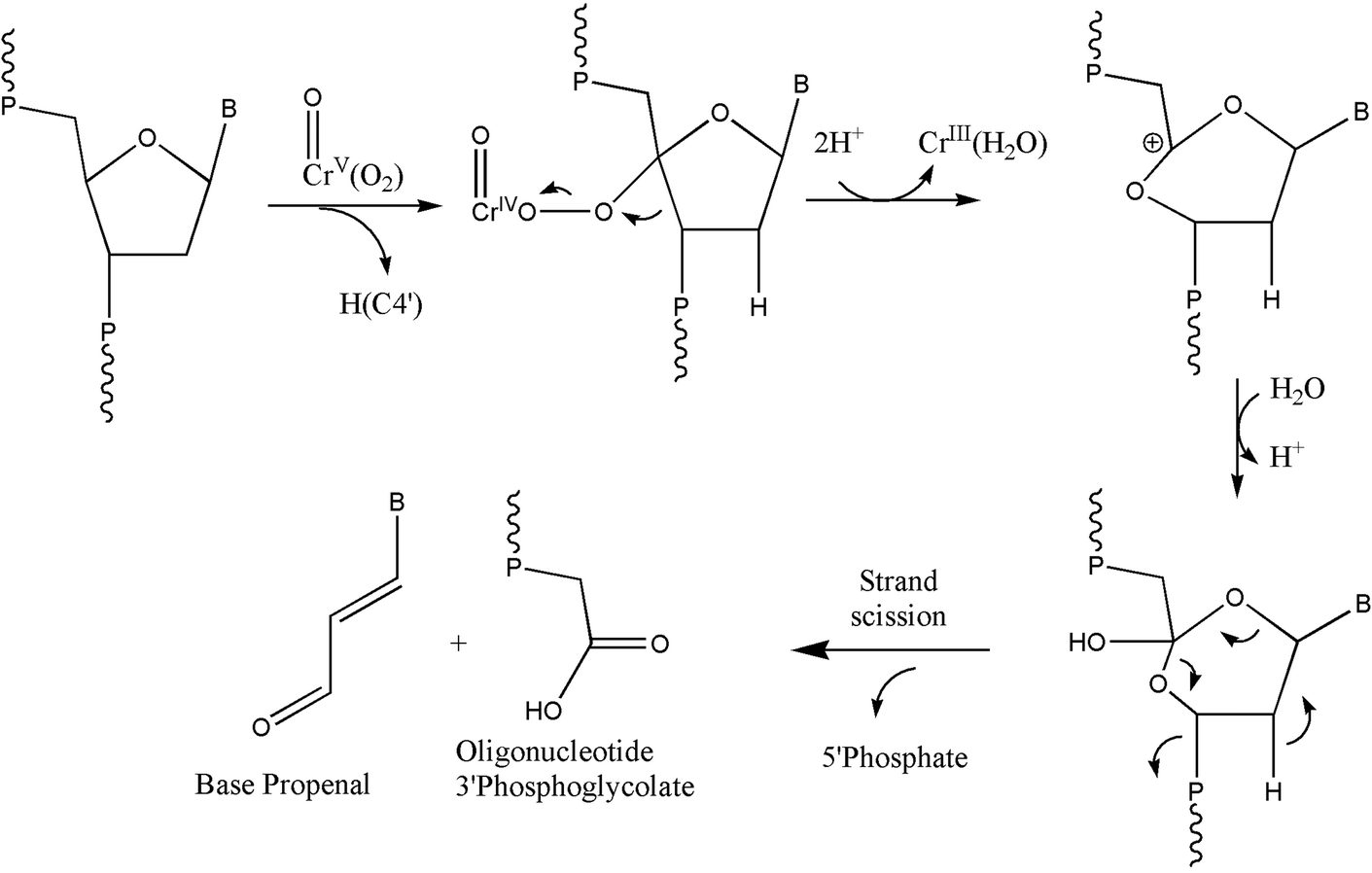 | ||
| Scheme 2 Oxidation of DNA through H4′ hydrogen abstraction by the peroxo-chromium(V) complexes. The scheme was adopted in part from earlier work.45,46 | ||
In addition to 5-MF and furfural, these reactions also generated other products, such as FAPy-G, 5-hydroxy-dC, and cytosine glycol, perhaps primarily due to the participation of the hydroxyl radical. The formation of FAPy-G is known to arise from hydroxyl radical attack at the C8 position of a guanine base, leading to the formation of 8-hydroxy-7,8-dihydroguanyl radical.47,48 Likewise, the modified cytosine products are due to the hydroxyl radical attack at the C5 and C6 double bond which subsequently generates alkali lability and alkali-induced strand scission.47,48 The remaining product, 5-hydroxy-dC, can form through two possible pathways.49 First, 5-hydroxy-dC can be formed by adding the hydroxyl radical to the C5 position of cytosine. The second pathway may be due to the dehydration of cytosine glycol, which can be formed through the oxidation of cytosine's C5–C6 double bond in cytosine. However, cytosine glycol is chemically unstable and can easily undergo decomposition via dehydration or deamination to 5-hydroxy-dC, 5-hydroxyuracil, and uracil glycol; this route is unlikely since uracil glycol and 5-hydroxyuracil were not observed in the products of our reactions.
