
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of nascent molecular electronic devices from gold nanoparticles and terminal alkyne functionalised monolayer films
Henrry M.
Osorio
ab,
Pilar
Cea
ab,
Luz M.
Ballesteros
ab,
Ignacio
Gascón
ab,
Santiago
Marqués-González
c,
Richard J.
Nichols
d,
Francesc
Pérez-Murano
e,
Paul J.
Low
cf and
Santiago
Martín
*ag
aDepartamento de Química Física, Facultad de Ciencias, Universidad de Zaragoza, 50009, Spain. E-mail: smartins@unizar.es
bInstituto de Nanociencia de Aragón (INA) y Laboratorio de Microscopias Avanzadas (LMA), Edificio i+d Campus Rio Ebro, Universidad de Zaragoza, C/Mariano Esquillor, s/n, 50018 Zaragoza, Spain
cDepartment of Chemistry, University of Durham, Durham DH1 3LE, UK
dDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK
eInstituto de Microelectrónica de Barcelona (IMB-CNM, CSIC), Campus UAB, 08193 Bellaterra, Spain
fSchool of Chemistry and Biochemistry, University of Western Australia, 35 Stirling Highway, Crawley, Perth, 6009, Australia
gInstituto de Ciencias de Materiales de Aragón (ICMA), Universidad de Zaragoza-CSIC, 50009 Zaragoza, Spain
First published on 3rd July 2014
Abstract
A metal–molecule–GNP assembly has been fabricated using an acetylene-terminated phenylene–ethynylene molecular monolayer, namely 4-((4-((4-ethynylphenyl)ethynyl)phenyl)ethynyl)benzoic acid (HOPEA), sandwiched between a gold substrate bottom electrode and gold nanoparticle (GNP) top contact electrode. In the first stage of the fabrication process, a monolayer of directionally oriented (carboxylate-to-gold) HOPEA was formed onto the bottom electrode using the Langmuir–Blodgett (LB) technique. In the second stage, the gold-substrate supported monolayer was incubated in a solution of gold nanoparticles (GNPs), which resulted in covalent attachment of the GNPs on top of the film via an alkynyl carbon–Au σ-bond thereby creating the metallic top electrode. Adsorption of the GNPs to the organic LB film was confirmed by both UV-vis absorption spectroscopy and X-ray photoemission spectroscopy (XPS), whilst the contact angle showed changes in the physical properties of the film surface as a result of top-coating of the LB film with the GNPs. Importantly, surface-enhanced Raman scattering (SERS) confirmed the covalent attachment of the metal particles to the LB film by formation of Au–C σ-bonds via a heterolytic cleavage of the alkyne C–H bond. Electrical properties of these nascent metal–molecule–GNP assemblies were determined from I–V curves recorded with a conductive-AFM in the Peak Force Tunneling AFM (PF-TUNA™) mode. The I–V curves obtained from these structures rule out the formation of any significant number of short-circuits due to GNP penetration through the monolayer, suggesting that this strategy of self-assembly of GNPs to alkyne-terminated monolayers is an effective ‘soft’ procedure for the fabrication of molecular junctions without damaging the organic layer.
Introduction
Molecular electronics research has rapidly developed over the last few years as techniques necessary to measure the electronic properties of molecules as either single entities or in small ensembles in various two- and three-electrode test platforms have been realized.1–3 These test-bed junctions have revealed the critical roles played by not only the chemical structure of the backbone of the molecular component but also the nature of the electrode–molecule contact.4,5 Despite the substantial number of fundamental problems that have been faced by this emerging area of science, such as the fluctuations in electrode–molecule contacts and hence coupling, quantum interference effects and the nature of the tunnelling to hopping transition,6 and challenges for the imminent future including progressing device designs beyond the mimicry of conventional circuits, the manipulation of electron spin as well as charge transport designs and gated charge transport,7 it is now possible to realistically contemplate molecular electronics as a potential, albeit future, technology with which to augment present-day silicon microelectronics technology.8Building on the success of measurements of molecular electronic characteristics in test platforms, the next significant challenge to be addressed before molecular electronics can be considered a truly viable and scalable technology is the construction of robust and reliable metal–molecule contacts. A wide variety of molecular functional groups have been tested as contacting groups for various substrates (principally gold), including thiols,9,10 amines,11,12 carboxylic acids,12–14 dithiocarboxylic acids,15 esters,16 pyridyl,17–19 cyano,20,21 isocyano,22,23 isothiocyanato,24 nitro,20 methylselenide,25 methylthiol,25 dimethylphosphine,25 trimethylsilane,26–31 fullerenes,32,33etc. However, most of these metal–molecule contacts exhibit fluxional functional-group to surface bonds leading to stochastic on–off switching, structural rearrangements of the leads, multiple distinct types of metal–molecule contact arising from molecule binding to different surface structures resulting in multiple and significantly different conductance signals, or high contact resistance.27,34–37
The second important challenge in molecular electronics is the fabrication of the top contact electrode in two terminal sandwich-based metal–organic monolayer–metal devices. The metallization of organic monolayers has been investigated for more than 30 years and, like the metal–molecule contact, is still not a well-resolved problem.37–40 A wide variety of techniques to deposit the top metal electrode have been described in the literature including direct and indirect evaporation,38,41–49 use of liquid metals,38,43,45,50,51 flip chip lamination,45,52 electrodeposition,53–55 and surface-diffusion-mediated deposition.56 We have recently shown that adsorption of aurate ([AuCl4]−) onto an ammonium ion terminated Langmuir–Blodgett film followed by photoreduction is also a convenient, “soft” method of assembling metal–molecule–metal structures with potential applications in the scaleable fabrication of molecular electronic devices.57 The most significant problems in the deposition of the second electrode are those related to damage of the functional molecules during the metallization of a monolayer or penetration of the second metal through the organic film, which results in a short circuit, rendering the device unusable.37,40 In addition, in most of these methods the metal–molecule contact is rather weak, whilst robust metal–organic junctions are required to improve the device performance, reproducibility, and stability.
