
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Robust optical oxygen sensors based on polymer-bound NIR-emitting platinum(II)–benzoporphyrins†
L. H.
Hutter‡
a,
B. J.
Müller‡
b,
K.
Koren
c,
S. M.
Borisov
*b and
I.
Klimant
b
aDepartment of Biochemistry, University of Oxford, South Parks Road, OX1 3QU, UK
bInstitute for Analytical Chemistry and Food Chemistry, Graz University of Technology, Stremayrgasse 9, A-8010 Graz, Austria. E-mail: sergey.borisov@tugraz.at
cMarine Biological Laboratory, University of Copenhagen, Strandpromenaden 5, DK-3000 Helsingør, Denmark
First published on 18th July 2014
Abstract
Several advanced optical oxygen sensor materials are presented. They are based on bright NIR-emitting platinum(II)–benzoporphyrins covalently incorporated into a variety of polymeric matrices. The dye–polymer conjugates are prepared either via Suzuki coupling of the brominated porphyrins to the styrene backbone or via co-polymerisation of the monomers with monostyryl porphyrin derivative. Importantly, in both strategies a highly stable C–C bond is obtained. The resulted materials benefit from excellent photophysical properties of the benzoporphyrin dyes (high brightness, emission in the NIR part of the spectrum) and high stability of the covalently grafted materials due to complete suppression of dye migration and leaching. This is demonstrated to be particularly important for operation of the sensors in harsh conditions e.g. during steam sterilization where the materials based on non-covalently grafted dyes showed significant drift of their calibration. Additionally, we present a new synthetic method for preparation of analytically pure benzoporphyrins via simple 1-step template condensation which a promising alternative to the commonly used Lindsey method.
Introduction
Optical oxygen sensors constitute widely-used analytical tools which are applied, for example, for the study of cellular function,1,2 in medical diagnostics,3,4 marine biology,5 food packaging6,7 and for process monitoring in industry and biotechnology.8 In a general layout a phosphorescent indicator dye is physically entrapped into a matrix material which is a polymer (polystyrene, poly(methylmethacrylate), silicone, or ethylcellulose and their derivatives9,10) or a sol–gel.11,12 The matrix acts as a solvent and a support for the dye, as a permeation-selective barrier for the interfering chemical species and it also controls oxygen permeability. A suitable matrix can enhance both the specificity and selectivity of the sensor as well as its stability over time. A variety of oxygen indicators has been developed over the past decades13 since the photophysical properties of the dyes have to be adjusted for a particular application. The development of dyes that are excitable in the red and NIR part of the electromagnetic spectrum has been of particular interest in recent years.14 These indicator dyes are well suited for measurements in autofluorescent or highly scattering media, such as in vivo samples in many bioanalytical applications. Oxygen indicators based on platinum(II) and palladium(II) complexes of porphyrin ketones15 and porphyrin lactones,16 and particularly of pi-extended benzo- and naphthoporphyrins17–19 and related compounds20 were found to be particularly promising for sensing applications. With the exception of water-dispersible oxygen-sensitive dendrimers (representing self-contained analytical tools primarily for imaging applications)21,22 the above indicators are physically entrapped in a suitable polymeric matrix. Major restrictions of this approach are determined by the level of chemical and physical interaction between the indicator and its local environment. Low solubility of the indicator in the sensor matrix gives rise to leaching and aggregation; both processes seriously compromise the performance of an optical oxygen sensor over time. A lipophilic indicator will unlikely leach out of the sensor in aqueous environment but it may slowly migrate into the hydrophobic sensing support such as the very popular polyethylene terephthalate. Such migration processes are greatly accelerated at elevated temperatures, e.g. during steam sterilization of the sensors. Leaching can be particularly critical for materials with large surface to volume ratio, such as oxygen-sensitive nanoparticles. Covalent immobilisation of the indicators into polymeric matrices represents the optimal strategy to overcome the above limitations. Oxygen sensing materials based on covalently grafted iridium(III)23,24 ruthenium(II)25–28 and platinum(II) or palladium(II) complexes29–33 were reported. The meso-tetra(pentafluorophenyl) porphyrin (TFPP) complexes, particularly, can be easily modified via nucleophilic substitution of the p-fluorine atom of the pentafluorophenyl groups.29–31,33 Significantly higher stability of the new materials compared to the sensors obtained via physical entrapment of the dyes was demonstrated.31 To the best of our knowledge highly promising NIR indicators have not been covalently grafted into oxygen-sensing materials. A versatile immobilisation strategy used for TFPP derivatives is not feasible since the meso-tetra(pentafluorophenyl)-tetrabenzoporphyrin complexes does not form readily.34Herein, we present an efficient and versatile strategy towards advanced optical oxygen sensing materials based on platinum(II)-benzoporphyrin complexes covalently incorporated into a variety of relevant matrices. New brominated porphyrins can be either directly attached to the polymers or modified to give reactive monomers. Importantly, the synthetic strategy relies on Suzuki coupling which results in chemically stable C–C bond. It will be demonstrated how the covalent immobilisation improves the performance of the sensor materials compared to conventional sensors. In addition we will present a new efficient method for the synthesis of benzoporphyrins via template condensation which, compared to the previous reports, results in analytically pure products.
