 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Zwitterionic pyridinium derivatives of [closo-1-CB9H10]− and [closo-1-CB11H12]− as high Δε additives to a nematic host†
Jacek
Pecyna
a,
Damian
Pociecha
b and
Piotr
Kaszyński
*ac
aDepartment of Chemistry, Vanderbilt University, Nashville, TN 37235, USA. E-mail: piotr.kaszynski@vanderbilt.edu; Tel: +1-615-322-3458
bDepartment of Chemistry, University of Warsaw, Zwirki i Wigury 101, 02-089 Warsaw, Poland
cFaculty of Chemistry, University of Łódź, Tamka 12, 91403 Łódź, Poland
First published on 24th January 2014
Abstract
Substituted closo-carbaborate–pyridinium zwitterions were prepared in 35–50% yield by reacting 1-amino-closo-1-carbaborates with 4-alkoxypyrylium triflates. Two of the new materials, 1[6]d and 2[10]b, exhibit a high temperature SmA phase, whose stability is driven by dipolar interactions. Solution studies in a nematic host, ClEster, demonstrated high positive dielectric anisotropy of these new compounds (Δε ≈ +50) resulting from a longitudinal molecular dipole moment of about 20 D.
Introduction
Polar liquid crystals and additives enable electro-optical switching1 and are essential components of materials for liquid crystal display (LCD) applications.2 Recently, we have demonstrated that zwitterionic derivatives of the [closo-1-CB9H10]− cluster (A, Fig. 1) have high dielectric anisotropy Δε and are useful additives to nematic materials for LCD. In this context, we developed a synthetic methodology and prepared 1-sulfonium3,4 and 1-quinuclidinium,3 and also 10-sulfonium4–6 and 10-pyridinium7 zwitterions, compounds of the general structure IIA and IA. Some of them exhibit nematic behavior and Δε reaching a record high value of 113.5(!) in nematic solutions.7 The preparation of 1-pyridinium derivatives of the [closo-1-CB9H10]− (A) and [closo-1-CB11H12]− (B) clusters however was very inefficient due to the mechanistic issue in the former,8 and instability of the key intermediate in the latter case.9 Such pyridinium derivatives IIA and IIB (Fig. 1, Q = Pyr) are predicted to have significant longitudinal dipole moments, and, consequently, high positive Δε. In addition, they are expected to have lower melting points and be more soluble than the quinuclidinium analogues.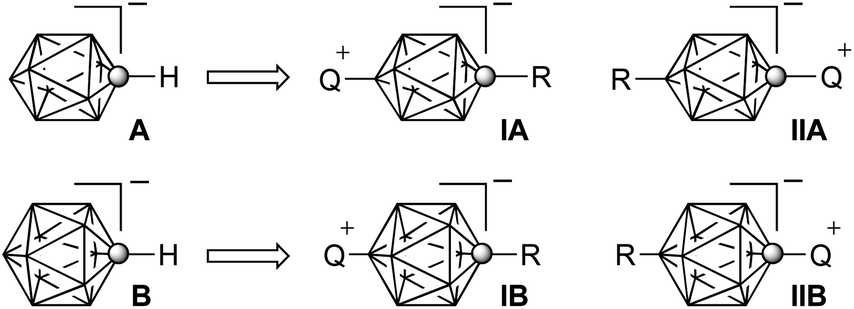 | ||
| Fig. 1 The structures of the [closo-1-CB9H10]− and [closo-1-CB11H12]− anions (A and B) and their zwitterionic 1,10-(IA, IIA) and 1,12-disubstituted (IB, IIB) derivatives. Q+ represents an onium fragment such as ammonium, sulfonium or pyridinium. Each vertex represents a BH fragment and the sphere is a carbon atom. | ||
Here we demonstrate a simple method for preparation of 1-pyridinium zwitterions of anions A and B and their use as high Δε additives to liquid crystal materials for LCD applications.