Prompted by the current landscape, in this contribution we explore the fabrication of a metallic electrode on top of a monolayer of a ‘wire-like’ bis(phenylene ethynylene) derivative, leading to the formation of robust and conducting metal–molecule–metal (or GNP) junctions contacted via a covalent Au–C bond which results in the easiest electron transfer between the metal and the organic monolayer. This work is inspired by the recent experiments of Maity et al.,58,59 who obtained acetylide-decorated Au clusters from arylacetylene (–C6H4–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–H) terminated organic compounds and Au nanoparticles weakly stabilized by polyvinylpyrrolidone (PVP). These authors have demonstrated that the terminal hydrogen in the alkyne (–CC–H) group is lost during this process with concomitant formation of an Au–CC σ-bond, accompanied by a weakening of the –CC– bond of the alkynyl group. The significant reduction of the pH value of the aqueous phase observed by these authors after the ligand exchange indicated that the ligation of the alkynyl group proceeds via deprotonation of the alkyne. This method obviates the need for the formation of aryl radicals (e.g. from diazonium compounds) or alkynyl anions (e.g. from abstraction of the alkynyl proton with a strong base) used in other methods for formation of metal–C bonds. Complementary DFT calculations concerning the adsorption of an ethynylbenzene radical on Au(111) have also shown that a strong covalent bond is formed with the surface upon removal of the terminal hydrogen-atom of the ethynyl group.60 In addition, Au–C covalent bonds have been recently studied in metal–molecule junctions,58,61,62 exhibiting high contact conductance resulting from the direct metal–carbon coupling,63–65 and prompting further consideration of this direct Au–C bond as a surface contacting group.
C–H) terminated organic compounds and Au nanoparticles weakly stabilized by polyvinylpyrrolidone (PVP). These authors have demonstrated that the terminal hydrogen in the alkyne (–CC–H) group is lost during this process with concomitant formation of an Au–CC σ-bond, accompanied by a weakening of the –CC– bond of the alkynyl group. The significant reduction of the pH value of the aqueous phase observed by these authors after the ligand exchange indicated that the ligation of the alkynyl group proceeds via deprotonation of the alkyne. This method obviates the need for the formation of aryl radicals (e.g. from diazonium compounds) or alkynyl anions (e.g. from abstraction of the alkynyl proton with a strong base) used in other methods for formation of metal–C bonds. Complementary DFT calculations concerning the adsorption of an ethynylbenzene radical on Au(111) have also shown that a strong covalent bond is formed with the surface upon removal of the terminal hydrogen-atom of the ethynyl group.60 In addition, Au–C covalent bonds have been recently studied in metal–molecule junctions,58,61,62 exhibiting high contact conductance resulting from the direct metal–carbon coupling,63–65 and prompting further consideration of this direct Au–C bond as a surface contacting group.
In the present work, an arylacetylene terminated molecule is used to fabricate and study metal–monolayer–metal junctions. A monolayer of an oligo (phenylene–ethynylene) (OPE) derivative, namely 4-((4-((4-ethynylphenyl)ethynyl)phenyl)ethynyl)benzoic acid (HOPEA), Fig. 1, assembled as a directionally oriented Langmuir–Blodgett (LB) monolayer66 has been incubated in a dispersion of unprotected gold nanoparticles (GNPs), resulting in chemisorption of GNPs to the LB film through Au–C bonds to give robust sandwich-like device structures in an experimentally simple fashion.
 | ||
| Fig. 1 Molecular structure of 4-((4-((4-ethynylphenyl)ethynyl)phenyl)ethynyl)benzoic acid (HOPEA). | ||
Results and discussion
Monomolecular LB films incorporating HOPEA were deposited onto quartz, mica, glass and gold substrates initially immersed in the water subphase that were withdrawn resulting in organized molecular films in which the carboxylate group is in contact with the substrate and the acetylenic (–CC–H) moiety is oriented towards the air.66 These films were incubated in a dispersion of freshly prepared GNPs for 0.5–4 hours. Immediately after the removal of the substrates from the incubation solution the films were thoroughly rinsed with Milli-Q® water to eliminate physisorbed GNPs from the film surface. Finally, the films were allowed to dry. The incubation time was optimized using quartz crystal microbalance (QCM) experiments (Fig. 2). The variation in the resonator frequency before and after the incubation process is indirectly related to the mass of GNPs incorporated on the QCM substrate by means of the Sauerbrey equation which establishes that:67 | (1) |
 | ||
| Fig. 2 Amount of gold deposited onto a HOPEA monomolecular film incubated in a dispersion of GNPs as a function of the incubation time. | ||
