 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Topotactic fluorination of strontium iron oxide thin films using polyvinylidene fluoride
T.
Katayama
a,
A.
Chikamatsu
*ab,
Y.
Hirose
abc,
R.
Takagi
a,
H.
Kamisaka
ab,
T.
Fukumura
ab and
T.
Hasegawa
abc
aDepartment of Chemistry, The University of Tokyo, Bunkyo-ku, Tokyo 113-0033, Japan. E-mail: chikamatsu@chem.s.u-tokyo.ac.jp; Fax: +81-3-5841-4603; Tel: +81-3-5841-4603
bCREST, Japan Science and Technology Agency (JST), Bunkyo-ku, Tokyo 113-0033, Japan
cKanagawa Academy of Science and Technology (KAST), Kawasaki, Kanagawa 213-0012, Japan
First published on 5th June 2014
Abstract
We report herein the topotactic fluorination of SrFeO3−δ thin films (δ ∼ 0, 0.5, 1) with polyvinylidene fluoride (PVDF). SrFeO3−xFx epitaxial thin films were obtained by fluorination at 150–270 °C, which is substantially lower than the reaction temperature for polycrystalline bulk samples prepared with PVDF. The fluorine content (x) of the film was widely varied by controlling the PVDF-treatment temperature and/or the amount of oxygen vacancies in the precursor film. The higher reactivity of the SrFeO2 and SrFeO2.5 thin films can be reasonably explained by a fluorine-diffusion mechanism via oxygen vacancies.
1. Introduction
Since the discovery of superconductivity (transition temperature of Tc = 46 K) in non-ordinary oxyfluorides Sr2CuO2F2+x,1 the replacement of O2− by F− in transition-metal oxides has attracted a great deal of attention as a chemical technique that can considerably modify the electronic properties of their mother compounds. Among these compounds, fluorine-substituted iron oxides have been a subject of intense study owing to their unique magnetic properties. For example, the partially F-substituted hexagonal perovskite, 15R-BaFeF0.2O2.42, shows drastically enhanced antiferromagnetic ordering with a Néel temperature (TN) of ≈ 700 K, which is close to the highest values ever reported for iron oxides.2 Such a high TN can be explained by local changes of the Fe–X–Fe (X = O, F) bond angle, bond length, as well as chemical reduction of iron.Iron oxyfluorides are often metastable and decompose at higher temperatures, making the development of low-temperature synthetic routes desirable. One promising method is a topotactic reaction employing a fluorination agent, polyvinylidene fluoride (PVDF), which is stable in air, and has a melting point of ∼170 °C.3 Fluorination using PVDF is advantageous for obtaining phase-pure oxyfluorides without metal fluoride impurities, which are frequently formed by other fluorinating agents such as F2 gas and NH4F.3 In addition, PVDF is a non-oxidizing reagent,4 in contrast to highly oxidizing F2 gas; therefore, metal ions are not oxidized during the fluorination reaction, but rather, reduced. Indeed, PVDF-promoted fluorination has been widely used to synthesize various species of iron oxyfluorides accompanied by a reduction in the Fe-oxidation state.5–13 For example, a perovskite oxide SrFeO3−δ (with Fe3+/4+) can be transformed to SrFeO2F (with Fe3+) by annealing with PVDF.5
It is expected that the reactivity of thin-film samples with PVDF should be much higher than that of bulk samples because thin films have larger surface areas and smaller volumes than bulk samples. More recently, the fluorination technique using PVDF was applied to SrFeO3−δ thin films, where a PVDF solution was spin-coated on the film and the film/polymer bilayer was annealed at 600 °C, though the SrFeO3−αFγ films thus obtained had relatively low fluorine contents (γ < 1) despite higher synthesis temperature than that for bulk SrFeO2F.14 Another important factor governing the reactivity of thin-film samples with PVDF is the amount of oxygen vacancies in the precursor oxides, because the incorporation of fluorine ions into the oxygen-vacancy sites would be faster than the actual replacement of oxygen by fluorine. In fact, bulk SrFeO2F can be obtained at 150 °C by fluorinating infinite-layer SrFeO2 with XeF2.15 Herein, we performed topotactic fluorine doping of SrFeOx (2 ≤ x < 3) thin films using PVDF. As a result, we succeeded in fabricating SrFeO3−xFx epitaxial thin films, in which the fluorine content (x) was controllable over a wide range (0.8 ≤ x ≤ ∼1.5) by means of the heat-treatment temperature. The fluorination reaction was conducted at 150–270 °C, which was much lower than the 400 °C reaction temperature reported for the bulk. We also found that the value of x is dependent on the amount of oxygen vacancies present in the precursor SrFeOx film.