Results and discussion
Synthesis
Compounds 1[n], 10-vertex derivatives of type IIA, and 2[n], 12-vertex derivatives of type IIB, were obtained by adapting a general method for converting pyrylium salts to N-substituted pyridinium derivatives10 and following a single example of using 4-alkoxypyrylium in this context.11 Thus, a reaction of 1-amino derivatives 3[n] and 4[n] with 4-alkoxypyrylium salts 5 in anhydrous THF gave 1[n] and 2[n], respectively, in 35–50% yields (Scheme 1). 4-Alkoxypyryliums are very rare,12 however triflate salts 5 were conveniently obtained by alkylation of 4H-pyran-4-one with appropriate alkyl triflate 6. In addition to three primary alkoxy derivatives 5a, 5b and 5d, the new method was demonstrated also for a secondary alkoxy derivative. Thus, (S)-2-octanol was converted to triflate 6c and subsequently to pyrylium salt 5c with apparent partial racemization (ee = 35%), as evident from the analysis of the pyridinium product 1[6]c. This indicates that the electrophilic O-alkylation of 4H-pyran-4-one with 6c proceeds through an ion pair and partial scrambling of the stereocenter. Triflate 6c was significantly more reactive than primary alkyl triflates and synthesis of 1[6]c and 2[6]c was performed at lower temperatures. | ||
| Scheme 1 Synthesis of 1[n] and 2[n]. Reagents and conditions: (i) CnH2n+1MgBr or CnH2n+1ZnCl, Pd(0); (ii) 60 °C, 1 h; (iii) THF, rt. | ||
The requisite alkyl amines 3[n] and 4[n] were obtained by alkylation of the corresponding iodo amines 7 and 8 under Pd-catalyzed coupling conditions using either RMgBr or RZnCl reagents. The synthesis of 10-hexyl amine 3[6] was reported previously13 and some 4[n] are reported elsewhere.9 The iodo amine 8 was prepared from iodo acid 9 following the procedure described for iodo amine 7 (Scheme 2),13 or alternatively obtained by iodination of [closo-1-CB11H11-1-NH3]− (10).9
 | ||
| Scheme 2 Synthesis of iodo amine 8. Reagents and conditions: (i) (COCl)2, CH2Cl2, rt; (ii) Me3SiN3, ZnCl2, CH2Cl2, 0 °C → rt; (iii) 80 °C, MeCN; (iv) t-BuOH, MeCN, 80 °C; (v) HCl, MeOH, rt; (vi) [NMe4]+ OH−, H2O; (vii) ICl, AcOH, 60 °C. | ||
Electronic absorption
Pyridinium derivatives 1[n] and 2[n] are colorless solids. Spectroscopic analysis demonstrated that the 12-vertex derivative 2[6]c is more transparent in the UV region than its analogue 1[6]c, however both compounds exhibit relatively strong π → π* absorption bands at λmax = 265 nm (calcd at 260 nm, ƒ = 0.16)14 for 2[6]c and at λmax = 282 nm (calcd at 309 nm, ƒ = 0.25) for 1[6]c (Fig. 2). The origin of this absorption is an efficient intramolecular charge transfer from the HOMO, localized on the cluster, to the LUMO on the pyridinium fragment as shown for 1[6]b and 2[6]b in Fig. 3.8 Interestingly, the HOMO of the latter has lower energy and significantly greater contribution from the B-alkyl chain than observed in the 10-vertex analogue 1[6]b. | ||
| Fig. 2 Electronic absorption spectra of 1[6]c and 2[6]c in MeCN. | ||
 | ||
| Fig. 3 B3LYP/6-31G(d,p) derived contours and energies of FMOs involved in low energy excitation in 1[6]b (left) and 2[6]b (right). | ||
Thermal analysis
All six compounds melt above 100 °C (Table 1). The lowest melting points were observed for the branched (2-octyloxy)pyridinium derivatives [6]c, which is in agreement with results obtained for bis-zwitterionic derivatives of the [closo-B10H10]2− cluster.15 It is considered that the branching methyl group close to the pyridinium ring disrupts efficient packing in the solid state driven by coulombic interactions.15 The highest melting point among the six compounds (216 °C) is exhibited by three-ring derivative 1[6]d. Data in Table 1 also suggest that derivatives of the [closo-1-CB9H10]− cluster (A) have lower melting points than the 12-vertex analogues (e.g.1[6]cvs.2[6]c), which is in agreement with general trends in mesogenic derivatives of 10- and 12-vertex carboranes.16,17| n | R | |||
|---|---|---|---|---|
| a Determined by DSC (5 K min−1) in the heating mode: Cr = crystal; SmA = smectic A; I = isotropic. b Decomp. c Cr–Cr transition at 117 °C (9.7 kJ mol−1). | ||||
| 1[n] | a | 6 | C5H11 | Cr 122 (33.7) I |
| c | 6 | CH(Me)C6H13 | Cr 101 (21.5) I | |
| d | 6 | CH2C6H10C5H11 | Cr 217 (33.5) SmA > 270 Ib | |
| 2[n] | b | 5 | C7H15 | Cr 205 (26.6) I |
| b | 10 | C7H15 | Crc 184 (16.2) SmA 200 (9.1) I | |
| c | 6 | CH(Me)C6H13 | Cr 130 (23.7) I | |
Polarizing optical microscopy (POM) and differential scanning calorimetry (DSC) revealed that 2[10]b and 1[6]d exhibit an enantiotropic SmA phase with the clearing temperature of 202 °C and above 270 °C, respectively (Table 1 and Fig. 4).