Results and discussion
Synthesis of the brominated Pt(II)–porphyrin indicators
Brominated Pt(II)–benzoporphyrins serve as a synthetic platform for the covalent immobilisation of the indicator dye in various polymer matrices via Suzuki coupling. Depending on immobilisation strategy either tetra- or mono-substituted derivates can be used (Fig. 1). The tetra-substituted Pt(II) benzoporphyrin was initially synthesized using an adapted standard protocol.35,36 This involved Lindsey-condensation37 of 4,5,6,7-tetrahydroisoindole with 4-bromobenzaldehyde yielding compound A1, the subsequent platination of the product (A2) and finally oxidation of the cyclohexene rings to yield the benzoporphyrin 3a. Surprisingly, the solubility of A1 was found to be rather low and the dye aggregated readily in organic solvents. On the other side, solubility of various metal-free tetraphenyltetracyclohexenoporphyrins, their Pt(II) complexes and the respective benzoporphyrins is known to be rather good.34,36 The bromine atoms in the para-position of the meso-substituted phenyl rings appear to promote aggregation of the dyes. Due to low solubility of A1 in organic solvents, purification of the product was challenging; hence the crude product was used in the next step after removal of the solvent.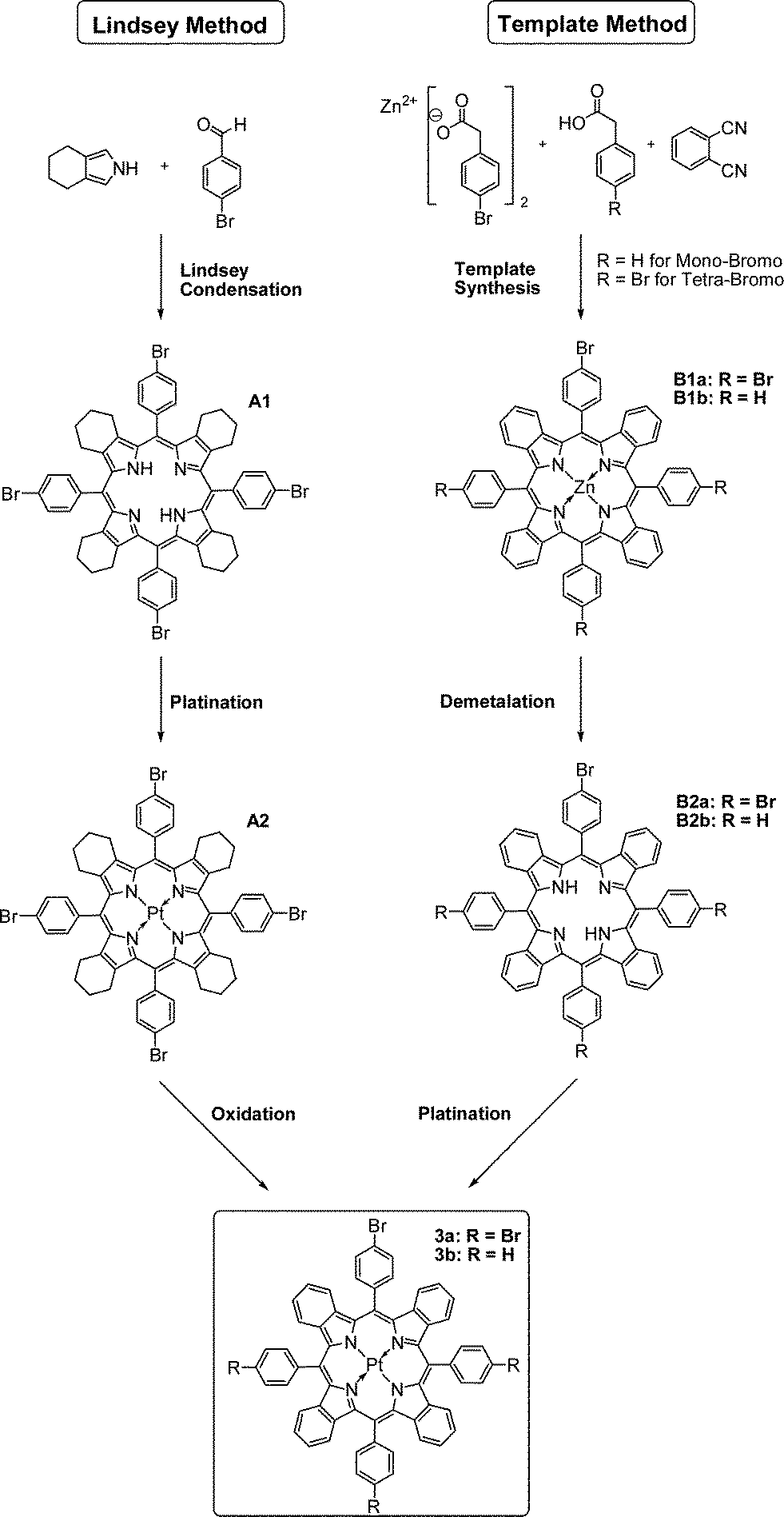 | ||
| Fig. 1 Synthetic approaches towards reactive bromo-modified Pt(II)–benzoporphyrins – commonly used Lindsey condensation (left) and a modified template-method (right). Tetra-substituted porphyrin PtTPTBPBr4 was synthesized using both methods. Mono-substituted dye PtTPTBPBr was prepared using template-method. | ||
In the second step, platination was performed in trimethylbenzene using Pt(C6H5CN)2Cl2 as a precursor complex, which is a modification of the method published previously.38 Trimethylbenzene allows for relatively high reaction temperatures (and therefore fast metalation) but in contrast to diphenylether can be easily removed under reduced pressure.
In the final step, the brominated benzoporphyrin 3a, was obtained via oxidation of A2 with 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ). Similarly to A1, the solubility of the Pt(II) benzoporphyrin was rather poor. The purification was possible only for diluted solutions.
One of the drawbacks of the above method is that it relies on 4,5,6,7-tetrahydroisoindole which is synthesised from not inexpensive ethyl isocyanoacetate. Template condensation represents an alternative method yielding Pt(II) benzoporphyrins in only 3 simple steps, starting from very cheap phthalimide and phenylacetic acid. However, the template condensation was reported to yield a number of benzyl-substituted side products which are extremely challenging to separate.38,39 Preliminary experiments indicated that the condensation of phthalimide with 4-bromophenylacetic acid instead of phenylacetic acid resulted in a low yield of the zinc benzoporphyrin. Herein we present a new efficient method to yield analytically pure benzoporphyrins (Fig. 1). By substituting phthalimide with dicyanobenzene, the reaction temperature could be reduced from 350 °C to 280 °C which greatly enhanced the yield of zinc tetra(4-bromophenyl)tetrabenzoporphyrin. The yield of the target compound after chromatographic purification was 6% which is rather good considering the low cost of the starting materials and simplicity of the procedure. Based on this novel protocol we were able to synthesize PtTPTBPBr4 (compound 3a) in only three steps. The Pt(II) complex can be conveniently obtained after demetalation of the respective zinc porphyrin and subsequent platination of the metal-free porphyrin. The 1H NMR analysis and mass spectroscopic investigation indicate that no other porphyrin derivatives are formed and the product is identical to that obtained via Lindsey method. Considering high potential of zinc40 and platinum(II) benzoporphyrins41 for energy conversion applications the new method is expected to be particularly promising for simple and cost-efficient synthesis of these sensitizers. Many other commercially available dicyanobenzenes and phenylacetic acid derivatives can be used to provide necessary functionalities.
Mono-brominated Pt(II) porphyrin (3b) is also of great practical interest for covalent immobilisation since it only has a single site suitable for modification and hence does not act as cross-linker when it becomes incorporated into a polymer. The respective zinc complex was obtained via the modified template condensation using a mixture of phenylacetic- and 4-bromophenylacetic acids. As determined by MALDI-TOF, this approach results in a mixture of products with a degree of bromo-substitution ranging from 0 to 2 (Fig. S6†). Chromatographic separation of these extremely similar compounds is virtually impossible. Due to bad ionization of the bromo-functionalized complexes, we were not able to reliably determine the contributions of the individual forms at this stage. However, as will be demonstrated in the following, a mixture of the mono-, dibromo- and unsubstituted porphyrins is suitable for preparation of soluble materials.