2. Experimental section
Synthesis
Three types of precursor SrFeOx films with different oxygen contents (x ≈ 2, 2.5, and 3) were prepared. Precursor SrFeO2.5 films were grown on SrTiO3 (001) (STO, Shinkosha Co.) substrates by a pulsed-laser deposition (PLD) technique, with a SrFeO3−δ ceramic pellet (20 mm in diameter and 5 mm in thickness, TOSHIMA Manufacturing Co.) used as a PLD target. The fourth harmonic of a Nd-YAG laser (wavelength λ = 266 nm) with an energy of 0.3 J cm−2 per shot and a repetition rate of 10/3 Hz was employed for ablation. The substrate temperature and oxygen partial pressure were kept at 700 °C and 7 × 10−5 mbar, respectively, during each deposition run, and coherent growth of the precursor SrFeO2.5 films on STO (001) substrates was confirmed. Oxidized SrFeOx (x ≈ 3) films with perovskite structure were fabricated by annealing SrFeO2.5 films at 700 °C in air for 2 h. Additional reduced SrFeOx (x ≈ 2) films with infinite-layer structures were obtained by heating SrFeO2.5 films with CaH2 (Wako Pure Chemical Industries, Ltd.) at 280 °C for 24 h in evacuated Pyrex tubes as described in ref. 16.The obtained precursor SrFeOx (x ≈ 2, 2.5, and 3) films were further subjected to fluorination with 0.1 g of PVDF (Fluorochem Ltd.) at temperatures (Tf) ranging from 100 to 450 °C for 24 h under an Ar gas flow of 70 cm3 min−1 (with the films covered by Al foil so as not to contact with PVDF directly). Typical film thickness, as measured using a stylus surface profiler, was ∼80 nm.
Characterization
Crystal structures of the SrFeO3−xFx films were obtained with an X-ray diffractometer employing Cu-Kα radiation and a transmission electron microscope (TEM). The chemical composition of the films was analysed by energy dispersive X-ray spectrometry (EDS) equipped with a scanning electron microscope in which the electron accelerating voltage was set at 2.5 keV to reduce the background signal from the substrate. The amount of fluorine was evaluated by nuclear reaction analysis (NRA) using the 19F(p,αγ)16O resonant nuclear reaction at 902 keV. In NRA measurements, a CaF2 single crystal was used as a reference for fluorine. The EDS and NRA results included experimental errors of ∼20%. The depth profiles of the chemical compositions were evaluated by X-ray photoemission spectroscopy (XPS) with Ar+-ion sputtering. The core levels of iron were also observed by XPS. The surface morphology was characterized by atomic force microscopy (AFM).3. Results and discussion
Crystal structure analysis
Fig. 1(a) shows the 2θ–θ X-ray diffraction (XRD) patterns of films obtained by fluorination of the SrFeO2.5 precursor film at Tf = 100–450 °C for 24 h. The XRD pattern of the SrFeO2.5 precursor film has also been included in the figure for comparison. The film treated with PVDF at 100 °C showed a diffraction peak at 2θ ≈ 45.8°, corresponding to the (002) reflection of the SrFeO2.5 structure, which means that PVDF did not react with the SrFeO2.5 film below 100 °C. The films fluorinated at 150–270 °C exhibited only the (002) diffraction peaks of the perovskite structure, indicating that SrFeO3−xFx can be obtained at 150–270 °C. Additionally, the position of the (002) peak was shifted to the lower-angle side (from 45.5° to 45.1°) on increasing Tf from 150 to 270 °C. At 300 °C, the diffraction peak of perovskite SrFeO3−xFx disappeared, and a peak assignable to SrF2 (002) appeared. Above 350 °C, the peaks corresponding to SrF2 (002) and Fe3O4 (002) evolved, reflecting the complete decomposition of perovskite SrFeO3−xFx into SrF2 and Fe3O4. | ||
| Fig. 1 (a) 2θ–θ X-ray diffraction patterns of the SrFeO2.5 precursor film and films fluorinated at 100–450 °C with PVDF for 24 h. (b) Logarithmic contour mapping in reciprocal space for asymmetric (103) peaks of the SrFeO3−xFx film on the SrTiO3 substrate fluorinated at 250 °C. (c) Lengths of a- and c-axes of SrFeO3−xFx films as a function of fluorination temperature. | ||
Fig. 1(b) shows an XRD reciprocal space map for asymmetric (103) diffraction of the SrFeO3−xFx film fluorinated at 250 °C. Notably, the qx value of the SrFeO3−xFx (103) peak coincides with that of STO (103). This implies that the a-axis of the SrFeO3−xFx film was completely locked to the STO lattice, even after treatment with PVDF. In other words, the perovskite-like cation network was maintained during the topotactic fluorination reaction promoted by PVDF.