 | ||
| Fig. 4 (Left) DSC trace of 2[10]b. The heating and cooling rates are 5 K min−1. (Right) The optical texture of 2[10]b obtained at 190 °C on cooling from the isotropic phase. | ||
XRD data
The formation of the SmA phase was confirmed by powder XRD measurements for 2[10]b. A diffractogram of the mesophase obtained at 195 °C showed a series of sharp commensurate reflections consistent with the lamellar structure with a layer spacing of 25.61 Å (Fig. 5). Considering the calculated molecular length of 31.75 Å,14 the observed layer spacing indicates 19% of interdigitation. The wide-angle region of the diffractogram shows an unsymmetric broad halo, which can be deconvoluted into two signals with the maxima 4.5 Å and 5.4 Å. The diffused signals correlate with the mean distance between the molten alkyl chains (former) and the mean separation between the carborane cages (latter). | ||
| Fig. 5 XRD pattern for 2[10]b at 195 °C. | ||
Temperature dependence studies demonstrated that the SmA phase has a negative thermal expansion coefficient, κ = −0.0030 (1) Å K−1, while the thermal expansion coefficient of the Cr2 phase is positive (κ = +0.00598 (3) Å K−1).14
Smectic behavior of boron cluster-derived mesogens is very rare16 even for polar compounds,6,7 and the observed high-temperature SmA phase for 2[10]b and 1[6]d results presumably from strong lateral dipole–dipole interactions of the zwitterions. This is supported by a comparison of 2[10]b with its non-polar isosteric analogue 11[10]b,7 a low temperature nematogen (TNI = 25 °C) derived from p-caraborane.

Binary mixtures
Three of the new compounds were investigated as additives to ClEster, which forms a nematic phase at ambient temperature characterized by a small negative Δε. Results demonstrated that the two-ring zwitterions are more soluble in the host than the three-ring derivative 1[6]d, and 2[10]b forms stable 6 mol% solutions in the host.14 Extrapolation of the virtual [TNI] for 2[10]b from the solution data gives the N–I transition at 82 ± 4 °C, which is significantly lower than the SmA–I transition at 202 °C. This difference further supports the notion that SmA stability originates from dipolar interactions between the zwitterions. The branched derivative 1[6]c significantly disrupts the nematic order of the host and its extrapolated [TNI] is below −100 °C.Dielectric measurements
Dielectric permittivity values change non-linearly with the concentration of the additives in ClEster as shown for 2[10]b in Fig. 6. This indicates some aggregation of the polar molecules in the solution, which is similar, albeit to lesser extent, to that previously observed for sulfonium (1-Sulf) and quinuclidinium (1-Quin) derivatives of type IIA.3 Therefore, dielectric parameters for the pure additives were extrapolated from dilute about 3 mol% solutions and results are shown in Table 2. | ||
| Fig. 6 Dielectric parameters as a function of concentration of 2[10]b in ClEster. | ||
Analysis of data in Table 2 demonstrates that all three pyridinium derivatives exhibit substantial dielectric anisotropy. Zwitterions 1[6]d and 2[6]b have Δε values 54 and 49, respectively, which, for comparable concentrations, are higher by about 10 than those for the previously investigated 1-Sulf and 1-Quin derivatives.3 The value Δε = 35 extrapolated for 1[6]c with the branched alkyl chain is the lowest in the series, which is presumably related to the low order parameter as evident from dramatic destabilization of the nematic phase of the host.