Photophysical properties
Fig. 2 compares the absorption and emission spectra of the new brominated porphyrin dyes with those of the non-substituted dye. Evidently, the substitution has a very minor effect on the spectral properties of the dyes. A small bathochromic shift of the Q-band is observed for PtTPTBPBr (3b) and PtTPTBPBr4 (3a) compared to PtTPTBP (1 and 5 nm, respectively). Similarly, the corresponding emission spectra shift bathochromically by 2 nm and 9 nm, respectively. Despite potential quenching of the “heavy” bromine atoms we did not observe any significant change in the luminescence decay times and luminescence quantum yields upon bromination (Fig. 2). This correlates well to the results of Zhao and co-workers who demonstrated that the heavy atom effect of the halogen atoms in structurally related BODIPY dyes is much less efficient if they are positioned far away from the π-conjugated core of the chromophore.42 | ||
| Fig. 2 Absorption (in dichloromethane) and emission spectra (in deoxygenated toluene) for the new Pt(II)-benzoporphyrin complexes. | ||
The photostability of the dye is of particular interest for practical applications; especially in those cases where high light densities are used or measurements are performed for prolong time. PtTPTBPF4 which has excellent photostability34 was used for comparison. The photostability of the new compounds was found to be similar to that of PtTPTBPF4.
Preparation of the porphyrin–polymer composites
In order to covalently couple the indicators to the sensor matrix, two different strategies were used (Fig. 3). Both rely on Suzuki coupling and result in chemically stable C–C bonds. In the first approach, polystyrene bearing different amounts of boronic acid residues (Table S1 ESI†) were prepared by co-polymerisation of styrene and 4-vinylphenylboronic acid (4). The tetra- or mono-brominated Pt(II) benzoporphyrin was attached to the polymer in one step. The limitation of the approach is that the amount of the incorporated boronic acid groups is not easy to control and is lower than theoretically expected (e.g. 0.07 instead of 0.1 mol%). In order to avoid cross-linking when using the tetra-brominated porphyrin it should be used in excess to saturate all the boronic acid groups. The unbound dye is then removed during purification of the polymer.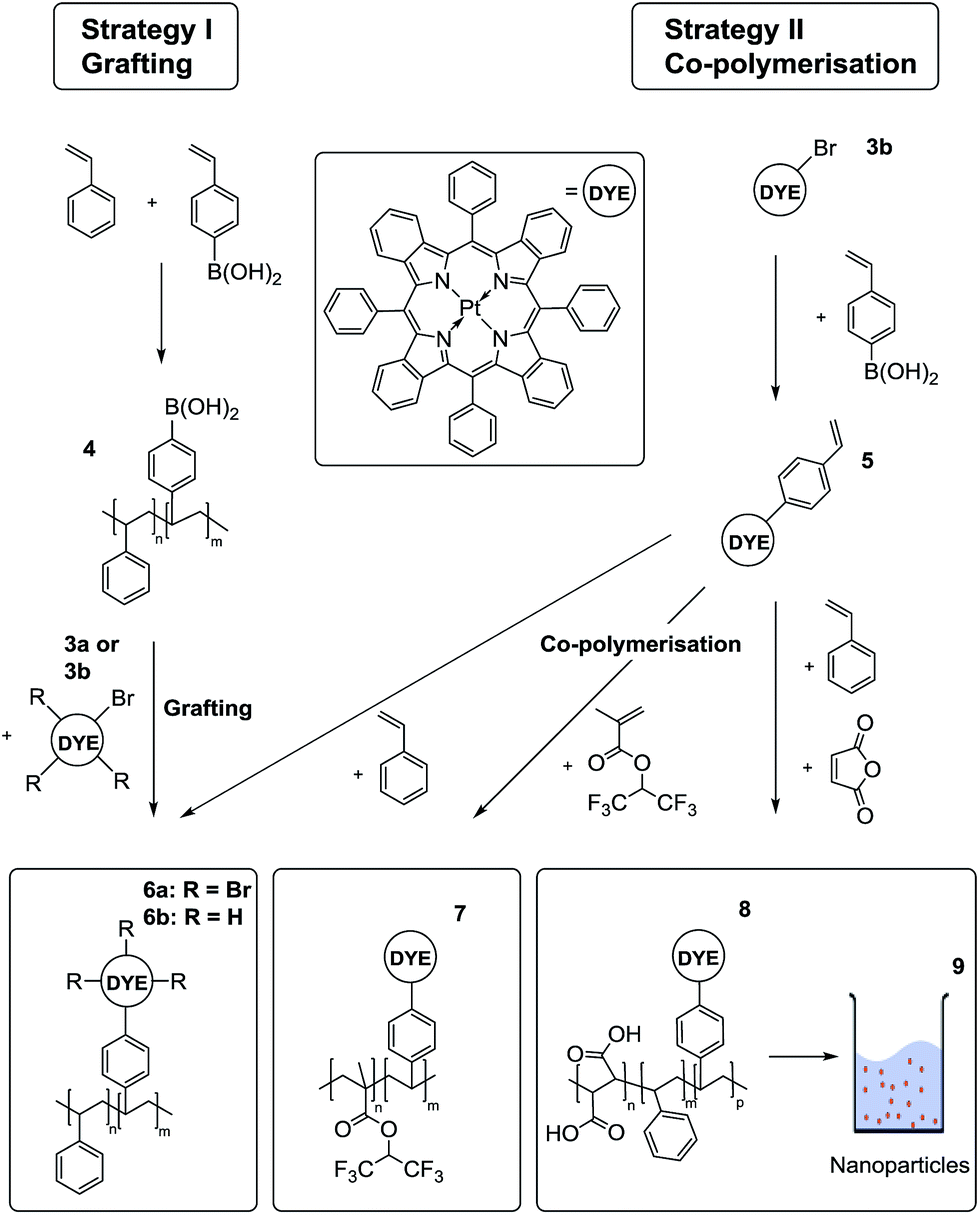 | ||
| Fig. 3 Two different strategies for covalent immobilisation of Pt(II)–benzoporphyrins based on C–C Suzuki-cross-coupling reaction. A co-polymer with boronic acid groups (4) can be used for covalent grafting of either using tetra- (3a) or monobromo- (3b) substituted benzopophyrin. In the second strategy, the indicator is modified with styryl groups (5) and subsequent polymerisation with a variety of monomers is performed (styrene (6b), 1,1,1,3,3,3-hexafluoroisopropylmethacrylate (7) or maleic anhydride and styrene (8)). The latter can be used for the preparation of water dispersible nanoparticles (9). | ||
In the second strategy, the mono-brominated dye was reacted with 4-vinylphenylboronic acid to produce reactive dye monomer (5). Using a standard protocol for Suzuki-coupling reactions, we were able to achieve acceptable yields of about 50% for the styryl-modified benzoporphyrin. The Heck reaction with the vinyl group of the boronic acid leads to the major side product. Due to the polarity of the boronic acid residue, the reactant as well as the side product could be readily removed by means of column chromatography. MALDI-TOF data (Fig. S7†) indicate a mixture of PtTPTBP, PtTPTBPStyr and PtTPTBPStyr2 (ratio 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100:55) which is likely to reflect the substitution pattern of the respective brominated complexes. Relatively high content of unmodified PtTPTBP and the di-substituted porphyrin in this mixture of products is not crucial for the subsequent co-polymerisation reactions since the unbound dye is removed in the purification process and the disubstituted porphyrin dye does cause significant cross-linking due to rather low concentrations used.
:100:55) which is likely to reflect the substitution pattern of the respective brominated complexes. Relatively high content of unmodified PtTPTBP and the di-substituted porphyrin in this mixture of products is not crucial for the subsequent co-polymerisation reactions since the unbound dye is removed in the purification process and the disubstituted porphyrin dye does cause significant cross-linking due to rather low concentrations used.
Oxygen-sensitive polymers were obtained by co-polymerisation of different monomers with the styrene-modified dye, using radical polymerisation with AIBN as initiator. Reactions towards material 6b and 8 where carried out without solvent resulting in polymers with high PDI values, ranging from 1.20 to 5.44. These PDI values are not likely to have major influence on the properties of the sensor. 1,1,1,3,3,3-Hexafluoroisopropyl methacrylate was polymerised in the solution of tetrahydrofuran (THF) as the dye is insoluble in the monomer. In all procedures, unbound indicator was removed via precipitation of the polymer in methanol. The precipitation procedure was repeated until no more indicator dye could be found in the solution. In the case of material 8, functionalized poly(styrene-co-maleic acid) particles were obtained by precipitation of a solution of the polymer in THF from water according to the reported procedure.43
Overall, the co-polymerisation approach is preferable to the first strategy (grafting) due to its high versatility: the approach can be used for numerous monomers and it warrants a higher degree of control over the amount of indicator incorporated into the polymer.
Properties of the sensing materials
Fig. 4 shows the effect of covalent immobilisation of the indicator on the calibration curve of the sensor (Stern–Volmer plot): PtTPTBPBr physically entrapped in polystyrene is used as a reference for the co-polymerised and grafted sensor materials. All three sensor materials possess similar phosphorescence lifetimes in the absence of oxygen. The sensitivities of all the materials are similar. It appears that the way of dye immobilisation only minor affects the calibration. The decay time plots (Fig. 4) are less linear compared to the luminescence intensity plots (Fig. S8†) which is a typical case for the optical oxygen sensors. In fact, a linear fit almost ideally describes the intensity plots (correlation coefficient r2 = 0.9997), but it is less adequate for the decay time plots (r2 = 0.997). The modified equation from the two-site model44 adapted for the decay time plots (eqn (1), ESI†) delivers satisfactory results (r2 = 0.9996).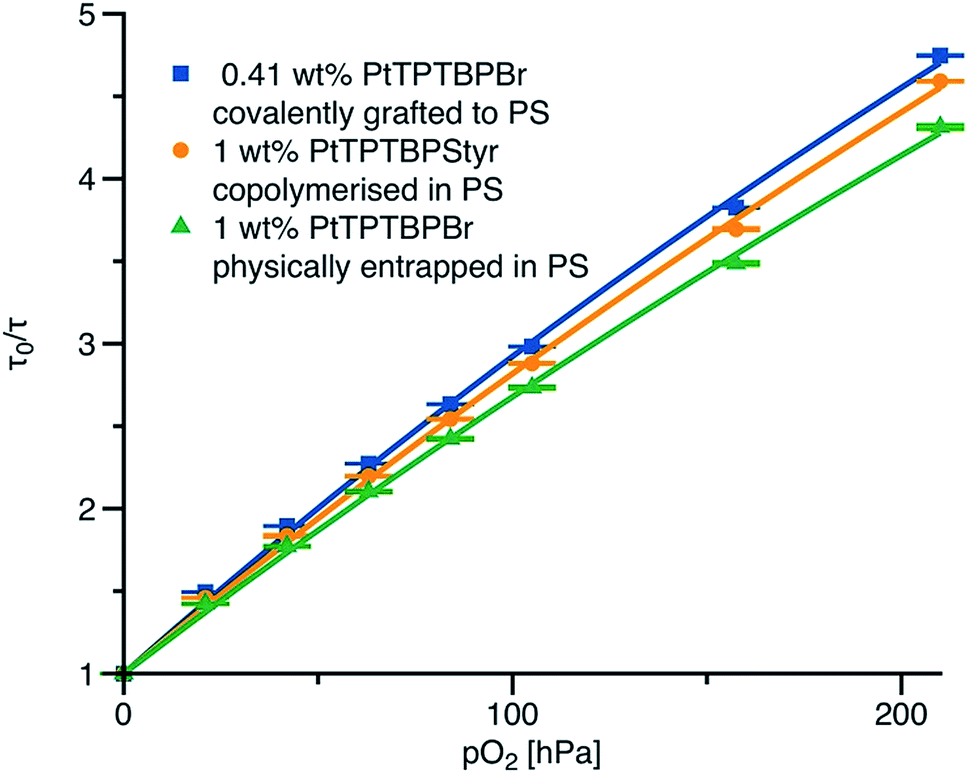 | ||
| Fig. 4 Stern–Volmer plots for polystyrene-based sensing materials at 25 °C. The fit is performed according to the eqn (1) (ESI†). The fit parameters are: f = 0.76, KSV1 = 0.0298 h Pa−1 for covalently grafted PtTPTBPBr; f = 0.82, KSV1 = 0.0249 h Pa−1 for co-polymerised PtTPTBPStyr and f = 0.82, KSV1 = 0.0228 h Pa−1 for physically entrapped PtTPTBPBr; KSV2 = 0.2KSV1 in all cases. | ||
The effect of dye loading on the luminescence lifetime for the grafted sensor materials is shown in Fig. 5. A typical oxygen sensor based on non-covalently entrapped indicator would contain about 1–1.5 wt% of an indicator dye. As can be seen, only slight decrease of the luminescence decay time is visible for a higher concentration of 1.8 wt%. However, the phosphorescence lifetimes become markedly shorter upon further increase of dye loading (50.7, 47.3 and 38.0 μs at 0.33, 1.82 and 6.92 wt%, respectively). This self-quenching is likely to be due to aggregation of the dye at higher dye loadings.
