 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stabilizing cations in the backbones of conjugated polymers†
Thomas P.
Voortman
ab,
Hilde D.
de Gier
b,
Remco W. A.
Havenith
bc and
Ryan C.
Chiechi
*ab
aStratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, 9747 AG Groningen, the Netherlands. E-mail: r.c.chiechi@rug.nl
bZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747 AG Groningen, the Netherlands
cGhent Quantum Chemistry Group, Department of Inorganic and Physical Chemistry, Ghent University, Krijgslaan 281, B-9000 Gent, Belgium
First published on 16th January 2014
Abstract
We synthesized a cross-conjugated polymer containing ketones in the backbone and converted it to a linearly conjugated, cationic polyarylmethine via a process we call “spinless doping” to create a new class of materials, conjugated polyions. This process involves activating the ketones with a Lewis acid and converting them to trivalent cations via the nucleophilic addition of electron-rich aryl moieties. Spinless doping lowers the optical band gap from 3.26 to 1.55 eV while leaving the intrinsic semiconductor properties of the polymer intact. Electrochemical reduction (traditional doping) further decreases the predicted gap to 1.18 eV and introduces radicals to form positive polarons; here, n-doping produces a p-doped polymer in its metallic state. Treatment with a nucleophile (NaOMe) converts the cationic polymer to a neutral, non-conjugated state, allowing the band gap to be tuned chemically, post-polymerization. The synthesis of these materials is carried out entirely without the use of Sn or Pd and relies on scalable Friedel–Crafts chemistry.
1 Introduction
The utility of conjugated polymers as electro-optical materials is their unique combination of semiconducting and mechanical properties; they can be processed from solution into devices with similar functionality to their more brittle inorganic counterparts. Devices like field-effect transistors and organic photovoltaics utilize conjugated polymers in their undoped, semiconducting state,1 while sensors and other devices utilize their doped, metallic state(s).2 Virtually all conjugated polymers can be converted from neutral intrinsic semiconductors to charged, metallic states by the addition (n-doping) or removal (p-doping) of electrons. These are redox processes that create the radical anions/cations (polarons) that serve as charge-carriers and that impart the dramatic increase in conductivity that is associated with doping. This process also introduces unpaired spins that create mid-gap states, significantly reducing the band-gap, and red-shifting the optical absorption (i.e., the optical properties are mainly affected by the creation of new states rather than the energies of the bands being shifted by the presence of charges). One exception is poly(aniline) (PANI), which can be converted to its metallic state(s) by protonation; however, this process too is the result of the spontaneous formation of unpaired spins.3 We previously reported an n-dopable conjugated polymer in which the influence of spin and charge could be separated.4 This polymer was based on a cross-conjugated polyketone that could be reduced (n-doped) into two distinct states, a normal, metallic, doped state and a charged, but semiconducting state. These states exhibited clear spectroscopic differences and differences in conductivity, however, they were transient, existing only while held at cathodic potentials under air-free conditions.This paper describes the stabilization of permanent charges via “spinless doping,” a process in which a cross-conjugated polyketone is converted to a linearly conjugated polymer that is a charged, intrinsic semiconductor by the addition of nucleophiles during post-polymerization modification. Unlike the previous polyketones, which were doped only transiently, these conjugated polyions (CPIs) are robust, air-stable, and exhibit dynamic band gaps. They separate the influences of the unpaired electrons that result from traditional redox doping from the inclusion of charges and show dramatic physical changes, including solubility, yet the position of the conduction band (determined electrochemically) remains almost unchanged from their cross-conjugated precursors. Although the polymers presented in this paper represent a proof-of-concept and have not yet been optimized for device applications, they are low band gap (∼1.5 eV) intrinsic semiconductors that are synthesized entirely without Pd or Sn (Scheme 1). The monomers are prepared by nucleophilic displacement and the polymers by the same Friedel–Crafts polycondensation that was initially used to prepare poly(ether ketone), an engineering plastic, on a pilot scale.5–7 (It was later replaced with a process allowing for better, higher-boiling solvents.8) Spinless doping is accomplished with Lewis acid activation and treatment with N,N-dimethylaniline. Thus, CPIs are built on scalable chemistry, which is an important feature of a conjugated polymer in the context of technological applications as an optoelectronic material;9,10 the lack of a scalable, industrially relevant synthetic route precludes commercial/technological applications.
 | ||
| Scheme 1 | ||
Structurally, the CPIs presented in this paper are polyarylmethines (PAMs); however, PAMs are traditional conjugated polymers that must be redox doped and are, therefore, not related to CPIs beyond structural similarities.11–16 Electronically, CPIs resemble conjugated polymers with trivalent atoms in the backbone such as boron17–21 or nitrogen.22 In fact, CPIs are isoelectronic with boron-containing polymers in that the carbocations possess an empty p-orbital, but they are charged because of the position of carbon in the periodic table. Nitrogen-containing polymers possess a full p-orbital, which means that they can potentially undergo spinless doping by protonation, however, the protonation of a full p-orbital will create an sp3 hybridized nitrogen, disrupting the conjugated backbone. There are also cation-containing polymers with imidizolium units in the backbone, however, these charges do not reside in the conjugation pathway of the backbone.23,24 Thus, while CPIs are structurally and electronically similar to PAMs and boron/nitrogen-containing polymers, they combine the properties of these different polymers in a unique way to form a new class of conjugated polymers with dynamically tunable band-gaps in the semi-conducting state (i.e., the conjugation length can be controlled post-synthetically and without redox) due to the presence of stable cations in the conjugated backbone.