Returning now to DNA cleavage, we noted extensive base-labile strand scissions from PAGE experiments. DNA oxidation may or may not be accompanied by strand scissions. For example, formations of apurinic and apyrimidinic sites due to the loss of purine and pyrimidine bases do not lead to strand scissions. Also, it is known that 8-oxo-dGuo and T base modifications do not induce alkali-labile DNA cleavage,50,51 hence such cleavages require further oxidation of 8-oxo-dGuo. Furthermore, oxidations by the hydroxyl radical also generated base-labile scissions. A largely random cleavage with a modest preference towards cleavage at G- and T-bases in the presence of a radical scavenger is also consistent with our observations of oxidation of dGuo to dGh and T base modifications.
Finally, how the DNA damage by hypervalent chromium species initiates carcinogenesis is yet to be fully understood, since many DNA damaging agents including the cross-linking agent cisplatin and the oxidative damaging agent bleomycin are well known anticancer drugs.52,53 In this regard, significant progress has been made in understanding the specific nature of the damage and cellular responses that link with the development of tumors. First, mere covalent binding to the phosphate moiety of the DNA shown earlier13 may not create a mutation on specific genes since the NER enzymes effectively remove chromium from phosphate.54 However, DNA mutations by chromium(VI), especially G to T transversions, are perhaps more relevant to cancer initiation and growth. In this regard, Henderson et al.55 have clearly demonstrated that hydantoin lesions are potent sources of replication errors. Therefore, the hydantoin formation observed in this study due to the oxidation of dGuo to dGh by the peroxo-chromium(V) species might lead to error-prone replications resulting in DNA mutation. Furthermore, induction p53-gene mutations have been suggested to result from the specific binding of Cr(V) at the G bases.56
In conclusion, the results presented here enabled comparison of the similarities and differences in DNA oxidation by oxo- and peroxo-chromium(V) complexes. It appears that oxo-chromium(V) oxidizes DNA by sugar oxidation through C1 and C5 2-deoxyribose positions, leading to the formation of 5-methylene-2-furanone and furfural, respectively. The peroxo-complex, on the other hand, transfers four electrons to 2′-deoxyguanosine, which yields dGh through the opening of the purine ring, while the oxochromium(V) only produced 8-OH-dG as the minor product. In addition, the peroxo-compound initiated oxidation through the C4′ position, yielding phosphoglycolate and base propenals. It also appears that both oxo- and perxo-chromium(V) complexes are somewhat selective in oxidizing at G bases. The combined data present evidence that oxo- and peroxo-chromium(V) complexes are highly capable of damaging DNA through multiple pathways. Some, if not all, may contribute to mutation during DNA replication processes.
Abbreviations
| Cr(V)-HEBA | Sodium bis(2-ethyl-2-hydroxybutanato)oxochromate |
| Cr(V)-BT | Bis(hydroxyethyl)amino-tris(hydroxymethyl)methaneoxochromate(V) |
| 5-OHdCyd | 5-Hydroxy-2′-deoxycytidine |
| dCydGl | 5,6-Dihydroxy-5,6-dihydro-2′-deoxycytidine |
| 8-oxoG | 8-Oxo-7,8-dihydro-2′-guanine |
| 8-oxodGuo | 8-Oxo-7,8-dihydro-2′-guanosine |
| Gh | Dehydroguanidinohydantoin |
| dGh | Deoxyribonucleoside derivative of 5-guanidinohydantoin |
| FAPy-G | 2,6-Diamino-5-formamido-4-hydroxypyrimidine |
| 5-MF | 5-Methylene-2-furanone |
| sp | 2-Imino-5,5′-spirodihydantoin |
Acknowledgements
We are grateful to Ms. Jennifer James for editing the manuscript and Ms. Ashley Merwin for technical assistance.References
- B. Sarkar, Metals and genetics, Metallomics: Int. Biomet. Sci., 2012, 4, 589–592 RSC.