UV-vis spectra were recorded before and after the incubation process in order to verify the assembly of GNPs onto the organic layer (Fig. 3). For the purpose of comparison, the inset of Fig. 3 shows the UV-vis spectrum of the aqueous dispersion of GNPs. The UV-vis spectrum of the pristine LB film features one absorption band centered at 280 nm, attributable to π–π* electronic transitions associated with the OPE backbone of HOPEA.66 The spectrum of the LB film after the incubation process features two absorption bands at 259 nm and at ca. 540 nm,68–71 revealing the incorporation of the GNPs onto the LB film. A significant red-shift of the surface plasmon resonance (SPR) band from 515 nm in the dispersion to 545 nm for the immobilized GNPs on the LB film surface is indicative of a change in the dielectric constant surrounding the nanoparticle. In addition, this shift in the maximum absorption position of the SPR band is consistent with an interaction of the GNPs and the organic film.61
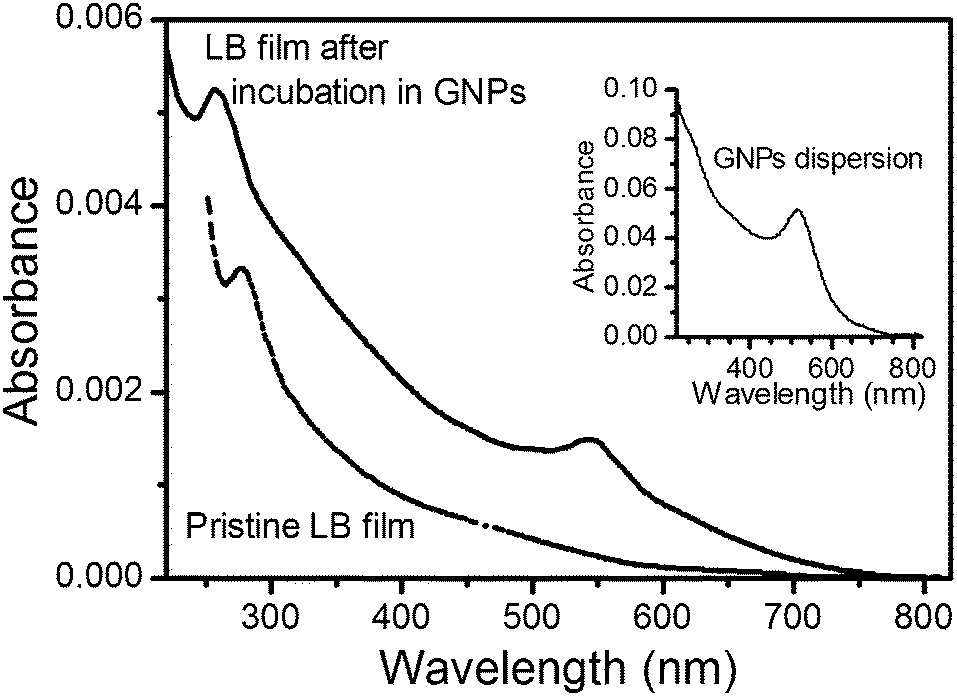 | ||
| Fig. 3 UV-vis spectrum of a pristine HOPEA monomolecular LB film and a spectrum of the same film after incubation in a dispersion of GNPs for 2.5 hours. The inset figure shows the UV-vis spectrum of the aqueous dispersion of GNPs. | ||
The assembly of GNPs onto the film has also been demonstrated by XPS. Fig. 4 shows the XPS spectrum of a HOPEA LB film on a glass substrate after the incubation process in a dispersion of GNPs. The Au4f region shows two peaks at 84.1 and 87.8 eV attributable to the presence of metallic gold.72–75
 | ||
| Fig. 4 XPS spectrum of Au4f photoelectrons of a HOPEA monomolecular LB film on a glass substrate after incubation in a dispersion of GNPs for 2.5 hours. | ||
However, neither QCM, UV-vis spectroscopy nor XPS results provide any information about the distribution, shape, or size of the GNPs on the surface of the film. To investigate these issues, Atomic Force Microscopy (AFM) and Scanning Electron Microscopy (SEM) images of LB films before and after incubation in a dispersion of GNPs were obtained (Fig. 5). The pristine HOPEA film shows a very homogeneous surface. In contrast, after incubation in the dispersion of GNPs both AFM and SEM images show the appearance of spots distributed all over the film revealing the presence of GNPs. The diameter of the GNPs immobilized on the monolayer surface determined by SEM was in the 15–30 nm range whilst the GNP height is in the 6–25 nm range as determined from AFM images. These compare with the 7–28 nm range of particle sizes determined for the initial dispersions by Dynamic Light Scattering (DLS) methods, see Experimental section.
 | ||
| Fig. 5 AFM (top) and SEM (bottom) images of a pristine HOPEA monomolecular LB film (left) deposited onto a mica substrate and the same LB film after incubation for 2.5 hours in a dispersion of GNPs (right). Images are 2 × 2 μm2 in size and the Z range of the AFM images is 30 nm. For a better view of the size and shape of the GNPs both AFM and SEM images have been magnified (500 × 500 nm). | ||
In addition, incubation of HOPEA monomolecular LB films caused significant modification to the appearance, physical characteristics, and composition of the film surface. Surface modification is verified by differences in contact angle measurements from pristine and incubated films of HOPEA in a dispersion of GNPs. The contact angle of water onto a pristine HOPEA LB film deposited onto a glass substrate is 60°, consistent with a well-formed hydrophobic LB film. After incubation of the HOPEA monomolecular LB film in a dispersion of GNPs for 2.5 h the contact angle is reduced to 43° indicating a more hydrophilic surface, with a contact angle value surprisingly close to the value associated with a bare gold substrate (39°).
The previous set of experiments demonstrates the assembly of GNPs onto the monomolecular film. Nevertheless, two further important questions concerning the strategy proposed in this paper to fabricate the top contact electrode in metal–organic monolayer–GNP sandwich structures have still to be addressed: (i) what is the nature of the gold–organic layer interaction? and (ii) does the fabrication of individual GNP electrodes induce the formation of short-circuits due to metallic contact between the bottom and top-contact electrodes or, in contrast, do the GNPs serve as well-behaved top contact electrodes?
As mentioned in the Introduction of this paper, it has been demonstrated58,59,75,76 that terminal alkynes react with GNPs with simultaneous abstraction of acetylenic hydrogen to give Au–CC stabilized structures.51,68,69 With a view to exploring related processes between the terminal alkyne moiety in surface bound HOPEA films and the deposited GNPs vibrational spectroscopy is a particularly useful tool, with several Raman and SERS studies61,63,65,77 having shown evidence for the formation of a carbon–gold covalent bond in related systems. Fig. 6 shows the Raman spectra of HOPEA in the solid state as a powder, as well as the SERS spectrum of a HOPEA pristine LB film deposited on silver mirrors and the spectrum after the incubation procedure in a dispersion of GNPs. All spectra show three major vibrational bands at 2216, 1598, and 1131 cm−1. The highest wavenumber band is associated with the localized vibrational motion of the internal alkyne moiety; the band at 1598 cm−1 is assigned to the symmetric stretch of the three aromatic rings along the long axis of the molecule, and the one at 1131 cm−1 arises from symmetric C–H bending of the phenyl C–H bonds (there are 12 phenyl C–H bonds in HOPEA).78,79 This band at ca. 1131 cm−1 is also present in an analogous compound to HOPEA without the terminal acetylene group, i.e., (4-[4-(phenylethynyl)-phenylethynyl benzoic acid).79 The spectrum of the HOPEA LB film after the incubation process exhibits a low intensity band at 399 cm−1 indicative of the presence of a covalent Au–C σ-bond.56 Such a band is not present in the Raman spectrum of HOPEA powder, nor in the spectra of pristine HOPEA LB films. In addition, the small band at 2110 cm−1, due to the terminal CC group, in the powder Raman spectrum and the SERS spectrum of the HOPEA pristine film is broadened and downshifted to 1990 cm−1 after incubation of the films in a dispersion of GNPs, also indicative of the formation of a –CC–Au bond.80
 | ||
| Fig. 6 Raman spectrum of HOPEA powder, SERS spectrum of a pristine HOPEA monomolecular LB film, and SERS spectrum of a HOPEA monomolecular LB film after incubation in a dispersion of GNPs. | ||
The electrical characteristics of the structures fabricated as described above were determined from collection and analysis of current–voltage (I–V) curves arising from charge flow across the organic films. Analysis of the I–V curves not only gives information concerning the conductance in the metal–monolayer–GNP assemblies but can also be used to rule out the formation of electrical short-circuits by incursion of the GNPs through the organic film that eventually lead to direct metal-to-metal contacts. As described in a previous paper, I–V curves were recorded with a conductive-AFM (Bruker ICON) in the Peak Force Tunneling AFM (PF-TUNA™) mode.57 This operation mode of the AFM is a valuable method for conductivity mapping of delicate samples since it avoids lateral forces that may otherwise damage the tip coating and the soft sample surface. This mode also facilitates the use of cantilevers with low spring constant. The peak force tunneling AFM used here combines a conducting AFM tip and associated low-noise current amplifier with a “tapping” mode AFM to probe current flow through these metal–molecule–GNP junctions. Nevertheless a compromise has to be reached with respect to the set-point force before recording the I–V curves. This compromise involves not applying a too high peak force during the measurements, which would result in large deformation of the monolayer underlying the GNPs, or too little force which would result in inadequate contact between the tip and the surface leading to ineffective electrical probing. The deformation or damage of the monolayer has been investigated as a function of the tip loading force (set-point force) to determine the most suitable set-point force compromise. Fig. 7 shows a c-AFM image of a HOPEA LB film after the incubation process in a GNP dispersion using a set-point force of 3.35 nN. Four gold nanoparticles labeled as GNP1, GNP2, GNP3, and GNP4 are clearly visible in this image. In the set-point force range between 3.35 and 10 nN, the section analysis shows practically constant heights of 25, 19.5, 13.5, and 11.5 nm for GNP1, GNP2, GNP3, and GNP4, respectively. The practically constant GNP height in this set-point force range indicates that no significant deformation of the monolayer occurs and that the GNPs are not substantially pushed into the monolayer by the AFM tip. If the set-point force is increased up to 16.75 nN, the section analysis of the GNPs gives heights of 21, 16.2, 10 and 9 nm for GNP1, GNP2, GNP3, and GNP4 respectively. These reduced height values reveal deformation of the monolayer for this applied set-point force. A further increase in the deformation of the monolayer occurs when a set-point force of 25.2 nN is applied since the section analysis shows heights of 18.8, 14.3, 8.8, and 7.4 nm for GNP1, GNP2, GNP3 and GNP4, respectively. Moreover, if the set-point force is tuned down to low values, e.g. to 6.7 nN, the section analysis of these GNPs shows heights of 24.2, 19.1, 13 and 11.3 nm. This restoration of height values indicates that the deformation produced in the monolayer after applying a high set-point force (25.2 nN) is elastic and does not induce permanent damage of the organic layer.
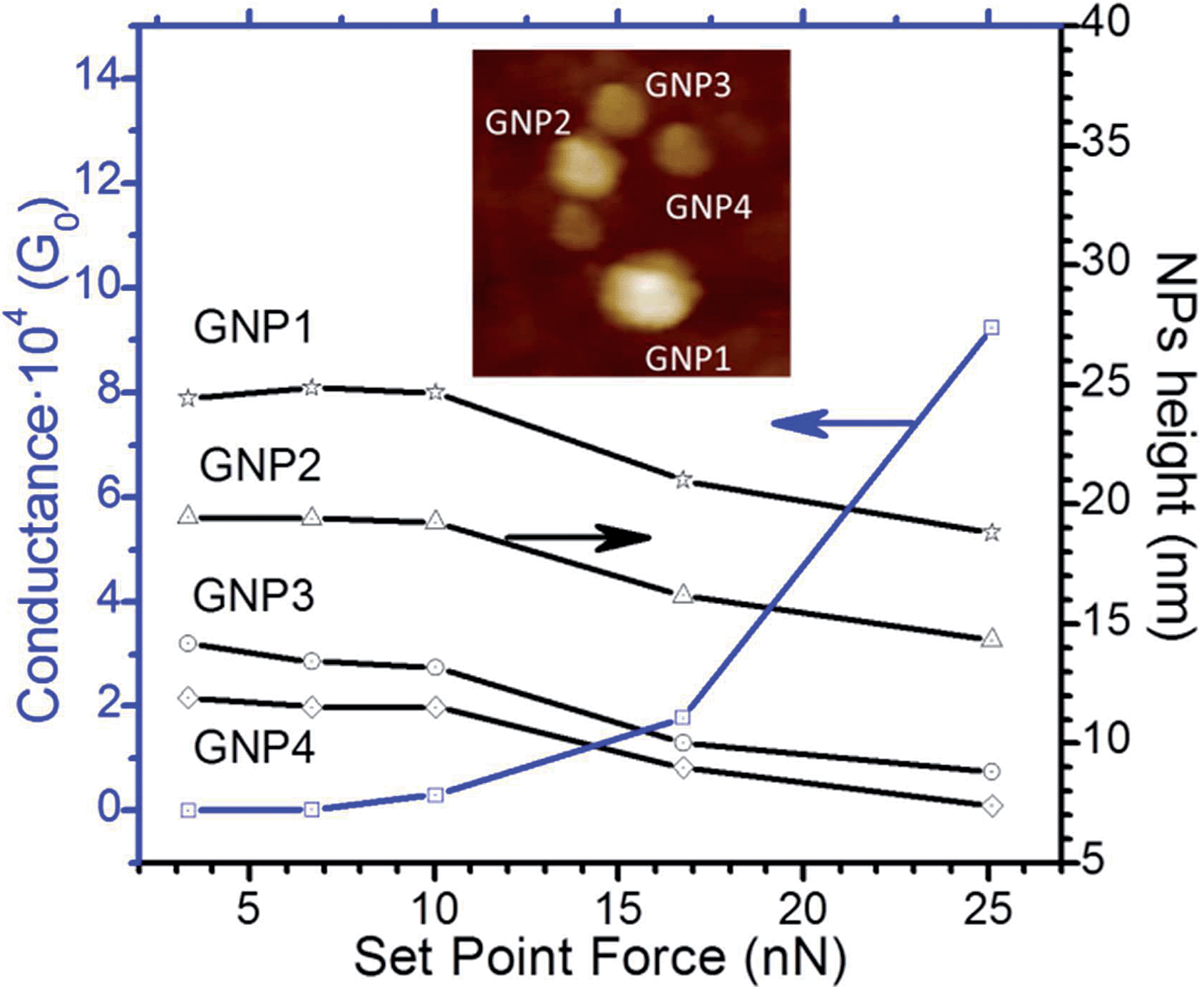 | ||
| Fig. 7 Height of GNPs determined with the c-AFM at the indicated set-point forces together with the average conductance values measured by locating the tip of the c-AFM on the indicated GNPs. The inset top image shows a representative example of a 350 × 350 nm2 image where GNPs can be clearly distinguished and was used to position the c-AFM tip onto the GNPs; the Z range is 50 nm. | ||
Having evaluated the influence of the applied set-point force on the monolayer, the I–V curves were recorded. To record these current–voltage curves, the c-AFM tip was located on the nanoparticle and a bias voltage was applied between the sample and the tip and tip voltage was swept (±1.1 V) with the LB-coated Au substrate set to ground. When a set-point force of 3.35 nN was used any resulting current which might have flowed is below the detection limit (see conductance versus force data in Fig. 7), whilst for a set-point force between 3.35 and 10 nN the I–V curves show low conductance. These results suggest that when low set-point forces are used to record the I–V curves, the electrical contact between the tip and the GNP is not sufficient to support measureable electrical current flow. Nevertheless, when a higher set-point force (between 16.7 and 25.1 nN) was applied, the I–V curves show a significant conductance (Fig. 7), revealing that for these set-point forces there is a good electrical contact between the tip and the GNP, while no damage to the monolayer occurs. Fig. 8 shows a representative I–V curve of all the curves (ca. 550) recorded on different GNPs when a set-point force of 16.7 nm was applied. The bottom inset of Fig. 8 shows the conductance histogram built from all the experimental data (10 I–V curves on each of different GNPs) in the −0.45 to 0.45 V ohmic region for each of the 550 I–V curves obtained experimentally by placing the AFM tip on top of different GNPs applying a set-point force of 16.7 nN. Different GNPs yield different I–V curves whose slope (and thereby conductance of the device) increases with the GNP diameter. All the curves measured exhibit the typical shape observed for metal–molecule–metal junctions, with a linear section only at relatively low bias voltages and increasing curve gradients at higher bias. Importantly, only curves with this behavior were observed, both over GNPs and on the organic monolayer not covered by GNPs (top inset of Fig. 8), and no low resistance traces characteristic of metallic short circuits were obtained over a wide range of set-point forces which rules out the presence of short-circuits confirming that robust and reliable top-contacts have been constructed without damaging the underlying organic monolayer film.
 | ||
| Fig. 8 Representative I–V curve obtained experimentally by positioning the c-AFM tip on top of a GNP when a set-point force of 16.7 nN was applied. Inset bottom: conductance histogram built from all the experimental data from −0.45 to 0.45 V for each I–V curve obtained (ca. 550 curves). Inset top: representative I–V curve obtained by positioning the c-AFM tip on the organic monolayer not covered by GNPs when a set point-force of 6.7 nN was applied; if higher set-point forces are applied damage on the LB film is produced. | ||
Experimental
In a previous paper we reported the synthesis of HOPEA as well as the optimum conditions needed to fabricate well-ordered, defect-free and directionally oriented LB films of this compound on mica, gold and quartz substrates.66 The same protocols have been used in this contribution to fabricate Langmuir–Blodgett (LB) films incorporating HOPEA. A Nima Teflon trough with dimensions (720 × 100) mm2, which was housed in a constant temperature (20 ± 1 °C) clean room, was used to fabricate the films. A Wilhelmy paper plate pressure sensor was used to measure the surface pressure (π) of the monolayers. The subphase was a pH 9 NaOH aqueous solution (Milli-Q water, resistivity 18.2 MΩ cm). A 1 × 10−5 M solution of HOPEA in hexane–ethanol (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) (both solvents purchased from Aldrich and used as received; purity HPLC grade 99% and >99.5%, respectively) was spread onto the aqueous surface. The spreading solvent was allowed to completely evaporate from the surface of the subphase over a period of at least 20 minutes before compression of the monolayer commenced at a constant sweeping speed of 0.015 nm2 per molecule per min. The solid substrates used for the transfer were cleaned carefully as described elsewhere.81,82 The monolayers were deposited by the vertical dipping method onto several substrates at a constant surface pressure of 18 mN m−1 and a speed of 3 mm min−1.
:1) (both solvents purchased from Aldrich and used as received; purity HPLC grade 99% and >99.5%, respectively) was spread onto the aqueous surface. The spreading solvent was allowed to completely evaporate from the surface of the subphase over a period of at least 20 minutes before compression of the monolayer commenced at a constant sweeping speed of 0.015 nm2 per molecule per min. The solid substrates used for the transfer were cleaned carefully as described elsewhere.81,82 The monolayers were deposited by the vertical dipping method onto several substrates at a constant surface pressure of 18 mN m−1 and a speed of 3 mm min−1.
The dispersion of GNPs was prepared by adding rapidly 0.5 mL of a 1.0 × 10−3 M NaBH4 aqueous solution to 30 mL of a 1.0 × 10−5 M HAuCl4 aqueous solution with vigorous stirring at 2 °C using an ice-water bath. The hydrodynamic diameter of these GNPs was found to be in the 7–28 nm range, as determined by Dynamic Light Scattering (DLS). DLS measurements were carried out using a NanoZS ZEN3600 instrument from Malvern. Incubation of the LB films in the dispersion of GNPs (at 2 °C) took place immediately after mixing the reactants.
UV-vis spectra were acquired on a Varian Cary 50 spectrophotometer and recorded using a normal incident angle with respect to the film plane. Atomic force microscopy (AFM) experiments to study the topography of the monolayers were performed by means of a Multimode 8 AFM system from Veeco, in tapping mode. The data were collected with a scan rate of 1 Hz and under ambient air conditions by using a silicon cantilever provided by Bruker, with a force constant of 40 mN m−1 and operating at a resonant frequency of 300 kHz. Scanning electron microscopy (SEM) images were obtained with a JEOL JSM 6400 microscope. X-ray photoelectron spectroscopy (XPS) spectra were acquired on a Kratos AXIS ultra DLD spectrometer with a monochromatic Al Kα X-ray source (1486.6 eV) using a pass energy of 20 eV. The photoelectron take off angle was 90° with respect to the sample plane. To provide a precise energy calibration, the XPS binding energies were referenced to the C1s peak at 284.6 eV. Quartz crystal microbalance (QCM) measurements were carried out using a Stanford Research System instrument and employing AT-cut, α-quartz crystals with a resonant frequency of 5 MHz having circular gold electrodes patterned on both sides. Contact angle experiments were performed with a commercial optical tensiometer Theta Lite from Attension. Raman and surface-enhanced Raman scattering (SERS) spectra were collected using a Confocal Raman Imaging from Witec, model Alpha300M+ with an excitation wavelength of 633 nm. Silver islands (thickness 9.1 nm) were prepared in an Edwards model 306 vacuum coater from a resistively heated tungsten boat. The substrates were Zuzi glass microscope slides cleaned in piranha solution for 30 min (3:1 97% H2SO4:30% H2O2), rinsed with deionized water, and dried in a stream of N2. Care: piranha solutions are exceptionally corrosive and highly oxidizing. Contact between piranha solutions and organic materials is considered extremely hazardous and must be avoided. During film deposition, the background pressure was maintained at 5 × 10−7 Torr, and the deposition rate (0.02 nm3 s−1) was monitored on an Electron Beam Evaporator Auto 500 from BOC Edwards. After deposition, annealing was performed at 200 °C for 60 minutes. The conducting-AFM (c-AFM) measurements were performed with a Bruker ICON microscope under humidity control, ca. 30%, with a N2 flow, in the Peak Force Tunneling AFM (PF-TUNA™) mode, and employing a PF-TUNA™ cantilever from Bruker (coated with Pt/Ir 20 nm, ca. 25 nm radius, 0.4 N m−1 spring constant and 70 kHz resonance frequency).
Conclusions
In this contribution, GNPs have been assembled onto an organic LB film to give arrays of metal–molecule–GNP devices. Formation of covalent Au–C σ-bonds through a simple wet-chemical procedure has been demonstrated by SERS whilst I–V curves ruled out the presence of short-circuits. The terminal alkyne moiety therefore appears to be a suitable functional group for the subsequent deposition of metal nanoparticles which can serve as the top contact electrode. The electrical measurements averaged over multiple gold–molecule–GNP assemblies show the suitability of this approach of forming C–Au bonds to the top GNP contact. This could be a useful approach for future nano-electronic assemblies which exploit more robust C–Au junctions rather than the typically more fluxional chemisorption chemical bonds used in conventional self-assembly. If needed these GNPs could be employed as seeds for deposition of a contiguous metal film using complementary methods such as electroless metal deposition subsequently leading to a controlled preparation of the top contact electrode in metal–monolayer–metal devices with a significant inhibition in the formation of short-circuits.Acknowledgements
P.C., I.G., F.P.-M., and S.M. are grateful for financial assistance from Ministerio de Economía y Competitividad from Spain and fondos FEDER in the framework of projects CTQ2012-33198, CSIC10-4E-805, and CSD2010-00024. P.C., F.P.-M. and S.M. are also grateful to the European SUDOE TRAIN2 project and the Nanolito network through the MAT2011-13099-E project funded by the Ministry of Economy of Spain for supporting this collaborative work. P.C., I.G. and S.M. acknowledge support from DGA and Fondos FEDER for funding Platon research group. H.M.O. and S.M. also thank their JIUZ-2013-CIE-05 grant. R.J.N., P.J.L. and S.M.-G. thank EPSRC for funding, and P.J.L. also gratefully acknowledges support from the Australian Research Council and the award of a Future Fellowship [FT120100073]. H.M.O. is grateful for financial assistance from Secretaría Nacional de Educación Superior, Ciencia, Tecnología e Innovación from Ministerio de Educación (Ecuador). The authors also thank I. Echaniz, G. Antorrena, I. Rivas, R. Valero, and C. Cuesta for technical support.Notes and references
- D. Xiang, H. Jeong, T. Lee and D. Mayer, Adv. Mater., 2013, 25, 4845–4867 CrossRef CAS PubMed.
- M. Tsutsui and M. Taniguchi, Sensors, 2012, 12, 7259–7298 CrossRef CAS PubMed.
- B. Branchi, F. Simeone and M. Rampi, Top. Curr. Chem., 2012, 313, 85–119 CrossRef CAS.
- C. C. Jia and X. F. Guo, Chem. Soc. Rev., 2013, 42, 5642–5660 RSC.
- M. Kiguchi and K. Satoshi, ChemPhysChem, 2012, 13, 1116–1126 CrossRef CAS PubMed.
- M. Ratner, Nat. Nanotechnol., 2013, 8, 378–381 CrossRef CAS PubMed.
- Editorial, Nat. Nanotechnol., 2013, 8, 385–389 CrossRef PubMed.
- Editorial, Nat. Nanotechnol., 2013, 8, 377 CrossRef PubMed.
- L. J. Richter, C. S.-C. Yang, P. T. Wilson, C. A. Hacker, R. D. van Zee, J. J. Stapleton, D. L. Allara, Y. Yao and J. M. Tour, J. Phys. Chem. B, 2004, 108, 12547–12559 CrossRef CAS.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- M. S. Hybertsen, L. Venkataraman, J. E. Klare, A. Cwhalley, M. L. Steigerwald and C. Nuckolls, J. Phys.: Condens. Matter, 2008, 20, 374115 CrossRef PubMed.
- A. Villares, D. P. Lydon, P. J. Low, B. J. Robinson, G. J. Ashwell, F. M. Royo and P. Cea, Chem. Mater., 2008, 20, 258–264 CrossRef CAS.
- S. Martin, W. Haiss, S. Higgins, P. Cea, M. C. Lopez and R. J. Nichols, J. Phys. Chem. C, 2008, 112, 3941–3948 CAS.
- L. M. Ballesteros, S. Martín, J. Cortés, S. Marqués-González, S. J. Higgins, R. J. Nichols, P. L. Low and P. Cea, Chem.–Eur. J., 2013, 19, 5352–5363 CrossRef CAS PubMed.
- Y. Xing, T. H. Park, R. Venkatramani, S. Reinan, D. N. Beratan, M. J. Therien and E. Borguet, J. Am. Chem. Soc., 2010, 132, 7946–7956 CrossRef CAS PubMed.
- A. Villares, D. P. Lydon, B. J. Robinson, G. Ashwell, F. M. Royo, P. J. Low and P. Cea, Surf. Sci., 2008, 602, 3683–3687 CrossRef CAS PubMed.
- B. Q. Xu and N. J. Tao, Science, 2003, 301, 1221–1223 CrossRef CAS PubMed.
- S. Y. Quek, M. Kamenetska, M. L. Stigerwald, H. J. Choi, S. G. Louie, M. S. Hybertsen, J. B. Neaton and L. Venkataraman, Nat. Nanotechnol., 2009, 4, 230–234 CrossRef CAS PubMed.
- W. Hong, D. Z. Manrique, P. Moreno-García, M. Gulcur, A. Mishchenko, C. J. Lambert, M. R. Bryce and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 2292–2304 CrossRef CAS PubMed.
- L. A. Zotti, T. Kirchner, J. C. Cuevas, F. Pauli, T. Huhn, E. Scheer and A. Erbe, Small, 2010, 6, 1929–1535 CrossRef PubMed.
- A. Mishchenko, L. A. Zotti, D. Vonlanthen, M. Burkle, F. Pauly, J. C. Cuevas, M. Mayor and T. Wandlowski, J. Am. Chem. Soc., 2011, 133, 184–187 CrossRef CAS PubMed.
- L. Venkataraman, J. E. Klare, I. W. Tam, C. Nuckolls, M. S. Hybertsen and M. N. Steigerwald, Nano Lett., 2006, 6, 458–462 CrossRef CAS PubMed.
- B. Kim, J. M. Beebe, Y. Jun, X. Y. Zhu and C. D. Frisbie, J. Am. Chem. Soc., 2006, 128, 4970–4971 CrossRef CAS PubMed.
- C. H. Ko, M. J. Huang, M. D. Fu and C. H. Chen, J. Am. Chem. Soc., 2010, 132, 756–764 CrossRef CAS PubMed.
- Y. S. Park, A. C. Whalley, M. Kamenetska, M. L. Steigerwald, M. S. Hybertsen, C. Nuckolls and L. Venkataraman, J. Am. Chem. Soc., 2007, 15768–15769 CrossRef CAS PubMed.
- G. Pera, S. Martin, L. M. Ballesteros, A. J. Hope, P. J. Low, R. J. Nichols and P. Cea, Chem.–Eur. J., 2010, 16, 13398–13405 CrossRef CAS PubMed.
- S. Marqués-González, D. S. Yufit, J. A. K. Howard, S. Martín, H. M. Osorio, V. M. García-Suárez, R. J. Nichols, S. J. Higgins, P. Cea and P. J. Low, Dalton Trans., 2013, 42, 338–341 RSC.
- N. Katsonis, A. Marchenko, S. Taillemite, D. Fichou, G. Chouraqui, C. Aubert and M. Malacria, Chem.–Eur. J., 2003, 9, 2574–2581 CrossRef CAS PubMed.
- N. Katsonis, A. Marchenko, D. Fichou and N. Barret, Surf. Sci., 2008, 602, 9–16 CrossRef CAS PubMed.
- A. Nion, N. Katsonis, A. Marchenko, C. Aubert and D. Fichou, New J. Chem., 2013, 37, 2261–2265 RSC.
- E. G. Petrov, A. Marchenko, O. Kapitanchuk, N. Katsonis and D. Fichou, Mol. Cryst. Liq. Cryst., 2014, 589, 3–20 CrossRef CAS.
- C. A. Martin, D. Ding, J. K. Sorensen, T. Bjornholm, J. M. van Ruitenbeek and H. S. J. van der Zant, J. Am. Chem. Soc., 2008, 130, 13198–13199 CrossRef CAS PubMed.
- J. Fock, J. K. Sorensen, E. Loertscher, T. Vosch, C. A. Martin, H. Riel, K. Kilsa, T. Bjornholm and H. S. J. van der Zant, Phys. Chem. Chem. Phys., 2011, 13, 2236–2241 RSC.
- G. K. Ramachandran, T. J. Hopson, A. M. Rawlett, L. A. Nagahara, A. Primak and S. M. Lindsay, Science, 2003, 300, 1413–1416 CrossRef CAS PubMed.
- Z. K. Keane, J. W. Ciszek, J. M. Tour and D. Natelson, Nano Lett., 2006, 6, 1518–1521 CrossRef CAS PubMed.
- S. Yasuda, S. Yoshida, J. Sasaki, Y. Okutsu, T. Nakamura, A. Taninaka, O. Takeuchi and H. Shigekawa, J. Am. Chem. Soc., 2006, 128, 7746–7747 CrossRef CAS PubMed.
- D. Vuillaume, Proc. IEEE, 2010, 1–12 Search PubMed.
- H. Haick and D. Cahen, Acc. Chem. Res., 2008, 41, 359–366 CrossRef CAS PubMed.
- H. B. Akkerman and B. De Boer, J. Phys.: Condens. Matter, 2008, 20, 013001 CrossRef.
- D. Vuillaume, C. R. Phys., 2008, 9, 78–94 CrossRef CAS PubMed.
- L. Costelle, M. T. Räisänen, J. T. Joyce, C. Silien, L.-S. Johansson, J. M. Campbell and J. Räisänen, J. Phys. Chem. C, 2012, 116, 22602–22607 CAS.
- R. Lovrinčić, O. Kraynis, R. Har-Lavan, A.-E. Haj-Yahya, W. Li, A. Vilan and D. Cahen, J. Phys. Chem. Lett., 2013, 4, 426–430 CrossRef.
- R. L. McCreery and A. J. Bergren, Adv. Mater., 2009, 21, 4303–4322 CrossRef CAS.
- R. M. Metzger, T. Xu and I. R. Peterson, J. Phys. Chem. B, 2001, 105, 7280–7290 CrossRef CAS.
- R. T. W. Popoff, K. L. Kavanagh and H.-Z. Yu, Nanoscale, 2011, 3, 1434–1445 RSC.
- A. Scott, D. B. Janes, C. Risko and M. A. Ratner, Appl. Phys. Lett., 2007, 91, 033508 CrossRef PubMed.
- Z. Shi, P. Lu and A. V. Walker, Langmuir, 2012, 28, 16909–16916 CrossRef CAS PubMed.
- A. V. Walker, T. B. Tighe, O. M. Cabarcos, M. D. Reinard, B. C. Haynie, S. Uppili, N. Winograd and D. L. Allara, J. Am. Chem. Soc., 2004, 126, 3954–3963 CrossRef CAS PubMed.
- Z. Zhu, D. L. Allara and N. Winograd, Appl. Surf. Sci., 2006, 252, 6686–6688 CrossRef CAS PubMed.
- R. Har-Lavan, O. Yaffe, P. Joshi, R. Kazaz, H. Cohen and D. Cahen, AIP Adv., 2012, 2, 012164–012213 CrossRef PubMed.