2 Experimental
2.1 General
All reagents and solvents were purchased from commercial sources and used without further purification unless otherwise indicated. NMR spectra were measured using a Varian AMX400 (400 MHz) instrument at 25 °C. FT-IR spectra were recorded on a Nicolet Nexus FT-IR fitted with a Thermo Scientific Smart iTR sampler. GPC measurements were done on a Spectra Physics AS 1000 series machine equipped with a Viskotek H-502 viscometer and a Shodex RI-71 refractive index detector. The columns (PLGel 5m mixed-C) (Polymer Laboratories) were calibrated using narrow disperse polystyrene standards (Polymer Laboratories). Samples were made in THF or CHCl3 at a concentration of ∼2.5 mg mL−1. UV/Vis measurements were carried out on a Jenway 6715 spectrometer in 1 cm fused quartz cuvettes with concentrations of 0.03–0.1 mg mL−1 in CH2Cl2. PFK and PFC were also measured in an acidic environment by dissolving a few drops of H2SO4 in CH2Cl2. EPR spectra were recorded on a Magnettech MiniScope MS400 using a quartz capillary at a concentration of 0.5–1 mM in THF and dichloroethane.2.2 Monomer
2.3 Polymers
2.4 Polymer-analogous reaction
2.5 Cyclic voltammetry
Cyclic voltammetry (CV) was carried out with a Autolab PGSTAT100 potentiostat in a three-electrode configuration where the working electrode was ITO-coated glass (7 mm × 50 mm × 0.7 mm, 10 Ω sq.−1, from PG&O), the counter electrode was a platinum wire, and the pseudo-reference was an Ag wire that was calibrated against ferrocene (Fc/Fc+). Polymer films of PFK and PFC were spin-coated at a concentration of ∼20 mg mL−1 from CH3Cl on pre-cleaned (soap, sonicate in DI H2O 2 × 5 min, acetone 10 min, isopropanol 10 min, dry at 110 °C 10 min) and plasma oxidized (5 min) ITO-coated glass at 500–1000 rpm with 200 rpm ramp for 1 minute. Prior to spin coating, the solutions were filtered through a Gelman GHP Acrodisc 0.45 μm membrane filter. Cyclic voltammograms were recorded at 200 mV s−1 with 0.1 M LiClO4 in either acetonitrile or propylene carbonate.2.6 DFT calculations
Geometries of PFC, PFC-Rad+, PFK, and PFK-H2+ were optimized using Density Functional Theory (DFT) (B3LYP/6-31G**) with GAMESS-UK26 without any symmetry constraints. For the radical PFC-Rad+, unrestricted B3LYP was used. Subsequently, the vertical excitation energies were calculated using time-dependent DFT (TD-DFT) (B3LYPg/6-31G**) with DALTON.27 The lowest 15 excited states were calculated for each system.3 Results and discussion
3.1 Spinless doping
We define spinless doping as the introduction of charges into the band structure of a conjugated polymer without redox and without the spontaneous formation of unpaired spins. Spinless doping is not simply the addition of charges, but specifically the introduction of charges in the conjugated backbone via closed-shell, two-electron processes. Therefore, any unpaired electrons in the band structure must form spontaneously; for example, by spin un-pairing due to internal redox.3 We distinguish between two types of spinless doping; transient, where charges are generated in situ and the resulting CPI cannot be isolated4,28,29 and permanent, where the charges are generated by stabilizing trivalent carbocations via addition/dehydration.Fig. 1 is a cartoon describing the differences between traditional redox doping and spinless doping. The left column shows the band structure of a CPI as cations are introduced into the backbone, in this case by masking/unmasking the cations with methoxide/acid, as is depicted for PFC in Scheme 1. Under acidic conditions, when there are no nucleophiles present to trap the cations, the polymer is fully conjugated and the band gap is minimized, as is shown by the red structure in the middle column. As the concentration of nucleophiles (OCH3−) increases, the cations are trapped as sp3 carbon centers (e.g., methyl ethers). This process decreases the average conjugation length and increases the band gap until the polymer is rendered completely non-conjugated, represented by the white structure in the middle column. Traditional redox doping, by contrast, begins from a semiconducting polymer. The purple structure in the middle column is formed by reduction, which introduces charges via the addition of single electrons. This redox process also lowers the band gap, but not by affecting the average conjugation length. Rather, as is shown in the right column, it creates mid-gap states that typically lead to band gaps in the near-IR.