Fig. 1(c) shows the plots of lengths of the a- and c-axes for the SrFeO3−xFx films as a function of Tf. The c-axis length increased from 3.989 to 4.022 Å upon increasing Tf from 150 to 270 °C in a nonlinear manner, whereas the a-axis length was essentially independent of Tf. In the case of bulk SrFeO2F, it was reported that the cell volume was greatly increased upon fluorination of precursor SrFeO3−δ because of the simultaneous occurrence of fluorine insertion and substitution.5,17 Therefore, in this case, the increase in the c-axis length suggests that similar fluorine insertion and substitution reactions take place.
Fig. 2(a) shows a wide-view cross-sectional TEM image of the SrFeO3−xFx film fluorinated at 270 °C. Neither segregated impurities nor amorphous phases were recognized from the TEM observations. Fig. 2(b) is a magnified view of the same image. The image clearly indicates a tetragonal perovskite structure with lattice constants of a ∼ 3.9 Å and c ∼ 4.0 Å, which is consistent with those obtained from XRD (a = 3.908 Å and c = 4.022 Å).
 | ||
| Fig. 2 Cross-sectional transition electron microscopy images of the SrFeO3−xFx film fluorinated at 270 °C with (a) a wide-range view and (b) a magnified view. | ||
Compositional analysis
To verify fluorine doping and to investigate the relationship between the doped fluorine content and Tf, EDS measurements were performed for O Kα, F Kα, and Fe Lα. Fig. 3(a) depicts the EDS spectra near the O Kα and F Kα peaks of the SrFeO2.5 precursor and SrFeO3−xFx films fluorinated at 150, 250, and 270 °C, where the spectral intensity was normalized by the area of the Fe Lα peak. As seen in the figure, the peak area of F Kα increased with increase in Tf, while that of O Kα showed a tendency to decrease. Fig. 3(b) shows the plots of the areas of the F Kα and O Kα peaks, SF and SO, respectively, against Tf. The SF/SO ratio of the film fluorinated at 250 °C is 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2, whereas that of the film fluorinated at 270 °C is enhanced to 2:1. This implies that the fluorine content of the SrFeO3−xFx films can be controlled by Tf.
:2, whereas that of the film fluorinated at 270 °C is enhanced to 2:1. This implies that the fluorine content of the SrFeO3−xFx films can be controlled by Tf.
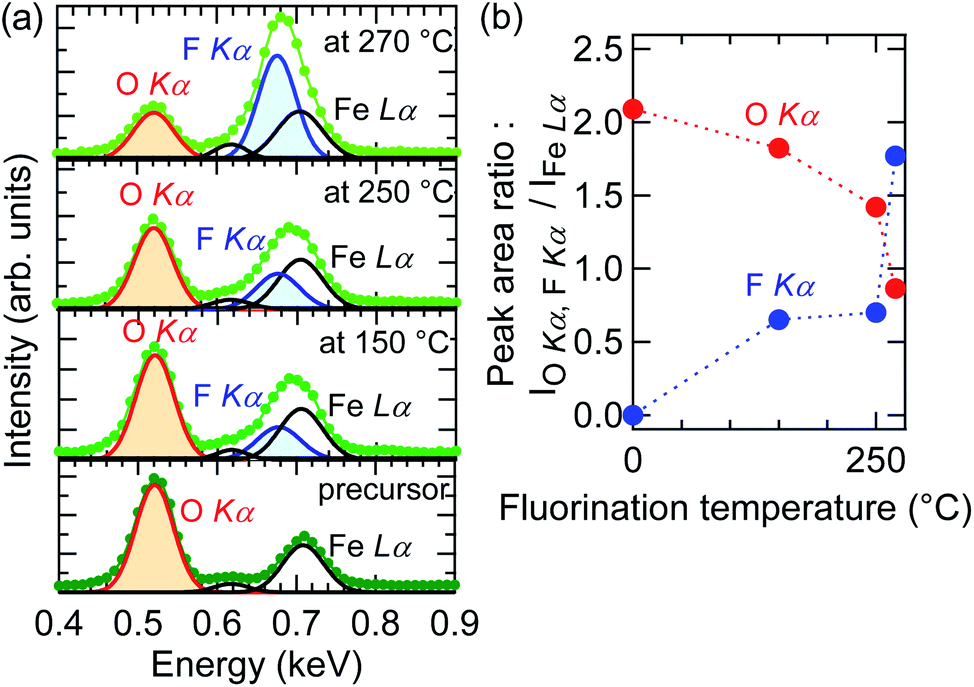 | ||
| Fig. 3 Energy dispersive X-ray spectra (EDS) near (a) O Kα, F Kα, and Fe Lα peaks of the SrFeO2.5 precursor film and SrFeO3−xFx films fluorinated at 150, 250, and 270 °C. (b) EDS peak area of F Kα and O Kα as a function of fluorination temperature. | ||
