 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extended conjugation in poly(triarylamine)s: synthesis, structure and impact on field-effect mobility†
Reiner Sebastian
Sprick
a,
Mario
Hoyos
c,
Marion Sofia
Wrackmeyer
a,
Adam Valentine
Sheridan Parry
a,
Iain Mark
Grace
b,
Colin
Lambert
b,
Oscar
Navarro
*d and
Michael Lewis
Turner
*a
aOrganic Materials Innovation Centre, School of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michael.turner@manchester.ac.uk; Fax: +44 0161 275 4273
bDepartment of Physics, Lancaster University, Lancaster, LA1 4YB, UK
cInstituto de Ciencia y Tecnología de Polímeros, ICTP-CSIC, c/Juan de la Cierva, 3, Madrid, 28006, Spain
dDepartment of Chemistry, University of Sussex, Brighton, BN1 9QJ, UK. E-mail: o.navarro@sussex.ac.uk; Fax: +44 01273 678734
First published on 12th June 2014
Abstract
Polytriarylamines with extended fused backbones are accessible by the coupling of anilines with dibromoarenes based on substituted indenofluorenes, diindenofluorenes, carbazoles and indolocarbazoles. The optical and electrochemical properties of these polymers show an increase in the HOMO energy levels and the onset of absorption on extending the length of the fused ring segment. The polymer derived from the indenofluorene unit shows the highest reported performance for a polytriarylamine in an OFET and this observation can be rationalized by DFT calculations of model oligomers that show higher calculated reorganization energies for the more extended diindenofluorene units.
Introduction
Semiconducting polymers are important components of the active layers of organic light-emitting diodes (OLEDs),1 photovoltaics (OPVs)2 and organic field-effect transistors (OFETs).3,4 These materials can be deposited from solution using conventional patterning processes such as screen, gravure or ink-jet printing and are compatible with fabrication on flexible, plastic substrates and the development of mechanically robust, conformable devices.5 Examples of p-type polymers include regioregular poly(3-hexylthiophene),6 poly(2,5-bis(3-alkylthiophen-2-yl)thieno[3,2-b]thiophenes)7 and recently very high performance donor–acceptor main chain polymers derived from diketopyrrolopyrrole units.8,9 Semicrystalline polymers with short intermolecular π–π stacking distances generally show the highest performance in OFETs, with charge mobilities of up 10 cm2 V−1 s−1.8 To achieve such high performance from semicrystalline polymer films requires optimized surface treatments and prolonged high temperature annealing to improve the molecular orientation and the domain morphology of the films. By contrast in OFET devices amorphous semiconducting polymers require little post-processing of the films beyond simple solvent removal but in general exhibit more moderate charge-carrier mobilities (<10−2 cm2 V−1 s−1). A recent report of an indenofluorene–phenanthrene copolymer (PIFPA, Fig. 1) showed a significant improvement in the measured mobility for an amorphous polymer, up to 0.3 cm2 V−1 s−1 in a bottom contact, top gate architecture.10 Polytriarylamines (PTAAs)11,12 are an important class of amorphous semiconducting polymers, as they show excellent ambient performance over extended periods of time,13 even in bottom gate device architectures where the channel is directly exposed to the atmosphere. This stable ambient performance and amorphous solid-state structure is desirable for use in chemical sensors and allows for simple and reproducible processing of PTAA thin-films when compared to the processes required for semicrystalline polymers.13 PTAAs have been employed as hole conductor materials for the active layer of photocopiers, laser printers,14 OLEDs,15 and vapour sensors.16,17 Furthermore, they have been used in electroluminescent devices,18 cathode components19 and as electroactive separator materials in rechargeable lithium batteries.20 | ||
| Fig. 1 Structures of PIFPA, PIFTAA and bridged oligoarenes. | ||
One of the major benefits of conjugated polymers as organic semiconductors is the ability to precisely tune the optoelectronic properties by modifying the backbone of the polymer. This precise control can be achieved by the incorporation of different co-monomers into the polymer main chain or by increasing the coplanarity of fused structures in the monomer units.2,5,7 For example, polymers derived from indenofluorene units (IF) show enhanced π-conjugation and more extensive π–π interactions leading to improved charge-carrier mobilities in OFETs when compared to those of the corresponding fluorene polymers.21 Similarly, incorporation of IF units into (bisthienyl)benzothiadiazole copolymers gave power conversion efficiencies in organic photovoltaic devices of up to 2.44%, whereas the corresponding diindenofluorene (DIF) copolymers gave improved efficiencies of up to 4.50%.22 In addition structurally related analogues of indenofluorenes, the indolocarbazole units, have been copolymerized with fluorenes,23 bithiophenes,24 and others units to tune the optoelectronic properties. The incorporation of repeating units with extended conjugation into the backbone of PTAAs, e.g. fluorene and indenofluorene (PIFTAA, Fig. 1), resulted in a significant improvement of the charge transport.11 Herein we present the use of the Buchwald–Hartwig polyamination to the synthesis and characterization of PTAAs incorporating bridged phenyl units IF,25DIF26 and ICz.27 By using this approach it is possible to obtain very rigid systems and the PTAAs derived from IF show the highest mobility in an OFET reported to date for this class of amorphous semiconducting polymer. The relationship between structure and performance in these materials was rationalized by DFT calculations of the electronic structure of oligomeric models.
Experimental
General remarks
All reagents were obtained from commercial sources and used as received, except for 2,4-dimethylaniline and 4-methoxy-2-methylaniline, these were distilled over KOH prior to usage under reduced pressure. Solvents were dried according to standard procedures or using an Innovative Technology PureSolv MD 7 system using aluminium oxide columns. Reactions were carried out under an argon atmosphere using standard Schlenk techniques.1H NMR spectra were recorded at 400 MHz on a Bruker Avance AV 400 MHz Ultrashield spectrometer in the solvent stated at 25 °C. Elemental analysis was performed using a Carlo Erba Instruments EA1108 elemental analyser. Palladium content was determined using a Thermo Scientific iCAP 6000 Series ICP Spectrometer. Bromine analysis was performed using a Metrohm 888 Titrando Autotitration system with a combined silver ring electrode after decomposition of the sample into solution. MALDI-TOF mass spectrometry was carried out using a Shimadzu Biotech AXIMA Confidence MALDI mass spectrometer in linear (positive) mode, referencing against PPG or PEG standards (4 k–12 kDa). Gel permeation chromatography was carried out in tetrahydrofuran (THF) at 35 °C using a Viscotek GPCmax VE2001 solvent/sample module with 2 × PL gel 10 μm Mixed-B and PL gel 500A columns, a Viscotek VE3580 RI detector and a VE 3240 UV-Vis multichannel detector. The flow rate was 1 mL min−1 and the system was calibrated with low polydispersity polystyrene standards in the range of 200 to 180 × 104 g mol−1 from Polymer Laboratories. The analysed samples contained n-dodecane as a flow marker. UV-Vis absorption spectra were recorded on a Varian Cary 5000 UV-Vis-NIR spectrophotometer in dichloromethane at room temperature. Fluorescence spectra were recorded on a Varian Cary Eclipse fluorimeter in dichloromethane solution at room temperature. Cyclic voltammetry was performed in dichloromethane solution scanning at 100 mV s−1 on a BASI Epsilon electrochemical workstation with a three-electrode cell, Ag/AgNO3 (in dichloromethane–acetonitrile 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) as reference electrode, platinum wire as counter electrode and working electrode, in nitrogen-purged, 0.1 M solution of tetrabutylammonium hexafluorophosphate [(C4H9)4N]PF6 as a supporting electrolyte at room temperature. Wafers with a 300 nm SiO2 layer on n++ Si were obtained from Si-Mat GmbH (Germany). Differential scanning calorimetry measurements were recorded using a Perkin Elmer Jade DSC instrument under nitrogen atmosphere, 5–10 mg of the sample was sealed in an aluminium pan with a crimping tool. The sample was heated from 30 °C to 220 °C or 350 °C at a heating rate of 10 °C min−1, held for 2 minutes at 220 °C or 350 °C and then cooled to 30 °C at a rate of 10 °C min−1. This cycle was repeated once. WAXS spectra were recorded on a Bruker AXS D8 Discover using the precipitated polymers or spin-coated on a WSi2–C coated silicon wafer.