 | ||
| Fig. 1 A cartoon depicting the mechanism of the change in band gap of CPIs as a function of treatment with methoxide/acid compared to traditional doping. Circles indicate the methine groups that separate the aromatic groups, which are represented by squares. Conjugation is indicated in red and purple. Diagrams of the valence and conduction bands are drawn to the left or right of each structure with an approximate band gap based on Fig. 4. Increasing the concentration of nucleophiles (methoxide) traps the methine cations as sp3 hybridized ethers, lowering the average conjugation length and producing a large band gap as indicated by the all-white structure. Traditional (oxidative) doping introduces both cations and unpaired spins, leading to the creation of mid-gap states and a low band-gap as indicated by the bottom, all-purple structure. | ||
One of the most important differences between the CPI depicted in red and the redox-doped polymer shown in purple, is that the CPI – despite bearing charges in the band structure – can still be redox doped (by reduction). We previously observed this process via spectroelectrochemistry in transient CPIs.4 In this paper, we rely on a combination of experiment and calculation to show that the same effect is present in PFC. However, proving that spinless doping does not induce spontaneous spin un-pairing and that CPIs are semiconductors is not straightforward. We measured the EPR spectra of both PFK and PFC and both gave no detectible signal, however, it is difficult to prove a negative. Likewise, conductivity alone is not evidence of semiconducting or metallic properties. Thus, the bulk of the Results and discussion describes the preparation and characterization of PFK and PFC and the characterization of the influences of spinless doping, which vary considerably from the well-known effects of traditional redox doping.30
3.2 Synthesis and characterization
We synthesized PFCvia a Lewis acid mediated polymer analogous reaction of N,N-dimethylaniline on the cross-conjugated fluorene-based precursor polymer PFK. This introduction of permanent cations is spinless doping; it is an electrophilic addition/dehydration, not a redox process. We prepared PFKvia the Friedel–Crafts polycondensation of fluorene 1 with terephthaloyl chloride mediated by AlCl3 and LiCl in dry CH2Cl2 according to Scheme 1 and described in more detail in the Experimental. A byproduct of the Friedel–Crafts polycondensation reaction is large quantities of AlOH3, which is a known flocculant and which can be challenging to separate from the desired polymer. Therefore, we attempted to minimize the AlCl3 loading as much as possible and increase reaction times under highly inert conditions. We varied the Lewis acid loading for PFK from three to ten equivalents without negatively affecting the degree of polymerization, provided the solvents and reagents were dried and the reactions were performed under strict anhydrous conditions. Lower Lewis acid loadings resulted in a better controlled reaction and an easier isolation of PFK, because even with three equivalents a near-equal amount of AlOH3 is formed per gram of polymer. Furthermore, we varied the polymerization time from one to three days under an Ar atmosphere. The lower Lewis acid loading and/or longer reaction times resulted in a trade-off between the degree of polymerization and polydispersity, where longer reaction times typically increase the polydispersity index (PDI) and peak molecular weight (Mp), but not the number average molecular weight (Mn).We determined Mn = 4000–5200, Mp = 8000 g mol−1, and PDI = 2.5–3.2 for PFK by gel permeation chromatography (GPC). See ESI† for a discussion of the batch-to-batch variability of Mn and Mp, the effects of reaction temperature, and a comparison against model compounds. As is almost always the case, these polymers have lower values of Mn than thermoplastics and block co-polymers and, thus, can be called oligomers by comparison. We prefer the term polymer because the electronic properties of conjugated polymers saturate after only a few repeats.31,32 Thus, these materials are optically and electronically equivalent from n ≈ 5–∞ and are, therefore, more appropriately called polymers in the context of this paper. (The mechanical properties, which do vary with molecular weight, do not affect the optical or electrochemical properties.) Moreover, our estimates of Mn are conservative and represent the lower bounds of the real molecular-weight distribution, rather than directly reporting the number produced by the GPC without interpretation. The most common method for determining Mn (using an optical detector) relies on dn/dc values to relate concentration to changes in the refractive index, which are then compared against a standard calibration curve. Using the common TriSEC method and dn/dc values calculated by the instrument yielded values of Mn ∼24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1. Since dn/dc values are virtually unknown for conjugated polymers, they cannot be verified against published values. Furthermore, volumetric/gravimetric errors, including the material that is removed by filtration, that precipitates during the measurement, or that aggregates in solution, causes a large increase in the reported values of Mn. This problem is exacerbated by rigid conjugated polymers where simply measuring a GPC at elevated temperatures to reduce aggregation can change Mn by an order of magnitude33 or introduce bi-modality, which renders Mn meaningless. From our conservative estimate of Mn and PDI, we determined Pn (the number of repeats) to be ∼9–40, which is relatively low, but not atypical for a conjugated polymer. Increasing the reaction temperature (see ESI†) increased Mp from 8000 to 31000 g mol−1, but increased the PDI.
000 g mol−1. Since dn/dc values are virtually unknown for conjugated polymers, they cannot be verified against published values. Furthermore, volumetric/gravimetric errors, including the material that is removed by filtration, that precipitates during the measurement, or that aggregates in solution, causes a large increase in the reported values of Mn. This problem is exacerbated by rigid conjugated polymers where simply measuring a GPC at elevated temperatures to reduce aggregation can change Mn by an order of magnitude33 or introduce bi-modality, which renders Mn meaningless. From our conservative estimate of Mn and PDI, we determined Pn (the number of repeats) to be ∼9–40, which is relatively low, but not atypical for a conjugated polymer. Increasing the reaction temperature (see ESI†) increased Mp from 8000 to 31000 g mol−1, but increased the PDI.