NRA measurements were conducted so as to quantitatively determine the fluorine content of the films. Fig. 4(a) shows the NRA spectrum of the SrFeO3−xFx film fluorinated at 250 °C. The γ-ray emitted by the nuclear reaction of 19F(p,αγ)16O was clearly observed in the SrFeO3−xFx film, and the x value was determined to be 0.92 ± 0.18. Fig. 4(b) shows the correlation between the c-axis length and x values in SrFeO3−xFx, where the x values were evaluated from EDS and NRA measurements independently, referred to as x(EDS) and x(NRA), respectively. For the evaluation of x(EDS), the relationship x = 3 × SF/(SF + a × SO) was used in an assumption of O:F = 3 − x:x, where the relative sensitivity factor (a) was estimated to be 0.989 based on a Monte Carlo simulation of electron trajectory in solids.18 As seen in Fig. 4(b), both x(EDS) and x(NRA) are in good agreement with each other, which indicates that the assumption mentioned above (O:F = (3 − x):x) is reasonable, although we cannot deny the possibility that a certain amount of oxygen vacancies remains in the film (in other words, the doped fluorine atoms were substituted for the oxygen sites of the perovskite lattice). Notably, the c-axis length of the SrFeO3−xFx film increased as the value of x increased, despite the fact that F− has a smaller ionic radius than O2−. This can be rationalized by taking the chemical reduction of the Fe ions into consideration. The shrinkage of cell volume associated with F− substitution is overwhelmed by cell expansion upon the reduction of Fe ions.5,17 The x value of the SrFeO3−xFx film fluorinated at 270 °C was ∼2, which is twice that of bulk SrFeO2F, though we cannot deny the possibility that the fluorine contents were overestimated because carbon impurities on the surface may adsorb fluorine ions during the PVDF treatment.
 | ||
| Fig. 4 (a) Nuclear reaction analysis (NRA) spectra of the SrFeO3−xFx film fluorinated at 250 °C. (b) c-Axis length dependence of x in the SrFeO3−xFx film, estimated from energy dispersive X-ray spectra (EDS) and NRA measurements. Different samples were used for NRA and EDS measurements. | ||
Because fluorination proceeds at the film surface, the population of fluorine ions is potentially higher closer to the surface. Fig. 5 shows the fluorine depth profile of the SrFeO3−xFx (x ≈ 1) film obtained at 250 °C, measured by XPS with Ar+-ion sputtering, where the peak area of F 1s relative to that of O 1s, AF/AO, at the surface (0 nm) was set to 1. Near the surface (0–15 nm) the AF/AO decreased with increasing depth, reaching ∼0.8 at 15 nm, suggesting the presence of impurities containing fluorine on the surface. The AF/AO value was virtually constant at 15–80 nm, and slightly increased near the interface of the film and the substrate. These results suggest that fluorine ions diffused not only in the vicinity of the surface, but also over the entire film. On the other hand, in the STO substrate region (>90 nm), fluorine was not detected, indicating that the diffusion of fluorine into the STO substrate is negligible.
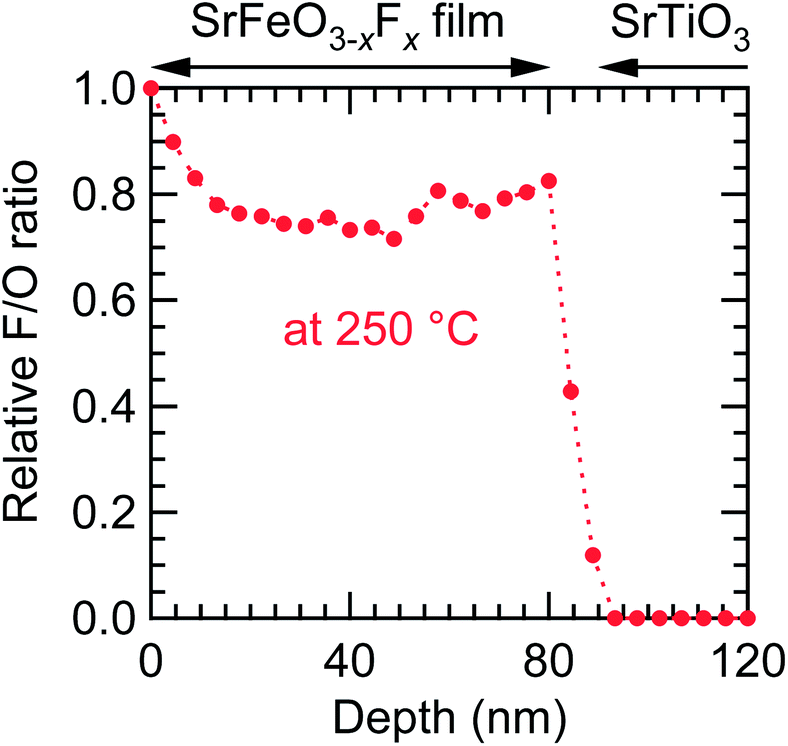 | ||
| Fig. 5 Fluorine depth profile of the SrFeO3−xFx (x ≈ 1) film fluorinated at 250 °C, obtained from the SrFeO2.5 precursor film, measured by X-ray photoemission spectroscopy with Ar+-ion sputtering. | ||
Valence of iron and surface morphology
Fig. 6 depicts the Fe 2p core-level XPS spectra of the SrFeO3−xFx films fluorinated at 150, 250, and 270 °C. Each spectrum showed Fe 2p3/2 and 2p1/2 peaks, and a satellite peak located between the Fe 2p1/2–Fe 2p3/2 doublet. Notably, the locations of the satellite peaks, which are known to be very sensitive to the oxidation state of Fe, differ from one sample to another. The satellite peaks in the SrFeO3−xFx films fluorinated at 150 and 250 °C were located at an Eb of ∼719 and ∼718 eV, respectively, which are equivalent to the peak in LaFeO3 with Fe3+ (Eb = 718.7 eV).19 That is, the valences of the Fe ions in the films are almost trivalent. Meanwhile, the satellite peak of the film fluorinated at 270 °C was located at an Eb of 712–719 eV, between the doublet peaks, suggesting that the Fe ion has a mixed valence state of Fe2+/Fe3+.20 The Fe2+/Fe3+ ratio was further evaluated by comparing the XPS data of the SrFeO3−xFx film fluorinated at 270 °C and the SrFeO2.5 film reported in ref. 21. The area-intensity of the Fe3+ satellite peak relative to the Fe 2p3/2 main peak for the fluorinated film was approximately half that for the SrFe3+O2.5 film, implying that ∼50% of Fe exists as Fe3+ in the SrFeO3−xFx film. Thus, we can roughly deduce the fluorine content (x) to be x ≈ 1.5.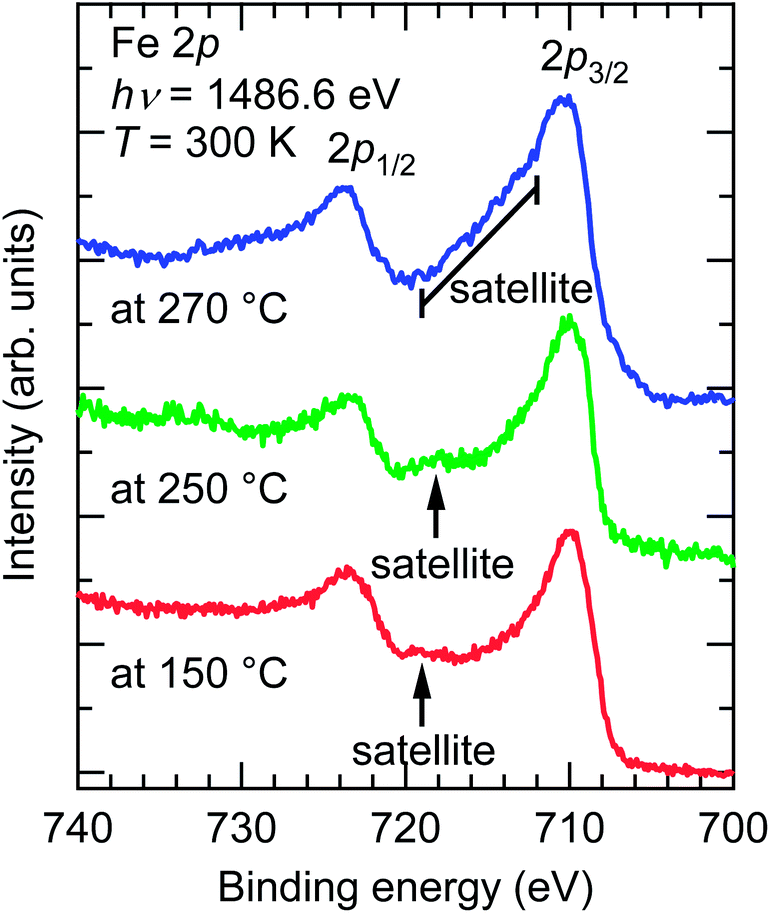 | ||
| Fig. 6 Fe 2p core-level X-ray photoemission spectra of SrFeO3−xFx films fluorinated at 150, 250, and 270 °C, obtained from SrFeO2.5 precursor films. | ||
Fig. 7 shows the AFM images of the SrFeO2.5 precursor film and the SrFeO3−xFx films fluorinated at 250 and 270 °C. The root mean square values of surface roughness were found to be 0.35, 0.64, and 0.81 nm, respectively, indicating that the fluorination process does not cause severe surface roughening.
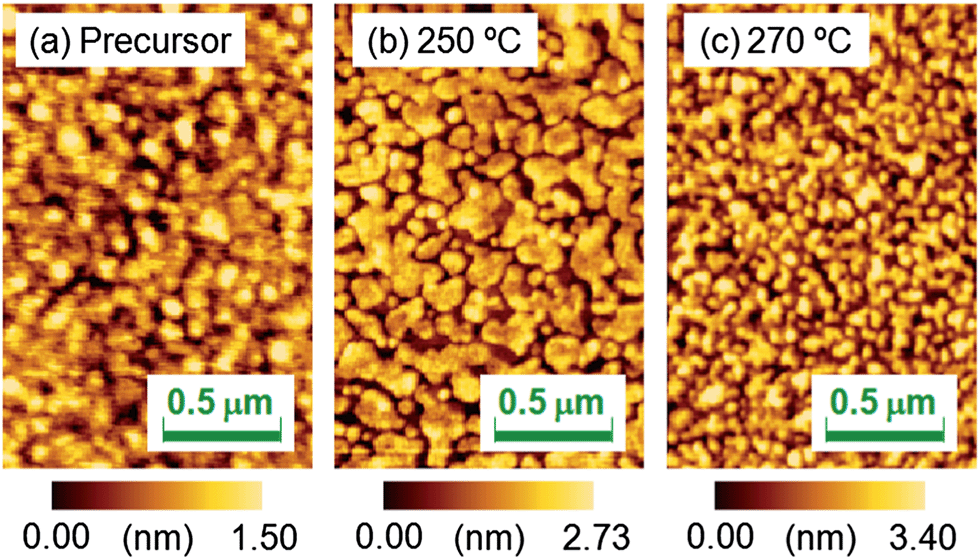 | ||
| Fig. 7 Atomic force microscopy images of the (a) SrFeO2.5 precursor film and SrFeO3−xFx films fluorinated at (b) 250 and (c) 270 °C. | ||
Dependence on oxygen vacancies of precursor films
The relationship between the fluorine content (x) and oxygen vacancies in the precursor films is another important consideration. Fig. 8(a) and (b) compare the XRD patterns of the oxidized SrFeOx (x ≈ 3) and reduced SrFeOx (x ≈ 2) precursor films fluorinated at 150 °C for 24 h. The oxidized and reduced precursor films show the (002) diffraction peaks of perovskite-type and infinite-layer structures with c = 3.835 and 3.490 Å, respectively. The c-axis length of the oxidized SrFeOx (x ≈ 3) film is close to that of the strained SrFeO3 film on the STO substrate, 3.823 Å.22 After treatment with PVDF, the film prepared from the SrFeOx (x ≈ 3) precursor showed two (002) diffraction peaks corresponding to c = 3.871 and 3.955 Å, whereas the film prepared from SrFeOx (x ≈ 2) exhibited one peak with c = 4.002 Å. Fig. 8(c) shows a plot of the c-axis lengths of the SrFeOx (x ≈ 2, 2.5, and 3) precursor and fluorinated films. As seen in Fig. 8(c), the c-axis length of the film fluorinated at 150 °C becomes longer as the oxygen content in the precursor SrFeOx film is decreased from x ≈ 3 to 2. Because the c-axis lengths of the fluorinated films reflect the fluorine content, this result suggests that precursor films containing more oxygen vacancies tend to incorporate more fluorine ions into the film. | ||
| Fig. 8 X-ray diffraction patterns of (a) oxidized SrFeOx (x ≈ 3) and (b) reduced SrFeOx (x ≈ 2) precursor films, and films fluorinated at 150 °C with PVDF for 24 h. (c) c-Axis lengths of SrFeOx (x ≈ 2, 2.5, and 3) precursor and fluorinated films. | ||
To investigate the origin of the two (002) peaks, the fluorine and oxygen depth profiles of the SrFeO3−xFx film obtained from the oxidized SrFeOx (x ≈ 3) precursor were measured in detail (Fig. 9). As seen from the figure, the AF/AOvs. depth plot shows two plateaus: ∼0.8 at 5–40 nm and ∼0.4 at 40–80 nm. These fluorine-rich and fluorine-poor regions correspond to the two (002) diffraction peaks at 2θ ≈ 45.9° and 47.0°, respectively (Fig. 8(a)). These results imply that the diffusion of fluorine ions is considerably slowed down as the concentration of oxygen vacancies decreases.
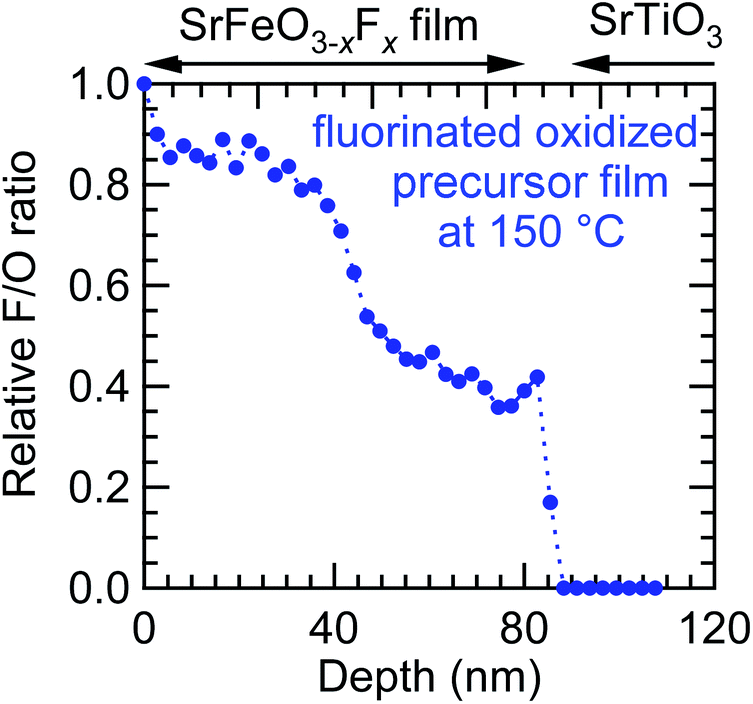 | ||
| Fig. 9 Fluorine depth profile of the SrFeO3−xFx film fluorinated at 150 °C obtained from the oxidized SrFeOx (x ≈ 3) precursor film by X-ray photoemission spectroscopy with Ar+-ion sputtering. | ||
Comparison with the diffusion mechanism
As stated above, the SrFeO3–xFx films were obtained at much lower temperatures (150–270 °C) than the bulk sample (400 °C).5 Now, we will discuss the difference in the reactivity between the thin film and bulk based on the diffusion equation.23 The fluorine ions were not diffused into the STO substrates (Fig. 5). In such a case, the relative fluorine concentration, C(x), is given by eqn (1): | (1) |
tanβn = L. Approximately, the value of D describes the overall shape of the C(x) curve, while k determines the C(x) value at x = 0. Fig. 10 shows the depth dependence of C(x) for a thin film with l = 80 nm, and for a bulk sample with l → ∞ at t = 24 h, where D was set to 3 × 10−15 cm2 s−1, so as to reproduce the fluorine depth profile measured by XPS (Fig. 5). In the case of the bulk sample, C(x) decreases with x in an exponential manner, with the diffusion length of 3 × 102 nm. That is, only fluorine diffuses into the surface regions, which means that the higher fluorine contents experimentally observed in thin films are attributable to smaller grain sizes, representing the maximum length of the diffusion path.
 | ||
| Fig. 10 Relative fluorine concentration (C(x)) vs. depth (x) curves at t = 24 h, calculated for a thin film with l = 80 nm and for a bulk sample with l → ∞. D and k were assumed to be 3 × 10−15 cm2 s−1 and 1 × 10−10 cm s−1, respectively. | ||
4. Conclusions
We have reported the successful synthesis of SrFeO3−xFx epitaxial thin films on STO substrates via topotactic fluorination of SrFeO3−δ precursor films using PVDF. The SrFeO3−xFx thin films were obtained at a lower temperature than polycrystalline bulk samples. Furthermore, the fluorine content (x) in the SrFeO3−xFx films was controllable by adjusting the fluorination temperature and/or the amount of oxygen vacancies in the precursor film. The higher fluorination reactivity in the SrFeO3−δ precursor film, compared with that observed in bulk samples, can be rationalized by taking smaller grain sizes, being the maximum length of the diffusion path, into account within the framework of the fluorine-diffusion model via oxygen vacancy.Acknowledgements
We thank Prof. Kimikazu Sasa, Mr Satoshi Ishii, Dr Hiroshi Naramoto, and Dr Daiichiro Sekiba of University of Tsukuba, and Prof. Katsuyuki Fukutani of the University of Tokyo for their assistance with the NRA measurements. This work was partially supported by the Murata Science Foundation, and the Nippon Sheet Glass Foundation for Materials Science and Engineering. The EDS measurements were conducted in the Research Hub for Advanced Nano Characterization, the University of Tokyo, supported by the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.Notes and references
- M. Al-Mamouri, P. P. Edwards, C. Greaves and M. Slaski, Nature, 1994, 369, 382 CrossRef CAS.