- D. M. Parkin, 14. Cancers attributable to occupational exposures in the UK in 2010, Br. J. Cancer, 2011, 105, S70–S72 CrossRef PubMed.
- S. Langard, One hundred years of chromium and cancer: A review of epidemiological evidence and selected case reports, Am. J. Ind. Med., 1990, 17, 189–214 CrossRef CAS.
- S. De Flora, et al., Genotoxicity of chromium compounds: A review, Mutat. Res., 1990, 238, 99–172 CrossRef CAS.
- X. Shi and N. S. Dalal, On the mechanism of the chromate reduction by glutathione: ESR evidence for the glutathionyl radical and an isolable Cr(V) intermediate, Biochem. Biophys. Res. Commun., 1988, 156, 137–152 CrossRef CAS.
- D. De Flora and K. E. Wetterhahn, Mechanisms of chromium metabolism and genotoxicity, Life Chem. Rep., 1989, 7, 169–244 Search PubMed.
- R. N. Bose, et al., Long-lived chromium(IV) and chromium(V) metabolites in the chromium(VI)-glutathione reaction: NMR, ESR, HPLC, and kinetic characterization, Inorg. Chem., 1992, 31, 1987–1994 CrossRef CAS.
- S. Moghaddas, et al., Mechanisms of formation and decomposition of hypervalent chromium metabolites in the glutathione-chromium(VI) reaction, J. Inorg. Biochem., 1995, 57, 135–146 CrossRef CAS.
- M. Reynolds, S. Armknecht, T. Johnston and A. Zhitkovich, Undetectable role of oxidative DNA damage in cell cycle, cytotoxic and clastogenic effects of Cr(VI) in human lung cells with restored ascorbate levels, Mutagenesis, 2012, 27, 437–443 CrossRef CAS PubMed.
- X. Shi, et al., Reaction of Cr(VI) with ascorbate and hydrogen peroxide generates hydroxyl radicals and causes DNA damage: Role of a Cr(IV)-mediated Fenton-like reaction, Carcinogenesis, 1994, 15, 2475–2478 CrossRef CAS PubMed.
- D. Y. Cupo and K. E. Wetterhahn, Modification of chromium(VI)-induced DNA damage by glutathione and cytochromes P-450 in chicken embryo hepatocytes, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 6755–6759 CrossRef CAS.
- Y. Chervona, A. Arita and M. Costa, Carcinogenic metals and the epigenome: understanding the effect of nickel, arsenic, and chromium, Metallomics, 2012, 4, 619–627 RSC.
- R. N. Bose, et al., Mechanisms of DNA damage by chromium(V) carcinogens, Nucleic Acids Res., 1998, 26, 1588–1596 CrossRef CAS PubMed.
- R. N. Bose, et al., Oxidative damage of DNA by chromium(V) complexes: Relative importance of base versus sugar oxidation, Nucleic Acids Res., 1999, 27, 2219–2226 CrossRef CAS PubMed.
- R. Marin, et al., Potentially deadly carcinogenic chromium redox cycle involving peroxochromium(IV) and glutathione, J. Am. Chem. Soc., 2010, 132, 10617–10619 CrossRef CAS PubMed.
- L. Joudah, et al., DNA oxidation by peroxo-chromium(V) species: oxidation of guanosine to guanidinohydantoin, Chem. Commun., 2002, 16, 1742–1743 RSC.
- A. Kortenkamp, et al., A role for molecular oxygen in the formation of DNA damage during the reduction of the carcinogen chromium(VI) by glutathione, Arch. Biochem. Biophys., 1996, 329, 199–207 CrossRef CAS PubMed.
- P. da Cruz Fresco and A. Kortenkamp, The formation of DNA cleaving species during the reduction of chromate by ascorbate, Carcinogenesis, 1994, 15, 1773–1778 CrossRef CAS PubMed.
- A. Kortenkamp et al. , Cytotoxic mutagenic and carcinogenic potential of heavy metals related to human environment, NATO-ASI S, Kluwer Academic Publishers, Dordrecht, 1996, pp. 15–34 Search PubMed.
- M. H. Dickman and M. T. Pope, Peroxo and superoxo complexes of chromium, molybdenum, and tungsten, Chem. Rev., 1994, 94, 569–584 CrossRef CAS.
- L. Zhang and P. A. Lay, EPR spectroscopic studies on the formation of chromium(V) peroxo complexes in the reaction of chromium(VI) with hydrogen peroxide, Inorg. Chem., 1998, 37, 1729–1733 CrossRef CAS.
- X. Shi and N. S. Dalal, On the hydroxyl radical formation in the reaction between hydrogen peroxide and biologically generated chromium(V) species, Arch. Biochem. Biophys., 1990, 277, 342–350 CrossRef CAS.
- L. Zhang and P. A. Lay, EPR Spectroscopic studies of the reactions of Cr(VI) with l-ascorbic acid, l-dehydroascorbic acid, and 5,6-O-isopropylidene-l-ascorbic acid in water. 1 Implications for chromium(VI) genotoxicity, J. Am. Chem. Soc., 1996, 118, 12624–12637 CrossRef CAS.
- J. Pourahmad and P. J. O'Brien, Biological reactive intermediates that mediate chromium(VI) toxicity, Adv. Exp. Med. Biol., 2001, 500, 203–207 CrossRef CAS.
- C. Grundmann and E. Kober, An improved synthesis of protoanemonin, J. Am. Chem. Soc., 1955, 77, 2332 CrossRef CAS.
- M. Krumpolc and J. Rocek, Synthesis of stable chromium(V) complexes of tertiary hydroxy acids, J. Am. Chem. Soc., 1979, 101, 3206–3209 CrossRef CAS.
- A. M. Maxam and W. Gilbert, Sequencing end-labeled DNA with base-specific chemical cleavages, Methods Enzymol., 1980, 65, 499–569 CrossRef CAS.
- K. A. Easom and R. N. Bose, Reactions of aminopolycarboxylic acids with high valent metal ions: EDTA assisted decomposition of carboxylato-bound chromium(V) complexes, Inorg. Chem., 1988, 27, 2331–2335 CrossRef CAS.
- P. A. Mazzer, et al., Mechanisms of DNA damage and insight into mutations by chromium(VI) in the presence of glutathione, J. Inorg. Biochem., 2007, 101, 44–55 CrossRef CAS PubMed.
- M. Berger and J. Cadet, Isolation and characterization of the radiation-induced degradation products of 2′-deoxyguanosine in oxygen-free aqueous solutions, Z. Naturforsch., 1985, 40B, 1519–1531 CAS.
- W. Chan, et al., Quantification of the 2-deoxyribonolactone and nucleoside 5′-aldehyde products of 2-deoxyribose oxidation in DNA and cells by isotope-dilution gas chromatography mass spectrometry: differential effects of y-radiation and Fe2+-EDTA, J. Am. Chem. Soc., 2010, 132, 6145–6153 CrossRef CAS PubMed.
- K. S. Lim, et al., In situ analysis of 8-oxo-7,8-dihydro-2′-deoxyguanosine oxidation reveals sequence and agent specific DNA damage, J. Am. Chem. Soc., 2012, 134, 18053–18064 CrossRef CAS PubMed.
- W. B. Mattes, J. A. Hartley and K. W. Kohn, DNA sequence selectivity of guanine–N7 alkylation by nitrogen mustards, Nucleic Acids Res., 1986, 14, 2971–2987 CrossRef CAS PubMed.
- H. Luo, et al., Chromium(IV)-mediated Fenton-like reaction causes DNA damage: Implication to genotoxicity of chromate, Ann. Clin. Lab. Sci., 1996, 26, 185–191 CAS.
- J. Cadet, et al., Hydroxyl radicals and DNA base damage, Mutat. Res., 1999, 424, 9–21 CrossRef CAS.
- S. Kawanishi, et al., The role of metals in site-specific DNA damage with reference to carcinogenesis, Free Radicals Biol. Med., 2002, 32, 822–832 CrossRef CAS.
- K. D. Sugden and K. E. Wetterhahn, Direct and Hydrogen Peroxide-Induced Chromium(V) Oxidation of Deoxyribose in Single-Stranded and Double-Stranded Calf Thymus DNA, Chem. Res. Toxicol., 1997, 10, 1397–1406 CrossRef CAS PubMed.
- K. D. Sugden, Formation of modified cleavage termini from the reaction of chromium(V)-with DNA, J. Inorg. Biochem., 1999, 77, 177–183 CrossRef CAS.
- X. Shi, et al., Cr(IV) causes activation of nuclear transcription factor-kappa B, DNA strand breaks and dG hydroxylation via free radical reactions, J. Inorg. Biochem., 1999, 75, 37–44 CrossRef CAS.
- G. S. Madugundu, et al., Radiation-induced formation of 2′,3′-dideoxyribonucleosides in DNA: a potential signature of low-energy electrons, J. Am. Chem. Soc., 2012, 134, 17366–17368 CrossRef CAS PubMed.
- J. Muller, V. Duarte, R. Hickerson and C. Burrows, Gel electrophoretic detection of 7,8-dihydro-8-oxoguanine and 7,8-dihydro-8-oxoadenine via oxidation by Ir(IV), Nucleic Acids Res., 1998, 26, 2247–2249 CrossRef CAS PubMed.
- C. Vialas, C. Claparols, G. Pratviel and B. Meunier, Guanine oxidation in double-stranded DNA by Mn-TMPyP/KHSO 5: 5,8-Dihydroxy-7,8-dihydroguanine residue as a key precursor of imidazolone and parabanic acid derivatives, J. Am. Chem. Soc., 2000, 122, 2157–2167 CrossRef CAS.
- K. S. Lim, et al., In situ analysis of 8-oxo-7,8-dihydro-2′-deoxyguanosine oxidation reveals sequence- and agent-specific damage spectra, J. Am. Chem. Soc., 2012, 134, 18053–18064 CrossRef CAS PubMed.
- J. N. Gremaud, B. D. Martin and K. D. Sugden, Influence of substrate complexity on the diastereoselective formation of spiroiminodihydantoin and guanidinohydantoin from chromate oxidation, Chem. Res. Toxicol., 2010, 23, 379–385 CrossRef CAS PubMed.
- S. M. Hecht, The chemistry of activated bleomycin, Acc. Chem. Res., 1986, 19, 383–391 CrossRef CAS.
- R. M. Burger, Cleavage of nucleic acids by bleomycine, Chem. Rev., 1998, 98, 1153–1169 CrossRef CAS PubMed.
- J. W. Sam, et al., Electrospray mass spectrometry of iron bleomycin: Demonstration that activated bleomycin is a ferric peroxide complex, J. Am. Chem. Soc., 1994, 116, 5250–5256 CrossRef CAS.
- C. J. Burrows and J. G. Muller, Oxidative nucleobase modifications leading to strand scission, Chem. Rev., 1998, 98, 1109–1152 CrossRef CAS PubMed.
- J. Hong, et al., Damaged products of cytosine in hydroxyl radical solution under UV-irradiation, Microchem. J., 2001, 68, 173–182 CrossRef CAS.
- P. M. Cullis, et al., Guanine radical cations are precursors of 7,8-dihydro-8-oxo-2′-deoxyguanosine but are not precursors of immediate strand breaks in DNA, J. Am. Chem. Soc., 1996, 118, 2775–2781 CrossRef CAS.
- M. J. Lutsig, et al., Synthesis of the diastereomers of thymidine glycol, determination of concentrations and rates of interconversion of their cis-trans epimers at equilibrium and demonstration of differential alkali lability within DNA, Nucleic Acids Res., 1992, 20, 4839–4845 CrossRef PubMed.
- N. J. Wheate, et al., The status of platinum anticancer drugs in the clinic and in clinical trials, Dalton Trans., 2010, 39, 8113–8127 RSC.
- S. M. Hecht, Bleomycine: New perspectives on the mechanism of action, J. Nat. Prod., 2000, 63, 158–168 CrossRef CAS PubMed.
- M. Reynolds, et al., Human nucleotide excision repair efficiently removes chromium-DNA phosphate adducts and protects cells against chromate toxicity, J. Biol. Chem., 2004, 279, 30419–30424 CrossRef CAS PubMed.
- P. T. Henderson, et al., The hydantoin lesions formed from oxidation of 7, 8-dihydro-8-oxoguanine are potent sources of replication errors in vivo, Biochem., 2003, 42, 9257–9262 CrossRef CAS PubMed.
- H. Arakawa, et al., Chromium(VI) induces both bulky DNA adducts and oxidative DNA damage at adenines and guanines in the p53 gene of human lung cells, Carcinogenesis, 2012, 33, 1993–2000 CrossRef CAS PubMed.
Footnote |
| † Current address: 1142 Kerber Pass, Chaska, MN 55318, USA. |
| This journal is © The Royal Society of Chemistry 2014 |