 | ||
| Fig. 5 Calibration plots (at 25 °C) for oxygen sensors based on PtTPTBPBr4 grafted to polystyrene (6a) for varying amount of the immobilised indicator. | ||
Importantly, the styryl-modified benzoporphyrin 5 can be easily incorporated in a variety of other polymeric matrixes. Hence, oxygen sensing materials covering a broad range of potential application fields can be produced (Fig. 6). For example, co-polymerisation with hexafluoroisopropyl methacrylate results in significantly more sensitive materials compared to polystyrene-based ones. Thus, this sensor is much better suitable for measurements at low oxygen. It should be mentioned that the same methodology can be adapted for grafting of analogous Pd(II) benzoporpyhrins which possess about 7 times longer phosphorescence decay times.34 The combination of Pd(II) benzoporphyrins and poly(hexafluoroisopropyl methacrylate) would be suitable for trace oxygen sensing.
 | ||
| Fig. 6 Stern–Volmer plots for sensing materials based on co-polymerised 5 at 25 °C. The fit parameters are: f = 0.90, KSV1 = 0.110 h Pa−1, KSV2 = 0.0041 h Pa−1 for poly(hexafluoroisopropylmethacrylate; f = 0.82, KSV1 = 0.0249 h Pa−1, KSV2 = 0.0050 h Pa−1 for polystyrene and f = 0.73, KSV1 = 0.0183 h Pa−1, KSV2 = 0.0050 h Pa−1 for PSMA particles. | ||
We also prepared a co-polymer of styrene and maleic acid with co-polymerised Pt(II) benzoporphyrin which was used to prepare nanoparticles via precipitation (9). The nanosensors (85 nm, PDI = 0.15) are negatively charged at neutral pH (Zeta potential – 40.9 mV) due to hydrolysis of maleic acid anhydride. Oxygen sensitivity is slightly lower than for the polystyrene materials (Fig. 6). Covalent grafting of the indicators in the nanoparticle-based sensors is of particular importance due to their high surface to volume ratio, and therefore intense interaction with the environment (e.g. proteins and other species in biological probes). Additionally relatively small diffusion distances in the nanoparticles can aggravate leaching of the non-covalently bound dyes. Our approach overcomes this limitation.
To assess the effect of covalent immobilisation on the long-term performance of the sensor and the ability of covalent immobilisation to suppress effects associated with migration of the indicator dye, we prepared sensor films on glass and polyethylene terephthalate (PET) support foil and exposed them to repeatable autoclave treatment for up to 15 hours at 135 °C. Fig. 7 shows luminescence lifetimes measured under nitrogen and air saturation for the sensor material based on compound 5 co-polymerised with polystyrene compared with corresponding data for compound 3a physically entrapped in polystyrene. Regardless of the support material the phosphorescence lifetimes under nitrogen are virtually not affected by high temperature and humidity. On the other hand, the decay times under air saturation for the physically entrapped indicator increase dramatically (13.0 to 23.8 μs after 15 h). This effect is caused by the migration of dye into less oxygen-permeable polymeric support. Such a change would result in ∼100% error in determination of pO2 at air saturation after 15 h of autoclavation. Although migration is very fast at elevated temperatures it is expected to be noticeable also at room temperature during prolong storage times. Obviously, the PET support is not suitable for the sensor film based on the physically entrapped dye. For comparison, only very minor increase of the decay time is observed when the non-covalently embedded dye material is coated on glass support. Notably, glass support is less flexible regarding processing and mechanical stability compared to PET. On the contrary, our new materials based on the covalently immobilised dye do not show any significant migration into either supports used. In fact only minor change in the decay times is observed even after 15 h of autoclavation which would produce and error of ∼18% in determination of pO2 at air saturation providing that the sensor is not recalibrated after sterilization. It should be mentioned that 30 min sterilization is typically sufficient and therefore the error in pO2 quantification is virtually negligible. Thus, the new materials are particularly promising for application in harsh conditions (elevated temperatures, steam sterilization).
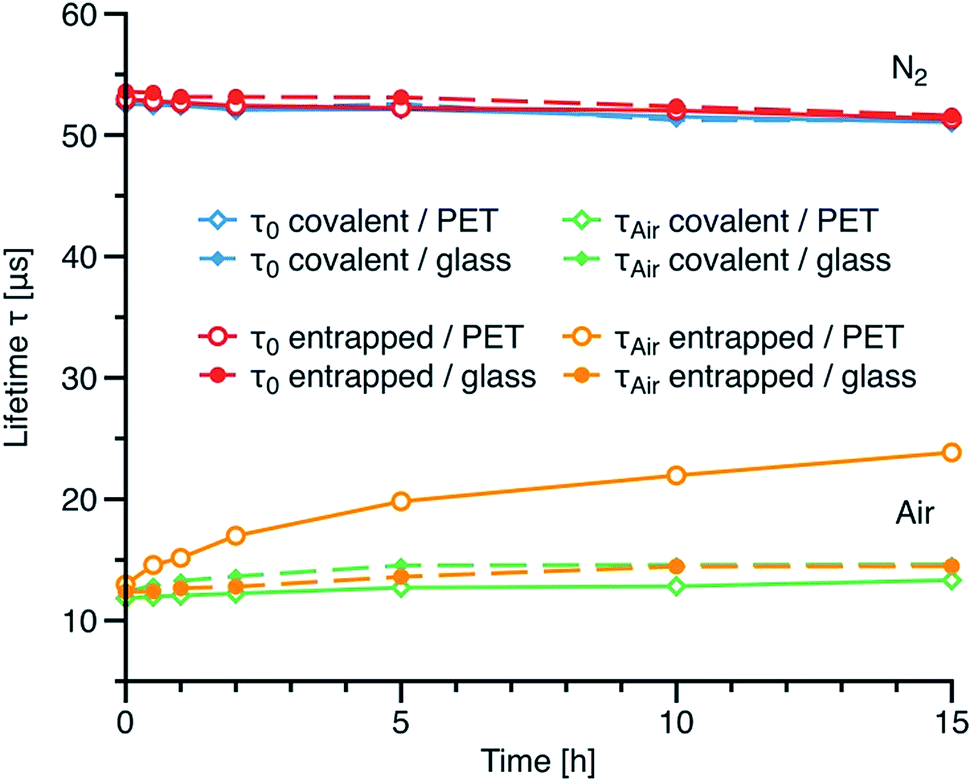 | ||
| Fig. 7 Influence of autoclavation (135 °C) on the luminescence decay times (at nitrogen and air saturation, measured at 25 °C) of the oxygen sensors based on 6b and on PtTPTBPBr4 physically entrapped in polystyrene. Two different supports (PET and glass) are compared. The same sensor foils were autoclaved repeatedly throughout the whole experiment. | ||
Suppressed migration of the covalently immobilised dye allowed us to directly apply the sensor composition to the PMMA fiber in order to obtain cheap and fast responding fiber-optic sensors (Fig. 8). The physically entrapped indicator dyes would migrate into the PMMA core over time and compromise sensor performance. However, in the case of sensor materials incorporating covalently immobilised indicator dyes, direct coating of the PMMA fiber allows for very thin sensor polymer layers without migration issues. The response times of such layers are very fast and are below 1 s.