To convert PFK to PFC, we redissolved the polymer in dry CH2Cl2 and performed a Lewis acid mediated electrophilic substitution on N,N-dimethylaniline of the activated ketones. However, when we performed the polymer analogous reaction method on PFK with three equivalents of AlCl3 and five equivalents of N,N-dimethylaniline for one day, the conversion was low. Therefore, we repeated the polymer analogous reaction with five equivalents of AlCl3 and ten equivalents of N,N-dimethylaniline for three days. Although polymer analogous reactions are never quantitative, the quality of the resulting PFC was unambiguously higher than the first attempt. This two-step conversion enabled us to qualitatively follow the conversion of the polymer analogous reaction spectroscopically. To render PFC sufficiently soluble in organic solvents, and to prevent overly strong binding of counter-ions, we isolated PFC as the BF4− salt by first trapping free PFC with methoxide to form the non-conjugated methyl ether (i.e., with CH3O− as shown in Scheme 1), dissolving it in aqueous HBF4, and repeatedly extracting it with CH2Cl2.
We encountered difficulties obtaining clean 1H-NMR spectra of PFC due to problems with locking and shimming (see ESI† for examples) possibly due to the combination of the ionic and polymeric nature of CPIs driving aggregation in organic solvents (PFC is sparingly soluble in CDCl3). Thus, we followed the process of spinless doping by FT-IR analysis, which clearly shows two carbonyl modes present in PFK at 1654 cm−1 and 1725 cm−1. Fig. 2 compares the FT-IR spectra of PFK to PFC. The intensities of these (presumed) ketone stretches decrease markedly as the ketones of PFK are converted to cations in PFC. We correlated these bands with absorption bands in the UV-Vis, allowing us to follow the process of spinless doping semi-quantitatively and, importantly, proving that the post-polymerization modification proceeds in high yield. The FT-IR spectra of model ketones and cations (single repeat-units) are shown in the ESI† to highlight the shifts that occur upon polymerization, and the very different spectra arising from the small-molecule analogs and PFC/PFK.
 | ||
| Fig. 2 FT-IR spectra showing PFK before (white) and after post-polymerization modification which converts it to PFC (light gray) by spinless doping with near completion, as indicated by the strong reduction of the carbonyl resonance mode at 1654 cm−1 and the complete disappearance of the carbonyl mode at 1725 cm−1, as highlighted by dashed lines. | ||
3.3 Band gap measurements
Fig. 3 and 4 show the absorption spectra of PFK and PFC respectively. The absorption spectra of PFK are shown in the neutral, cross-conjugated, semiconducting form and in the charged, transient spinless doped, protonated state (the solid and dashed lines in Fig. 3 respectively). Protonation shifts the band gap of PFK from 3.2 to 2.3 eV in a process analogous to that shown in Fig. 1 except that instead of converting between sp2 methylium cations and sp3 alcohols, the cations are converting cross-conjugation to linear conjugation. We measured the absorption spectra of as-prepared PFC and in acidified CH2Cl2 (the dashed and dotted lines in Fig. 4 respectively). By comparing the FT-IR spectra to the UV-Vis spectra, we ascribe the shoulder in the absorption spectrum of PFC at ∼430 nm to cations resulting from the protonation of residual ketone (similar to protonated PFK). This peak only appears in acidified CH2Cl2, which also has the effect of red-shifting λmax by 48 nm and lowering the band gap from 1.77 to 1.55 eV. This shift is the result of maximizing the average conjugation length by converting the small amount of residual ketones from cross-conjugated carbonyls to conjugated methines and increasing the delocalization of the permanent carbocations, as is depicted in Fig. 1 as the all-red structure. We also measured PFC-OMe (the solid line in 5) by trapping PFC with methoxide, corresponding to the all-white structure in Fig. 1. The absorption spectrum of PFC-OMe cuts off at 400 nm (∼3 eV) and shows no signs of residual cations (or protonated residual ketone). These absorption spectra demonstrate the dynamism of the band gap of PFC and its dependence on the number of conjugating cationic methines. This tunability is a unique property of CPIs that results in changes to the hybridization of carbon atoms in the backbone and should not be confused with the protonic acid doping of PANI. | ||
| Fig. 3 Absorption spectra of PFK in neutral and acidified CH2Cl2 depicting a bathochromic shift of λmax by 120 nm upon transient acid doping, turning the color of the polymer from yellow to deep red. We ascribe λmax of PFK in neutral CH2Cl2 to weakly interacting fluorene units through cross-conjugating ketones. After the transient acid doping the polymer becomes fully conjugated resulting in the observed bathochromic shift. | ||
 | ||
| Fig. 4 Absorption spectra of PFC as trapped as the methyl ether PFC-OMe (CH2Cl2; solid line), the BF4− salt (CH2Cl2; dashed line), and the fully protonated state (acidified CH2Cl2; dotted line). We ascribe λmax of PFC-OMe to the absorption of fluorene chromophores not conjugated to each other. The BF4− salt of PFC has a λmax at ∼540 nm and a small shoulder at ∼435 nm, indicating that the residual ketones are only sparsely protonated by HBF4. In acidified CH2Cl2 the residual ketones are protonated resulting in an additional bathochromic shift of ∼48 nm, the appearance of an additional peak at ∼430 nm, and broadening of the absorption tail (i.e., lowering of the band gap). | ||
To further elucidate the effect of spinless doping we measured cyclic voltammograms (CV spectra) of spin-cast films of PFK and PFC on ITO, which served as the working electrode. We used Pt wire as a counter electrode and an Ag/Ag+ pseudo reference electrode calibrated with ferrocene. Measurements of PFK were straightforward; we cast films from CHCl3 and measured them in degassed CH3CN. We chose LiClO4 as an electrolyte to facilitate the movement of cations in and out of the films as they were cycled. Measurements on PFC, by contrast, were confounded by wetting issues. PFC is only sparingly soluble in CHCl3, giving very thin films, while thicker films cast from THF did not adhere to the electrode. Due to the ionic backbone, PFC is readily (and violently exothermically) soluble in acetonitrile, thus we performed the CV measurements in propylene carbonate (PC), however, LiClO4 is considerably less soluble in PC. Worse, films of PFC tend to de-wet in PC and float off of the electrode, particularly when reduced. These experimental complexities forced us to perform CV measurements under different conditions for PFC and PFK, which can complicate direct comparison.
The results of the CV measurements are shown in Fig. 5 and 6. PFK shows two reduction waves at −1.7 V and −1.9 V (taken from the second scan) and a semi-reversible re-oxidation wave at ∼−1.1 V with a shoulder at ∼−0.8 V. We could not suppress charging effects sufficiently to resolve both re-oxidation waves clearly; neutral PFK is insulating enough that upon re-oxidation, charges become trapped in the film and their sudden expulsion gives rise to a small, aberrant peak (i.e., retarded re-oxidation). We observed the exact same effect in previous studies on n-dopable polyketones.4 The intensity of the redox waves appears to grow upon increasing the number of cycles suggesting retarded switching, which we ascribe to the reorganization of the polymer film as counterions diffuse into it. Larger shifts have been observed for n-type polymers,34 but it is observed in thin films of conjugated polymers more frequently than it is reported, as the tendency is to show only one exemplary trace.
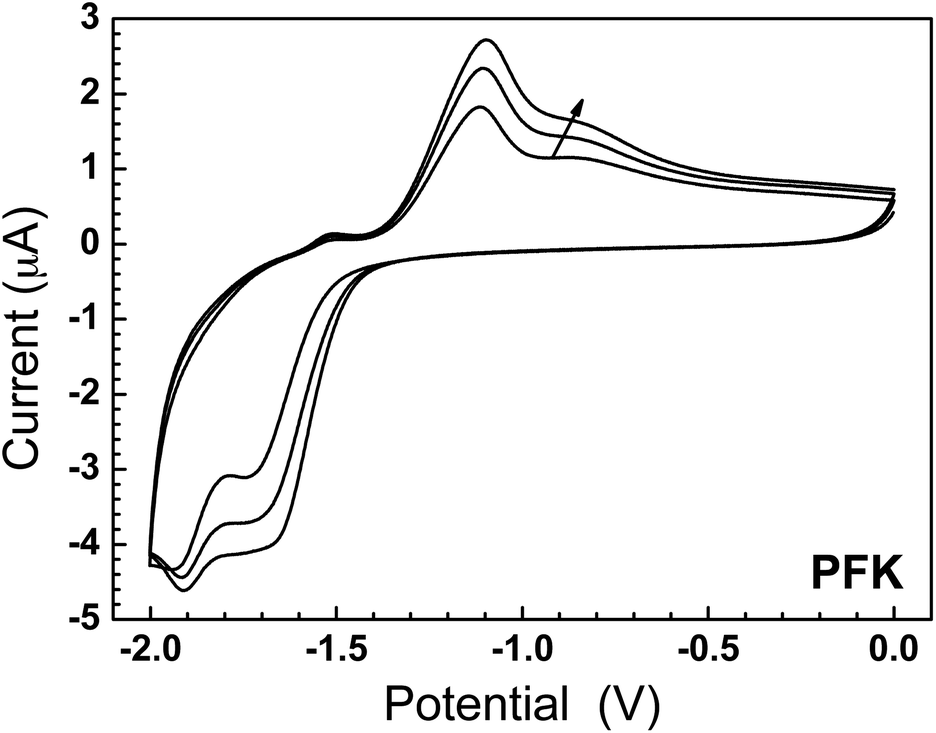 | ||
| Fig. 5 Cyclic voltammograms of a thin film of PFKversus Ag/Ag+ on an ITO working electrode with a Pt wire counter electrode immersed in 0.1 mM LiClO4 in acetonitrile at 200 mV s−1. The two reduction waves (−1.65 and −1.90 V) are indicative of the cross-conjugated ketones being reduced sequentially to form first a radical anion and then a closed-shell anion, i.e., a transient CPI. The re-oxidation waves are partially obscured by the sudden release of trapped charge, the results from the insulator–metal–semiconductor transition, and are typical of cross-conjugated polyketones. | ||
 | ||
| Fig. 6 Cyclic voltammograms of a thin film of PFCversus Ag/Ag+ on an ITO working electrode with a Pt wire counter electrode immersed in 0.1 mM LiClO4 in propylene carbonate at 200 mV s−1. The broad, featureless reduction (∼−2 V) and re-oxidation (∼−0.7 V) waves are indicative of traditional redox doping/de-doping of the band structure of a semiconducting conjugated polymer. In this case, the polymer is being n-doped (reduced), to a p-doped (radical cationic) state because the semiconducting form comprises closed-shell cationic charges. | ||
The reduction wave of PFC (Fig. 6), taken from the second scan, lies at −1.7 V. The entire reduction wave gradually shifts to more negative potentials and broadens to one feature upon increasing the number of scans until it stabilizes at ∼−2.0 V. PFC re-oxidizes at a potential of ∼−0.8 V. These results indicate that the cations in PFC are more difficult to reduce than the neutral state of PFK, demonstrating the remarkable stability of charges in CPIs. Not only are they delocalized into a band structure, but the electron-donating nature of the pendant dimethylaniline groups adds further stabilization. Qualitatively, the CV spectra of PFK show the expected two-electron reduction of cross-conjugated polyketones, while the CV spectra of PFC have the broad features that are typical of the reduction (or oxidation) of a band structure. If PFC comprised isolated cations in a “string of pearls”, we would expect sharp reduction peaks.
From the optical and electrochemical data, we calculated the positions of the valence and conduction bands (i.e., the bands derived from the HOMO and LUMO of the monomers, respectively) of PFK to be −5.83 and −2.57 eV, respectively. The values of PFC are −4.14 and −2.61 eV, demonstrating that spinless doping – in this case – slightly lowered the conduction band and significantly raised the valence band. This observation implies that the electron-rich dimethylaniline moieties destabilize the valence band more than the charges (which reside in the valence band) stabilize the conduction band. In other words, the dimethylaniline moieties impart more donor character than the cations impart acceptor character. The observed decrease in the band gap in PFC as compared to PFK is, therefore, principally a combination of the conversion from cross- to linear-conjugation and the destabilization of the valence band. This result is in contrast to traditional redox doping, which shifts the bands only slightly in energy and which instead lowers the band gap by creating mid-gap states that result from the presence of unpaired spins.30
3.4 DFT calculations
An ideal experiment to prove that PFC is an intrinsic semiconductor would be to measure the absorption spectra as a function of reduction (spectroelectrochemistry). The expectation is for the absorption band to shift from the visible into the near IR, exactly as most normal conjugated polymers behave.4,34 Unfortunately, the poor mechanical stability of films of PFC at cathodic potentials precluded the collection of usable absorption data. Thus, we turned to DFT calculations for insight. The experimental design is as follows; we have absorption data for PFK in the neutral and protonated form, absorption data for PFC, and electrochemical data for PFK and PFC. From these data we calculated the optical band gaps and reduction onsets, from which we estimate the absolute positions of the valence and conduction bands. If we can replicate the optical gaps using DFT calculations, we can conclude that they behave as “normal” conjugated polymers in that their properties are predictable and that, for example, PFC does not undergo PISUM. We then calculate the expected optical band gap of the redox-doped state of PFC that is formed upon reduction, i.e., we deliberately add an un-paired spin. (Interestingly, this is a case of n-doping to produce a p-doped conjugated polymer). If that band gap is significantly red-shifted, it is strong evidence that PFC is both spinless and is an intrinsic semiconductor because it means that our model systems are accurate reflections of the materials that we are measuring. The structures of protonated PFK (PFK–H+) and the reduced form of PFC (PFC-Rad), which we measured, and the isolated model dimers PFC2+, PFC-Rad+, and PFK-H2+, which we calculated, are shown in Scheme 2. The isolated model dimer of PFK is the deprotonated (neutral ketone) form of PFK–H2+. | ||
| Scheme 2 | ||
The calculated spectrum for PFK under neutral conditions comprises a minimum band gap (Eoptg) of 3.41 eV and a main peak (λmax) at 3.55 eV. These calculated peaks are blue-shifted from the experimental data by ∼0.3 eV because we performed the calculations on isolated model dimers (without counterions), but they otherwise fit the experimental data perfectly, with the exception of an under-estimation of Eoptg for PFK-H+. When protonated, Eoptg and λmax decrease to 2.04 eV for PFK-H2+, which underestimates the experimental values likely because, experimentally, PFK-H+ is in equilibrium and never fully protonated. These data are shown in detail in the ESI†. We calculated Eoptg = 1.83 eV and λmax = 2.33 eV for PFC2+ as a model for PFC in its fully cationic state. These calculations correspond to the experimentally-derived gaps in the fully protonated state of PFC (in acidified CH2Cl2), i.e., when all of the residual ketones are protonated and the average conjugated length of PFC is maximized. We calculated Eoptg = 1.18 eV for PFC-Rad+ as a model for PFC in the redox-doped state, where one cationic site per repeat unit is replaced by a radical. That value corresponds to a maximum absorption of 1030 nm, which is well into the near IR and in close agreement with our previous transient-doping study on a related polyketone (Fig. 7).4 The experimental and calculated data are summarized in Table 1.
 | ||
| Fig. 7 The absorption spectrum of PFC (black; left axis) plotted with the predicted spectra of PFC (blue; right axis) and PFC-Rad (red; right axis). The predicted spectrum for PFC-Rad shows the emergence of a band at 1050 nm, which is in close agreement with the transient, open-shelled (redox doped) CPIs measured previously.4 | ||
| E optg exp. | E optg calc. | E valence exp. | E conduction exp. | |
|---|---|---|---|---|
| PFK | 3.26 | 3.41 | −5.83 | −2.57 |
| PFK-H+ (PFK-H2+) | 2.30 | 2.04 | — | — |
| PFC (PFC2+) | 1.55 | 1.83 | −4.14 | −2.61 |
| PFC-Rad (PFC-Rad) | — | 1.18 | — | — |
The close agreement between DFT calculations and experimental values demonstrates that CPIs behave as normal, intrinsic semiconducting polymers; their electronic properties are completely predictable. Thus, CPIs are in fact simply an intrinsic semiconductor polymer with charges in the valence band (i.e., conjugated backbone). These data are very strong evidence that spinless doping does not lead to spontaneous spin un-pairing; if spontaneous spin un-pairing occurred, the experimental and calculated band gaps would not be in such close agreement. Moreover, the deliberate addition of unpaired electrons would not lead to a dramatic lowering of the gap.
4 Conclusions
The polymers presented in this paper are proofs of concept for the stabilization of closed-shell cations in the backbones of conjugated polymers. These charges are added via spinless doping, which chemically converts a cross-conjugated precursor polymer to a conjugated polyion; a conjugated, semiconducting polymer bearing charges in the conjugated backbone. These polymers effectively disassociate charge and spin. In traditional redox doping, polarons, which comprise both spin and charge, are created in the band structure, shrinking the band gap by creating mid-gap states. Similarly, the charges in CPIs have little effect on the absolute positions of the valence and conduction bands, which are influenced more strongly by the electron-rich groups used to stabilize the cations. However, in the absence of spin, no mid-gap states are created, thus CPIs can still undergo traditional redox doping to create (in principle) positive polarons (p-doping) through electrochemical reduction. Although we were unable to observe this phenomenon optically, DFT calculations show that, indeed, CPIs behave as normal conjugated polymers and that traditional redox doping does lead to a p-doped polymer with a significantly reduced band gap.The key feature of CPIs is that their electronic states can be altered by chemical modification and/or redox. Unlike PANI, they are readily processible and do not undergo PISUM. A fully cationic CPI can be rendered non-conjugated by treatment with a base or nucleophile and the band gap was tuned by exposure to acid. This phenomenon is unique to CPIs and has implications for patterning, sensing, tunable absorption, and any application that takes advantage of the dynamic, post-synthetic modification of the band gap of a semiconducting polymer. The conversion of the cross-conjugated precursor polymer to the CPI via spinless doping also inverts the solubility by turning a hydrophobic backbone into a charged, hydrophilic backbone. In this paper that transformation rendered the CPI sparingly soluble in CH3Cl (and only as the BF4− salt), but further manipulation of the pendant groups will allow for tunable orthogonal solubility.
Unlike virtually all light-harvesting polymers, CPIs are synthesized entirely without the use of Sn or Pd, relying on inherently scalable chemistry. (The poly[3-alkylthiophene]s and poly[alkoxy phenylene–vinylene]s are notable exceptions and, not coincidentally, have been among the most commercially successful conjugated polymers.) Yet, even PFC has a band gap of 1.55 eV, which can be tuned further by altering the backbone and the aryl groups used to stabilize the cations. While this paper only characterizes the influences of spinless doping, we are confident that, through the same synthetic tailoring that is commonplace in traditional conjugated polymer chemistry, CPIs will afford a variety of new conjugated materials with potential applications in organic-electronic devices.
Acknowledgements
This work is part of the research program of the Foundation for Fundamental Research on Matter (FOM), which is part of the Netherlands Organization for Scientific Research (NWO). This is a publication by the FOM Focus Group “Next Generation Organic Photovoltaics”, participating in the Dutch Institute for Fundamental Energy Research (DIFFER). The authors acknowledge the Zernike Institute for Advanced Materials of the University of Groningen for additional financial support (“Dieptestrategie” program). RCC also acknowledges the European Research Council for the ERC Starting Grant 335473 (MOLECSYNCON) and RWAH also acknowledges Prof. Dr R. Broer (University of Groningen, the Netherlands) for fruitful discussions.References
- X. Guo, M. Baumgarten and K. Müllen, Prog. Polym. Sci., 2013, 33(12), 1832–1908 CrossRef PubMed.
- J. Huang, S. Virji, B. H. Weiller and R. B. Kaner, J. Am. Chem. Soc., 2003, 125, 314–315 CrossRef CAS PubMed.
- F. Wudl, R. O. Angus, F. L. Lu, P. M. Allemand, D. Vachon, M. Nowak, Z. X. Liu, H. Schaffer and A. J. Heeger, J. Am. Chem. Soc., 1987, 109, 3677–3684 CrossRef CAS.
- R. C. Chiechi, G. Sonmez and F. Wudl, Adv. Funct. Mater., 2005, 15, 427–432 CrossRef CAS.
- I. Goodman, J. E. McIntryre and W. Russell, Brit. Patent, 971, 227, 1964.
- K. J. Dahl, V. Jansons and S. Moore, Pat. No. 4,808,693, 1989.
- J. Yang, Ph.D. thesis, Virginia Tech, 1998.
- M. Kutz, Applied Plastics Engineering Handbook, William Andrew, 2011 Search PubMed.
- B. Azzopardi, C. J. M. Emmott, A. Urbina, F. C. Krebs, J. Mutale and J. Nelson, Energy Environ. Sci., 2011, 4, 3741 Search PubMed.
- H. Spanggaard and F. C. Krebs, Sol. Energy Mater. Sol. Cells, 2004, 83, 125–146 CrossRef CAS PubMed.
- H. Braunling, R. Becker and G. Blochl, Synth. Met., 1991, 42, 1539–1547 CrossRef.
- H. Braunling, G. Blochl and R. Becker, Synth. Met., 1991, 41, 487–491 CrossRef CAS.
- H. Braunling, R. Becker and G. Blochl, Synth. Met., 1993, 55, 833–838 CrossRef.
- W. Chen and S. Jenekhe, Macromolecules, 1995, 28, 454–464 CrossRef CAS.
- W. Chen and S. Jenekhe, Macromolecules, 1995, 28, 465–480 CrossRef CAS.
- S. Jenekhe, Nature, 1986, 322, 345–347 CrossRef CAS.
- N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 5112–5113 CrossRef CAS.
- N. Matsumi, K. Naka and Y. Chujo, J. Am. Chem. Soc., 1998, 120, 10776–10777 CrossRef CAS.
- R. Corriu, T. Deforth, W. Douglas, G. Guerrero and W. Siebert, Chem. Commun., 1998, 963–964 RSC.
- H. Kobayashi, N. Sato, Y. Ichikawa, M. Miyata, Y. Chujo and T. Matsuyama, Synth. Met., 2003, 135, 393–394 Search PubMed.
- A. Sundararaman, M. Victor, R. Varughese and F. Jäkle, J. Am. Chem. Soc., 2005, 127, 13748–13749 CrossRef CAS PubMed.
- M. Bernius, M. Inbasekaran, E. Woo, W. Wu and L. Wujkowski, J. Mater. Sci.: Mater. Electron., 2000, 11, 111–116 CrossRef CAS.
- M. Toba, T. Nakashima and T. Kawai, Macromolecules, 2009, 42, 8068–8075 CrossRef CAS.
- M. Toba, T. Nakashima and T. Kawai, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1895–1906 CrossRef CAS.
- N. Giuseppone, G. Fuks and J.-M. Lehn, Chem. – Eur. J., 2006, 12, 1723–1735 CrossRef CAS PubMed.
- M. F. Guest, I. J. Bush, H. J. J. Van Dam, P. Sherwood, J. M. H. Thomas, J. H. Van Lenthe, R. W. A. Havenith and J. Kendrick, Mol. Phys., 2005, 103, 719–747 CrossRef CAS.
- K. Aidas, C. Angeli, K. L. Bak, V. Bakken, R. Bast, L. Boman, O. Christiansen, R. Cimiraglia, S. Coriani, P. Dahle, E. K. Dalskov, U. Ekström, T. Enevoldsen, J. J. Eriksen, P. Ettenhuber, B. Fernández, L. Ferrighi, H. Fliegl, L. Frediani, K. Hald, A. Halkier, C. Hättig, H. Heiberg, T. Helgaker, A. C. Hennum, H. Hettema, E. Hjertenaes, S. Høst, I.-M. Høyvik, M. F. Iozzi, B. Jansík, H. J. A. Jensen, D. Jonsson, P. Jørgensen, J. Kauczor, S. Kirpekar, T. Kjaergaard, W. Klopper, S. Knecht, R. Kobayashi, H. Koch, J. Kongsted, A. Krapp, K. Kristensen, A. Ligabue, O. B. Lutnaes, J. I. Melo, K. V. Mikkelsen, R. H. Myhre, C. Neiss, C. B. Nielsen, P. Norman, J. Olsen, J. M. H. Olsen, A. Osted, M. J. Packer, F. Pawlowski, T. B. Pedersen, P. F. Provasi, S. Reine, Z. Rinkevicius, T. A. Ruden, K. Ruud, V. V. Rybkin, P. Sałek, C. C. M. Samson, A. S. de Merás, T. Saue, S. P. A. Sauer, B. Schimmelpfennig, K. Sneskov, A. H. Steindal, K. O. Sylvester-Hvid, P. R. Taylor, A. M. Teale, E. I. Tellgren, D. P. Tew, A. J. Thorvaldsen, L. Thøgersen, O. Vahtras, M. A. Watson, D. J. D. Wilson, M. Ziolkowski and H. Ågren, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013 DOI:10.1002/wcms.1172.
- K. Sugiyasu, C. Song and T. M. Swager, Macromolecules, 2006, 39, 5598–5600 CrossRef CAS.
- P. A. Peart and J. D. Tovar, J. Org. Chem., 2010, 75, 5689–5696 CrossRef CAS PubMed.
- T. A. Skotheim, R. L. Elsenbaumer and J. R. Reynolds, Handbook of Conducting Polymers, CRC Press, New York, 2007 Search PubMed.
- Q. Wang, Y. Qu, H. Tian, Y. Geng and F. Wang, Macromolecules, 2011, 44, 1256–1260 CrossRef CAS.
- B. Liu and S. K. Dishari, Chem. – Eur. J., 2008, 14, 7366–7375 CrossRef CAS PubMed.
- J. Li, Y. Zhao, H. S. Tan, Y. Guo, C.-A. Di, G. Yu, Y. Liu, M. Lin, S. H. Lim, Y. Zhou, H. Su and B. S. Ong, Sci. Rep., 2012, 2, 754 Search PubMed.
- A. K. Agrawal and S. A. Jenekhe, Chem. Mater., 1996, 8, 579–589 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tc32204a |
| This journal is © The Royal Society of Chemistry 2014 |