 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Air-stable photoconductive films formed from perylene bisimide gelators†
Emily R.
Draper
a,
James J.
Walsh
b,
Tom O.
McDonald
a,
Martijn A.
Zwijnenburg
c,
Petra J.
Cameron
d,
Alexander J.
Cowan
*b and
Dave J.
Adams
*a
aDepartment of Chemistry, University of Liverpool, Crown Street, Liverpool, L69 7ZD, UK. E-mail: d.j.adams@liverpool.ac.uk
bStephenson Institute for Renewable Energy, University of Liverpool, Peach Street, Liverpool, L69 7ZF, UK. E-mail: a.j.cowan@liverpool.ac.uk
cDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK
dDepartment of Chemistry, University of Bath, 1 South, Bath, BA2 7AY, UK
First published on 28th May 2014
Abstract
We show that amino acid-PBIs can form one-dimensional structures at high pH and then gels at low pH. Both the dried solutions and dried gels are photoconductive. Interestingly, photoconductivity of these materials requires that the incident light has a wavelength shorter than 400 nm, in stark contrast with the absorption maxima of the PBIs. The photoconductivity correlates with the formation of the perylene radical anion, which is unusually highly stable in air for many hours.
Introduction
π-conjugated systems are used extensively in electronic devices, including solar cells and light emitting diodes.1 Such devices are normally formed using deposition or coating routes that can lead to the formation of aggregates with different size and shape distributions, which can influence the electronic properties. Self-assembly is a simple method of organizing optoelectronically active π-conjugated molecules in a defined manner with precise control at both the nano- and micro-scale.2–6 One self-assembly route is to exploit low molecular weight gelators (LMWG).2 Gelation using suitably designed LMWG results from self-assembly of the gelator into well-defined one-dimensional structures, which then can entangle or cross-link. When the LMWG contain π-conjugated groups, the result of this assembly is the stacking of these groups, which can be exploited to form conductive pathways for electronic devices.2 The assembly of LMWG based around perylene bisimides (PBI) is of great interest from the perspective of electronic materials, since PBIs are n-type materials.7–10 PBIs have strong absorption, long fluorescence lifetimes and high quantum yields. There are a growing number of reports showing the formation of self-assembled wires and fibers from PBI-based gelators. PBIs can also be (photo)conductive and have been suggested as candidates for many organic electronic devices;11–13 for example as alternative electron acceptors in solar cells7,14,15 or in field-effect transistors.16 PBIs are readily sequentially reduced to the radical anion and dianion. Significant work has been done on the formation of one-dimensional structures based on PBIs,9,10,17,18 for example showing supramolecular chirality19 and the formation of liquid crystallinity.20 A key point is that the optoelectronic performance of such materials can depend not only on the chemical structure, but also on the morphology and uniformity of the aggregates formed.21,22 For PBIs, the aggregates formed can be controlled by functional group, solvent, or concentration.21,23 Exciton diffusion has been shown to occur in one-dimension in PBI-aggregates.24 The majority of the reported PBI-based LMWG gel organic solvents, due to the high hydrophobicity of the perylene group. Examples have been used in light harvesting and also in bulk heterojunctions, formed from self-sorted gel fibers.25 Recently, PBI-based hydrogelators have been reported,26 for example, an amino acid-based PBI was shown to form photoconductive xerogels.27 The LUMO level of an aspartic acid-based PBI has been suggested to be suitable for use as an electron acceptor in a solar cell; however the reduced PBI solution formed was highly sensitive to the presence of O2, which is not promising for OPV applications. It is therefore highly desirable to have reduced PBI species in thin films which are resistant to anion oxidation by O2.28Results and discussion
Here we discuss the photoconductivity of four PBIs LMWG, prepared by the reaction of 3,4:9,10-perylenetetracarboxyldianhydride with a number of amino acids following literature procedures for the coupling of amino acids to the anhydrides.28–301 and 2 (Fig. 1) have been previously reported.29,30 We focus on the L-alanine (1), L-histidine (2), L-phenylalanine (3) and L-valine (4) functionalized PBIs (Fig. 1). | ||
| Fig. 1 Structures of the PBIs used in this study. | ||
1–4 all form deep red solutions at high pH (≈pH 10) at a concentration of 5 mg mL−1 (photographs are shown in Fig. 2a for 1; data for 2–4 are shown in Fig. S7–S9, ESI†). Viscosity measurements of solutions of 1–4 at high pH show them to be shear thinning (Fig. S10†), suggesting that structures present are aligning at high shear causing the lower viscosity. Solutions of 3 have much higher viscosity than the other LMWG. We have previously noted that certain hydrophobic LMWG with similar molecular structures form worm-like micelles at high pH,31,32 leading to viscous solutions. Hence, it is likely that worm-like micelles are formed at high pH from 1–4. Indeed, the dried solutions generally showed the presence of long one-dimensional structures by SEM (Fig. 2b and S7–S9†). Dried solutions of 3 however only showed disordered structures (Fig. S8†).
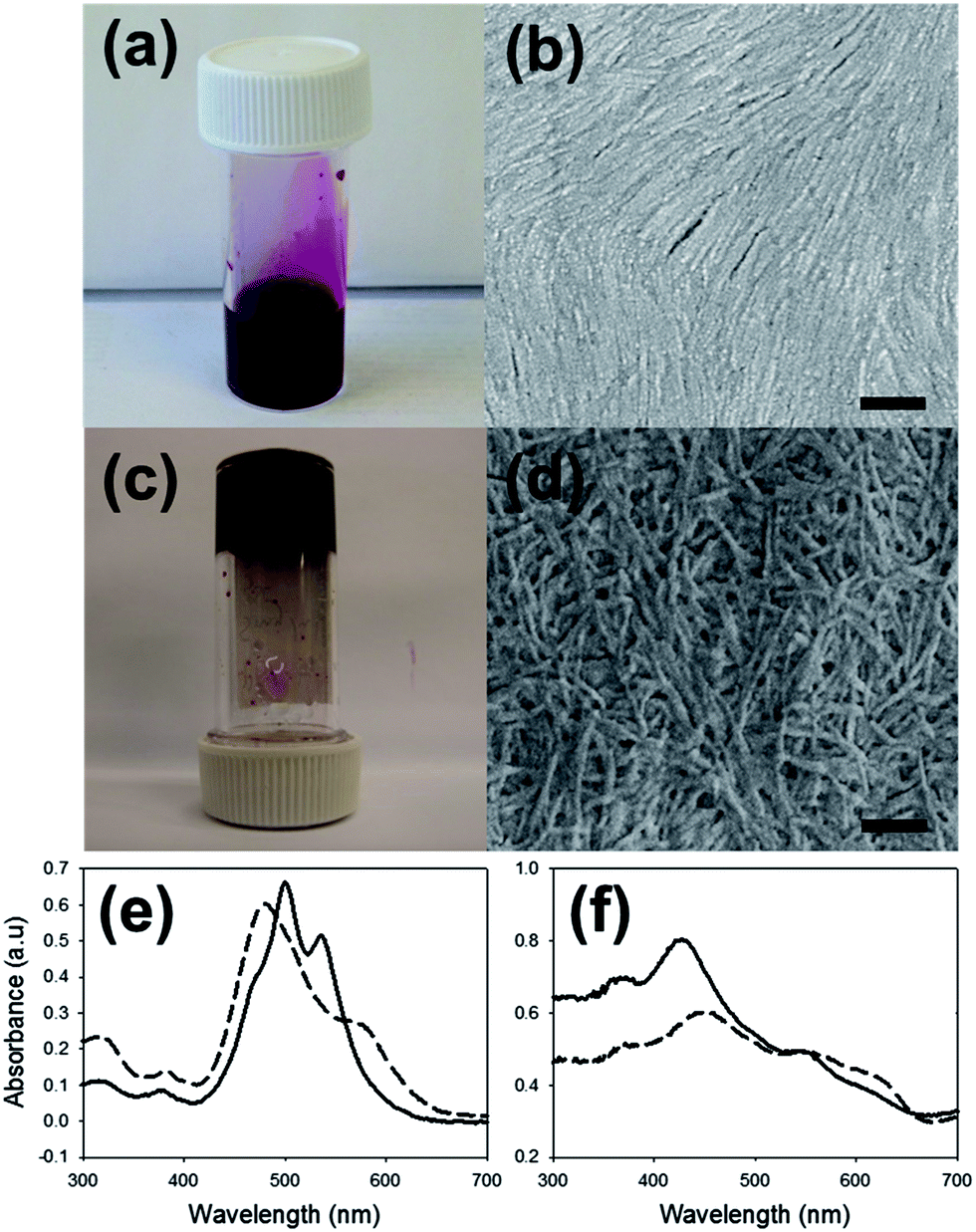 | ||
| Fig. 2 (a) Photograph of a solution of 1 at pH 10 at a concentration of 5 mg mL−1. (b) SEM image of structures formed on drying the solution shown in (a). (c) A gel formed on acidification of the solution of 1 in (a). (d) SEM image of structures formed on drying the gel shown in (c). For (b) and (d), the scale bar represents 2 μm. (e) UV-Vis spectra of the solution (—) and gel (- - -). (f) UV-Vis spectra of the dried solution and the dried gel. | ||
The viscous solutions formed self-supporting dark red gels on lowering the pH (again, photographs are shown in Fig. 2c for 1; data for 2–4 in Fig. S7–S9†). We adjusted the pH by adding glucono-δ-lactone, which hydrolyses slowly to gluconic acid,33 resulting in a homogeneous pH drop for LMWG.34 On gelation, 1, 2, and 4 form transparent gels while 3 forms gels which are turbid. The rheological properties of these gels are similar for those formed from related LMWG,27 with the storage modulus (G′) and loss modulus (G′′) being only weakly dependent on frequency (Fig. S15–S18†). SEM of the dried gels (xerogels) showed the presence of thin entangled fibers. SEM of the dried solutions showed similar structures, with greater alignment of the fibrous structures than the corresponding xerogels. In all cases, powder X-ray diffraction (pXRD) data shows both the dried solutions and xerogels contain a low degree of crystallinity (Fig S11–S14†). The most intense peak in all samples is at 2θ = ∼25.5°, corresponding to approximately 3.5 Å, arising from π–π stacking.
The absorption spectra of these materials in solution (5 mg mL−1) are typical of PBI-LMWG. UV-Vis data at high pH (Fig. 2e and S20–S22†) showed a shoulder at 470 nm, and peaks at 490 nm and 540 nm, attributed to the 0–0 and 0–1 vibronic bands of the S0 → S1 transitions.35 The ratio of the peak intensities at 490 nm and 540 nm indicates a significant degree of aggregation.26,36 In the gel state, the UV-Vis data showed strong absorption at 470 nm and 590 nm, with the peak at 470 nm now being dominant (Fig. 2e and S20–S22†). This change in relative intensity suggests a change in the aggregation of the perylenes.26 The spectra for both the solution and gel showed peaks at 325 nm and 380 nm, corresponding to the electronic S0 → S2 transition.35 Fluorescence data showed fine structure emission with maxima at 540 nm and 595 nm for the solution on excitation at 365 nm or 490 nm (Fig. S22†). The fluorescence intensity of the gel is significantly lower due to self-quenching, with the peak maxima slightly shifted to shorter wavelength, again due to a change in aggregation.
PBIs are well known to be photoconductive, with a number of photoconductive LMWG systems having been reported.26,27,37 To investigate the photoconductivity of these materials, we dried both the solution and gel phases. Films were readily obtained from both the solutions and gels of 1, 2 and 4 simply by drying in air (final water content ∼6 wt% by TGA). For 3, drying led to an inhomogenous film that did not adhere well to the substrate (Fig. S23†), correlating with the SEM data. There are fundamental changes in the UV-Vis spectra on drying. For both dried solutions and xerogels, the UV-Vis data showed an increase of absorbance in the UV region where there is a change in the intensity ratio of the peaks at 375 nm and those in the region of 470–590 nm. This suggests a difference in the arrangement of the perylene aggregates upon drying. The macroscopic conductivity of the dried solutions and gels was measured both in the dark and under illumination with a xenon lamp. Both showed Ohmic response and a significantly increased current under illumination, assigned to the samples becoming photoconductive (see Fig. S24–27†), with symmetric data during the voltage sweep. In general, the dried solutions were more conductive than the xerogels. 1 showed the highest current under illumination of the LMWG, and the photo-response was significantly greater for the dried solution than for the dried gel. 3 showed a very weak response, which we attribute primarily to the poor film quality.