:1) as reference electrode, platinum wire as counter electrode and working electrode, in nitrogen-purged, 0.1 M solution of tetrabutylammonium hexafluorophosphate [(C4H9)4N]PF6 as a supporting electrolyte at room temperature. Wafers with a 300 nm SiO2 layer on n++ Si were obtained from Si-Mat GmbH (Germany). Differential scanning calorimetry measurements were recorded using a Perkin Elmer Jade DSC instrument under nitrogen atmosphere, 5–10 mg of the sample was sealed in an aluminium pan with a crimping tool. The sample was heated from 30 °C to 220 °C or 350 °C at a heating rate of 10 °C min−1, held for 2 minutes at 220 °C or 350 °C and then cooled to 30 °C at a rate of 10 °C min−1. This cycle was repeated once. WAXS spectra were recorded on a Bruker AXS D8 Discover using the precipitated polymers or spin-coated on a WSi2–C coated silicon wafer.
Materials
6,6′,12,12′-Tetra-n-octyl-6,12-dihydroindeno [1,2b] fluorene,25,28 6,6,12,12,15,15-hexa-n-butyl-12,15-dihydro-6H-cyclopenta[1,2-b:5,4-b′]difluorene,26 2,7-dibromocarbazole,29 3,9-dibromo-5,11-di-n-octylindolo[3,2-b]carbazole30 and (IPr)Pd(allyl)Cl31 were synthesized using literature procedures.Polymerization procedure A
A Schlenk tube was charged with KOtBu and flame dried with a heat gun under vacuum. After cooling to room temperature the tube was filled with argon and the dibromoarene and NHC–Pd complex were added. The tube was then degassed for 10 min under reduced pressure and purged with argon. Toluene (anhydrous and degassed), end-capper (p-bromoanisole, 50 mmol L−1 toluene stock solution) and the aniline were added and the reaction mixture was heated to 105 °C for the time specified. After cooling to room temperature the crude product was precipitated into methanol, filtered off and dried. The crude polymer was filtered over an n-hexane wet silica pad using dichloromethane as eluent. The filtrate was extracted with an aqueous sodium N,N-diethyldithiocarbamate solution (0.1 M), then water and dried over MgSO4 and filtered. After evaporation of the solvent, the polymer was precipitated from a concentrated dichloromethane solution into methanol and filtered off. An extraction with a Soxhlet apparatus was performed. The polymer was then precipitated from a concentrated solution into methanol, filtered off and dried.Polymerization procedure B
A Schlenk tube was charged with KOtBu and flame dried with a heat gun under vacuum. After cooling to room temperature the tube was filled with argon and the dibromoarene was added. The tube was then degassed for 10 min under reduced pressure and purged with argon. Toluene (anhydrous and degassed) and end-capper (p-bromoanisole, 50 mmol L−1 toluene stock solution) were added and the reaction mixture was heated to 105 °C for 10 minutes. The aniline and (IPr)Pd(allyl)Cl (dissolved in toluene) were added and the reaction mixture was kept for the time specified at 105 °C. After cooling to room temperature the crude product was precipitated into methanol, filtered off and dried. The crude polymer was filtered over a silica pad (wetted with n-hexane) using dichloromethane as eluent. The filtrate was extracted with an aqueous sodium N,N-diethyldithiocarbamate solution (0.1 M), then water and dried over MgSO4 and filtered. After evaporation of the solvent, the polymer was precipitated from a concentrated dichloromethane solution into methanol and filtered off. An extraction with a Soxhlet apparatus was performed. The polymer was then precipitated from a concentrated solution into methanol, filtered off and dried.Poly[[(2,4-dimethylphenyl)imino][6,6′,12,12′-tetra-n-octyl-6,12-di-hydroindeno[1,2b]fluorene-2,8-diyl] (1)
Following polymerization procedure A; KOtBu (224 mg, 2 mmol), (IPr)Pd(allyl)Cl (10 mg, 3 mol%), p-bromoanisole (0.070 mL of a 50 mmol L−1 stock solution in toluene, 0.67 mol%), 2,8-dibromo-6,6′,12,12′-tetra-n-octyl-6,12-dihydroindeno[1,2b]fluorene (430 mg, 0.5 mmol), 2,4-dimethylaniline (0.062 mL, 0.5 mmol) and toluene (3 mL) were heated for 30 minutes. After work-up and Soxhlet extraction with methanol, acetone, 2-butanone and dichloromethane 0.149 g (36%) of the title compound as a yellow solid were obtained from the dichloromethane fraction. 1H NMR (400 MHz, C6D6, δ): 7.75 (s, 2H, indenofluorene-H), 7.64 (d, J = 8.0 Hz, 2H, indenofluorene-H), 7.55 (s, 2H, indenofluorene-H), 7.34 (d, J = 8.5 Hz, 1H, Ar-H), 7.22 (d, J = 8.0 Hz, 2H, indenofluorene-H), 7.05–7.00 (m, 2H, phenyl-H), 2.25 (s, 3H, phenyl-CH3), 2.19 (s, 3H, phenyl-CH3), 2.11–2.01 (m, 8H, alkyl-H), 1.28–1.12 (m, 48H, alkyl-H), 0.90 (t, 12H, alkyl-CH3). Anal. calcd for (C60H87N)n: C, 87.63; H,10.66; N,1.70%; found: C, 86.62; H,10.14; N,1.74; Br, trace (<0.1); Pd, trace (<0.1)%.Poly[[(2-methyl-4-methoxyphenyl)imino][6,6′,12,12′-tetra-n-octyl-6,12-dihydroindeno[1,2b]fluorene-2,8-diyl] (2)
Following polymerization procedure A; KOtBu (224 mg, 2 mmol), (IPr)Pd(allyl)Cl (10 mg, 3 mol%), p-bromoanisole (0.075 mL of a 50 mmol L−1 stock solution in toluene, 0.71 mol%), 2,8-dibromo-6,6′,12,12′-tetra-n-octyl-6,12-dihydroindeno[1,2b]fluorene (430 mg, 0.5 mmol), 4-methoxy-2-methylaniline (0.065 mL, 0.5 mmol) and toluene (3 mL) were heated for 20 minutes. After work-up and Soxhlet extraction with methanol, acetone, 2-butanone and dichloromethane 178 mg (42%) of the title compound as a yellow solid were obtained from the dichloromethane fraction. 1H NMR (400 MHz, C6D6, δ): 7.77 (s, 2H, indenofluorene-H), 7.66 (d, J = 8.5 Hz, 2H, indenofluorene-H), 7.58 (s, 2H, indenofluorene-H), 7.33 (d, J = 8.5 Hz, 1H, phenyl-H), 7.22 (d, J = 8.5 Hz, 2H, indenofluorene-H), 6.90 (d, J = 2.5 Hz, 1H, phenyl-H), 6.82–6.77 (m, 1H, phenyl-H), 3.37 (s, 3H, phenyl-OCH3), 2.24 (s, 3H, phenyl-CH3), 2.20–1.99 (m, 8H, alkyl-H), 1.27–1.12 (m, 48H, alkyl-H), 0.91 (t, J = 7.0 Hz, 12H, alkyl-CH3). Anal. calcd for (C60H78NO)n: C, 85.96; H, 10.46; N, 1.46%; found C, 83.81; H, 10.46; N, 1.72, Br, trace; Pd, trace (<0.1)%.Poly[[(2,4-dimethylphenyl)imino][6,6′,12,12′,15,15′-hexa-n-butyl-12,15-dihydro-6H-cyclopenta[1,2-b:5,4-b′]difluorene-2,10-diyl] (3)
Following polymerization procedure B; KOtBu (224 mg, 2 mmol), 2,10-dibromo-6,6′,12,12′,15,15′-hexa-n-butyl-12,15-dihydro-6H-cyclopenta[1,2-b:5,4-b′]difluorene (418 mg, 0.5 mmol), p-bromoanisole (0.075 mL of a 50 mmol L−1 stock solution in toluene, 0.375 mol%), toluene (3 mL, anhydrous and degassed), 2,4-dimethylaniline (0.065 mL, 0.5 mmol) and (IPr)Pd(allyl)Cl (12 mg in 0.5 mL toluene, 4 mol%) were heated for 25 minutes at 105 °C. After work-up and Soxhlet extraction with methanol, acetone, 2-butanone and dichloromethane 193 mg (49%) of the title compound as a dark yellow powder were obtained from the dichloromethane fraction. 1H NMR (400 MHz, C6D6, δ): 7.95 (s, 2H, difluorene-H), 7.79 (s, 2H, difluorene-H), 7.65 (d, J = 8.0 Hz, 2H, difluorene-H), 7.43 (s, 2H, difluorene-H), 7.31–7.25 (m, 3H, difluorene-H and phenyl-H), 6.98–6.93 (m, 2H, phenyl-H), 2.19 (s, 3H, phenyl-CH3), 2.15 (s, 3H, phenyl-CH3), 2.10–1.86 (m, 12H, alkyl-H), 1.18–0.87 (m, 24H, alkyl-H), 0.68 (t, J = 7.0 Hz, 6H, alkyl-CH3), 0.60 (t, J = 7.0 Hz, 12H, alkyl-CH3). Anal. calcd for (C59H75N)n: C, 88.77; H, 9.47; N, 1.75%; found C, 87.77; H, 9.09; N, 1.85%, Br, none found; Pd, none found.Poly[[(2-methyl-4-methoxyphenyl)imino][6,6′,12,12′,15,15′-hexa-n-butyl-12,15-dihydro-6H-cyclopenta[1,2-b:5,4-b′]difluorene-2,10-diyl] (4)
Following polymerization procedure B; KOtBu (224 mg, 2 mmol), 2,10-dibromo-6,6′,12,12′,15,15′-hexa-n-butyl-12,15-dihydro-6H-cyclopenta[1,2-b:5,4-b′]difluorene (418 mg, 0.5 mmol), toluene (3 mL, anhydrous and degassed), p-bromoanisole (0.075 mL of a 50 mmol L−1 stock solution in toluene, 0.375 mol%), 4-methoxy-2-methylaniline (0.065 mL, 0.5 mmol) and (IPr)Pd(allyl)Cl (4 mol%, 12 mg in 0.5 mL toluene) were heated for 11 minutes at 105 °C. After work-up and Soxhlet extraction with methanol, acetone, 2-butanone and dichloromethane 119 mg (30%) of the title compound as a dark yellow powder were obtained from the dichloromethane fraction. 1H NMR (400 MHz, C6D6, δ): 7.97 (s, 2H, difluorene-H), 7.80 (s, 2H, difluorene-H), 7.66 (d, J = 8.0 Hz, 2H, difluorene-H), 7.46 (s, 2H, difluorene-H), 7.30–7.24 (m, 3H, difluorene-H and phenyl-H), 6.85 (d, J = 2.5 Hz, 1H, phenyl-H), 6.75 (dd, J = 2.5 Hz and 8.5 Hz, 1H), 3.33 (s, 3H, phenyl-OCH3), 2.17 (s, 3H, phenyl-CH3), 2.14–1.86 (m, 12H, alkyl-H), 1.20–0.84 (m, 24H, alkyl-H), 0.68 (t, J = 7.0 Hz, 6H, alkyl-CH3), 0.60 (t, J = 7.0 Hz, 12H, alkyl-CH3). Anal. calcd for (C59H75NO)n: C, 87.03; H, 9.28; N, 1.72%; found C, 85.30; H, 8.58; N, 2.14%, Br, none found; Pd, none found.Poly[[(2,4-dimethylphenyl)imino][(n-octyl)carbazole-2,7-diyl] (5)
Following polymerization procedure A; KOtBu (898 mg, 8 mmol), 2,7-dibromo-N-(n-octyl)carbazole (874 mg, 2 mmol), 1,3-bis(2,6-di-i-propylphenyl)imidazol-2-ylidene(1,4-naphthoquinone)palladium(0) dimer (52 mg, 2 mol%), were added to a Schlenk flask inside a glove-box. 2,4-Dimethylaniline (0.25 mL, 1 mmol) and toluene (12 mL, anhydrous and degassed) were added and the reaction mixture was heated to 105 °C for 24 hours. End-capping was performed for 2.5 hours. After work-up and Soxhlet extraction with methanol, acetone, hexane and 2-butanone 260 mg (33%) of the title compound as a yellow solid were obtained from the 2-butanone fraction. 1H NMR (500 MHz, C6D6, δ): 7.85 (d, J = 8.5 Hz, 2H, carbazole-H), 7.37 (d, J = 8.0 Hz, 1H, phenyl-H), 7.29–7.21 (m, 4H, carbazole-H), 7.02–6.92 (m, 2H, phenyl-H), 3.58–3.43 (m, 2H, alkyl-H), 2.28 (s, 3H, alkyl-CH3), 2.18 (s, 3H, alkyl-CH3), 1.39–1.29 (m, 2H, alkyl-H), 1.28–1.18 (m, 2H, alkyl-H), 1.14–1.05 (m, 2H, alkyl-H), 1.03–0.95 (m, 2H, alkyl-H), 0.90 (t, J = 7.0 Hz, 3H, alkyl-CH3), 0.87–0.78 (m, 4H, alkyl-H). Anal. calcd for (C28H34N2)n: C, 84.37; H, 8.60; N, 7.03%; found: C, 83.85; H, 8.43; N, 6.66%; Br, none found; Pd, none found.Poly[[(2-methyl-4-methoxyphenyl)imino][(n-octyl)carbazole-2,7-diyl] (6)
Following polymerization procedure A; KOtBu (449 mg, 4 mmol), 2,7-dibromo-N-(n-octyl)carbazole (437 mg, 1 mmol), (IPr)Pd(allyl)Cl (24 mg, 4 mol%), 2-methoxy-2-methylaniline (0.130 mL, 1 mmol), toluene (6 mL, anhydrous and degassed) and p-bromoanisole (0.075 mL of a 50 mmol L−1 stock solution in toluene, 0.125 mol%) were heated to 105 °C for 24 hours. End-capping was performed for 2.5 hours. After work-up and Soxhlet extraction with methanol, acetone and 2-butanone, 264 mg (60%) of the title compound as a yellow solid were obtained from the 2-butanone fraction. 1H NMR (500 MHz, C6D6, δ): 7.88 (d, J = 8.5 Hz, 2H, carbazole-H), 7.37 (d, J = 9.0 Hz, 1H, phenyl-H), 7.31–7.24 (m, 4H, carbazole-H), 6.88 (d, J = 3.0 Hz, 1H, phenyl-H), 6.77 (dd, J = 9.0 Hz and 3.0 Hz, 1H, phenyl-H), 3.61–3.50 (m, 2H, alkyl-H), 3.36 (s, 3H, phenyl-OCH3), 2.27 (s, 3H, phenyl-CH3), 1.41–1.33 (m, 2H, alkyl-H), 1.28–1.18 (m, 2H, alkyl-H), 1.14–1.05 (m, 2H, alkyl-H), 1.04–0.97 (m, 2H, alkyl-H), 0.95–0.83 (m, 7H, alkyl-H and alkyl-CH3). Anal. calcd for (C28H34N2O)n: C, 81.12; H, 8.27; N, 6.76%; found: C, 80.78; H, 8.11; N, 6.59; Br, none found; Pd, trace (<0.1)%.Poly[[(2,4-dimethylphenyl)imino][5,11-di-n-octylindolo[3,2-b]carbazole-3,9-diyl] (7)
Following polymerization procedure B; KOtBu (224 mg, 2 mmol), 3,9-dibromo-5,11-di-n-octylindolo[3,2-b]carbazole (319 mg, 0.5 mmol), toluene (3 mL, anhydrous and degassed), 2,4-dimethylaniline (0.065 mL, 0.5 mmol), (IPr)Pd(allyl)Cl (4 mol%, 12 mg in 0.5 mL toluene) were heated for 22 hours at 105 °C. End-capping was performed for 2.5 hours. After work-up and Soxhlet extraction with methanol, acetone, 2-butanone and THF 79 mg (26%) of the title compound as a dark yellow solid were obtained from the THF fraction. 1H NMR (400 MHz, C6D6, δ): 8.18 (d, J = 8.0 Hz, 2H, indolocarbazole-H), 7.98 (s, 2H, indolocarbazole-H), 7.50 (d, J = 8.0 Hz, 1H, phenyl-H), 7.52–7.48 (m, 4H, indolocarbazole-H), 7.09–7.05 (m, 2H, phenyl-H), 3.93–3.88 (m, 4H, alkyl-H), 2.38 (s, 3H, phenyl-CH3), 2.23 (s, 3H, phenyl-CH3), 1.76–1.52 (m, 4H, alkyl-H), 1.24–1.04 (m, 20H, alkyl-H), 0.87 (t, J = 7.0 Hz, 6H, alkyl-CH3). Anal. calcd for (C42H53N3)n: C, 83.09; H, 8.91; N, 7.00%; found: C, 82.74; H, 9.13; N, 6.43%; Br, none found; Pd, none found.Poly[[(2-methyl-4-methoxyphenyl)imino][5,11-di-n-octylindolo[3,2-b]carbazole-3,9-diyl] (8)
Following polymerization procedure B; KOtBu (224 mg, 2 mmol), 3,9-dibromo-5,11-di-n-octylindolo [3,2-b]carbazole (319 mg, 0.5 mmol), toluene (3 mL, anhydrous and degassed), 4-methoxy-2-methylaniline (0.065 mL, 0.5 mmol) and (IPr)Pd(allyl)Cl (4 mol%, 12 mg in 0.5 mL toluene) were heated further 24 hours at 105 °C. End-capping was performed for 2.5 hours. After work-up and Soxhlet extraction with methanol, acetone, 2-butanone and THF 115 mg (37%) of the title compound as a dark brownish yellow solid were obtained from the THF fraction. 1H NMR (400 MHz, C6D6, δ): 8.20 (d, J = 8.5 Hz, 2H, indolocarbazole-H), 7.99 (s, 2H, indolocarbazole-H), 7.50 (d, J = 8.5 Hz, 1H, phenyl-H), 7.46–7.40 (m, 4H, phenyl-H), 6.95 (d, J = 2.0 Hz, 1H, phenyl-H), 6.84 (dd, J = 8.5, 2.0 Hz, 1H, phenyl-H), 3.97–3.86 (m, 4H, alkyl-H), 3.40 (s, 3H, phenyl-OCH3), 2.38 (s, 3H, phenyl-CH3), 1.77–1.51 (m, 4H, alkyl-H), 1.31–1.00 (m, 20H, alkyl-H), 0.87 (t, J = 7.0 Hz, 6H, alkyl-CH3). Anal. calcd for (C42H53N3O)n: C, 81.91; H, 8.67; N, 6.82%; found: C, 81.29; H, 8.76; N, 6.64%; Br, none found; Pd, none found.Results and discussion
Synthesis
A series of six new PTAAs (1–4, 7 and 8) was synthesized by the polyamination of commercially available 2,4-dimethylaniline and 2-methyl-4-methoxyaniline with the dibromoarene monomers shown in Fig. 2, using a palladium/N-heterocyclic carbene catalytic system based on either (IPr)Pd(allyl)Cl or [(IPr)Pd(naphthoquinone)]2 (IPr = bis(2,6-diisopropylphenyl)imidazol-2-ylidene) catalysts.32,33 In addition derivatives of the 3,7-linked carbazole based polymers 5 and 6 (ref. 32) were prepared for comparison with n-octyl chains instead of the branched 2-ethyl hexyl chains reported previously. High molecular weight polymers of the less soluble diindenofluorene and indolocarbazole monomers were obtained by adding the catalyst after homogenization of the reaction mixture at 105 °C. For the carbazole and indolocarbazole monomers higher catalyst loadings of 4 mol% resulted in higher molecular weight polymers than those obtained with a lower loading of 2 mol%.32 | ||
| Fig. 2 Synthesis of polytriarylamines. Reaction conditions 1 and 2: 3 mol% (IPr)Pd(allyl)Cl, 4 eq. KOtBu, toluene. 3 and 4: 4 mol% (IPr)Pd(allyl)Cl, 4 eq. KOtBu, toluene. 5: 4 mol% [(IPr)Pd(1,4-naphthoquinone)]2 4 eq. KOtBu, toluene. 6–8: 4 mol% (IPr)Pd(allyl)Cl, 4 eq. KOtBu, toluene. | ||
The molecular weight of all polymers was controlled for optimum performance in OFETs, as previous work has shown that it is critical to avoid bimodal molecular weight distributions in PTAAs to be used in organic electronics.34,35 Control was achieved by the use of shorter reaction times for polymer 1–4 and an in situ end-capping by the monofunctional 4-bromoanisole to limit the molecular weight of the step-growth polymerization.34,35 All polymers were purified by filtration over silica and extraction with sodium diethyldithiocarbamate solution to remove palladium catalyst residue10 followed by Soxhlet extraction to remove small molecules and oligomers giving number average molecular weights (Mn) in the range of 5100–21500 g mol−1 (Table 1). Lower molecular weights were obtained for the carbazole derived polymers (5–8) and similar effects have been observed previously.32 Due to the extensive work-up the isolated yields of the recovered polymers were generally <60%. In the cases of polymers 4, 5 and 7 a larger proportion of the material was lower molecular weight and this was removed during the Soxhlet extraction procedure.
