
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceLight switchable optical materials from azobenzene crosslinked poly(N-isopropylacrylamide)-based microgels†
Qiang Matthew
Zhang
,
Xue
Li
,
Molla R.
Islam
,
Menglian
Wei
and
Michael J.
Serpe
*
Department of Chemistry, University of Alberta, Edmonton, AB T6G 2G2, Canada. E-mail: michael.serpe@ualberta.ca; Fax: +1 780 492 8231; Tel: +1 780 492 5778
First published on 23rd July 2014
Abstract
4,4′-Di(acrylamido)-azobenzene was used as a crosslinker in poly(N-isopropylacrylamide)-based microgels. The microgels were subsequently used to fabricate microgel-based optical materials (etalons), which exhibited optical properties that were switchable upon exposure to UV irradiation. We also show that the extent of the response depended on the UV exposure time. These materials could find applications for controlled/triggered drug delivery, as well as in various optical applications.
Stimuli-responsive polymers, or “smart materials”, have attracted great attention in the natural sciences within the last few decades.1–4 These macromolecules/materials undergo physical and/or chemical changes in response to small variations in their environment. This interesting behavior has revolutionized the way we think about drug delivery, and has led to many advances in various fields of material science, chemistry, engineering, and physics.2,5,6 Of the various stimuli that can be used to excite these systems, light has attracted much interest due to its ability to locally excite materials from remote locations.7,8 Additionally, light induced excitation of materials can easily be started/stopped by simply switching the excitation source on/off, while the magnitude of the response can be tuned by modulating the excitation source intensity, and/or wavelength. As a result, these materials have been used for drug delivery, as nanoreactors, artificial muscles, and motors.7–12
Most previously reported photoresponsive materials have relied on photochromic molecules.1 These molecules undergo a reversible isomerization upon irradiation with specific wavelengths of light. This process is capable of not only altering the molecular structure of the materials, but is often accompanied by a local polarity change as well as a color change in the photochromic units. Several photochromic molecules have been used in the design of light responsive polymers, including azobenzene, spiropyran, dithienylethene, diazonaphthoquinone, and stilbene;13,14 the most widely used being azobenzene. Azobenzene-containing molecules are well known to undergo trans to cis photoisomerizations and vice versa, which depend on the irradiation wavelength.15 It is also notable that the isomerization results in a strong polarity change in the photochromic unit. The polarity change is a direct consequence of the change in the dipole moment from 0 Debye for the trans-isomer to 3 Debye for the cis-isomer.16 Therefore, the photoisomerization from trans to cis increases the hydrophilicity of materials.17 Azobenzene-containing polymers have been actively investigated for applications in optical data storage, nonlinear optical devices, sensors and actuators, as well as materials suitable for photo-fabrication/processing.18 Additionally, it has been shown that the photo-isomerization can lead to material deformations.19–21 For example, azobenzene photoisomerization in elastomers can yield up to a 20% change in their critical dimension.22 Microgels, which are colloidally stable hydrogel particles in the size range of 100 nm to ∼2 μm in diameter, have also been made responsive to a number of stimuli by addition of functional monomers23 or nanoparticles to their structure.24,25 In this submission, azobenzene-containing microgels were synthesized, and used to fabricate optical materials (etalons); we show that the device's optical properties depend on the wavelength of light irradiation.
The structure of an etalon can be seen in Scheme 1, and was fabricated following our previously published painting protocol.26,27 In short, a concentrated microgel solution was painted on an Au-coated glass substrate, followed by rinsing away the excess microgels that were not attached directly to the Au. Finally, the microgel layer was dried, and a subsequent layer of Au deposited on the microgel layer. These devices show visible color and unique multipeak reflectance spectra. The position and order of the peaks can be predicted from eqn (1):26,27
λ = 2nd![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ/m cosθ/m | (1) |
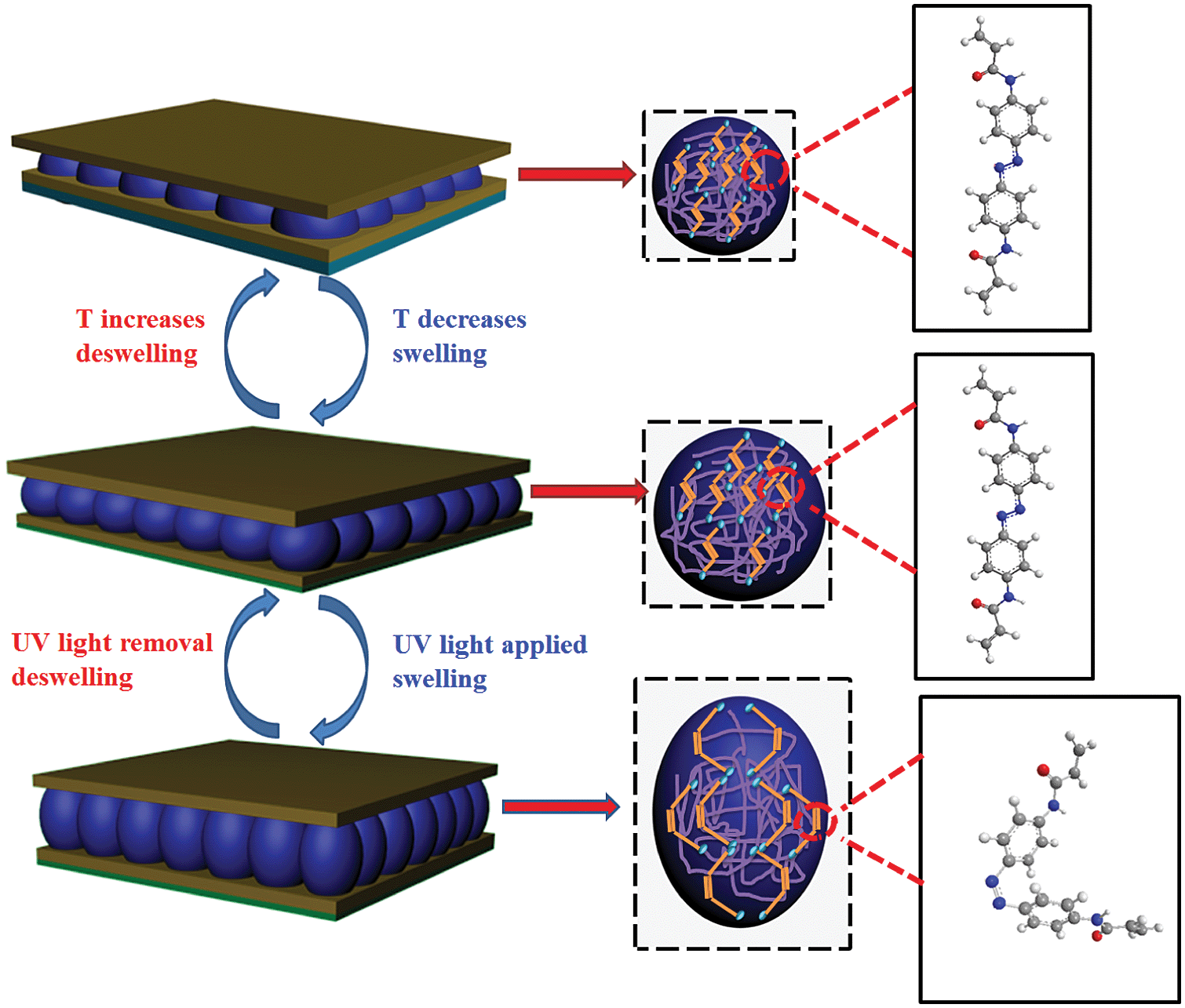 | ||
| Scheme 1 Schematic structure and proposed response mechanism for etalons fabricated by sandwiching a monolayer of azobenzene crosslinked microgels between two reflective Au mirrors on a glass substrate. (Far right) cis–trans isomerization of 4,4′-di(methacrylamido)-azobenzene. | ||
In the present work, azobenzene groups were incorporated into microgels as a crosslinker via copolymerization of the azobenzene crosslinker 4,4′-di(acrylamido)-azobenzene (DAAB) with N-isopropylacrylamide (NIPAm) and N,N′-methylenebis(acrylamide) (BIS) (see ESI†). The azobenzene crosslinker DAAB was synthesized by the reaction of acryloyl chloride and (E)-4-((4-aminophenyl)diazenyl)benzenamine, as shown in the ESI,† and was characterized by 1H NMR and 13C NMR (Fig. S1 and S2†). This particular azobenzene was chosen due to the rigidity of the amide bond, which can effectively transfer the photoisomerization into conformational changes of the microgel's polymer network. Furthermore, an azobenzene crosslinker was used to translate the photoisomerization to the polymer network to yield a microgel response – in the absence of the crosslinker there is no response. Additionally, without a covalent bond crosslinking the azobenzene groups and the polymer network no photoisomerization induced microgel response is expected. As noted above, the microgels were composed of two crosslinkers; DAAB, and BIS. The total percent of the crosslinker (mol/mol) was kept constant at 5%. The microgels are denoted as MG-X, where X represents the percent of azobenzene crosslinker, either 0.5% or 1.0%. The content limit of azobenzene crosslinker in microgel is 1%. When it is above 1%, the reaction generates bulk hydrogels.
Transmission electron microscope (TEM) images of the as synthesized microgels are shown in Fig. 1. As can be seen, in both cases, the microgels are spherical, with an average dry diameter of 380 ± 10 nm (via analysis of the microscope images). The microgel diameter was also characterized by dynamic light scattering (DLS) (Fig. S3 and S4†). The DLS revealed a hydrodynamic radius (Rh) of 1310 nm for MG-1.0%, and 1005 nm for MG-0.5% in their swollen state (at 20 °C). Furthermore, DLS revealed a Rh of 390 nm for MG-1.0%, and 380 nm for MG-0.5% in their collapsed state (at 46 °C). The microgels lower critical solution temperature (LCST) was also determined to be ∼30.5 °C for MG-0.5% and ∼29.2 °C for MG-1.0%. For comparison, the microgel without azobenzene crosslinker (MG-0%) was prepared under the same polymerization conditions, and showed a hydrodynamic radius (Rh) of 660 nm and 442 nm in the swollen and collapsed state respectively, and LCST of around 32.5 °C. As can be seen, the hydrodynamic diameter at low temperature (fully swollen) increased with increasing azobenzene crosslinker content. While this is the case, both MG-1% and MG-0.5% collapsed to a very similar diameter when the temperature was increased to above the LCST. This observation was attributed to the DAAB (longer compared to BIS)28 allowing the microgels to swell more. Conversely, the longer DAAB crosslinker has little effect on the microgel diameter when they are fully collapsed. Finally, the decrease in LCST with increased DAAB azobenzene composition can be attributed to enhanced hydrophobicity at high DAAB composition, reinforcing the relatively hydrophobic deswollen state.
 | ||
| Fig. 1 TEM micrographs of (a) microgel-1%, and (b) MG-0.5%. | ||
Microgel-based etalons were subsequently constructed from the synthesized microgels, and their responsivity to irradiation investigated. A SEM image of the resultant etalon is shown in the ESI,† which clearly shows the microgel layer under the Au overlayer. Initial experiments were focused on characterizing etalon thermoresponsivity, to confirm the basic responsivity/function of the devices. To accomplish this, MG-1% microgels were used to make etalons, which were immersed in water; the resulting reflectance spectrum is shown in Fig. 2. As can be seen, a characteristic multipeak reflectance spectrum was observed, with peaks at 490 nm and 638 nm at 25 °C. From the relative positions of the reflectance peaks, and using eqn (1), the order (m) of each peak can be calculated. The peak at 638 nm is m = 3 (noted as λ3), and λ4 is 490 nm. When conducting these experiments, it is important to compare the same order peaks before and after introduction of a stimulus. When the temperature was increased to 30 °C, λ3 exhibited a blue shift of 33 nm, while it exhibits an additional ∼70 nm shift (100 nm total shift) when the temperature was increased to 35 °C. The λ4 peak blue shifts into ultraviolet region at 35 °C (<400 nm), which can no longer be detected by our spectrometer. These peak shifts are a direct result of the thermoresponsivity of the NIPAM containing microgels, which collapse at elevated temperature, decreasing the distance between the etalons two gold mirrors. The blue shifts can be predicted from eqn (1).29,30
 | ||
| Fig. 2 Reflectance spectra for a MG-1% etalon in water at the indicated temperatures. | ||
Since the basic thermal response of the pNIPAm microgel-based devices was confirmed, the etalons response to UV irradiation was subsequently characterized. As can be seen in Scheme 1, when the azobenzene is exposed to UV (UVP B-100 AP, 365 nm, 100 W, 20 cm from the etalon), it undergoes a trans–cis isomerization, which causes the microgels to swell/deform as detailed in Scheme 1; this swelling response has been previously observed by Wang et al.31 To further characterize the UV response, we performed DLS on microgels in solution before and after UV exposure, and found the average diameter to increase from 832 nm ± 15 nm to 903 nm ± 23 nm after UV exposure (these values are the average of three measurements at 30 °C, data in ESI†). We also characterized the surface morphology of the etalon before and after exposure to UV. This was done via atomic force microscopy (AFM, tapping mode, Asylum Research, Santa Barbara, CA) in liquid at 25 °C, and the results are shown in Fig. 3. As can be seen, the microgels are difficult to observe before UV exposure, but become more obvious after UV exposure due to their swelling. Therefore an increase in the etalons mirror–mirror distance was predicted, along with a spectral red shift. The optical response of the etalons, constructed from both MG-0.5% and MG-1%, to UV exposure is shown in Fig. 4. For these experiments, the etalons were secured in a Petri dish, which was subsequently filled with H2O at the indicated temperatures. As can be seen, in all cases at the λ3 peak clearly red-shifts after being exposed to UV for the indicated elapsed times. Specifically, λ3 for MG-1% at 30 °C shifted 32 nm after 30 min exposure, and an additional 35 nm after 90 min further exposure. We point out that while the photoisomerization of the azobenzene moieties should be complete within several minutes, the microgel/etalon response is significantly slower.
 | ||
| Fig. 3 AFM images of microgel-1% etalon (a and b) before and (c and d) after UV stimulation. (a and c) 2-D and (b and d) 3-D images. | ||
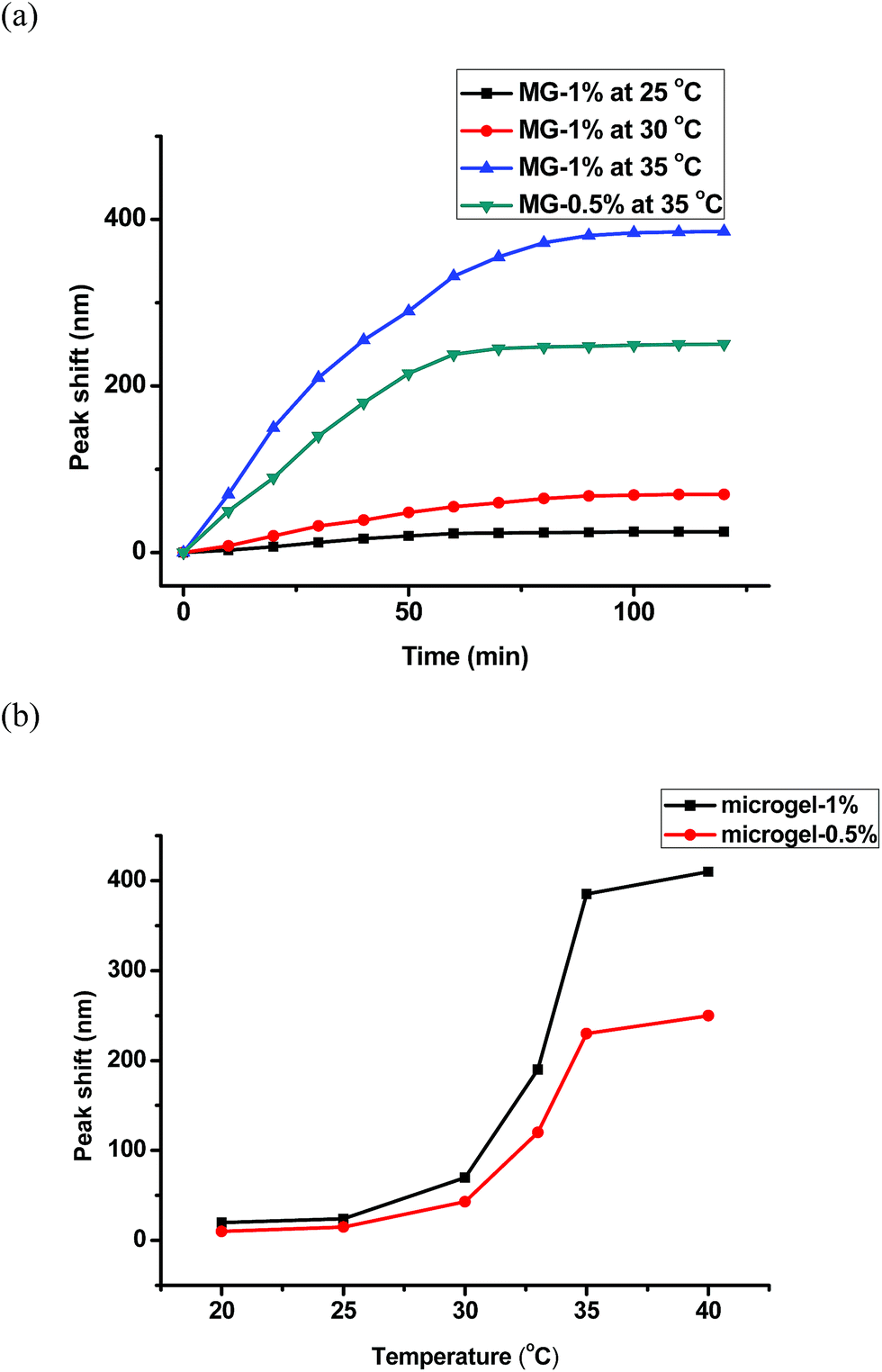 | ||
| Fig. 4 (a) λ3 peak shift for the indicated microgel-based etalons and temperatures as a function of UV exposure time; (b) total peak shifts (λ3-final − λ3-initial) for the indicated microgels exposed to UV irradiation at various solution temperatures. | ||
The extent of the spectral shift has been shown to be related to both the amount of azobenzene crosslinker in the microgel, and the temperature of the solution. For example, etalons composed of MG-0% showed no response to UV irradiation (Fig. S6†), while etalons composed of MG-0.5% and MG-1% showed total spectral shifts of 250 nm and 380 nm, respectively, at 35 °C. Furthermore, as can be seen from both Fig. 4a and b, as the temperature of the solution the etalon is immersed in is increased, the extent of spectral shift increases. For example, as the solution temperature was increased from 20 °C to 40 °C, the peak shift extent changed from 25 nm to 405 nm for etalons composed of MG-1%, while the shifts increased from 11 nm to 265 nm for etalons composed of MG-0.5% at the same temperatures. We attribute this phenomenon to the fact that the microgels deswell at increasing temperature. When this occurs, the polymer chains have more free volume available to expand. Hence, when the azobenzene groups are photoisomerized, the polymer is more free to expand, causing the microgels, and hence the etalons, to be more responsive to light. The reflectance spectra of the etalon at low temperature, high temperature, and high temperature after UV exposure can be seen in ESI.† These spectra show a significant blue shift in the reflectance peak as the temperature is increased followed by a red shift after UV exposure at high temperature. We point out that the total response we observe here (maximum: 408 nm) is much larger than others have observed previously.32–35 Importantly, the response to UV is reversible, i.e., once the UV is removed, the spectrum of etalon returns to its initial state after the etalon has been stored.
Finally, we wanted to show that the optical properties of the etalons could be systematically tuned as a function of UV exposure time. For these studies, the etalons were immersed in H2O at 40 °C and exposed to 2 min intervals of UV irradiation. The results are shown in Fig. 5, which show that after exposure of the etalons to UV irradiation for 2 min (symbols on plot), the reflectance peaks red shift, and stabilize, until it is again exposed to another 2 min UV irradiation. This can be continued for a number of cycles for both etalons composed of MG-0.5%, and MG-1.0%. The ability to cause the etalons to systematically change solvation state in response to UV light can be used to expand on our controlled drug delivery platform,36 which is currently underway.
 | ||
| Fig. 5 Peak shifts for the indicated microgel-based etalons as a function of time after sequential 2 min UV exposures (indicated by symbols). | ||
In summary, an azobenzene-containing crosslinker was synthesized, and used to synthesize photoresponsive pNIPAm-based microgels and microgel-based etalons. Etalons response to UV irradiation, as a function of temperature, was characterized, and was shown to exhibit a dramatic dependence on temperature. Finally, we show that the extent of the response could be controlled in a systematic fashion by controlling the extent of UV exposure.
Acknowledgements
MJS acknowledges funding from the University of Alberta (the Department of Chemistry and the Faculty of Science), the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI), the Alberta Advanced Education & Technology Small Equipment Grants Program (AET/SEGP) and Grand Challenges Canada. MJS acknowledges Mark McDermott for the use of the thermal evaporator.Notes and references
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- M. A. C. Stuart, W. T. S. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS PubMed.
- P. J. Glazer, J. Leuven, H. An, S. G. Lemay and E. Mendes, Adv. Funct. Mater., 2013, 23, 2964–2970 CrossRef CAS.
- C. Weder, Nature, 2009, 459, 45–46 CrossRef CAS PubMed.
- H. Therien-Aubin, Z. L. Wu, Z. H. Nie and E. Kumacheva, J. Am. Chem. Soc., 2013, 135, 4834–4839 CrossRef CAS PubMed.
- J. K. Liu, S. H. Wen, Y. Hou, F. Zuo, G. J. O. Beran and P. Y. Feng, Angew. Chem., Int. Ed., 2013, 52, 3241–3245 CrossRef CAS PubMed.
- A. Priimagi, G. Cavallo, A. Forni, M. Gorynsztejn-Leben, M. Kaivola, P. Metrangolo, R. Milani, A. Shishido, T. Pilati, G. Resnati and G. Terraneo, Adv. Funct. Mater., 2012, 22, 2572–2579 CrossRef CAS.
- L. C. Yin, H. Y. Tang, K. H. Kim, N. Zheng, Z. Y. Song, N. P. Gabrielson, H. Lu and J. J. Cheng, Angew. Chem., Int. Ed., 2013, 52, 9182–9186 CrossRef CAS PubMed.
- P. Baumann, V. Balasubramanian, O. Onaca-Fischer, A. Sienkiewicz and C. G. Palivan, Nanoscale, 2013, 5, 217–224 RSC.
- S. J. Leung and M. Romanowski, Adv. Mater., 2012, 24, 6380–6383 CrossRef CAS PubMed.
- T. Ikeda and T. Ube, Mater. Today, 2011, 14, 480–487 CrossRef CAS.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- Y. Zhao, J. Mater. Chem., 2009, 19, 4887–4895 RSC.
- K. G. Yager and C. J. Barrett, J. Photochem. Photobiol., A, 2006, 182, 250–261 CrossRef CAS PubMed.
- G. S. Kumar and D. Neckers, Chem. Rev., 1989, 89, 1915–1925 CrossRef CAS.
- W. Jiang, G. Wang, Y. He, X. Wang, Y. An, Y. Song and L. Jiang, Chem. Commun., 2005, 3550–3552 RSC.
- D. R. Wang and X. G. Wang, Prog. Polym. Sci., 2013, 38, 271–301 CrossRef CAS PubMed.
- M. Yamada, M. Kondo, J. I. Mamiya, Y. L. Yu, M. Kinoshita, C. J. Barrett and T. Ikeda, Angew. Chem., Int. Ed., 2008, 47, 4986–4988 CrossRef CAS PubMed.
- Y. L. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS PubMed.
- N. Ishii, J. Mamiya, T. Ikeda and F. M. Winnik, Chem. Commun., 2011, 47, 1267–1269 RSC.
- H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS.
- H. Z. An, M. E. Helgeson and P. S. Doyle, Adv. Mater., 2012, 24, 3838–3844 CrossRef CAS PubMed.
- T. Hellweg, Angew. Chem., Int. Ed., 2009, 48, 6777–6778 CrossRef CAS PubMed.
- T. Hellweg, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1073–1083 CrossRef CAS.
- C. D. Sorrell and M. J. Serpe, Adv. Mater., 2011, 23, 4088–4092 CrossRef CAS PubMed.
- C. D. Sorrell, M. C. Carter and M. J. Serpe, Adv. Funct. Mater., 2011, 21, 425–433 CrossRef CAS.
- J. X. Jiang, F. Su, A. Trewin, C. D. Wood, N. L. Campbell, H. Niu, C. Dickinson, A. Y. Ganin, M. J. Rosseinsky and Y. Z. Khimyak, Angew. Chem., Int. Ed., 2007, 46, 8574–8578 CrossRef CAS PubMed.
- S. Seiffert, Angew. Chem., Int. Ed., 2013, 52, 11462–11468 CrossRef CAS PubMed.
- S. Nayak and L. A. Lyon, Chem. Mater., 2004, 16, 2623–2627 CrossRef CAS.
- Y. B. Li, Y. N. He, X. L. Tong and X. G. Wang, J. Am. Chem. Soc., 2005, 127, 2402–2403 CrossRef CAS PubMed.
- L. Hu and M. J. Serpe, Chem. Commun., 2013, 49, 2649–2651 RSC.
- L. Hu and M. J. Serpe, ACS Appl. Mater. Interfaces, 2013, 5, 11977–11983 CAS.
- L. Hu and M. J. Serpe, J. Mater. Chem., 2012, 22, 8199–8202 RSC.
- M. R. Islam and M. J. Serpe, Macromolecules, 2013, 46, 1599–1606 CrossRef CAS.
- Y. Gao, G. P. Zago, Z. Jia and M. J. Serpe, ACS Appl. Mater. Interfaces, 2013, 5, 9803–9809 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tc00653d |
| This journal is © The Royal Society of Chemistry 2014 |