However, in all cases, despite the expected correlation between the absorption spectrum of the perylene group and the proposed photoconductivity, both the dried gel and solution remain highly resistive when irradiated with light above 400 nm. Instead, a significant photocurrent was only induced when irradiated with wavelengths shorter than 400 nm (Fig. 3a and S28–S30†). The onset wavelength of the photoresponse of the dried solution and xerogel varied. For 1, 2 and 4, the xerogel resistance decreased significantly with irradiation of wavelengths shorter than 400 nm, whereas the dried solutions only became active when irradiated at wavelengths of 375 nm or shorter. Due to the poor film quality, we did not attempt to collect this data for 3. Irradiation of samples of 1, 2 and 4 with 365 nm LED irradiation again showed a significant decrease in resistance, indicating photoconductivity. For 1 and 4, the dried solution was found to be approximately one order of magnitude less resistive under irradiation than the xerogel (Fig. 3b). This may be due to the packing in the solution versus gel state, but could also be due to differences in fiber thicknesses, density, and orientation. However, the photocurrents measured in our two electrode experiment were consistently greater with both the dried solutions and xerogels of 1 than for 2 and 4, indicating a greater degree of photoconductivity. The conductivity for PBIs has been related to well-ordered π-stacking and the morphology has also been shown to be key.21 The UV-Vis and pXRD data for 1, 2 and 4 are similar; hence, we ascribe the higher conductivity of 1 to differences in fiber morphology over molecular packing.
 | ||
| Fig. 3 (a) Plot of photocurrent (at 2 V potential difference) against irradiation wavelength for a dried solution of 1 overlaid with the UV-Vis spectrum. (b) Plots of I–V curves for (⋯) a dried solution of 1 and (- - -) a xerogel for 1 on irradiation at 365 nm. Inset is the data for the xerogel (- - -) under irradiation and (—) in the dark. (c) Photoresponse for a xerogel of 1, initially in the dark, then irradiated with 365 nm light, followed by the lamp being turned off. (d) Transient photoresponse for a dried solution of 1 by turning on and off the 365 nm light for multiple cycles. | ||
Focusing on 1, on irradiating a dried solution in a two electrode cell (2 V) with 365 nm light, the photocurrent increases with time (Fig. 3c). On switching off the light, photocurrent persists for typically 1–8 hours, with the decay shown in Fig. 3c being fitted to a single exponential process, with a lifetime of ca. 5300 s. This indicates that the photo-induced conductivity is remarkably long-lived in air. There is variability from sample to sample, due to differences in film thicknesses and fiber density and orientation, but the films show lifetimes consistently >3000 s. Switching the light on and off demonstrated the stability of the films (Fig. 3d). Similar behavior was observed for the xerogel (Fig. S31†).
On irradiation with 365 nm light, both the xerogel (Fig. 4a) and dried solution (Fig. 4b) were also observed to change color. This color change was reversible over several hours. Monitoring this process by UV-Vis-NIR spectroscopy for the xerogel, we observed the formation of a new absorption feature with maxima at 735 nm, 820 nm and 1000 nm (Fig. 4c), in good agreement with data for the formation of the radical anion38 formed by chemical reduction. For the dried solution, similar absorptions were observed, albeit with a higher relative intensity, in addition to an increase in the relative intensity of the shoulder at 615 nm (Fig. 4d), as expected for the dianion.28 We confirmed that these peaks were characteristic of these species by reduction of 1 in solution using sodium dithionite (Fig. 4e and S38†).39 On the basis of these data, we suggest that the higher anion concentration in the dried solution explains the higher conductivity of the dried film over the dried gel. The possible presence of the dianion in the dried solution may also contribute, but the overlap of the spectral features with the ground state and anion prevent definitive assignment. We again highlight however that this apparent higher conductivity could also be due to differences in the density of the film, orientation of fibres or fibre thickness (see above). Simultaneously measuring the UV-Vis spectrum and photocurrent after switching off the LED showed that the initial decay was similar (Fig. 4f), and that the conductivity was essentially zero at the point where approximately 20% of the intensity for the peak at 730 nm remained. This is unsurprising; for conductivity a contiguous percolation pathway is required, and hence bulk conductivity is expected to decay before the presence of any conductive species.
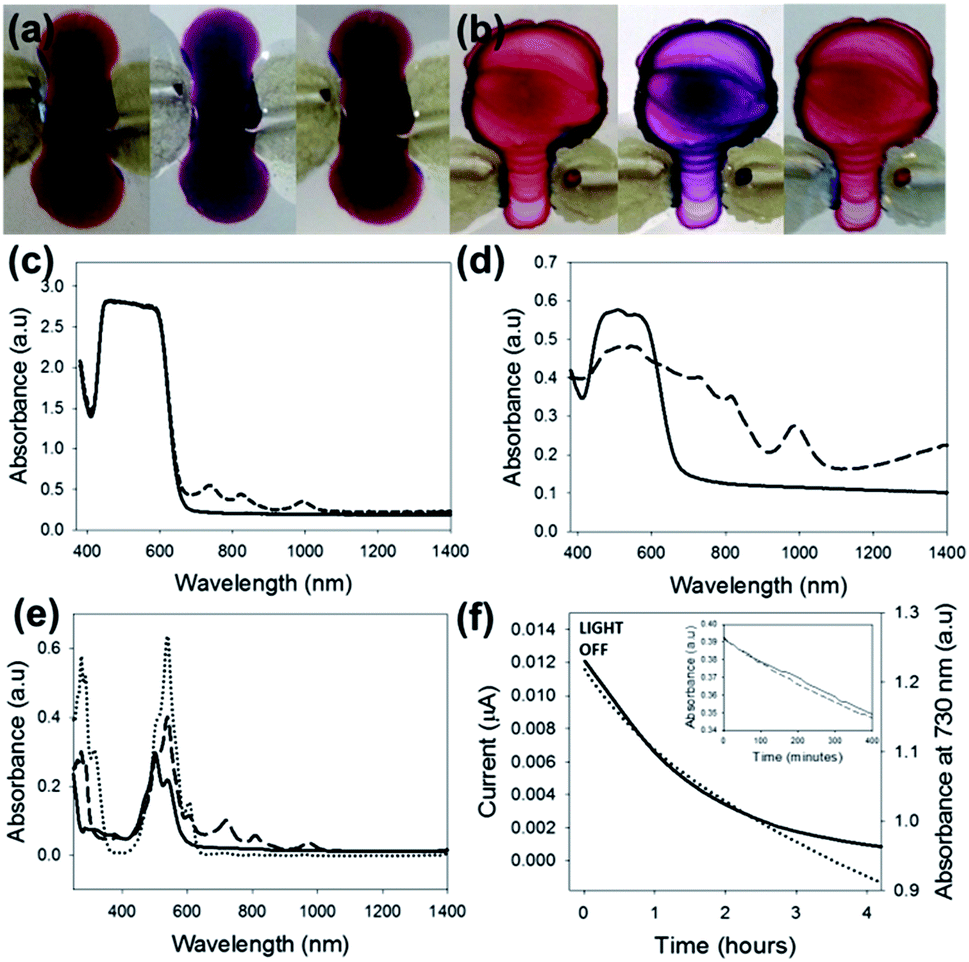 | ||
| Fig. 4 (a) On irradiation with 365 nm light, a xerogel of 1 (left) changes color (middle). After the light is turned off, the original color slowly returns (right), photograph after 18 hours. (b) A similar effect is observed for the dried solution. (c) UV-Vis-NIR spectra for a xerogel of 1 (—), and for the same film 1 minute after irradiation with 365 nm light (- - -). (d) UV-Vis-NIR spectra for a dried solution of 1 (—), and for the same film 1 minute after irradiation with 365 nm light (- - -). (e) UV-Vis-NIR showing absorbance of 1 in solution after addition of various amounts of sodium dithionite. Solid line represents before sodium dithionite was added, dashed line is after 20 mg of sodium dithionite was added (radical anion of 1) and the dotted line is after 40 mg of sodium dithionite was added (dianion). (f) Comparison of rate of decrease in conductivity and absorbance at 730 nm. Inset: change of absorbance at 730 nm with time after the 365 nm LED was turned off in air (- - -) and under argon (—). | ||
The insensitivity of the spectral features of the decay to O2 was further demonstrated by comparing the decay in air compared to that under argon. The rate of decay was very similar in both cases (Fig. 4f, inset). Importantly, no changes in the UV-Vis spectra were observed when the samples were irradiated at 450 nm (see for example Fig. S37†), and hence the lack of photocurrent when irradiated at higher wavelengths can be ascribed to the lack of formation of either radical anion or dianion. The correlation between conductivity and the presence of the radical anion is expected. For example, a thin film of a PBI has been shown to form the radical anion by a self-doping mechanism on dehydration in air.15 A concomitant increase in conductivity was observed. What are perhaps surprising is both the wavelength dependence of the conductivity found here and the O2 tolerance of the photoconductivity. The lowest energy required for light to be absorbed in a material is referred to as the optical gap. Absorption of light of this energy results in the formation of excitons; excited electron–hole pairs bound through electrostatic interaction. Photoconductivity, however, requires not neutral excitons but free charge carriers (free electrons, free holes, or both), where the label free signifies that the charge carriers are not bound together in an exciton and hence able to contribute to electronic conductivity. The minimum energy needed to generate such free charge carriers is the transport (or quasiparticle) gap. This transport gap is always larger or equal to the optical gap, with the difference between the two equal to the exciton binding energy Eebe; the amount of energy by which excitons are stabilized with respect to free electrons and holes. Eebe can be very small, e.g. in elemental and compound semiconductors, but for organic materials is typically in the order of tenths of an eV. Hence, we ascribe the difference between absorption and photoconductivity onset to the Eebe, the extra energy required to form free electrons and holes from excitons.
An enhancement in conductivity for a PBI has been found previously on generation of the radical anion using hydrazine vapor.40 Elsewhere, the conductivity of PBI fibers was increased in the presence of diethylamine.26 In both these cases, a strong electron donor was present. In our work, no such donor is present, explaining the greater Eebe here. The fate of the cation is not clear. Whilst there is significant data available on the radical anion and dianion for a range of PBIs, there is less on the radical cation. Data has been reported for bay-substituted PBIs,41–43 with radical cations absorbing in the near-IR (NIR) region, however direct comparison to our materials is difficult. We do observe a broad peak above 1200 nm in the UV-Vis spectrum for the dried solution of 1 after illumination with 365 nm light (Fig. 4d) that on the basis of the solution phase chemical reduction experiment is not assigned to the radical anion or dianion (Fig. 4e). This feature grows in under UV illumination with a maximum at ∼1700 nm, and decays at the same rate as the radical anion. It is possible that the NIR absorption is due to the radical cation, although this would represent a remarkably long-lived charge separated state. Alternatively, it may be assignable to a morphological change following irradiation, and further studies are ongoing.
The slow rate of decay of the photocurrent is surprising. The PBI radical anion in solution can be used as a sensitive probe for O2.28,44 Our materials behave similarly (Fig. S38†). However, when dried as a film, it is clear that the O2 sensitivity is significantly reduced. Indeed, all of the data above are for measurements carried out in air. Under an argon atmosphere, the rate of decrease was similar to that in air; this shows that O2 has little effect on the recovery of the dried sample (Fig. 4f). An issue with many n-type semiconductors is the lack of stability in air. For example, photoconductive PBI-based nanofibers have been shown to be highly sensitive to the presence of O2,37 where the photoconductivity was found to be three times higher under argon than under air. This was ascribed to high surface area available due to the morphology and the scavenging ability of O2. However, in other cases, the morphology can result in a kinetic barrier to the intrusion of water or O2, for example by using PBIs containing perfluorinated substituents.45,46 It has also been reported that aggregation stabilizes the radical anion. When incorporated in a film, the stability of the radical anion to oxygen was found to be higher, taking 20 minutes to be re-oxidized, as opposed to being immediately re-oxidized when in homogeneous solution.47 This was attributed to slow diffusion of O2 into the film. The slow rate of decay in our systems is significantly longer than for this system.47 It is unclear as to whether this is due to film thickness, morphology, or a result of the decreased diffusion rates of O2 through the films.
Conclusions
In conclusion, PBIs are of increasing interest for applications in organic electronics, including as alternative acceptor materials in OPV.7 Morphological control of such materials is likely to be critical for controlling their use. Here, we report self-assembling PBIs, which form long 1D structures that are photoconductive. The conductivity correlates with the presence of the radical anion, which is found to be remarkably long lived (on a timescale of hours), even in air and in the presence of water. The different molecules use here show different behaviour. For LMWG, small changes in molecular structure can have profound effects on the self-assembly and this is again exemplified here for 1–4. It is clear from the SEM images that the more ordered structures formed by 1 lead to higher photoconductivity; the reasons why 1 forms such structures as compared to 2–4 is currently under investigation. The materials presented here show markedly different conduction properties to those in the literature. Typically, the photoconductivity decays very rapidly (sub second timescales), and the decay is further accelerated by O2. The materials presented here maintain their photoconductivity for several hours after the light is turned off due to the high stability of the radical anion. It should also be noted that the gels and solutions presented here contain well defined 1D structures, which will be invaluable in a wide range of applications.Acknowledgements
We thank the EPSRC for a DTA (ERD). We thank Prof. K. Durose and Dr R. Treharne (SIRE) for providing access to the UV-Vis-NIR spectrometer. MAZ and AJC acknowledge the EPSRC for Fellowships (EP/I004424/1) and (EP/K006851/1) respectively.Notes and references
- A. Facchetti, Chem. Mater., 2010, 23, 733–758 CrossRef.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- G. Koshkakaryan, P. Jiang, V. Altoe, D. Cao, L. M. Klivansky, Y. Zhang, S. Chung, A. Katan, F. Martin, M. Salmeron, B. Ma, S. Aloni and Y. Liu, Chem. Commun., 2010, 46, 8579–8581 RSC.
- L. M. Klivansky, D. Hanifi, G. Koshkakaryan, D. R. Holycross, E. K. Gorski, Q. Wu, M. Chai and Y. Liu, Chem. Sci., 2012, 3, 2009–2014 RSC.
- J. Zhao, J. I. Wong, J. Gao, G. Li, G. Xing, H. Zhang, T. C. Sum, H. Y. Yang, Y. Zhao, S. L. Ake Kjelleberg, W. Huang, S. C. Joachim Loo and Q. Zhang, RSC Adv., 2014, 4, 17822–17831 RSC.
- J. Zhao, J. I. Wong, C. Wang, J. Gao, V. Z. Y. Ng, H. Y. Yang, S. C. J. Loo and Q. Zhang, Chem. – Asian J., 2013, 8, 665–669 CrossRef CAS PubMed.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- S. Yagai, M. Usui, T. Seki, H. Murayama, Y. Kikkawa, S. Uemura, T. Karatsu, A. Kitamura, A. Asano and S. Seki, J. Am. Chem. Soc., 2012, 134, 7983–7994 CrossRef CAS PubMed.
- D. Görl, X. Zhang and F. Würthner, Angew. Chem., Int. Ed., 2012, 51, 6328–6348 CrossRef PubMed.
- F. Wurthner, Chem. Commun., 2004, 1564–1579 RSC.
- Y. Che, H. Huang, M. Xu, C. Zhang, B. R. Bunes, X. Yang and L. Zang, J. Am. Chem. Soc., 2010, 133, 1087–1091 CrossRef PubMed.
- C. Huang, S. Barlow and S. R. Marder, J. Org. Chem., 2011, 76, 2386–2407 CrossRef CAS PubMed.
- C. Huang, M. M. Sartin, N. Siegel, M. Cozzuol, Y. Zhang, J. M. Hales, S. Barlow, J. W. Perry and S. R. Marder, J. Mater. Chem., 2011, 21, 16119–16128 RSC.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024–1026 RSC.
- T. H. Reilly, A. W. Hains, H.-Y. Chen and B. A. Gregg, Adv. Energy Mater., 2012, 2, 455–460 CrossRef CAS.
- J. M. Mativetsky, E. Orgiu, I. Lieberwirth, W. Pisula and P. Samorì, Adv. Mater., 2014, 26, 430–435 CrossRef CAS PubMed.
- J. K. Gallaher, E. J. Aitken, R. A. Keyzers and J. M. Hodgkiss, Chem. Commun., 2012, 48, 7961–7963 RSC.
- L. Tian, R. Szilluweit, R. Marty, L. Bertschi, M. Zerson, E.-C. Spitzner, R. Magerle and H. Frauenrath, Chem. Sci., 2012, 3, 1512–1521 RSC.
- P. K. Sukul, P. K. Singh, S. K. Maji and S. Malik, J. Mater. Chem. B, 2013, 1, 153–156 RSC.
- R. A. Cormier and B. A. Gregg, J. Phys. Chem. B, 1997, 101, 11004–11006 CrossRef CAS.
- Y. Sun, C. He, K. Sun, Y. Li, H. Dong, Z. Wang and Z. Li, Langmuir, 2011, 27, 11364–11371 CrossRef CAS PubMed.
- Y. Chen, Y. Feng, J. Gao and M. Bouvet, J. Colloid Interface Sci., 2012, 368, 387–394 CrossRef CAS PubMed.
- K. Balakrishnan, A. Datar, T. Naddo, J. Huang, R. Oitker, M. Yen, J. Zhao and L. Zang, J. Am. Chem. Soc., 2006, 128, 7390–7398 CrossRef CAS PubMed.
- H. Marciniak, X.-Q. Li, F. Würthner and S. Lochbrunner, J. Phys. Chem. A, 2010, 115, 648–654 CrossRef PubMed.
- K. Sugiyasu, S.-i. Kawano, N. Fujita and S. Shinkai, Chem. Mater., 2008, 20, 2863–2865 CrossRef CAS.
- A. Datar, K. Balakrishnan and L. Zang, Chem. Commun., 2013, 49, 6894–6896 RSC.
- S. Roy, D. Kumar Maiti, S. Panigrahi, D. Basak and A. Banerjee, RSC Adv., 2012, 2, 11053–11060 RSC.
- L. Zhong, F. Xing, W. Shi, L. Yan, L. Xie and S. Zhu, ACS Appl. Mater. Interfaces, 2013, 5, 3401–3407 CAS.
- W. Tuntiwechapikul, T. Taka, M. Béthencourt, L. Makonkawkeyoon and T. Randall Lee, Bioorg. Med. Chem. Lett., 2006, 16, 4120–4126 CrossRef CAS PubMed.
- Y. Xu, S. Leng, C. Xue, R. Sun, J. Pan, J. Ford and S. Jin, Angew. Chem., Int. Ed., 2007, 46, 3896–3899 CrossRef CAS PubMed.
- L. Chen, G. Pont, K. Morris, G. Lotze, A. Squires, L. C. Serpell and D. J. Adams, Chem. Commun., 2011, 47, 12071–12073 RSC.
- L. Chen, T. O. McDonald and D. J. Adams, RSC Adv., 2013, 3, 8714–8720 RSC.
- Y. Pocker and E. Green, J. Am. Chem. Soc., 1973, 95, 113–119 CrossRef CAS.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- S. Akimoto, A. Ohmori and I. Yamazaki, J. Phys. Chem. B, 1997, 101, 3753–3758 CrossRef CAS.
- C. Backes, C. D. Schmidt, K. Rosenlehner, F. Hauke, J. N. Coleman and A. Hirsch, Adv. Mater., 2010, 22, 788–802 CrossRef CAS PubMed.
- Y. Che, X. Yang, G. Liu, C. Yu, H. Ji, J. Zuo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2010, 132, 5743–5750 CrossRef CAS PubMed.
- R. O. Marcon and S. Brochsztain, J. Phys. Chem. A, 2009, 113, 1747–1752 CrossRef CAS PubMed.
- E. Shirman, A. Ustinov, N. Ben-Shitrit, H. Weissman, M. A. Iron, R. Cohen and B. Rybtchinski, J. Phys. Chem. B, 2008, 112, 8855–8858 CrossRef CAS PubMed.
- Y. Che, A. Datar, X. Yang, T. Naddo, J. Zhao and L. Zang, J. Am. Chem. Soc., 2007, 129, 6354–6355 CrossRef CAS PubMed.
- M. J. Ahrens, M. J. Tauber and M. R. Wasielewski, J. Org. Chem., 2006, 71, 2107–2114 CrossRef CAS PubMed.
- A. S. Lukas, Y. Zhao, S. E. Miller and M. R. Wasielewski, J. Phys. Chem. B, 2002, 106, 1299–1306 CrossRef CAS.
- M. Berberich, A.-M. Krause, M. Orlandi, F. Scandola and F. Würthner, Angew. Chem., Int. Ed., 2008, 47, 6616–6619 CrossRef CAS PubMed.
- I.-S. Shin, T. Hirsch, B. Ehrl, D.-H. Jang, O. S. Wolfbeis and J.-I. Hong, Anal. Chem., 2012, 84, 9163–9168 CAS.
- B. A. Jones, M. J. Ahrens, M.-H. Yoon, A. Facchetti, T. J. Marks and M. R. Wasielewski, Angew. Chem., Int. Ed., 2004, 43, 6363–6366 CrossRef CAS PubMed.
- J. H. Oh, S. Liu, Z. Bao, R. Schmidt and F. Würthner, Appl. Phys. Lett., 2007, 91, 212107 CrossRef PubMed.
- R. O. Marcon and S. Brochsztain, Langmuir, 2007, 23, 11972–11976 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The details of the synthesis, characterization data for all compounds, rheological data, addition photographs, UV-Vis and fluorescence data, SEMs, and conductivity data. See DOI: 10.1039/c4tc00744a |
| This journal is © The Royal Society of Chemistry 2014 |