| Polymer | Yielda % | M n /g mol−1 | M w /g mol−1 | M w/Mnb |
|---|---|---|---|---|
| a Isolated yield. b Calculated from gel-permeation chromatography (GPC). Measurements carried out using THF as the eluent and calibrated vs. polystyrene standards. | ||||
| 1 | 36 | 12400 |
16700 |
1.4 |
| 2 | 42 | 13400 |
23100 |
1.7 |
| 3 | 49 | 15000 |
24800 |
1.7 |
| 4 | 30 | 9300 | 14600 |
1.6 |
| 5 | 33 | 5100 | 9000 | 1.8 |
| 6 | 60 | 8200 | 16700 |
2.0 |
| 7 | 26 | 7800 | 11800 |
1.5 |
| 8 | 44 | 11800 |
19000 |
1.6 |
However, the influence of the polymer molecular weight is not expected to be significant for these polymers, as previous work has shown that in polymers with a Mn above 5000 g mol−1 the influence is only marginal.36 The new polymers are soluble in common organic solvents (CHCl3, CH2Cl2, THF and toluene) and were characterized by 1H NMR spectroscopy, MALDI-TOF mass spectrometry, elemental analysis and their physical properties examined by UV-Vis spectroscopy, photoluminescence and cyclic voltammetry.
MALDI-TOF MS analysis
Polymers 1–8 were analyzed by MALDI-TOF mass spectrometry. This proved to be much simpler for polymers containing indenofluorene (1, 2), diindenofluorene (3, 4) and indolocarbazole moieties (7, 8) in the backbone than for the biphenyl series reported earlier.32 The absorption maxima of these polymers are much further from the wavelength of the laser used for matrix desorption, resulting in less photoinduced fragmentation of the polymer main-chains than previously observed.The MALDI-TOF mass spectrum of polymer 1 (Fig. 3) shows two main series of peaks associated with the expected repeating unit terminated by either anilines (H-[XY]n-X) or anisole and anilines (H-[XY]n-XA). Similar results were obtained for the methoxy derivatives 2, 3 and 4 (see ESI†). For polymers 7 and 8 where the end-capping was performed at the end of the reaction, predominantly aniline terminated main-chains with no anisole end-capped polymers are observed. End-capping of the N–H functionalities at the end of the polymerization was ineffective, as we also have found previously in polymerizations of biphenyls and fluorenes.33 Elemental analysis of the polymers revealed only trace levels of bromine or palladium were present in the polymers.
 | ||
| Fig. 3 MALDI-TOF MS spectrum of polymer 1 and possible structures. X = 2,4-dimethylaniline, Y = indenofluorene, A = anisole. | ||
Photophysical properties
The optical properties of the polymer 1 to 8 were studied by UV-Vis and photoluminescence measurements in dichloromethane solution (Fig. 4 and ESI†). The absorption maxima varied between 425 nm and 452 nm. The optical band gap was determined from the onset of the absorption spectra (see Table 2).37 The absorption maximum of 1 is observed at λmax = 443 nm and this represents a bathochromic shift of 23 nm compared to the previously reported fluorene derivative (λmax = 420 nm).32 The absorption maximum of 3 is bathochromically shifted by 9 nm over that of 1 (λmax = 452 nm), due to the extension of the π-conjugated system on replacing the indenofluorene unit with the diindenofluorene unit in the polymer backbone.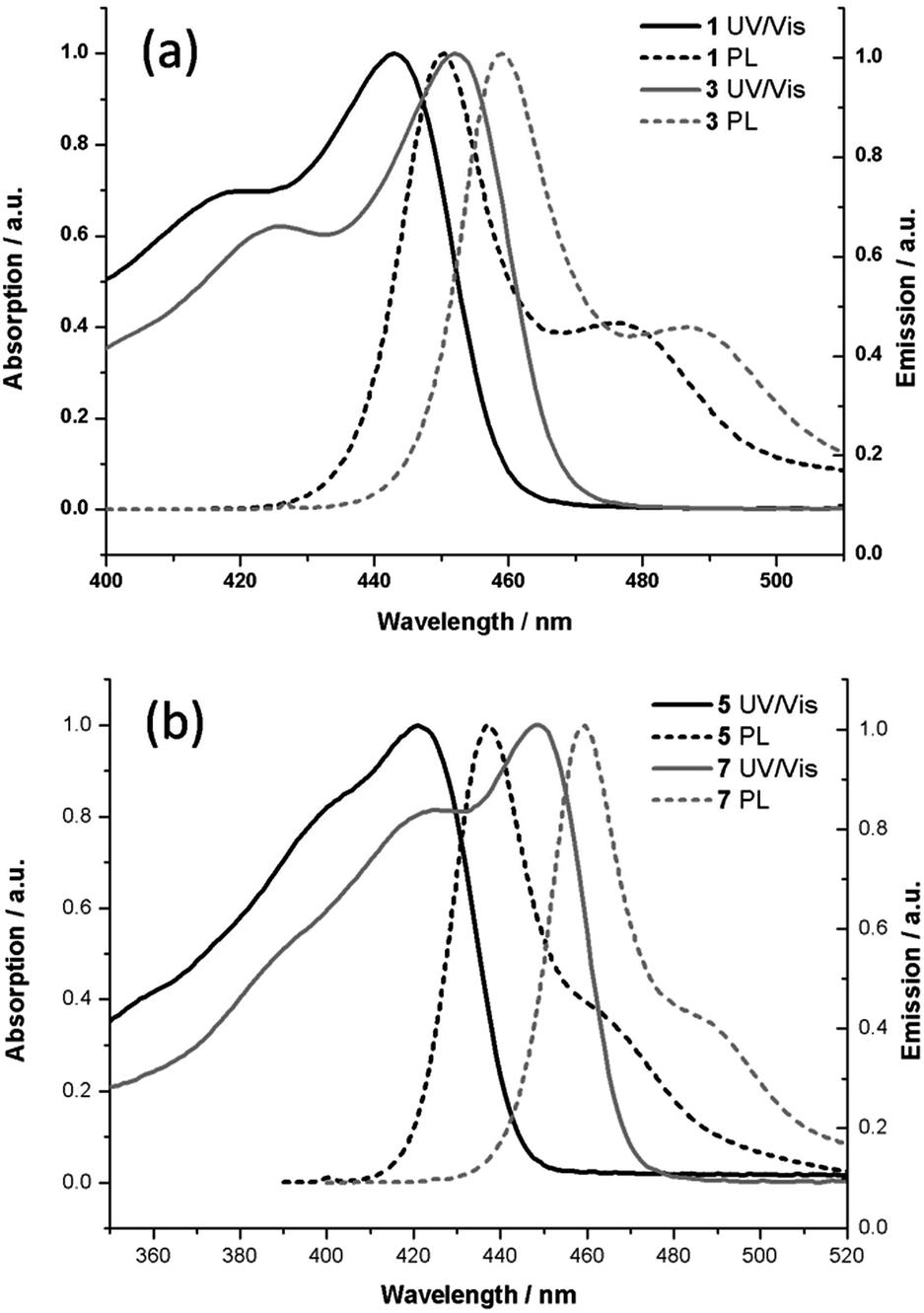 | ||
| Fig. 4 UV-Vis and PL spectra of polymer 1, 3 (a) and 5, 7 (b) in dichloromethane. | ||
| Polymer | λ max/nm | λ em/nm | λ g /nm | Stokes shiftb/nm | λ max film/nm | E g /eV | E 1/2/eV | E HOMO /eV | E LUMO /eV | T g °C−1 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Calculated from onset of absorption. b Difference between λmax and λem. c From optical absorption data. d Estimated vs. vacuum level from EHOMO = −1.4E1/2 – 4.6 eV.37 e Estimated from ELUMO = EHOMO + Eg f No glass transition observed. | ||||||||||
| 1 | 443 | 450 | 461 | 7 | 442 | 2.69 | 0.02 | −4.6 | −1.9 | 76 |
| 2 | 447 | 453 | 462 | 6 | 443 | 2.68 | 0.03 | −4.6 | −1.9 | 66 |
| 3 | 452 | 459 | 470 | 7 | 443 | 2.64 | 0.09 | −4.7 | −2.1 | —f |
| 4 | 453 | 460 | 470 | 7 | 445 | 2.64 | 0.09 | −4.7 | −2.1 | —f |
| 5 | 425 | 437 | 442 | 12 | 419 | 2.81 | −0.13 | −4.5 | −1.7 | 110 |
| 6 | 425 | 440 | 447 | 14 | 421 | 2.78 | −0.13 | −4.5 | −1.7 | 105 |
| 7 | 448 | 460 | 472 | 12 | 449 | 2.63 | −0.04 | −4.6 | −2.0 | 98 |
| 8 | 452 | 462 | 475 | 10 | 452 | 2.61 | −0.04 | −4.6 | −2.0 | 102 |
A similar effect was found for 7, which is bathochromically shifted by 23 nm when compared to 5 due to the extended conjugation of the indolocarbazole over that of the carbazole derivative. For the methoxy series (polymers 2, 4, 6, and 8) the absorption maxima are further red-shifted over those of the methyl series (polymers 1, 3, 5 and 7) as the electron-donating methoxy group raises the HOMO energy level. Small Stokes shifts are observed for all polymers and the vibrational fine structure of the PL spectra indicate that rotational freedom of the arene units in the polymer backbone is limited (Fig. 5).
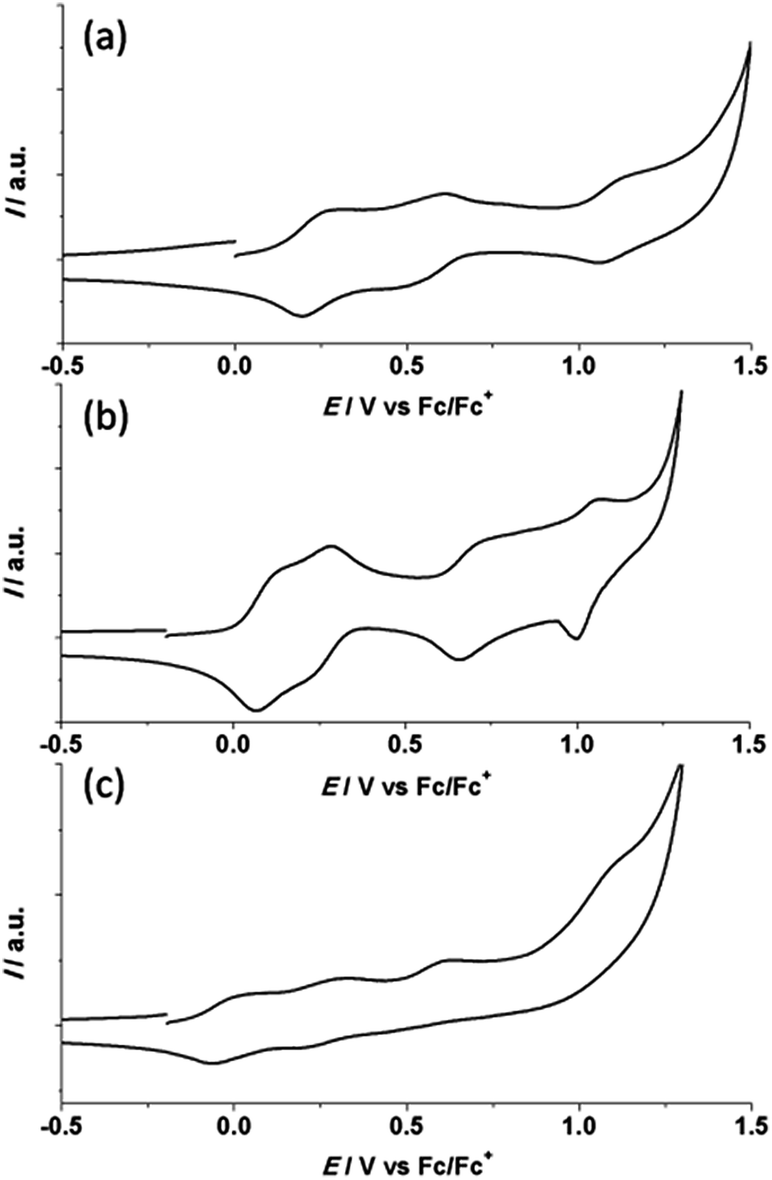 | ||
| Fig. 5 Cyclic voltammograms of 1 (a), 3 (b) and 7 (c) in dichloromethane containing 0.1 M [(C4H9)4N]PF6 at scan rate 100 mV s−1. | ||
The electronic structures of oligomers based on polymer 1–8 (n = 2, 3 and 4) were computed and the predicted HOMO/LUMO energy levels and the band gaps are collected in Table 3. Comparing these values to the experimental values shows that the general trends are consistent between theory and experiment. For the tetramers the HOMO is delocalized (see Fig. 6 and ESI†) over the arylamine segments but it does not extend on to the π-system of the terminal arenes possibly due to the high torsion angle between the main chain and these subunits or the fact that the molecule is no longer symmetric.
| Polymer | E HOMO /eV | E LUMO /eV | λ + /eV |
|---|---|---|---|
| a Negative value of ionization potential. b From Kohn–Sham calculations. c Reorganization energies for holes (λ+) of the tetramers. d Charged state of the tetramer could not be relaxed. | |||
| 1 | −4.8 | −2.1 | 0.09 |
| 2 | −4.8 | −2.1 | 0.15 |
| 3 | −4.7 | −2.3 | 0.15 |
| 4 | −4.7 | −2.3 | —d |
| 5 | −4.7 | −1.7 | 0.15 |
| 6 | −4.8 | −1.8 | 0.31 |
| 7 | −4.7 | −2.0 | 0.08 |
| 8 | −4.6 | −1.9 | 0.14 |
 | ||
| Fig. 6 Highest occupied molecular orbital distribution for tetramers of 1 (a), 3 (b) and 7 (c). | ||
Field-effect transistor results
Polymers 1–8 were used to fabricate OFETs, initially in the bottom-gate, top-contact architecture. Highly doped silicon substrates were used, which acted as the gate electrode with a 300 nm silicon oxide layer as the gate dielectric. In order to enhance charge transport32,38,39 the SiO2 layer was treated with n-octadecyltrichlorosilane (OTS) by spin-casting from 1,1,2-trichloroethylene (TCE) solution, using a modified method reported by Bao and co-workers.40The polymers (1–8) were deposited on the OTS-treated SiO2/Si substrates from TCE solution and the mobility in the saturation region was calculated from the transfer characteristics of the OFETs. All polymers gave statistically significant numbers of working devices for parameter extraction and the results are summarized in Table 4.
| Polymer | Hole-mobilitya/cm2 V−1 s−1 | I on/off | Hysteresisb/V | V th /V |
|---|---|---|---|---|
| a Calculated from an average of 3 best devices in the saturation regime (see ESI for more details). b Difference between forward and backward sweep. c Determined from the onset of the square root of ID characteristics for forward sweep. d Top-gate/bottom contact devices. | ||||
| 1 | 5.0 × 10−2 | 7 × 104 | 2 | −4 |
| 1 | 5.1 × 10−2 | 2 × 103 | <1 | −2 |
| 2 | 2.6 × 10−3 | 2 × 103 | 3 | −16 |
| 3 | 2.8 × 10−3 | 2 × 105 | 2 | −14 |
| 4 | 3.5 × 10−3 | 2 × 104 | 2 | −9 |
| 5 | 1.1 × 10−3 | 3 × 103 | 1 | −1 |
| 6 | 1.2 × 10−3 | 7 × 102 | 4 | −1 |
| 7 | 3.7 × 10−3 | 2 × 103 | 1 | −6 |
| 8 | 1.8 × 10−3 | 3 × 104 | 3 | −9 |
The highest hole mobility was observed for polymer 1 (5.0 × 10−2 cm2 V−1 s−1), and this is the highest mobility reported to date for polytriarylamine transistors.11 Top-gate bottom-contact devices were also fabricated for this polymer using poly(1,1,2,4,4,5,5,6,7,7-decafluoro-3-oxa-1,6-heptadiene) (CYTOP) as the gate dielectric, however, the configuration seems to have no influence on the observed mobility (5.1 × 10−2 cm2 V−1 s−1) as expected for amorphous semiconducting polymers. A typical transfer and output characteristic for polymer 1 is shown in Fig. 7. All other polymers showed more moderate hole mobilities on OTS-treated SiO2/Si substrates that ranged from 1.1 × 10−3 to 3.7 × 10−3 cm2 V−1 s−1, the hysteresis was low for all devices (<4 V), and the threshold voltages and on/off ratios are in the range previously reported for PTAAs. The hole-mobility of 2, 3 and 4 were significantly lower than those of 1. The carbazole containing polymers 5 and 6 gave very similar mobilities of ca. 1 × 10−3 cm2 V−1 s−1, which increased for the indolocarbazole derivatives 7 and 8, with 7 giving a higher performance at 3.7 × 10−3 cm2 V−1 s−1.
 | ||
| Fig. 7 (a) Transfer characteristics (VD = −60 V) and (b) output characteristics of polymer 1 in top-gate/bottom-contact device. | ||
Charge-transport in disordered amorphous materials is believed to occur via an intermolecular hopping mechanism and can therefore be described by the Marcus theory.41 The two main contributions for charge-transport in Marcus theory are the internal reorganization energy and the charge transfer integral. The influence of the charge transfer integral was found to be limited for analogous amorphous compounds42,43 and it has been demonstrated for tetraphenyldiamines that internal reorganization energies can be related to the observed hole mobilities.44
The relationship between structure and performance in OFET devices of polymers 1–8 was investigated by calculation of the optimized geometries and reorganization energies for oligomers based on the repeating units (n = 2, 3 and 4) of the polymers. Optimized geometries of oligomers show that the phenylene rings in the indenofluorene (1, 2) and the indolocarbazole (7, 8) units are coplanar. However, in the diindenofluorene unit in polymers 3 and 4 the phenylene rings are twisted with respect to each other. This distortion reduces the conjugation of the polymer backbone and increases the conformation rotational disorder of the backbone, both effects potentially having a negative impact on the expected hole mobility. Even though the trend in the absolute numbers does not correspond precisely with the measured mobility, the overall trend is that the polymers with the lowest calculated reorganization energies (1 and 7) showed the highest mobilities in OFET devices (Table 4).
The distorted diindenofluorene derivative 3 has a higher reorganization energy than 1 and a lower measured mobility in an OFET. For the carbazole and indolocarbazole series a correlation between the internal reorganization energies and the observed hole-mobility can be found, even though for the reorganization energy for 6 seems out of proportion with λ+ = 0.31 eV. However, precisely correlating the reorganization energies of the tetramers with the measured hole mobilities of the polymers is not possible as the polymers are polydisperse, end-groups are present and for ease of calculation the solubilising aliphatic side-chains were omitted.45
Differential scanning calorimetry (DSC) was used to analyse the thermal behavior of the polymers. Polymers 3 and 4 showed no glass-transition (Tg) on heating or cooling, whilst all other polymers exhibit a second order transition (see Table 2). Powders of polymer 1 isolated by precipitation from solution show a small melting isotherm at 138 °C in the first scan of the DSC (see ESI†). However, the polymer cooled into a glassy state showed no endothermic transitions in the second scan. Wide angle X-ray scattering (WAXS) of 1 isolated by precipitation from solution shows reflections associated with a low degree of crystallinity. WAXS of thin films of 1 deposited by spin-coating on a silicon substrate showed no reflections and was consistent with an amorphous film. It appears that polymer 1 may partially crystallize during precipitation from dichloromethane solution into a non-solvent such as methanol but this crystallization is not possible when the polymer is deposited as a thin film directly from a good solvent during spin-coating. Spin-coated thin films of 1 remain amorphous even by extensive thermal or solvent annealing and the partial crystalline character of the powders cannot be restored (see ESI†).
Conclusions
PTAAs with extended fused backbones were synthesized by the (NHC)–Pd catalysed C–N coupling of anilines and dibromoarenes. The optical and electrochemical properties of these polymers showed an increase in the HOMO energies and the onset of absorption on extending the conjugation from indenofluorene (1, 2) to diindenofluorene (3, 4) and from carbazole (5, 6) to diindolocarbazole (7, 8). The highest OFET performance was found for a polymer (1) derived from a dibromoindenofluorene and 2,4-dimethylaniline with hole mobilities of 5 × 10−2 cm2 V−1 s−1 in top- and bottom-gated devices. This is the highest reported performance for a polytriarylamine. The lower performance of more extended diindenofluorene polymers (3, 4) and polymer 2 was rationalized by DFT calculations of model oligomers that show higher reorganization energies for these polymer structures. Polymers derived from indolocarbazole units (7) have the lowest predicted reorganization energies of those in this study, suggesting that optimization of OFET fabrication for these materials could lead to further improvement in polytriarylamine device performance.Acknowledgements
M.L.T and O.N. acknowledge the EPSRC (EP/H004408/1) and the NSF (CHE 0924324) for financial support. R.S.S. acknowledges Gareth Smith for help with the MALDI-TOF MS measurements.Notes and references
- S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097–1116 CrossRef CAS PubMed.
- C. L. Chochos and S. A. Choulis, Prog. Polym. Sci., 2011, 36, 1326–1414 CrossRef CAS PubMed.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- A. Pron, P. Gawrys, M. Zagorska, D. Djurado and R. Demadrille, Chem. Soc. Rev., 2010, 39, 2577–2632 RSC.
- R. R. Søndergaard, M. Hösel and F. C. Krebs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 16–34 CrossRef.
- A. Marrocchi, D. Lanari, A. Facchetti and L. Vaccaro, Energy Environ. Sci., 2012, 5, 8457–8474 CAS.
- I. McCulloch, M. Heeney, C. Bailey, K. Genevicius, I. Macdonald, M. Shkunov, D. Sparrowe, S. Tierney, R. Wagner, W. Zhang, M. L. Chabinyc, R. J. Kline, M. D. McGehee and M. F. Toney, Nat. Mater., 2006, 5, 328–333 CrossRef CAS PubMed.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- S. Georgakopoulos, Y. Gu, M. M. Nielsen and M. Shkunov, Appl. Phys. Lett., 2012, 101, 213305 CrossRef PubMed.
- W. Zhang, J. Smith, R. Hamilton, M. Heeney, J. Kirkpatrick, K. Song, S. E. Watkins, T. Anthopoulos and I. McCulloch, J. Am. Chem. Soc., 2009, 131, 10814–10815 CrossRef CAS PubMed.
- I. D. V. Ingram, D. J. Tate, A. V. S. Parry, R. S. Sprick and M. L. Turner, Appl. Phys. Lett., 2014, 104, 153304 CrossRef PubMed.
- H. Kempa, K. Reuter, M. Bartzsch, U. Hahn, A. C. Hübler, D. Zielke, M. Forster and U. Scherf, in Proc. IEEE Polytronic Conference, 2005, pp. 67–71 Search PubMed.
- P. Reiss, E. Couderc, J. De Girolamo and A. Pron, Nanoscale, 2011, 3, 446–489 RSC.
- O. Solomeshch, V. Medvedev, P. R. Mackie, D. Cupertino, A. Razin and N. Tessler, Adv. Funct. Mater., 2006, 16, 2095–2102 CrossRef CAS.
- D. C. Wedge, A. Das, R. Dost, J. Kettle, M.-B. Madec, J. J. Morrison, M. Grell, D. B. Kell, T. H. Richardson and S. Yeates, Sens. Actuators, B, 2009, 143, 365–372 CrossRef PubMed.
- A. Das, R. Dost, T. Richardson, M. Grell, J. J. Morrison and M. L. Turner, Adv. Mater., 2007, 19, 4018–4023 CrossRef CAS.
- I. K. Yakushchenko, M. G. Kaplunov, O. N. Efimov, M. Y. Belov and S. N. Shamaev, Phys. Chem. Chem. Phys., 1999, 1, 1783–1785 RSC.
- J. K. Feng, Y. L. Cao, X. P. Ai and H. X. Yang, J. Power Sources, 2008, 177, 199–204 CrossRef CAS PubMed.
- S. L. Li, X. P. Ai, H. X. Yang and Y. L. Cao, J. Power Sources, 2009, 189, 771–774 CrossRef CAS PubMed.
- J. Kim, S. H. Kim, I. H. Jung, E. Jeong, Y. Xia, S. Cho, I.-W. Hwang, K. Lee, H. Suh, H.-K. Shim and H. Y. Woo, J. Mater. Chem., 2010, 20, 1577–1586 RSC.
- Q. Zheng, B. J. Jung, J. Sun and H. E. Katz, J. Am. Chem. Soc., 2010, 132, 5394–5404 CrossRef CAS PubMed.
- W.-Y. Lee, C.-W. Chen, C.-C. Chueh, C.-C. Yang and W.-C. Chen, Polym. J., 2008, 40, 249–255 CrossRef CAS.
- N. Blouin, A. Michaud, S. Wakim, P.-L. T. Boudreault, M. Leclerc, B. Vercelli, S. Zecchin and G. Zotti, Macromol. Chem. Phys., 2006, 207, 166–174 CrossRef CAS.
- S. Setayesh, D. Marsitzky and K. Müllen, Macromolecules, 2000, 33, 2016–2020 CrossRef CAS.
- Q. Zheng, S. K. Gupta, G. S. He, L.-S. Tan and P. N. Prasad, Adv. Funct. Mater., 2008, 18, 2770–2779 CrossRef CAS.
- Y. Wu, Y. Li, S. Gardner and B. S. Ong, J. Am. Chem. Soc., 2005, 127, 614–618 CrossRef CAS PubMed.
- C. Poriel, J.-J. Liang, J. Rault-Berthelot, F. Barrière, N. Cocherel, A. M. Z. Slawin, D. Horhant, M. Virboul, G. Alcaraz, N. Audebrand, L. Vignau, N. Huby, G. Wantz and L. Hirsch, Chem.–Eur. J., 2007, 13, 10055–10069 CrossRef CAS PubMed.
- F. Dierschke, A. C. Grimsdale and K. Müllen, Synthesis, 2003, 2470–2472 CAS.
- P.-L. T. Boudreault, S. Wakim, N. Blouin, M. Simard, C. Tessier, Y. Tao and M. Leclerc, J. Am. Chem. Soc., 2007, 129, 9125–9136 CrossRef CAS PubMed.
- O. Navarro and S. P. Nolan, Synthesis, 2006, 366–367 CAS.
- R. S. Sprick, M. Hoyos, J. J. Morrison, I. M. Grace, C. Lambert, O. Navarro and M. L. Turner, J. Mater. Chem. C, 2013, 1, 3327–3336 RSC.
- M. Hoyos, R. S. Sprick, C. Wang, M. L. Turner and O. Navarro, J. Polym. Sci. Part A: Polym. Chem., 2012, 50, 4155–4160 CrossRef CAS.
- S. Barard, M. Heeney, L. Chen, M. Cölle, M. Shkunov, I. McCulloch, N. Stingelin, M. Philips and T. Kreouzis, J. Appl. Phys., 2009, 105, 013701–013706 CrossRef PubMed.
- R. S. Sprick, M. Hoyos, O. Navarro and M. L. Turner, React. Funct. Polym., 2012, 72, 337–340 CrossRef CAS PubMed.
- M.-B. Madec, J. J. Morrison, M. Rabjohns, M. L. Turner and S. G. Yeates, Org. Electron., 2010, 11, 686–691 CrossRef CAS PubMed.
- B. D'Andrade, S. Datta, S. Forrest, P. Djurovich, E. Polikarpov and M. Thompson, Org. Electron., 2005, 6, 11–20 CrossRef PubMed.
- J. Veres, S. Ogier, G. Lloyd and D. de Leeuw, Chem. Mater., 2004, 16, 4543–4555 CrossRef CAS.
- M. Horie, Y. Luo, J. J. Morrison, L. Majewski, A. Song, B. R. Saunders and M. L. Turner, J. Mater. Chem., 2008, 18, 5230–5236 RSC.
- Y. Ito, A. A. Virkar, S. Mannsfeld, J. H. Oh, M. Toney, J. Locklin and Z. Bao, J. Am. Chem. Soc., 2009, 131, 9396–93404 CrossRef CAS PubMed.
- H. Gao, J. Mol. Struct., 2010, 962, 80–84 CrossRef CAS PubMed.
- S. F. Nelsen and F. Blomgren, J. Org. Chem., 2001, 66, 6551–6559 CrossRef CAS PubMed.
- B. C. Lin, C. P. Cheng and Z. P. M. Lao, J. Phys. Chem. A, 2003, 107, 5241–5251 CrossRef CAS.
- R. D. Hreha, C. P. George, A. Haldi, B. Domercq, M. Malagoli, S. Barlow, J.-L. Brédas, B. Kippelen and S. R. Marder, Adv. Funct. Mater., 2003, 13, 967–973 CrossRef CAS.
- S. Zade, Acc. Chem. Res., 2010, 44, 14–24 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer synthesis, details of DFT calculations, FET preparation and characterization, MALDI-TOF MS spectra, UV-Vis and PL spectra, FET characteristics, DSC traces, WAXS and further annealing experiments. See DOI: 10.1039/c4tc00871e |
| This journal is © The Royal Society of Chemistry 2014 |