- O. Yaffe, L. Scheres, L. Segev, A. Biller, I. Ron, E. Salomon, M. Giesbers, A. Kahn, L. Kronik, H. Zuilhof, A. Vilan and D. Cahen, J. Phys. Chem. C, 2010, 114, 10270–10279 CAS.
- S. Pookpanratana, J. W. F. Robertson, C. Jaye, D. A. Fischer, C. A. Richter and C. A. Hacker, Langmuir, 2013, 29, 2083–2091 CrossRef CAS PubMed.
- Y.-D. Chiu, W.-P. Dow, K. Krug, Y.-F. Liu, Y.-L. Lee and S.-L. Yau, Langmuir, 2012, 28, 14476–14487 CrossRef CAS PubMed.
- C. Silien and M. Buck, J. Phys. Chem. C, 2008, 112, 3881–3890 CAS.
- C. Silien, D. E. Lahaye, M. Caffio, R. Schaub, N. R. Champness and M. Buck, Langmuir, 2011, 27, 2567–2574 CrossRef CAS PubMed.
- A. P. Bonifas and R. L. McCreery, Nat. Nanotechnol., 2010, 5, 612–617 CrossRef CAS PubMed.
- S. Martin, G. Pera, L. M. Ballesteros, A. J. Hope, S. Marqués-González, P. J. Low, F. Pérez-Murano, R. J. Nichols and P. Cea, Chem.–Eur. J., 2014, 20, 3421–3426 CrossRef CAS PubMed.
- P. Maity, H. Tsunoyama, M. Yamauchi, S. Xie and T. Tsukuda, J. Am. Chem. Soc., 2011, 133, 20123–20125 CrossRef CAS PubMed.
- P. Maity, S. Takano, S. Yamazoe, T. Wakabayashi and T. Tsukuda, J. Am. Chem. Soc., 2013, 135, 9450–9457 CrossRef CAS PubMed.
- M. J. Ford, R. C. Hoft and A. McDonagh, J. Phys. Chem. B, 2005, 109, 20387–20392 CrossRef CAS PubMed.
- L. Laurentius, S. R. Stoyanov, S. Gusarov, A. Kovalenko, R. Du, G. P. Lopinski and M. T. McDermott, ACS Nano, 2011, 5, 4219–4227 CrossRef CAS PubMed.
- D. M. Shewchuk and M. T. McDermott, Langmuir, 2009, 25, 4556–4563 CrossRef CAS PubMed.
- Z. L. Cheng, R. Skouta, H. Vazquez, J. R. Widawsky, S. Schneebeli, W. Chen, M. S. Hybertsen, R. Breslow and L. Venkataraman, Nat. Nanotechnol., 2011, 6, 353–357 CrossRef CAS PubMed.
- T. Hines, I. Díez-Pérez, H. Nakamura, T. Shimazaki, Y. Asai and N. Tao, J. Am. Chem. Soc., 2013, 135, 3319–3322 CrossRef CAS PubMed.
- W. Hong, H. Li, S.-X. Liu, Y. Fu, J. Li, V. Kaliginedi, S. Decurtins and T. Wandlowski, J. Am. Chem. Soc., 2012, 134, 19425–19431 CrossRef CAS PubMed.
- L. M. Ballesteros, S. Martín, C. Momblona, S. Marqués-González, M. C. López, R. J. Nichols, P. J. Low and P. Cea, J. Phys. Chem. C, 2012, 116, 9142–9150 CAS.
- G. Sauerbrey, Z. Phys., 1959, 155, 206–222 CrossRef CAS.
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CAS.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215–4221 CrossRef CAS PubMed.
- H. Itoh, K. Naka and Y. Chujo, J. Am. Chem. Soc., 2004, 126, 3026–3027 CrossRef CAS PubMed.
- M. Treguer-Delapierre, J. Majimel, S. Mornet, E. Duguet and S. Ravaine, Gold Bull., 2008, 41, 195–207 CrossRef CAS.
- H.-G. Boyen, G. Kästle, F. Weigl, B. Koslowski, C. Dietrich, P. Ziemann, J. P. Spatz, S. Riethmüller, C. Hartmann, M. Möller, G. Schmid, M. G. Garnier and P. Oelhafen, Science, 2002, 297, 1533–1536 CrossRef CAS PubMed.
- M. M. Kemp, A. Kumar, S. Mousa, T.-J. Park, P. Ajayan, N. Kubotera, S. A. Mousa and R. J. Linhardt, Biomacromolecules, 2009, 10, 589–595 CrossRef CAS PubMed.
- G. Liu, E. Luais and J. J. Gooding, Langmuir, 2011, 27, 4176–4183 CrossRef CAS PubMed.
- C. Shan, H. Yang, D. Han, Q. Zhang, A. Ivaska and L. Niu, Biosens. Bioelectron., 2010, 25, 1070–1074 CrossRef CAS PubMed.
- R. Casado, M. Contel, M. Laguna, P. Romero and S. Sanz, J. Am. Chem. Soc., 2003, 125, 11925–11935 CrossRef CAS PubMed.
- Z. L. Cheng, R. Skouta, H. Vazquez, J. R. Widawsky, S. Schneebeli, W. Chen, M. S. Hybertsen, R. Breslow and L. Venkataraman, Nat. Nanotechnol., 2011, 6, 353–357 CrossRef CAS PubMed.
- A. Beeby, K. S. Findlay, P. J. Low, T. B. Marder, P. Matousek, A. W. Parker, S. R. Rutter and M. Towrie, Chem. Commun., 2003, 2406–2407 RSC.
- A. Villares, G. Pera, S. Martin, R. J. Nichols, D. P. Lydon, L. Applegarth, A. Beeby, P. J. Low and P. Cea, Chem. Mater., 2010, 22, 2041–2049 CrossRef CAS.
- W. Hong, H. Li, S.-X. Liu, Y. Fu, J. Li, V. Kaliginedi, S. Decurtins and T. Wandlowski, J. Am. Chem. Soc., 2012, 47, 19425–19431 CrossRef PubMed.
- P. Cea, J. P. Morand, J. S. Urieta, M. C. López and F. M. Royo, Langmuir, 1996, 12, 1541–1544 CrossRef CAS.
- S. Martín, P. Cea, C. Lafuente, F. M. Royo and M. A. C. López, Surf. Sci., 2004, 563, 27–40 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2014 |