Computational results for 1[6]b and 2[6]b in Table 3 indicate that the value and orientation of the calculated dipole moment for both series of pyridinium zwitterions are essentially the same: 20 D oriented about 6° relative to the long molecular axis. Consequently, assuming a typical order parameter S = 0.65 and the Kirkwood factor g = μeff2/μ2 = 0.50, the calculated dielectric anisotropy values in ClEster are Δε = 115 (ε|| = 140) for 1[6]b and Δε = 107 (ε|| = 130) for 2[6]b, according to the Maier–Meier relationship between molecular and bulk parameters of the nematic phase.18 Thus, the observed differences in the extrapolated dielectric parameters for the pyridinium zwitterions in Table 2 reflect different compatibility with the host: the degree of aggregation (Kirkwood factor g) and the impact on the order parameter (Sapp).
| Compound | μ ∣∣/D | μ ⊥/D | μ/D | β /° | Δα/Å3 | α avrg/Å3 |
|---|---|---|---|---|---|---|
| a Dipole moments and polarizability obtained at the B3LYP/6-31G(d,p) level of theory in ClEster dielectric medium. Polarizability values calculated from diagonal polarizability tensors were converted from a.u. to Å3 using the factor 0.1482. b Angle between the net dipole vectors μ and μ∣∣. | ||||||
| 1[6]b | 20.18 | 2.36 | 20.32 | 6.7 | 37.0 | 57.1 |
| 2[6]b | 20.03 | 2.26 | 20.15 | 6.4 | 33/4 | 59.1 |
| 1-Sulf | 16.10 | 2.95 | 16.37 | 10.4 | 24.8 | 53.3 |
| 1-Quin | 16.77 | 3.31 | 17.09 | 11.2 | 23.4 | 53.3 |
In comparison, the same calculations for 1-Sulf and 1-Quin, with assumed S = 0.65 and g = 0.50, give the significantly lower predicted values, Δε = 77 (ε|| = 95) and Δε = 81 (ε|| = 100), respectively. Trends in computational results are consistent with the extrapolated experimental dielectric parameters in Table 2.
Conclusions
We have developed a method for efficient preparation of two types of 1-pyridinium zwitterions derived from the [closo-1-CB9H10]− (A) and [closo-1-CB11H12]− anions (B) that exhibit mesogenic properties and are suitable for low concentration, high Δε additives to nematic hosts. The method appears to be general and opens access to a variety of derivatives of the general structure II where Q = 4-alkoxypyridine (Fig. 1). The method permits manipulation with the structure of the R group and the alkoxy substituent for tuning properties of the compounds, especially for improving solubility in the nematic hosts. Dielectric measurements indicate that the pyridinium zwitterions 1[n] and 2[n] are significantly more effective dipolar additives to nematic hosts than those previously investigated (1-Sulf and 1-Quin).Computational details
Quantum-mechanical calculations were carried out using Gaussian 09 suite of programs.19 Geometry optimizations for unconstrained conformers of 1[6]b and 2[6]b with the most extended molecular shapes were undertaken at the B3LYP/6-31G(d,p) level of theory using default convergence limits. The alkoxy group was set in all-trans conformation co-planar with the pyridine ring in the input structure. The orientation of the alkyl substituents on the alicyclic ring and carborane cage in the input structure was set according to conformational analysis of the corresponding 1-ethyl derivatives. No conformational search for the global minimum was attempted.Calculations in solvent media using the PCM model20 were requested with the SCRF (solvent = generic, read) keyword and eps = 3.07 and epsinf = 2.286 input parameters.
Electronic excitation energies for 1[6]b and 2[6]b in MeCN dielectric medium were obtained at the B3LYP/6-31G(d,p) level using the time-dependent DFT method21 supplied in the Gaussian package. Solvent calculations using the PCM model20 were requested with the SCRF (solvent = CH3CN) keyword. Selected molecular orbitals involved in these transitions are shown in Fig. S5 and S6.†
Experimental part
General
Reactions were carried out under Ar and subsequent manipulations were conducted in air. NMR spectra were obtained at 128 MHz (11B) and 400 MHz (1H) in CDCl3 or CD3CN. 11B chemical shifts were referenced to the solvent (1H) or to an external sample of B(OH)3 in MeOH (11B, δ = 18.1 ppm). Optical microscopy and phase identification were performed using a polarized microscope equipped with a hot stage. Thermal analysis was obtained using a TA Instruments DSC using small samples of about 0.5–1.0 mg.Binary mixture preparation
Solutions of the pyridinium derivatives 1[n] or 2[n] in the ClEster host (15–20 mg of the host) were prepared in an open vial. The mixture of the compound and host in CH2Cl2 was heated for 2 h at 60 °C to remove the solvent. The binary mixtures were analyzed by polarized optical microscopy (POM) to ensure that the mixtures were homogenous. The mixtures were then allowed to stand for 2 h at room temperature before thermal and dielectric measurements.Dielectric measurements
Dielectric properties of solutions of selected pyridinium 1[n] or 2[n] and 1-Quin in ClEster were examined with a Liquid Crystal Analytical System (LCAS – Series II, LC Vision, Inc.) using GLCAS software version 0.13.14, which implements literature procedures for dielectric constants.6 The homogenous binary mixtures were loaded into ITO electro-optical cells by capillary forces with moderate heating supplied by a heat gun. The cells (about 4 μm thick, electrode area 0.581 cm2 or about 10 μm thick, electrode area 1.000 cm2, and anti-parallel rubbed polyimide layer) were obtained from LC Vision, Inc. The filled cells were heated to an isotropic phase and cooled to room temperature before measuring dielectric properties. Default parameters were used for measurements: triangular shaped voltage bias ranging from 0.1–20 V at 1 kHz frequency. The threshold voltage Vth was measured at a 5% change. For each mixture the measurement was repeated 10 times for two independent cells. The results were averaged to calculate the mixture's parameters. Results are shown in Tables S4–S6.† All measurements were run at 25 °C. Error in concentration is estimated at about 1.5%. The resulting extrapolated values for pure additives are shown in Table 2.General procedure for preparation of pyridinium derivatives 1[n] and 2[n]
A mixture of amine 3[n] or 4[n] (1 mmol) and the appropriate crude pyrylium triflate 5 [freshly prepared from 4H-pyran-4-one (1.2 mmol) and alkyl triflate 6] in THF (1 mL) under Ar was stirred overnight at room temperature. The solvent was evaporated to give a dark solid. Pure product 1[n] or 2[n] was obtained as a white crystalline solid in 34–51% yield by column chromatography (CH2Cl2/hexane, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) followed by recrystallization from iso-octane/toluene and then EtOH.
ε) 245 (4.06), 282 (4.16). Anal. calcd for C20H42B9NO: C, 58.61; H, 10.33; N, 3.42. Found: C, 58.57; H, 10.26; N, 3.46%.
ε) 264 (4.47). Anal. calcd for C20H44B11NO: C, 55.42; H, 10.23; N, 3.23. Found: C, 55.71; H, 10.17; N, 3.28%.
:1) followed by recrystallization from iso-octane/toluene and then EtOH.
ε) 245 (4.06), 282 (4.16). Anal. calcd for C20H42B9NO: C, 58.61; H, 10.33; N, 3.42. Found: C, 58.57; H, 10.26; N, 3.46%.
ε) 264 (4.47). Anal. calcd for C20H44B11NO: C, 55.42; H, 10.23; N, 3.23. Found: C, 55.71; H, 10.17; N, 3.28%.
General methods for preparation of pyrylium salts 5
General methods for preparation of alkyl triflates 6
Preparation of 1-decyl-12-(4-heptyloxyphenyl)-p-carborane (11[10]b)
A solution of 1-decyl-12-(4-hydroxyphenyl)-p-carborane (12[10], 125 mg, 0.318 mmol), heptyl tosylate (104 mg, 0.382 mmol), K2CO3 (132 mg, 0.956 mmol) and NBu4Br (10 mg, 0.032 mmol) in anhydrous CH3CN (5 mL) was refluxed overnight. The mixture was cooled down to room temperature and filtered. The residue was washed with CH2Cl2 (3 × 10 mL), dried (Na2SO4) and evaporated to dryness. The crude product was purified by column chromatography (hexane/CH2Cl2, 2:1) to give 160 mg of 11[10]b as a colorless liquid, which was crystallized from CH3CN containing a few drops of EtOAc at −80 °C: 1H NMR (CDCl3, 400 MHz) δ 0.88 (t, J = 7.1 Hz, 6H), 1.0–2.6 (br m, 10H), 1.12–1.42 (m, 24H), 1.64 (pseudo t, J = 8.1 Hz, 2H), 1.73 (quint, J = 6.6 Hz, 2H), 3.87 (t, J = 6.5 Hz, 2H), 6.65 (d, J = 9.0 Hz, 2H), 7.09 (t, J = 9.0 Hz, 2H); 11B NMR (CDCl3, 128 MHz) δ −12.3 (d, J = 164 Hz). Anal. calcd for C25H50B10O: C, 63.25; H, 10.62. Found: C, 63.99; H, 10.73%.
1-Decyl-12-(4-methoxyphenyl)-p-carborane (11[10]e)
A solution of (4-methoxyphenyl)-p-carborane23 (200 mg, 0.80 mmol) in Et2O (3 mL) was treated with BuLi (0.8 mL, 1.90 mmol, 2.5 M in hexanes) at −78 °C. The mixture was stirred for 15 minutes and then warmed up to room temperature and stirred for 2 h. THF (1.0 mL) was added to the mixture and the solution was stirred for 1 h at room temperature. The reaction was cooled to 0 °C and a solution of 1-iododecane (236 mg, 0.88 mmol) in Et2O was added to the mixture. The reaction was allowed to warm up to room temperature and stirred overnight. The solvent was removed in vacuo and the residue was dissolved in CH2Cl2 and passed through a short silica gel plug. The solvent was evaporated and the resulting mixture was separated by column chromatography (hexane). The desired product 11[10]e was isolated as the second fraction (122 mg, 40% yield) as a colorless film. The product was crystallized from CH3CN containing a few drops of EtOAc and then cold EtOAc containing a few drops of hexane at −80 °C: mp 38–40 °C (no mesophase was detected upon cooling to −20 °C); 1H NMR (CDCl3, 400 MHz) δ 0.88 (t, J = 7.1 Hz, 3H), 1.0–2.6 (br m, 10H), 1.12–1.35 (m, 16H), 1.65 (pseudo t, J = 8.1 Hz, 2H), 3.74 (s, 3H), 6.67 (d, J = 9.0 Hz, 2H), 7.11 (d, J = 9.0 Hz, 2H); 11B NMR (CDCl3, 128 MHz) δ −12.3 (d, J = 164 Hz); anal. calcd for C19H38B10O: C, 58.42; H, 9.81. Found: C, 58.84; H, 9.39%.The first oily fraction (27 mg), obtained in the chromatographic separation of 11[10]e, was identified as 1-decyl-12-(3-decyl-4-methoxyphenyl)-p-carborane which apparently resulted from ortho-lithiation of (4-methoxyphenyl)-p-carborane under the reaction conditions: 1H NMR (CDCl3, 400 MHz) δ 0.88 (t, J = 6.9 Hz, 3H), 0.89 (t, J = 7.7 Hz, 3H), 1.05–1.35 (m, 30H), 1.44–1.52 (m, 2H), 1.64 (br t, J = 8.2 Hz, 2H), 2.48 (t, J = 7.7 Hz, 2H), 3.74 (s, 3H), 6.58 (d, J = 8.7 Hz, 1H), 6.94 (d, J = 2.6 Hz, 1H), 6.98 (dd, J1 = 8.6 Hz, J2 = 2.6 Hz, 1H); 11B NMR (CDCl3, 128 MHz) δ −12.3 (d, J = 163 Hz); HRMS, calcd for C29H59B10O: m/z = 533.5496, found m/z = 533.5507.
1-Decyl-12-(4-hydroxyphenyl)-p-carborane (12[10])
A solution of 1-decyl-12-(4-methoxyphenyl)-p-carborane (11[10]e, 187 mg, 0.478 mmol) in CH2Cl2 (5 mL) was treated with BBr3 (360 mg, 1.44 mmol, 1.0 M in CH2Cl2) at 0 °C and the reaction was allowed to warm up to room temperature and stirred overnight. Water (10 mL) was added to the mixture and the organic layer was separated, dried (Na2SO4) and evaporated to dryness to give the crude product as a colorless film, which was purified by column chromatography (CH2Cl2) to give 145 mg (78% yield) of a white solid. The product was recrystallized at −80 °C from CH3CN containing a few drops of EtOAc and then EtOAc containing a few drops of hexane to give phenol 12[10] as a colorless film: 1H NMR (CDCl3, 400 MHz) δ 0.88 (t, J = 7.1 Hz, 3H), 1.0–2.6 (br m, 10H), 1.10–1.30 (m, 16H), 1.64 (pseudo t, J = 8.1 Hz, 2H), 4.71 (s, 1H), 6.59 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.8 Hz, 2H); 11B NMR (CDCl3, 128 MHz) δ −12.3 (d, J = 164 Hz). Anal. calcd for C18H36B10O: C, 57.41; H, 9.64. Found: C, 57.18; H, 9.33%.Acknowledgements
This work was supported by the NSF grant DMR-1207585. We are grateful to Professor Roman Dąbrowski of Military University of Technology (Warsaw, Poland) for the gift of ClEster. We thank Ms. Amanda Doody for help with chiral HPLC analysis of 1[6]c.References
- L. M. Blinov and V. G. Chigrinov, Electrooptic Effects in Liquid Crystal Materials, Springer-Verlag, New York, 1994 Search PubMed.
- P. Kirsch and M. Bremer, Angew. Chem., Int. Ed., 2000, 39, 4216 CrossRef CAS.
- B. Ringstrand, P. Kaszynski, A. Januszko and V. G. Young, Jr, J. Mater. Chem., 2009, 19, 9204 RSC.
- J. Pecyna, B. Ringstrand, M. Bremer and P. Kaszynski, submitted.
- J. Pecyna, R. P. Denicola, B. Ringstrand, A. Jankowiak and P. Kaszynski, Polyhedron, 2011, 30, 2505 CrossRef CAS PubMed.
- B. Ringstrand and P. Kaszynski, J. Mater. Chem., 2011, 21, 90 RSC.
- B. Ringstrand and P. Kaszynski, J. Mater. Chem., 2010, 20, 9613 RSC.
- B. Ringstrand, P. Kaszynski and A. Franken, Inorg. Chem., 2009, 48, 7313 CrossRef CAS PubMed.
- J. Pecyna, B. Ringstrand, S. Pakhomov, A. G. Douglass and P. Kaszynski, in preparation.
- A. R. Katritzky, Tetrahedron, 1980, 36, 679 CrossRef CAS.
- H. Ishino, S. Tokunaga, H. Seino, Y. Ishii and M. Hidai, Inorg. Chem., 1999, 38, 2489 CrossRef CAS.
- P. Mäding, J. Steinbach and B. Johannsen, J. Labelled Compd. Radiopharm., 2000, 43, 565 CrossRef.
- B. Ringstrand, H. Monobe and P. Kaszynski, J. Mater. Chem., 2009, 19, 4805 RSC.
- For details see the ESI.†.
- A. Jankowiak, A. Baliński, J. E. Harvey, K. Mason, A. Januszko, P. Kaszyński, V. G. Young, Jr and A. Persoons, J. Mater. Chem. C, 2013, 1, 1144 RSC.
- P. Kaszynski, in Boron Science: New Technologies & Applications, ed. N. Hosmane, CRC Press, 2012, p. 305 Search PubMed.
- B. Ringstrand, A. Jankowiak, L. E. Johnson, P. Kaszynski, D. Pociecha and E. Górecka, J. Mater. Chem., 2012, 22, 4874 RSC.
- W. Maier and G. Meier, Z. Naturforsch., A: Astrophys., Phys. Phys. Chem., 1961, 16, 262 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- M. Cossi, G. Scalmani, N. Rega and V. Barone, J. Chem. Phys., 2002, 117, 43 CrossRef CAS PubMed.
- R. E. Stratmann, G. E. Scuseria and M. J. Frisch, J. Chem. Phys., 1998, 109, 8218 CrossRef CAS PubMed.
- S. Wang and A. Zhang, Org. Prep. Proced. Int., 2008, 40, 293 CrossRef CAS.
- M. A. Fox, J. A. H. MacBride, R. J. Peace and K. Wade, J. Chem. Soc., Dalton Trans., 1998, 401 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Additional synthesis and characterization details for intermediates 4[6], 5, 6, and 8, ee measurement details, additional XRD data, partial TD-DFT output, and archive of calculated equilibrium geometries for 1[6]b and 2[6]b. See DOI: 10.1039/c3tc32351j |
| This journal is © The Royal Society of Chemistry 2014 |