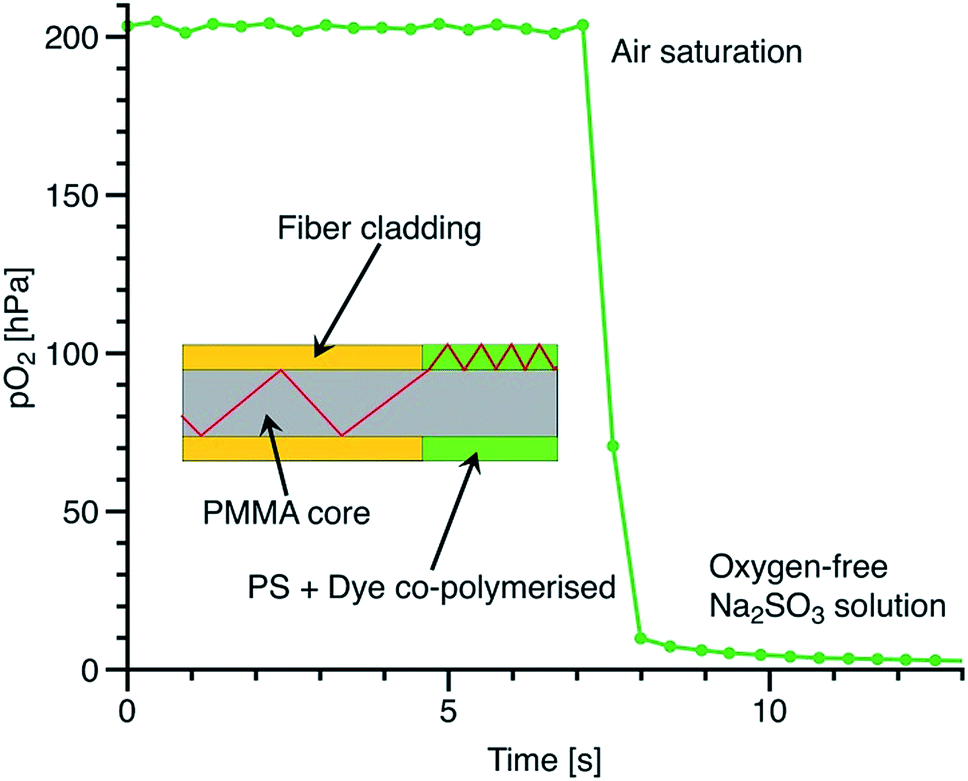 | ||
| Fig. 8 Cross-section and dynamic response of the fiber-optic oxygen sensor. The excitation light is coupled into a thin sensor layer coated onto the core of a PMMA fiber. | ||
Conclusions
We presented a simple and versatile strategy for the preparation of high performance oxygen-sensing materials. These materials rely on the NIR emitting bright and photostable Pt(II) benzoporphyrins covalently immobilised into different polymeric matrices. The new sensors overcome the limitations of their predecessors based on physically entrapped dyes. Migration and leaching are completely eliminated which not only ensures the reliable operation of the sensors in harsh conditions (such as elevated temperatures) but also enables higher flexibility of sensor formats (e.g. fast responding fiber-optic sensors based on PMMA fibers). We also reported a novel straightforward and inexpensive synthetic route to the template-directed synthesis of (bromo-substituted) Pt(II)–benzoporphyrins. The new method not only allows preparation of reactive bromo-substituted porphyrins available for Suzuki coupling, but also other numerous porphyrins which can be used for a variety of emerging applications such as triplet–triplet annihilation upconversion.Experimental
Materials
Ethyl isocyanoacetate, 1-nitro-1-cyclohexene, 2,3-dicyano-5,6-dichlorobenzoquinone (DDQ), boron-trifluorid-diethyletherate (BF3·Et2O), 1,2,4-trimethylbenzene (TMB), benzonitrile, potassium carbonate (K2CO3), styrene, 1,1,1,3,3,3-hexafluoroisopropyl methacrylate, phenylacetic acid, 4-bromophenylacetatic acid, 1,2-dicyanobenzene and water-free dichloromethane (DCM) were purchased from Aldrich. Zinc 4-bromophenylacetate was obtained as a white precipitate in an exchange reaction between zinc acetate and 4-bromophenylacetic acid. 4-Bromobenzaldehyde, 4-vinylboronic acid, tetrakis(triphenylphosphine)palladium(0) (Pt(PPh3)4) and methanesulfonic acid were from ABCR. Maleic anhydrid and sodium sulfite (Na2SO3) were obtained from Fluka. Azobisisobutyronitrile (AIBN), aluminium oxide (neutral, 50–200 μm) (Al2O3) and polystyrene (PS) (Mw = 260000 g mol−1) were purchased from Acros. Poly(ethylene terephthalate) (PET) support Melinex 505 was obtained from Pütz. Nitrogen (99.999% purity) was obtained from Air Liquide. Hydrochloric acid 37% (HCl), sodium sulfate anhydrous (Na2SO4) and all other used solvents were from VWR. Zinc(II) oxide (ZnO) and silica-gel 60 (0.063–0.200 mm) were from Merck. Platinum(II) chloride (PtCl2) was obtained from ChemPur. PMMA fibers were obtained from Ratioplast. HPLC grade chloroform, ethyl acetate and water-free dimethylformamide (DMF) were obtained from Roth. 4,5,6,7-Tetrahydroisoindole was synthesized as described elsewhere.36 The precursor Pt(C6H5CN)Cl2 was obtained by stirring PtCl2 in boiling benzonitrile for one hour and precipitating the resulted product with hexane. The yellow product is filtrated, washed with hexane and dried at 60 °C.
Synthetic procedures
000 M−1 cm−1 for the Soret band at 440 nm. UV-Vis (acetone): λmax (rel. int.) = 440 (1.00), 535 (0.09), 583 (0.06), 624 (0.07), 688 (0.08) nm.
000 M−1 cm−1 for the Soret band at 440 nm) was dissolved in TMB (60 ml) and heated to 150 °C. Pt(C6H5CN)Cl2 (90 mg, 0.192 mmol) was pre-dissolved in a small volume of TMB and was added to the reaction mixture. The formed HCl was removed by bubbling N2 through the reaction mixture. The reaction progress was monitored via UV-Vis spectra. After 1 h, the reaction was cooled and the insoluble products (metallic platinum) removed by filtration. The filtrate was refluxed with 45 mg Pt(C6H5CN)Cl2. After the completion of the reaction, 50 ml hexane were added and the product was purified via chromatography on silica-gel (eluent: hexane/DCM, 2/1, v/v). Product fractions were determined via UV-Vis absorption spectra (see Fig. S1, ESI†). Yield: 52 mg, 51%. 1H NMR (300 MHz, CDCl3) δ 7.92 (d, J = 8.2 Hz, 8H), 7.81 (d, J = 8.1 Hz, 8H), 2.36–2.30 (m, 16H), 1.54–1.50 (m, 16H). MALDI: m/z: [M+] calc. for C60H48Br4N4Pt: 1340.0232; found, 1340.0132. UV-Vis (acetone): λmax (rel. int.) = 410 (1.00), 523 (0.07), 558 (0.09) nm.
710 g mol−1, Mw = 404020 g mol−1, Mz = 1218120 g mol−1, PDI = 4.05.
000 M−1 cm−1 at 430 nm in DCM = 0.41 wt%. Yield: 589 mg, 72%. GPC data: Mn = 118950 g mol−1, Mw = 457810 g mol−1, Mz = 1320760 g mol−1, PDI = 3.85.
000 M−1 cm−1 for the Soret band at 430 nm in DCM = 1.0 wt%. Yield: 340 mg, 60%. GPC data: Mn = 39790 g mol−1, Mw = 92470 g mol−1, Mz = 187850 g mol−1, PDI = 2.32.
000 M−1 cm−1 at 430 nm in THF = 0.64 wt%. Yield: 202 mg, 20%. GPC data: Mn = 57000 g mol−1, Mw = 68500 g mol−1, Mz = 83220 g mol−1, PDI = 1.20.
000 M−1 cm−1 at 430 nm in DCM = 0.56 wt%. Yield: 340 mg, 60%. GPC data: Mn = 46050 g mol−1, Mw = 250500 g mol−1, Mz = 888800 g mol−1, PDI = 5.44.
Autoclavation experiments
100 mg of the sensor material PS co-polymerised with compound 5 or 1 mg PtTPTBPBr4 (3a) and 100 mg PS were dissolved in 1.0 g HPLC grade chloroform. These sensor “cocktails” were coated onto PET and microscopy slides using a 25 μm coating knife. The sensors were dried at 60 °C for 12 h. The sensors were autoclaved (Sanoclav from Wolf, Germany) at 135 °C for 15 h and phosphorescence lifetimes were determined under air and nitrogen in regular intervals.Photostability
Dye solutions in water-free DMF were illuminated with and LED array (617 nm) at the following settings: 7.0 W, 7.88 V, 882 mA (photon flux: 5500 μmol s−1 m−2). After each measurement the cuvette was shortly unsealed and shaken to ensure oxygen saturation in the sample. The degradation was determined by calculating the average value of the three maximum absorption points in the Q-band.Measurements
1H NMR spectra were recorded on a 300 MHz instrument from Bruker. MALDI-TOF mass spectra were recorded on a Micromass TofSpec 2E in refectron mode at an accelerating voltage of +20 kV. Boronic acid content was determined using elemental analyses on a Spectro Ciros Vision ICP-OES after microwave assisted digestion on a Muliwave 3000 from Anton Paar (Austria). Absorption measurements were performed on a Cary 50 UV-Vis spectrophotometer from Varian. Luminescence spectra were acquired on a Fluorolog-3 luminescence spectrometer (Horiba). Relative luminescence quantum yields were determined according to Crosby and Demas45 using platinum(II) tetraphenyltetrabenzoporphyrin (Φ = 0.51)34 as a reference. The samples were de-oxygenated in a screw-cap cuvette (Hellma) by bubbling argon through the solutions for 25 min. GPC measurements for determination of molecular weights and the PDI of the polymers were carried out in tetrahydrofuran (THF) using a Merck Hitachi L6000 pump, separation columns of Polymer Standards Service (5 μm grade size) and a refractive-index detector from Wyatt Technology. For calibration Polystyrene Standards purchased from Polymer Standard Service were used. Particle size and zeta potential were measured using a Zetasizer Nano-ZS from Malvern Instruments.Calibration of the polystyrene-based sensors (3 sensors were measured to obtain average values) was performed in the frequency-domain using a SR830 DSP lock-in amplifier (Stanford Research Systems). Excitation light from a 435 nm LED obtained from Roithner, Austria was filtered through a BG12 filter obtained from Schott, Germany. The excitation light was sinusoidally modulated at a frequency of 5 kHz and guided to the sensor film using a bifurcated fiber bundle. The emitted light was guided back to a photomultiplier unit (H5701-02) obtained from Hamamatsu, Japan. The emission light was filtered through an RG 9 filter obtained from Schott. Calibration of all other sensors and the stability tests were performed using a Firesting oxygen meter from Pyroscience (Germany). The modulation frequency of 4 kHz was used. The gas mixtures were adjusted with a custom-built gas-mixing device based on mass-flow controllers from MKS (http://www.mksinst.com) and Voegtlin (Switzerland, http://www.red-y.com) by mixing nitrogen and compressed air. The temperature was kept constant at 25 °C with a HAAKE DC50 or ThermoHAAKE K10 thermostat.
Acknowledgements
The support from Josefine Hobisch and Robert Saf (Institute of Chemistry and Technology of Materials, TU Graz), Herbert Motter and Eveline Maier (Institute of Analytical Chemistry and Food Chemistry) is gratefully acknowledged. The work was financially supported by ERC Project “Oxygen” – grant number 267233.References
- R. I. Dmitriev and D. B. Papkovsky, Cell. Mol. Life Sci., 2012, 69, 2025–2039 CrossRef CAS PubMed.
- D. B. Papkovsky and R. I. Dmitriev, Chem. Soc. Rev., 2013, 42, 8700–8732 RSC.
- S. Schreml, R. J. Meier, O. S. Wolfbeis, T. Maisch, R.-M. Szeimies and M. Landthaler, et al. , Exp. Dermatol., 2011, 20, 550–554 CrossRef PubMed.
- J. Lecoq, A. Parpaleix, E. Roussakis, M. Ducros, Y. G. Houssen and S. A. Vinogradov, et al. , Nat. Med., 2011, 17, 893–898 CrossRef CAS PubMed.
- M. Staal, S. M. Borisov, L. F. Rickelt, I. Klimant and M. Kühl, J. Microbiol. Methods, 2011, 85, 67–74 CrossRef CAS PubMed.
- A. Hempel, M. G. O'Sullivan, D. B. Papkovsky and J. P. Kerry, LWT – Food Sci. Technol., 2013, 50, 226–231 CrossRef CAS PubMed.
- A. Mills, Chem. Soc. Rev., 2005, 34, 1003–1011 RSC.
- O. Kohls and T. Scheper, Sens. Actuators, B, 2000, 70, 121–130 CrossRef CAS.
- O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657–2669 RSC.
- X. Wang and O. S. Wolfbeis, Chem. Soc. Rev., 2014, 43, 3666–3761 RSC.
- B. D. MacCraith and C. McDonagh, J. Fluoresc., 2002, 12, 333–342 CrossRef CAS.
- B. Korzeniowska, R. Nooney, D. Wencel and C. McDonagh, Nanotechnology, 2013, 24, 442002 CrossRef CAS PubMed.
- M. Quaranta, S. M. Borisov and I. Klimant, Bioanal. Rev., 2012, 4, 115–157 CrossRef PubMed.
- H. Xiang, J. Cheng, X. Ma, X. Zhou and J. J. Chruma, Chem. Soc. Rev., 2013, 42, 6128 RSC.
- D. B. Papkovsky, G. V. Ponomarev, W. Trettnak and P. O'Leary, Anal. Chem., 1995, 67, 4112–4117 CrossRef CAS.
- G. Khalil, M. Gouterman, S. Ching, C. Costin, L. Coyle and S. Gouin, et al. , J. Porphyrins Phthalocyanines, 2002, 6, 135–145 CrossRef CAS.
- S. A. Vinogradov and D. F. Wilson, J. Chem. Soc. Perkin Trans., 1995, 103–111 RSC.
- M. A. Filatov, S. Baluschev, I. Z. Ilieva, V. Enkelmann, T. Miteva and K. Landfester, et al. , J. Org. Chem., 2012, 77, 11119–11131 CrossRef CAS PubMed.
- S. M. Borisov, G. Nuss and I. Klimant, Anal. Chem., 2008, 80, 9435–9442 CrossRef CAS PubMed.
- S. M. Borisov, G. Zenkl and I. Klimant, ACS Appl. Mater. Interfaces., 2010, 2, 366–374 CAS.
- I. Dunphy, S. A. Vinogradov and D. F. Wilson, Anal. Biochem., 2002, 310, 191–198 CrossRef CAS.
- T. V. Esipova, A. Karagodov, J. Miller, D. F. Wilson, T. M. Busch and S. A. Vinogradov, Anal. Chem., 2011, 83, 8756–8765 CrossRef CAS PubMed.
- M. C. DeRosa, D. J. Hodgson, G. D. Enright, B. Dawson, C. E. B. Evans and R. J. Crutchley, J. Am. Chem. Soc., 2004, 126, 7619–7626 CrossRef CAS PubMed.
- M. C. DeRosa, P. J. Mosher, G. P. A. Yap, K.-S. Focsaneanu, R. J. Crutchley and C. E. B. Evans, Inorg. Chem., 2003, 42, 4864–4872 CrossRef CAS PubMed.
- B. Lei, B. Li, H. Zhang, L. Zhang and W. Li, J. Phys. Chem. C, 2007, 111, 11291–11301 CAS.
- Z. Wang, A. r. McWilliams, C. E. B. Evans, X. Lu, S. Chung and M. a. Winnik, et al. , Adv. Funct. Mater., 2002, 12, 415–419 CrossRef CAS.
- H. Zhang, B. Lei, W. Mai and Y. Liu, Sens. Actuators, B, 2011, 160, 677–683 CrossRef CAS PubMed.
- H. Zhang, B. Li, B. Lei, W. Li and S. Lu, Sens. Actuators, B, 2007, 123, 508–515 CrossRef CAS PubMed.
- Y. Tian, B. R. Shumway, W. Gao, C. Youngbull, M. R. Holl and R. H. Johnson, et al. , Sens. Actuators, B, 2010, 150, 579–587 CrossRef CAS PubMed.
- Y. Tian, B. R. Shumway and D. R. Meldrum, Chem. Mater., 2010, 22, 2069–2078 CrossRef CAS PubMed.
- K. Koren, S. M. Borisov and I. Klimant, Sens. Actuators, B, 2012, 169, 173–181 CrossRef CAS PubMed.
- M. Obata, N. Matsuura, K. Mitsuo, H. Nagai, K. Asai and M. Harada, et al. , J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 663–670 CrossRef CAS PubMed.
- S. M. Borisov, P. Lehner and I. Klimant, Anal. Chim. Acta, 2011, 690, 108–115 CrossRef CAS PubMed.
- S. M. Borisov, G. Nuss, W. Haas, R. Saf, M. Schmuck and I. Klimant, J. Photochem. Photobiol., A, 2009, 201, 128–135 CrossRef CAS PubMed.
- O. S. Finikova, A. V. Cheprakov, I. P. Beletskaya, P. J. Carroll and S. A. Vinogradov, J. Org. Chem., 2004, 69, 522–535 CrossRef CAS PubMed.
- C. Borek, K. Hanson, P. I. Djurovich, M. E. Thompson, K. Aznavour and R. Bau, et al. , Angew. Chem., Int. Ed., 2007, 46, 1109–1112 CrossRef CAS PubMed.
- J. Lindsey, H. Hsu and I. Schreiman, Tetrahedron Lett., 1986, 27, 4969–4970 CrossRef CAS.
- S. M. Borisov and I. Klimant, Dyes Pigm., 2009, 83, 312–316 CrossRef CAS PubMed.
- K. Ichimura, M. Sakuragi, H. Morii, M. Yasuike, M. Fukui and O. Ohno, Inorg. Chim. Acta, 1991, 182, 83–86 CrossRef CAS.
- X. Cui, J. Zhao, P. Yang and J. Sun, Chem. Commun., 2013, 49, 10221 RSC.
- M. J. Currie, J. K. Mapel, T. D. Heidel, S. Goffri and M. A. Baldo, Science, 2008, 321, 226–228 CrossRef CAS PubMed.
- J. Zhao, W. Wu, J. Sun and S. Guo, Chem. Soc. Rev., 2013, 42, 5323–5351 RSC.
- S. M. Borisov, T. Mayr, G. Mistlberger, K. Waich, K. Koren and P. Chojnacki, et al. , Talanta, 2009, 79, 1322–1330 CrossRef CAS PubMed.
- E. R. Carraway, J. N. Demas, B. A. DeGraff and J. R. Bacon, Anal. Chem., 1991, 63, 337–342 CrossRef CAS.
- G. A. Crosby and J. N. Demas, J. Phys. Chem., 1971, 75, 991–1024 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc00983e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |