
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpectral shifting and NIR down-conversion in Bi3+/Yb3+ co-doped Zn2GeO4
Guojun
Gao
a,
Mingying
Peng
bc and
Lothar
Wondraczek
*ac
aOtto Schott Institute of Materials Research, University of Jena, 07743 Jena, Germany. E-mail: lothar.wondraczek@uni-jena.de
bState Key Laboratory of Luminescent Materials and Devices, School of Materials Science and Technology, South China University of Technology, 510641 Guangzhou, China
cThe Chinese-German Research Center for Photonic Materials and Devices at South China University of Technology, 510641 Guangzhou, China
First published on 14th August 2014
Abstract
We report on spectral modification through NIR down-conversion (DC) photoluminescence (PL) in Yb3+-Bi3+-co-doped Zn2GeO4. Energetic downshifting (DS) of UV-A irradiation occurs via intrinsic luminescence of the high-bandgap semiconductor Zn2GeO4 as well as via active Bi3+ centres. In parallel, both species act as sensitizers for Yb3+, strongly extending its excitation region to ∼500 nm. In the absence of Bi3+, band-to-band absorption of Zn2GeO4 in the UV region results in PL at ∼475–625 nm. Doping with Yb3+ initiates energy transfer from trapped defect states to two neighbouring Yb3+ ions in a cooperative DC process, resulting in Yb3+-related photoemission at ∼1000 nm. The introduction of Bi3+ into Zn2GeO4:Yb3+ greatly extends the absorption band to the visible blue region. Then, energy transfer also occurs through cooperative DC from Bi3+ to Yb3+. As a result, a strong increase in the absolute Yb3+-related PL intensity is observed. This enables ultra-efficient harvesting of UV-A to visible radiation for energy conversion processes.
Introduction
The spectral conversion of incident sunlight through photoluminescence has been proposed to enhance the efficiency of various solar energy harvesting processes.1–7 For example, in the case of crystalline silicon solar cells, a large fraction of the incoming solar energy is lost either because photons do not surpass the energy gap which is necessary to generate a photoelectron (NIR-tail of the solar spectrum), or because their excess energy causes thermalization or electron–hole recombination (near-UV to green part of the solar spectrum). Conversion strategies consequently follow some straightforward schemes: for example, in up-conversion processes (UC), two or more low-energy photons are used to generate one photon of higher energy. In down-shifting (DS) or quantum cutting (QC) processes, on the other side, a high-energy photon is converted into one (DS) or more (QC) photons of lower energy.1,2,8–12 In the latter, a quantum efficiency which is theoretically higher than unity can be achieved. As a concept, all three processes enable to adjust the incoming solar spectrum through increasing the number of photons within a certain spectral region. Whether or not this is advantageous for a specific solar energy conversion process, however, depends on many other factors, including quantum efficiency, secondary absorption processes, converter properties, system design and cost.Here, we concentrate on the model system of Zn2GeO4:Yb3+ and its further sensitization with Bi3+ co-dopants. Yb3+ has a rather simple band structure with the single excited state of 2F5/2 at an energy of ∼1 eV. Yb3+ photoluminescence therefore occurs at a wavelength of ∼1000 nm, which is very close to the maximum photoelectronic conversion efficiency of silicon. Hence, trivalent ytterbium is an often-sought emitter for solar spectral conversion through QC or DS.1–4 For this, a suitable sensitizer is required which adds favourable absorption properties to the emission behaviour of Yb3+. Most of the trivalent rare earth (RE3+) ions, e.g., Pr3+, Er3+, Nd3+, Ho3+, Tb3+, Tm3+ and Dy3+ have been considered for this purpose.1,2,8–19 However, the parity-forbidden 4fn → 4fn transitions in these ions typically result in weak and narrow absorption bands which stand in contrast to the wish for efficient (broadband) harvesting of sunlight. As alternatives, ions such as Ce3+, Eu2+, Bi3+ and Mn2+, and/or combination with intrinsic absorption of the host itself (e.g., YVO4 and ZnO) have therefore been studied for Yb3+ activation.20–24
The orthogermanate of zinc, Zn2GeO4, is a wide-bandgap semiconductor with a gap-energy of ∼4.68 eV.25 Without further dopants, it exhibits broadband luminescence in the blue to green spectral region which originates from the recombination of native structural defects.26,27 Emission from Zn2GeO4 occurs at about twice the energy which is necessary to excite Yb3+: 2F7/2 → 2F5/2. This means that it can be used to sensitize Yb3+. On the other hand, absorption covers only the UV region of ∼250 to 350 nm, which prevents efficient solar harvesting. To overcome this, we introduce trivalent bismuth as further activator into the Zn2GeO4 host. Bi3+ has a 6s2 electronic configuration with ground state 1S0 and the excited state 6s6p which splits into the 3P0, 3P1, 3P2 and 1S1 levels. The emission band of Bi3+ is typically located in the blue or green, originating from the ligand-field dependent relaxation of 3P1 → 1S0.28–36 Although the absorption band of 1S0 → 3P1 is spin-forbidden, it shows reasonably high oscillator strength due to spin–orbit coupling between the 3P1 and the 1P1 level.12,35 Similar to Zn2GeO4, the absorbed energy of Bi3+ can probably be transferred to Yb3+ through DC processes. Introduction of Bi3+ into Zn2GeO4:Yb3+ is therefore expected to enable broadband activation of Yb3+ NIR photoluminescence.
Experimental section
Powder samples and compacted pellets with nominal compositions of Zn1.998−xGeO4:Yb0.0023+Bix3+ (x = 0, 0.006, 0.02, 0.06 and 0.18 and is the number of Zn2+ which is displaced by Bi3+ in Zn2GeO4) were synthesized through conventional high-temperature solid state reaction. For that, ZnO, GeO2, Bi2O3 and Yb2O3 (≥4 N) were used as starting materials. Stoichiometric batches of ∼2 g were thoroughly mixed in an agate mortar, pre-calcined at 900 °C for 4 h in air, again ground in an agate mortar, pressed into pellets and finally fired in air at 1300 °C for 12 h.20 Blank (Zn2GeO4) and singly-doped (Zn1.998GeO4:Bi0.02) samples were prepared in the same way for reference.Powder X-ray diffraction (XRD) patterns were obtained on a Siemens Kristalloflex D500 diffractometer over a 2θ range of 10–70°. UV-vis diffuse reflectance (DR) spectra were recorded with a double-beam photospectrometer (Cary 5000) over the spectral range of 200 to 800 nm with 1 nm step size. Static photoluminescence (PL) and PL excitation (PLE) spectra and dynamic decay curves were collected at room temperature with a high-resolution spectrofluorometer (Horiba Jobin Yvon Fluorolog FL3-22), using a static 450 W Xe lamp and a pulsed 75 W Xe flashlamp as excitation sources, respectively. NIR PL was observed with an InP/InGaAs-based thermoelectrically cooled photomultiplier tube (NIR-PMT, Hamamatsu H10330A-75). PLE spectra were measured between 250 to 500 nm with a step size of 1 nm. PL spectra were recorded between 390 and 700, and 930 and 1200 nm with a step size of 1 nm. PLE spectra were corrected over the lamp intensity with a silicon photodiode. PL spectra were corrected with the spectral response of employed PMT.
Results and discussion
Fig. 1a displays ex situ powder XRD patterns of as-prepared Zn1.998−xGeO4:Yb0.0023+Bix3+ for varying values of x. As the major crystal phase in all samples, we identify rhombohedral Zn2GeO4 (JCPDS card no. 00-013-0687). A minor amount of secondary Bi4(GeO4)3 (cubic I![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3d, JCPDS card no. 01-084-0505) can also be indexed at x > 0.06. The unit cell crystal structure of Zn2GeO4 and Bi4(GeO4)3 are shown in Fig. 1b and c, respectively. The crystal structure of willemite-type Zn2GeO4 belongs to space group R
3d, JCPDS card no. 01-084-0505) can also be indexed at x > 0.06. The unit cell crystal structure of Zn2GeO4 and Bi4(GeO4)3 are shown in Fig. 1b and c, respectively. The crystal structure of willemite-type Zn2GeO4 belongs to space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) (no. 148). It is comprised of corner-sharing [ZnO4] and [GeO4] tetrahedra (Fig. 1b).20,25 While the incorporation of Bi3+ into the crystal lattice is expected to result in a change of lattice parameters and in decreasing symmetry (assumedly due to the parallel formation of oxygen vacancies which is necessary for charge compensation), we do not observe a notable shift in the diffraction peaks. This indicates the very low solubility of Bi3+ ions in Zn2GeO4 which can be understood on the basis of the large difference in ionic radii between Bi3+ and Zn2+ or Ge4+. The ionic radii of Zn2+ and Ge4+ in fourfold coordination are 0.60 and 0.39 Å, respectively.37 There is no clear data available on fourfold-coordinated Bi3+, though. For fivefold coordination, it is 0.96 Å, a significant 60% higher than the IVZn2+ radius. The appearance of the secondary crystal phase is a consequence of this low solubility of Bi3+ ions in Zn2GeO4. Eulytite-type Bi4(GeO4)3 is composed of [BiO6] octahedra and [GeO4] tetrahedra (Fig. 1c).38 It is known as scintillator material and – due to the VIBi3+-site – is an excellent host for optically active dopants.38,39
(no. 148). It is comprised of corner-sharing [ZnO4] and [GeO4] tetrahedra (Fig. 1b).20,25 While the incorporation of Bi3+ into the crystal lattice is expected to result in a change of lattice parameters and in decreasing symmetry (assumedly due to the parallel formation of oxygen vacancies which is necessary for charge compensation), we do not observe a notable shift in the diffraction peaks. This indicates the very low solubility of Bi3+ ions in Zn2GeO4 which can be understood on the basis of the large difference in ionic radii between Bi3+ and Zn2+ or Ge4+. The ionic radii of Zn2+ and Ge4+ in fourfold coordination are 0.60 and 0.39 Å, respectively.37 There is no clear data available on fourfold-coordinated Bi3+, though. For fivefold coordination, it is 0.96 Å, a significant 60% higher than the IVZn2+ radius. The appearance of the secondary crystal phase is a consequence of this low solubility of Bi3+ ions in Zn2GeO4. Eulytite-type Bi4(GeO4)3 is composed of [BiO6] octahedra and [GeO4] tetrahedra (Fig. 1c).38 It is known as scintillator material and – due to the VIBi3+-site – is an excellent host for optically active dopants.38,39
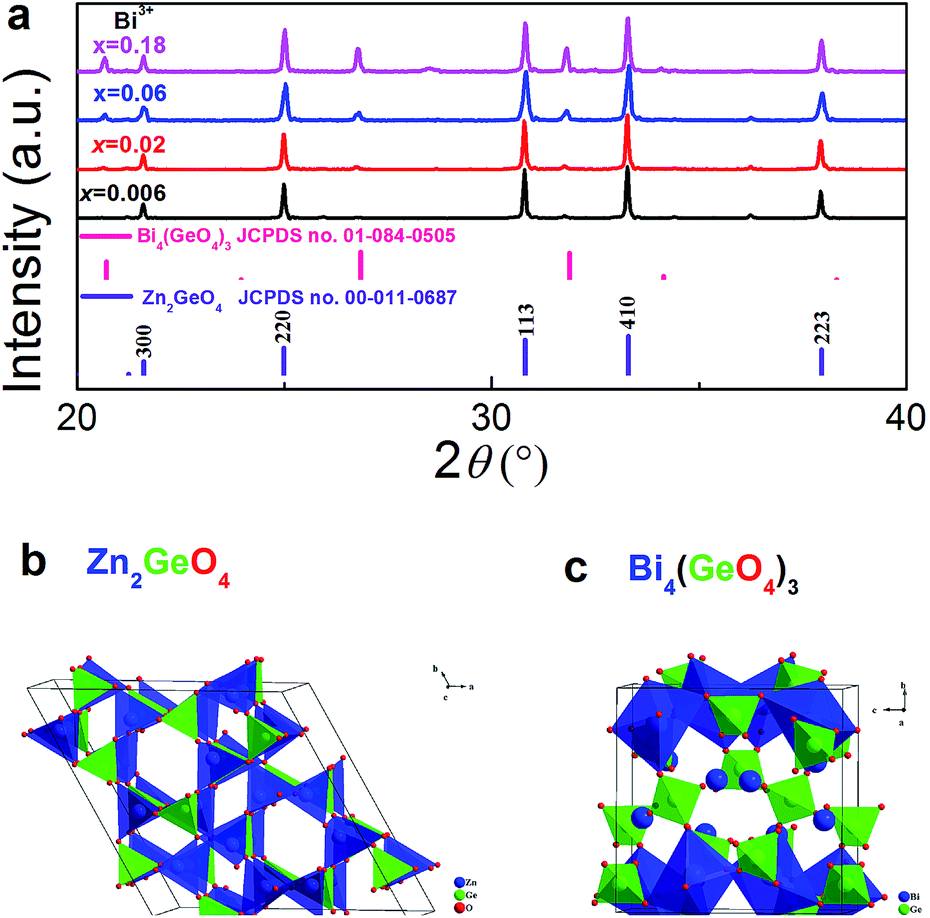 | ||
| Fig. 1 (a) Ex situ powder XRD patterns of Zn1.998−xGeO4:Yb0.0023+Bix3+ (x = 0, 0.006, 0.02, 0.06 and 0.18) in dependence of Bi3+ doping concentration. Tabulated standard diffraction patterns of Zn2GeO4 and Bi4(GeO4)3 are shown for reference. The unit cell crystal structure of (b) Zn2GeO4 and (c) Bi4(GeO4)3 with [ZnO4]/[BiO6] and [GeO4] tetrahedra is also shown. | ||
Fig. 2 exemplarily shows DR spectra of blank Zn2GeO4 and Bi3+ (x = 0.02) singly-doped Zn2GeO4. The blank Zn2GeO4 sample exhibits a broad absorption band in the UV region, i.e., from ∼200 to 360 nm. This absorption band can be deconvoluted in at least two contributions: an intense band at ∼200–290 nm and a weaker shoulder at ∼290–360 nm. The intense absorption band is ascribed to electronic transitions from the valence band to the conduction band of Zn2GeO4 host, whereas the origin of the weaker shoulder is not clear.20 From the absorption edge in UV region at ∼263 nm, we estimate a band-gap energy of ∼4.71 eV which is good accordance with reported data.25,40 Addition of Bi3+ adds a strong yellow absorption band (originating from the allowed transition of 1S0 → 3P1 in trivalent bismuth), shifting the UV-edge to ∼450 nm.
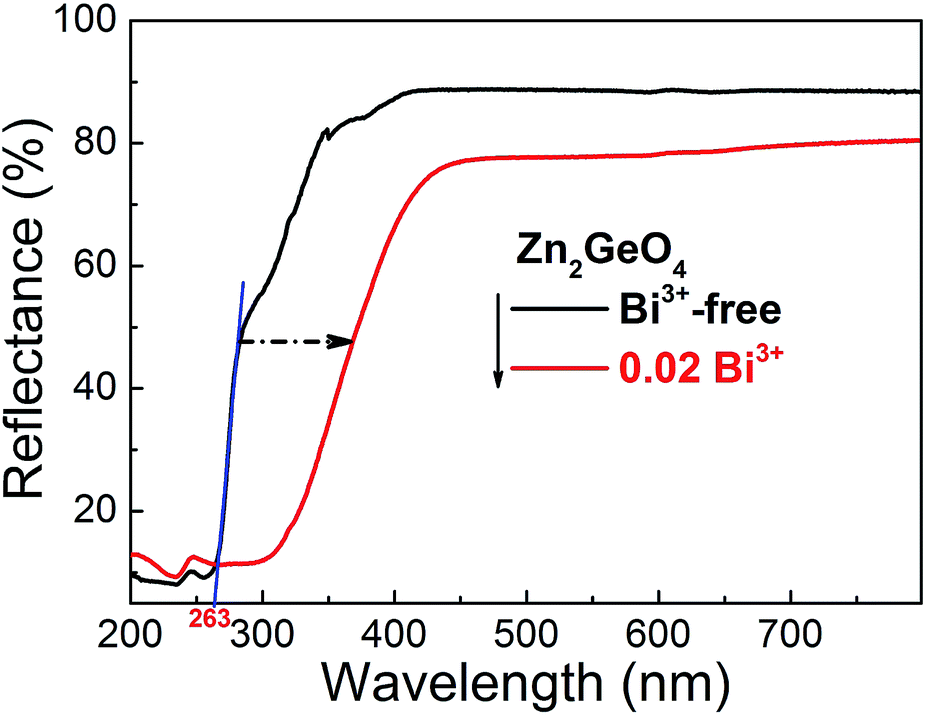 | ||
| Fig. 2 UV-vis diffuse relectance spectra of blank Zn2GeO4 (black line) and Bi3+ (x = 0.02) singly doped Zn2GeO4 (red line). The band gap value of Zn2GeO4 is derived by the intersection of tangent (blue line) with abscissa. The blue tangent is shown as a guide for the eye to highlight the band-gap of Zn2GeO4. | ||
As already mentioned, Zn2GeO4 is a self-activated phosphor. In Fig. 3a and b, we show the room-temperature PLE and PL spectra of blank Zn2GeO4. PLE occurs in a strong band from 250 to 280 nm and another, weaker band from 280 to 375 nm, which is consistent with the DR spectra shown in Fig. 2. The excitation maximum locates at ∼270 nm which correspond-well with the band gap energy of pure Zn2GeO4. Under UV excitation at 270 nm, PL occurs in the spectral region of ∼475–625 nm with a maximum at ∼540 nm with a full width at half maximum (FWHM) of ∼1647 cm−1 (∼48 nm) (Fig. 3b). This PL band is assigned to radiative defect-recombination, i.e., a donor (VO˙ and Zni˙) and an acceptor defect (VZn′ and ionized VGe).27 As for effective PL lifetime τ1/e, we find a range < 3 μs (that is, below the pulse length of the employed Xe flash lamp).
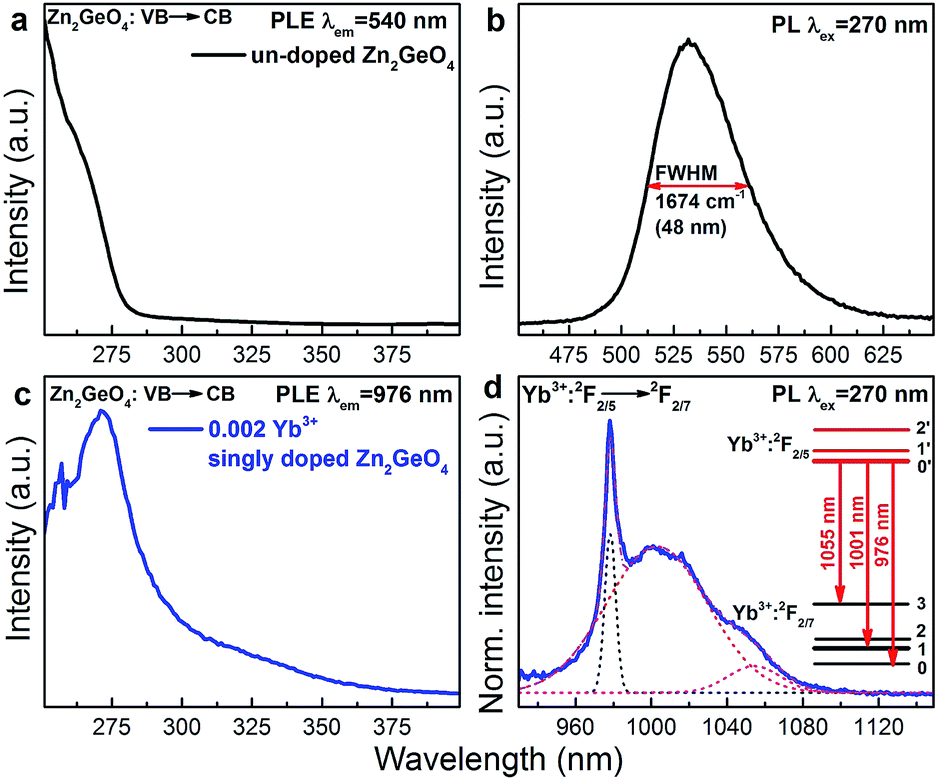 | ||
| Fig. 3 (a) Room-temperature PLE (λem = 540 nm) and (b) PL (λex = 270 nm) spectra for greenish PL in pure Zn2GeO4. (c) PLE (λem = 976 nm) and (d) PL (λex = 270 nm) spectra of Yb3+:2F2/5 → Yb3+:2F2/7 emission in Yb3+ singly doped Zn2GeO4. The inset of (d) presents the Stark splitting energy levels of Yb3+ from the best fit of the PL spectrum of Yb3+. The dashed and solid lines in (d) represent the individual Gaussian functions and total fit of the spectra, respectively, as used for deconvolution. | ||
Room-temperature PLE and PL spectra of Yb3+ in Yb3+ singly-doped Zn2GeO4 are provided in Fig. 3c and d. The PLE spectrum is very similar to the spectrum of pure Zn2GeO4 (Fig. 3a) and also corresponds to the DR spectrum (Fig. 2), showing the aforementioned two contributions at 250–290 nm and 290–375 nm. This directly evidences PL activation of Yb3+ through energy transfer from the Zn2GeO4 host. The corresponding NIR PL covers a broad spectral region of 930–1100 nm with a sharp peak at 976 nm and two red-shifted shoulders. The PL spectrum can best be deconvoluted into three Gaussian functions with maxima at 976, 1001 and 1055 nm (as shown in Fig. 3d). Since neither Bi3+ nor the Zn2GeO4 host are expected to contribute to the NIR emission at this excitation band, the observed NIR PL is assigned to the transition from the lowest stark level of Yb3+:2F2/5 to the three different stark levels of the ground state of Yb3+:2F2/7 (inset of Fig. 3d). The occurrence of Yb3+-related PL after excitation into the conduction band of Zn2GeO4 confirms existence of energy transfer from Zn2GeO4 to Yb3+.
Fig. 4a–c present room-temperature PLE and PL spectra of Bi3+ in Zn1.998−xGeO4:Yb0.0023+Bix3+ for varying values of x. In the presence of Bi3+, an additional PL band appears in the spectral range of 390 to 500 nm (maximum at 420 nm, FWHM ∼4325 cm−1, excitation at 350 nm). This is ascribed to the transition of Bi3+: 3P1 → 1S0 (Fig. 4c). The Stokes-shift of this Bi3+-related PL is relatively low, i.e., ∼5200 cm−1.29 With increasing Bi3+ concentration, the intensity of the Bi3+-related PL is found to decrease as a result of concentration quenching effect (inset of Fig. 4c).41,42 The corresponding PLE spectra exhibit two distinct bands, i.e., from 250 to 290 nm and from 310 to 390 nm, with peaks at ∼250 and 345 nm, respectively (Fig. 4a). The high-energy part is ascribed to the extrinsic PLE band of the aforementioned transition from the valence to the conduction band of Zn2GeO4, which suggests the energy transfer from the Zn2GeO4 host to Bi3+. The latter contribution is assigned to the intrinsic transition of Bi3+: 1S0 → 3P1. Under extrinsic excitation through the Zn2GeO4 host (270 nm), the overlapping PL bands the Bi3+ active center and the Zn2GeO4 host itself can be observed simultaneously with peaks at ∼450 and ∼525 nm, respectively (Fig. 4b and d). The overall PL spectrum therefore spans the very broad spectral region of 390–650 nm with a FWHM of ∼4020 cm−1 (∼160 nm). The maximum PL intensity of Bi3+: 3P1 → 1S0 is found for x = 0.06, whereas PL from the Zn2GeO4 host is quenched with increasing concentration of Bi3+, again confirming the occurrence of energy transfer from Zn2GeO4 to Bi3+. It is further observed that under extrinsic excitation through Zn2GeO4, the PL peak which is attributed to Bi3+: 3P1 → 1S0 shifts to longer wavelength, i.e., from ∼420 to 450 nm, as compared to intrinsic excitation of Bi3+ centers. This evidences the occurrence of energy transfer from Zn2GeO4 to Bi3+ through the defect level of Zn2GeO4 (VO˙ and Zni˙). As with the intrinsic luminescence from the Zn2GeO4 host, the effective lifetime of the Bi3+: 3P1 → 1S0 emission is <3 μs.
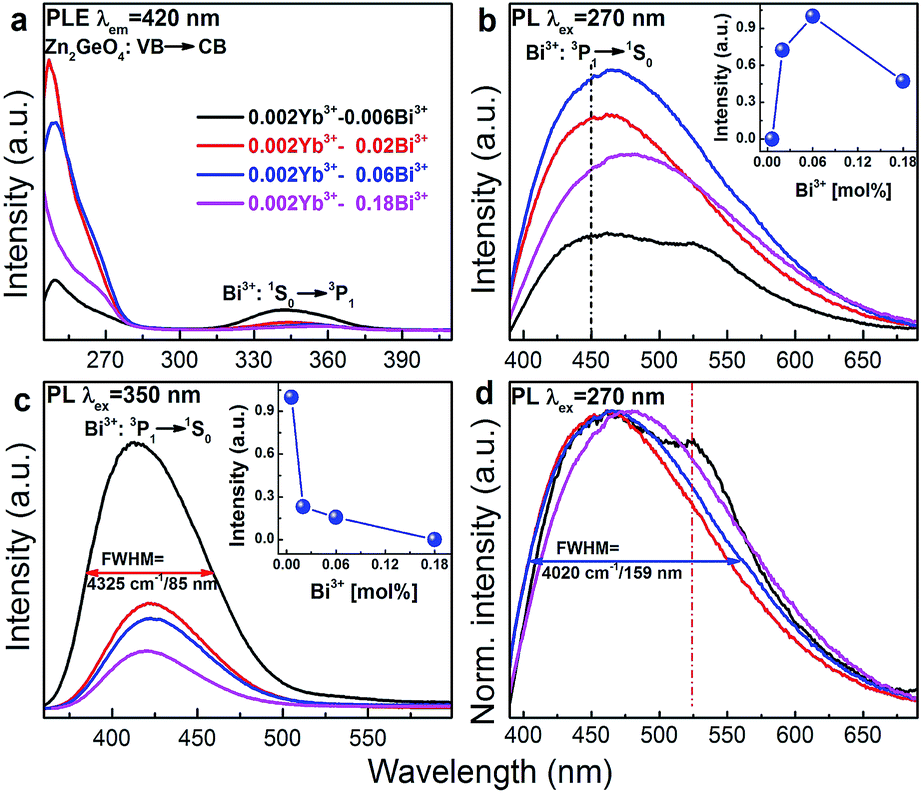 | ||
| Fig. 4 Room-temperature PLE (λem = 420 nm), PL ((b) λex = 270 nm and (c) λex = 350 nm) and (d) normalized PL (λem = 270 nm; normalized to the PL peak at 450 nm) spectra of Zn1.998−xGeO4:Yb0.0023+Bix3+ (x = 0.006, 0.02, 0.06 and 0.18) as a function of Bi3+ doping concentration. The insets of (b) and (d) show PL peak intensity in Zn1.998−xGeO4:Yb0.0023+Bix3+ phosphors versus Bi3+ doping concentration. The drawn lines in the inset of (b) and (c) are to guide the eye. | ||
Room-temperature PLE (monitoring Yb3+: 2F5/2 → 2F7/2 PL at 976 nm) and PL (through extrinsic excitation through the Zn2GeO4 host at 280 nm and through the Bi3+ band at 365 nm, respectively) spectra of Zn1.998−xGeO4:Yb0.0023+Bix3+ are shown in Fig. 5a–c. The PLE bands of the Zn2GeO4 host (250 to 315 nm) as well as of the Bi3+ active center (345 to 450 nm) can be identified in the PLE spectra of Yb3+ (Fig. 5a). This clearly confirms that energy transfer occurs from those entities to Yb3+. The typical Yb3+-related NIR emission can consequently be observed either through excitation of the Zn2GeO4 host (270 nm) or through excitation of Bi3+: 1S0 → 3P1 (365 nm, Fig. 5b and c). When excited at 270 nm, the introduction of a small amount of Bi3+ (x = 0.06) strongly enhances the PL intensity of Yb3+ (here: more than 12 times, Fig. 5b), whereas higher Bi3+ concentration results is quenching of the Yb3+-related PL. Similarly, when excited at 365 nm (i.e., on the Bi3+ band), the PL intensity of Yb3+ attains a maximum for x = 0.06 (Fig. 5c).
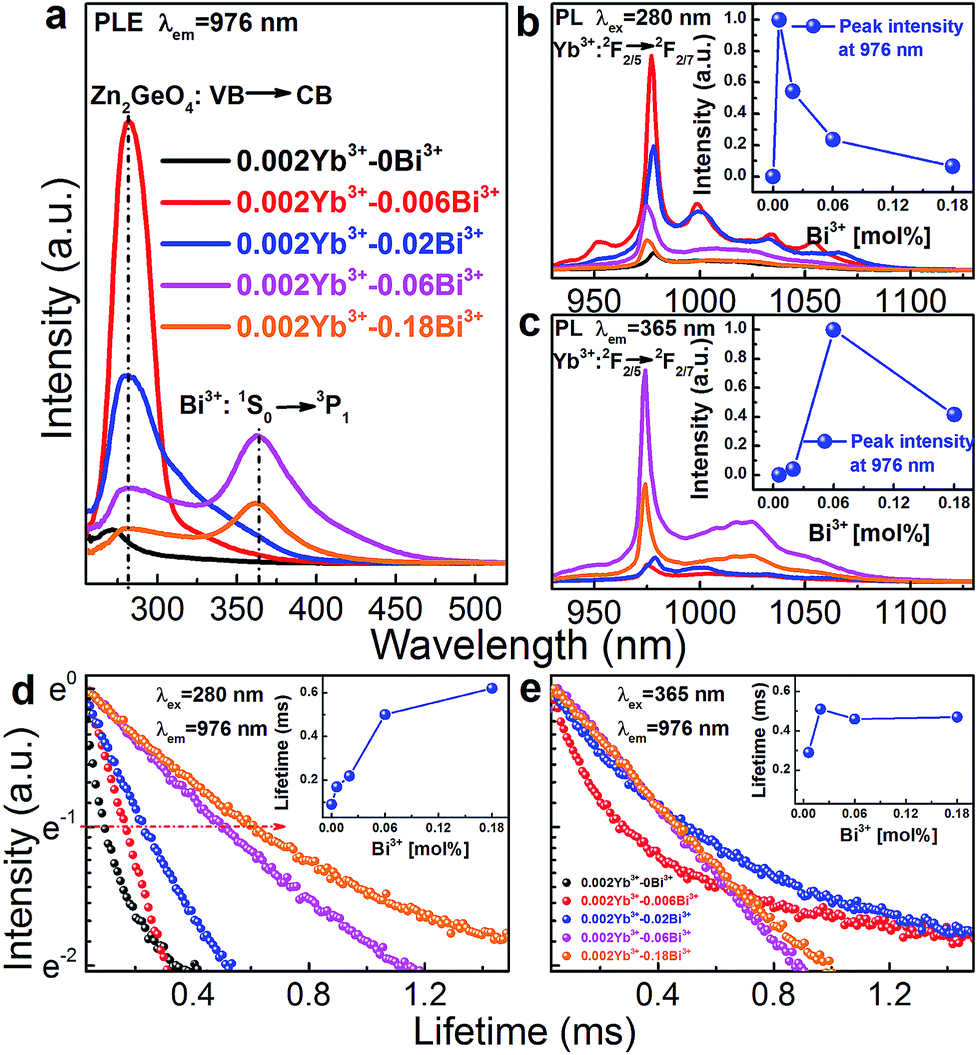 | ||
| Fig. 5 Room-temperature PLE (λem = 976 nm) and PL (upon excitation of (b) Zn2GeO4 host at λex = 280 nm and (c) of Bi3+ at λex = 365 nm) spectra for NIR PL from Yb3 emission in Zn1.998−xGeO4:Yb0.0023+Bix3+ (x = 0, 0.006, 0.02, 0.06 and 0.18) phosphors as a function of Bi3+ concentration. Insets (b) and (c) show PL peak intensity of Yb3+ in Zn1.998−xGeO4:Yb0.0023+Bix3+ phosphors in dependence of Bi3+ concentration. Normalized room-temperature decay curves for NIR PL from Yb3 at 976 nm under pulsed excitation of at (d) 280 nm (Zn2GeO4 host) and (e) 365 nm (Bi3+). Inset of (d) and (e) shows effective lifetime τ1/e for NIR PL from Yb3 emission as a function of Bi3+ concentration. The lines in the insets of (b), (c), (d) and (e) are guides for the eye. | ||
Fig. 5d and e represent normalized decay curves of Yb3+: 2F5/2 → 2F7/2 PL at λem = 976 nm for excitation through the Zn2GeO4 host and through Bi3+, respectively. All decay curves deviate from a single-exponential form, which is related to the different mechanisms of energy transfer, and eventually also to the presence of various types of Bi3+-related emission species (where the above-noted optimal dopant concentration coincides with the onset of Bi4(GeO4)3 formation (Fig. 1)). The effective lifetime τ1/e of Yb3+: 2F5/2 → 2F7/2 PL increases with increasing Bi3+ concentration from ∼90 to 620 μs for excitation through the host (Fig. 5d). When excited through Bi3+ species, it first increases from 290 to 510 μs for a Bi3+ concentration up to (x = 0.02). For higher Bi3+ dopant concentration, a saturation is found (Fig. 5e and inset of Fig. 5e).
The overall scheme of spectral conversion in Zn2GeO4:Bi3+,Yb3+ is illustrated in Fig. 6 and compared to the solar irradiance spectrum. Without Bi3+, only photons in the spectral region below 350 nm (which accounts for only a small portion of the total solar irradiance) can be harvested. In this case, conversion occurs through intrinsic emission as well as through Yb3+ emission centers in the spectral regions of 475–625 nm (green) and 930–1100 nm (NIR). The introduction of Bi3+ strongly enhances the spectral harvesting efficiency through extending the absorption band to up to 450 nm. DS PL from both Bi3+ and Zn2GeO4 then spans a broad region in the blue-to-red spectral range. Through energy transfer from Zn2GeO4 to Yb3+via Bi3+ as well as directly from Bi3+ to Yb3+, notable conversion to the NIR region can be obtained. In this way, Yb3+-emission is activated over the excitation region of 250–500 nm.
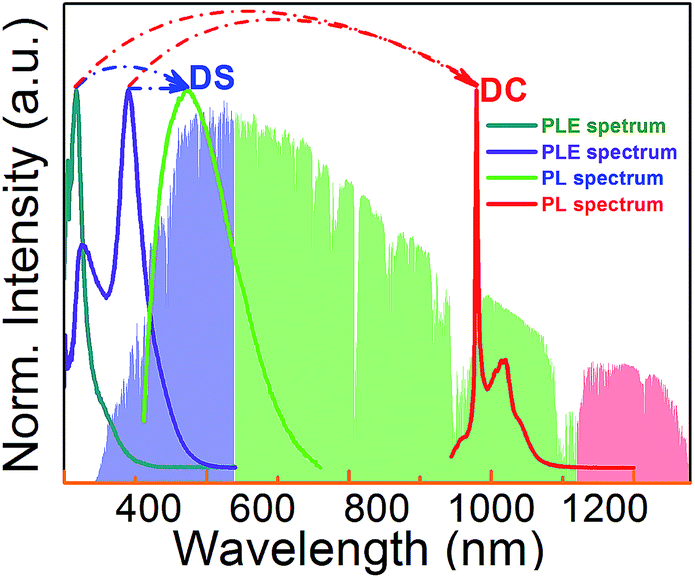 | ||
| Fig. 6 PLE spectrum of Yb3+ in Yb3+ singly (cyan line) and Yb3+/Bi3+ co-doped (violet line) Zn2GeO4, and PL spectrum of Zn1.998−xGeO4:Yb0.0023+Bi0.063+ phosphors in visible (green line) and NIR (red line) region under excitation at 280 nm. The standard solar spectrum for air mass 1.5 according to ASTM G173 - 03(2012) is shown in the background for reference. | ||
This process is summarized in Fig. 7 which depicts the energy level diagrams of the Zn2GeO4 host together with the Bi3+ and Yb3+ emission centers. For Yb3+ singly-doped Zn2GeO4, the electrons are excited from valence to conduction band, leaving hole centers at the valence band under excitation at 270 nm. These defects are subsequently trapped through non-radiative processes.20,26,27 Their recombination is accompanied by the intrinsic PL emission of the Zn2GeO4 host at ∼ twice the energy-gap of Yb3+: 2F7/2 → 2F5/2. As a result, besides PL emission, the trapped energy can also be transferred to two neighboring Yb3+ entities, resulting in NIR emission at ∼1000 nm. Due to the absence of an intermediate energy level at ∼1000 nm, the energy transfer from Zn2GeO4 host to Yb3+ is understood as a second order cooperative DC process. With the introduction of Bi3+, the absorption band extends to the blue region. Electrons which are excited into the conduction band of Zn2GeO4 can relax to the defect level of Zn2GeO4 (VO˙ and Zni˙) and then to the excited level of Bi3+: 3P1. Alternatively, the excited electrons at conduction band of Zn2GeO4 can directly relax to the excited level of Bi3+: 3P1 through a multi-phonon-assisted relaxation process. At a small Stokes shift of the Bi3+: 3P1 level, DS PL from Bi3+ can be observed at an emission energy which is a little bit more than twice the absorption energy of Yb3+: 2F7/2 → 2F5/2. Similar to host excitation, also here, energy can be transferred to two neighboring Yb3+ in a cooperative DC process to yield Yb3+ emissions at ∼1000 nm. Under excitation at 365 nm, the electrons are excited from the ground state of Bi3+: 1S0 to the excited state of Bi3+: 3P1. The following processes are the same as just described.
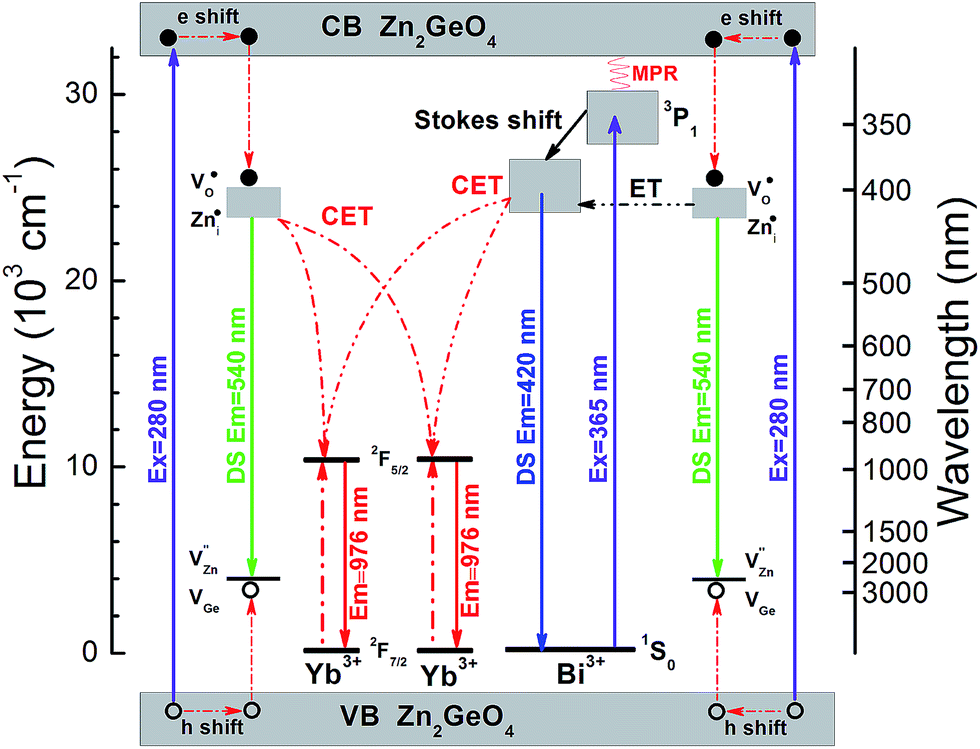 | ||
| Fig. 7 Energy level diagrams of Zn2GeO4 host, Bi3+ and Yb3+, and schematic illustration of the energy transfer mechanisms from the Zn2GeO4 host to Yb3+, from the Zn2GeO4 host to Yb3+via Bi3+ and from Bi3+ to Yb3+ (CET: cooperative energy transfer; ET: energy transfer; MET: multi-phonon-assisted energy transfer). | ||
Conclusions
In conclusion, we discussed the spectral properties of the high-bandgap semiconductor Zn2GeO4 co-doped with Yb3+ and Bi3+ as a model system for ultra-efficient conversion of the UV-A/blue part of the solar spectrum to the NIR spectral region. We have shown that Yb3+-related PL can be activated through intrinsic absorption of the Zn2GeO4 lattice itself, and through Bi3+ centers. Energy transfer then occurs via cooperative DC from trapped defect states and from the 3P1 level of Bi3+. This results in a strong increase in the absolute intensity of Yb3+-related PL. The material enables ultra-efficient harvesting of UV-A to visible radiation for energy conversion processes which rely on NIR irradiation such at c-Si photovoltaics and various photochemical processes.Acknowledgements
The authors gratefully acknowledge financial support from the German Science Foundation (DFG) through grant no. WO 1220/2-2 and the Department of Education of Guangdong Province (grant no. 2013gjhz0001).Notes and references
- X. Huang, S. Han, W. Huang and X. Liu, Chem. Soc. Rev., 2012, 42, 173–201 RSC.
- Q. Y. Zhang and X. Y. Huang, Prog. Mater. Sci., 2010, 55, 353–427 CrossRef CAS PubMed.
- J. de Wild, A. Meijerink, J. K. Rath, W. G. J. H. M. van Sark and R. E. I. Schropp, Energy Environ. Sci., 2011, 4, 4835–4848 CAS.
- B. M. van der Ende, L. Aarts and A. Meijerink, Phys. Chem. Chem. Phys., 2009, 11, 11081–11095 RSC.
- L. Wondraczek, M. Batentschuk, M. A. Schmidt, R. Borchardt, S. Scheiner, B. Seemann, P. Schweizer and C. J. Brabec, Nat. Commun., 2013, 4, 2047 Search PubMed.
- M. Peng and L. Wondraczek, J. Mater. Chem., 2009, 19, 627–630 RSC.
- Q. Xia, M. Batentschuk, A. Osvet, P. Richter, D.-P. Häder, J. Schneider, C. J. Brabec, L. Wondraczek and A. Winnacker, Opt. Express, 2013, 21, A909–A916 CrossRef PubMed.
- D. Chen, Y. Wang and M. Hong, Nano Energy, 2012, 1, 73–90 CrossRef CAS PubMed.
- T. Trupke, M. A. Green and P. Würfel, J. Appl. Phys., 2002, 92, 1668–1674 CrossRef CAS PubMed.
- G. Gao and L. Wondraczek, Opt. Mater. Express, 2013, 3, 633–644 CrossRef CAS.
- D. Yu, S. Ye, M. Peng, Q. Zhang and L. Wondraczek, Appl. Phys. Lett., 2012, 100, 191911 CrossRef PubMed.
- D. Yu, X. Huang, S. Ye, M. Peng, Q. Y. Zhang and L. Wondraczek, Appl. Phys. Lett., 2011, 99, 161904 CrossRef PubMed.
- J. Zhou, Y. Teng, S. Zhou and J. Qiu, Int. J. Appl. Glass Sci., 2012, 3, 299–308 CrossRef CAS PubMed.
- R. Zhou, Y. Kou, X. Wei, C. Duan, Y. Chen and M. Yin, Appl. Phys. B, 2012, 107, 483–487 CrossRef CAS.
- L. Aarts, B. van der Ende, M. F. Reid and A. Meijerink, Spectrosc. Lett., 2010, 43, 373–381 CrossRef CAS.
- J. J. Eilers, D. Biner, J. T. van Wijngaarden, K. Krämer, H.-U. Güdel and A. Meijerink, Appl. Phys. Lett., 2010, 96, 151106 CrossRef PubMed.
- Q. Y. Zhang, G. F. Yang and Z. H. Jiang, Appl. Phys. Lett., 2007, 91, 051903 CrossRef PubMed.
- B. M. van der Ende, L. Aarts and A. Meijerink, Adv. Mater., 2009, 21, 3073–3077 CrossRef CAS PubMed.
- H. Lin, D. Chen, Y. Yu, A. Yang and Y. Wang, Opt. Lett., 2011, 36, 876–878 CrossRef CAS PubMed.
- G. Gao and L. Wondraczek, J. Mater. Chem. C, 2013, 1, 1952–1958 RSC.
- Y. Li, J. Wang, W. Zhou, G. Zhang, Y. Chen and Q. Su, Appl. Phys. Express, 2013, 6, 082301 CrossRef.
- X. Y. Huang and Q. Y. Zhang, J. Appl. Phys., 2010, 107, 063505 CrossRef PubMed.
- X. Wei, S. Huang, Y. Chen, C. Guo, M. Yin and W. Xu, J. Appl. Phys., 2010, 107, 103107 CrossRef PubMed.
- M. Balestrieri, G. Ferblantier, S. Colis, G. Schmerber, C. Ulhaq-Bouillet, D. Muller, A. Slaoui and A. Dinia, Sol. Energy Mater. Sol. Cells, 2013, 117, 363–371 CrossRef CAS PubMed.
- C. Yan, N. Singh and P. S. Lee, Appl. Phys. Lett., 2010, 96, 053108 CrossRef PubMed.
- G. Anoop, K. M. Krishna and M. K. Jayaraj, J. Electrochem. Soc., 2008, 155, J7–J10 CrossRef CAS PubMed.
- Z. Liu, X. Jing and L. Wang, J. Electrochem. Soc., 2007, 154, H500–H506 CrossRef CAS PubMed.
- G. Blasse and B. C. Grabmaier, Luminescent materials, Springer-Verlag, 1994 Search PubMed.
- G. Blasse and A. Bril, J. Chem. Phys., 1968, 48, 217–222 CrossRef CAS PubMed.
- M. Peng, G. Dong, L. Wondraczek, L. Zhang, N. Zhang and J. Qiu, J. Non-Cryst. Solids, 2011, 357, 2241–2245 CrossRef CAS PubMed.
- M. Ilmer, B. C. Grabmaier and G. Blasse, Chem. Mater., 1994, 6, 204–206 CrossRef CAS.
- F. Kang, X. Yang, M. Peng, L. Wondraczek, Z. Ma, Q. Zhang and J. Qiu, J. Phys. Chem. C, 2014, 118, 7515–7522 CAS.
- W. Xu, M. Peng, Z. Ma, G. Dong and J. Qiu, Opt. Express, 2012, 20, 15692–15702 CrossRef CAS PubMed.
- N. Niu, F. He, S. Gai, C. Li, X. Zhang, S. Huang and P. Yang, J. Mater. Chem., 2012, 22, 21613–21623 RSC.
- U. Rambabu and S.-D. Han, Ceram. Int., 2013, 39, 701–708 CrossRef CAS PubMed.
- M. Peng, B. Sprenger, M. A. Schmidt, H. Schwefel and L. Wondraczek, Opt. Express, 2010, 18, 12852–12863 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- R. Nitsche, J. Appl. Phys., 1965, 36, 2358–2360 CrossRef CAS PubMed.
- Y. T. Arslanlar, Z. Kotan, R. Kibar, A. Canımoğlu and N. Can, Spectrosc. Lett., 2013, 46, 590–596 CrossRef CAS.
- Q. Liu, Y. Zhou, J. Kou, X. Chen, Z. Tian, J. Gao, S. Yan and Z. Zou, J. Am. Chem. Soc., 2010, 132, 14385–14387 CrossRef CAS PubMed.
- G. Gao, R. Meszaros, M. Peng and L. Wondraczek, Opt. Express, 2011, 19, A312–A318 CrossRef PubMed.
- G. Gao, S. Reibstein, E. Spiecker, M. Peng and L. Wondraczek, J. Mater. Chem., 2012, 22, 2582–2588 RSC.
| This journal is © The Royal Society of Chemistry 2014 |