- M. Sturza, H. Kabbour, S. Daviero-Minaud, D. Filimonov, K. Pokholok, N. Tiercelin, F. Porcher, L. Aldon and O. Mentré, J. Am. Chem. Soc., 2011, 133, 10901 CrossRef CAS PubMed.
- P. R. Slater, J. Fluorine Chem., 2002, 117, 43 CrossRef CAS.
- Y. Kobayashi, M. Tian, M. Eguchi and T. E. Mallouk, J. Am. Chem. Soc., 2009, 131, 9849 CrossRef CAS PubMed.
- F. J. Berry, R. Heap, Ö. Helgason, E. A. Moore, S. Shim, P. R. Slater and M. F. Thomas, J. Phys.: Condens. Matter, 2008, 20, 215207 CrossRef.
- F. J. Berry, F. C. Coomer, C. Hancock, Ö. Helgason, E. A. Moore, P. R. Slater, A. J. Wright and M. F. Thomas, J. Solid State Chem., 2011, 184, 1361 CrossRef CAS PubMed.
- F. J. Berry, X. Ren, R. Heap, P. Slater and M. F. Thomas, Solid State Commun., 2005, 134, 621 CrossRef CAS PubMed.
- R. Heap, P. R. Slater, F. J. Berry, O. Helgason and A. J. Wright, Solid State Commun., 2007, 141, 467 CrossRef CAS PubMed.
- Ö. Helgason, Hyperfine Interact., 2008, 184, 143 CrossRef.
- O. Clemens, M. Kuhn and R. Haberkorn, J. Solid State Chem., 2011, 184, 2870 CrossRef CAS PubMed.
- F. J. Berry, A. F. Bowfield, F. C. Coomer, S. D. Jackson, E. A. Moore, P. R. Slater, M. F. Thomas, A. J. Wright and X. Ren, J. Phys.: Condens. Matter, 2009, 21, 256001 CrossRef PubMed.
- F. J. Berry, X. Ren, R. Heap, P. Slater and M. F. Thomas, J. Phys. Chem. Solids, 2008, 69, 2032 CrossRef CAS PubMed.
- C. A. Hancock, T. Herranz, J. F. Marco, F. J. Berry and P. R. Slater, J. Solid State Chem., 2012, 186, 195 CrossRef CAS PubMed.
- E. J. Moon, Y. Xie, E. D. Laird, D. J. Keavney, C. Y. Li and S. J. May, J. Am. Chem. Soc., 2014, 136, 2224 CrossRef CAS PubMed.
- C. K. Blakely, J. D. Davis, S. R. Bruno, S. K. Kraemer, M. Zhu, X. Ke, W. Bi, E. E. Alp and V. V. Poltavets, J. Fluorine Chem., 2014, 159, 8 CrossRef CAS PubMed.
- S. Inoue, M. Kawai, Y. Shimakawa, M. Mizumaki, N. Kawamura, T. Watanabe, Y. Tsujimoto, H. Kageyama and K. Yoshimura, Appl. Phys. Lett., 2008, 92, 161911 CrossRef PubMed.
- I. D. Brown and D. Altermatt, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1985, 41, 244 CrossRef.
- D. Drouin, A. R. Couture, D. Joly, X. Tastet, V. Aimez and R. Gauvin, Scanning, 2007, 29, 92 CrossRef CAS PubMed.
- H. Wadati, D. Kobayashi, H. Kumigashira, K. Okazaki, T. Mizokawa, A. Fujimori, K. Horiba, M. Oshima, N. Hamada, M. Lippmaa, M. Kawasaki and H. Koinuma, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 035108 CrossRef.
- T. Yamashita and P. Hayes, Appl. Surf. Sci., 2008, 254, 2441 CrossRef CAS PubMed.
- A. Chikamatsu, T. Matsuyama, Y. Hirose, H. Kumigashira, M. Oshima and T. Hasegawa, J. Electron Spectrosc. Relat. Phenom., 2012, 184, 547 CrossRef CAS PubMed.
- H. Yamada, M. Kawasaki and Y. Tokura, Appl. Phys. Lett., 2002, 80, 622 CrossRef CAS PubMed.
- E. Fischer, L. Joshua and J. L. Hertz, Solid State Ionics, 2012, 218, 18 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |