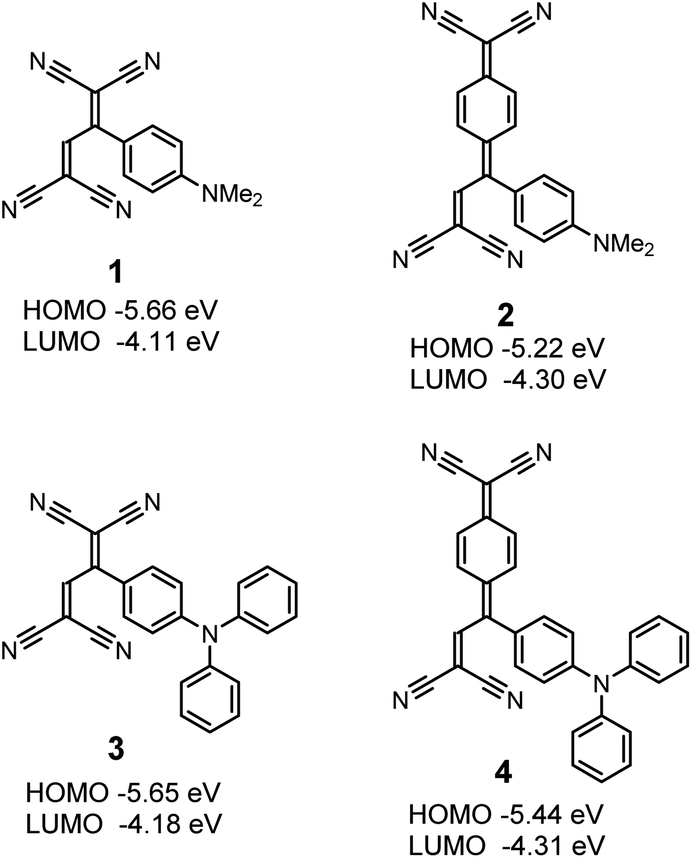
Novel design of organic donor–acceptor dyes without carboxylic acid anchoring groups for dye-sensitized solar cells†
Received
2nd November 2013
, Accepted 27th December 2013
First published on 6th January 2014
Abstract
Organic donor–acceptor dyes, formed by a high-yielding [2 + 2] cycloaddition–retroelectrocyclisation process between aniline-substituted alkynes and tetracyanoethylene (TCNE) or 7,7,8,8-tetracyanoquinodimethane (TCNQ), were employed as novel photosensitizers without carboxylic acid anchoring groups in dye-sensitized solar cells (DSSCs). The efficient adsorption of the donor–acceptor dyes onto TiO2 was confirmed by UV-vis and IR spectroscopies. The photovoltaic performances of the DSSCs suggested that the triphenylamine derivatives 3 and 4 provide higher current densities (Jsc) as compared to the corresponding dimethylaniline counter molecules 1 and 2. This was mainly due to the excellent charge-separation efficiencies and lower charge-recombination rates of the triphenylamine moieties. It was also found that the devices sensitized by the TCNQ-adducted dyes 2 and 4 display open-circuit voltages (Voc) higher than those of the TCNE-adducted counter dyes 1 and 3. All these results were reasonably explained by the J–V curve fitting based on the equivalent-circuit model as well as the comparison between the absorption and incident-photon-to-current-conversion efficiency (IPCE) spectra.
Introduction
Dye-sensitized solar cells (DSSCs) have attracted increasing attention due to their significant advantages of low cost, ease of manufacturing, and flexibility as compared to conventional silicon solar cells.1–6 The main component of DSSCs is the photosensitizer, which absorbs sunlight and generates electric charges. The high-performance sensitizers reported so far are Ru complexes, which show the maximum power conversion efficiencies (PCEs) exceeding 11%.7–9 However, the development of metal-free organic sensitizers has been ongoing because their physical properties, such as molar extinction coefficients and absorption ranges, can be controlled by their molecular design. The chemical structures of most efficient organic sensitizers consist of donor–π–acceptor systems bearing carboxylic acid-based anchoring groups at the acceptor site.10 The conventional synthetic routes of the donor–π–acceptor moieties are based on metal-catalysed cross-coupling reactions, and the most common acceptor/anchoring group is cyanoacrylic acid. In other words, there is a limitation when designing new acceptor units.11–18
Recently, a more straightforward synthetic approach to donor–π–acceptor systems was introduced. It is based on the formal [2 + 2] cycloaddition–retroelectrocyclisation reaction between activated alkynes and electron-poor alkenes.19–21 The alkyne activation is achieved by the substitution with electron-donating aromatic amines, allowing for the smooth progress of the reaction at room temperature in a click chemistry fashion.22–25 The common electron-poor alkenes in this reaction are tetracyanoethylene (TCNE) and 7,7,8,8-tetracyanoquinodimethane (TCNQ). The resulting TCNE-adducted structures are donor-substituted 1,1,4,4-tetracyanobuta-1,3-dienes (TCBDs), and the TCNQ-adducts are donor-substituted cyclohexa-2,5-diene-1,4-diylidene-expanded TCBDs (Fig. 1). The important features of these chromophores include intramolecular charge-transfer interactions, potent redox activities, and nonplanar molecular arrangements. It is expected that the last feature contributes to the suppression of dye aggregation in the DSSCs. In addition, the chromophores do not possess any carboxylic acid groups. However, it was recently reported that dicyanomethylene-based compounds, such as TCNE and TCNQ, form surface complexes with TiO2.26,27 This result suggests the possibility that polycyanated derivatives can efficiently adsorb onto TiO2. It is interesting to examine if this is also true for the tetracyanated donor–acceptor compounds. We now present a new design concept for organic sensitizers and report for the first time a detailed study of DSSCs based on donor-substituted TCBDs. Careful selection of both the donor and acceptor groups eventually led to the improvement of the photovoltaic performances.
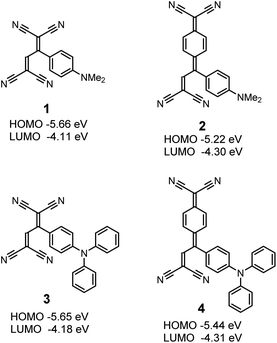 |
| | Fig. 1 Chemical structures and energy levels of organic donor–acceptor dyes 1–4. | |
Experimental section
Materials
All the reagents were purchased from Kanto, Tokyo Kasei, Wako, and Aldrich. 2-(4-(Dimethylamino)phenyl)buta-1,3-diene-1,1,4,4-tetracarbonitrile (1),28 2-(4-(3,3-dicyano-1-(4-(dimethylamino)phenyl)allylidene)cyclohexa-2,5-dien-1-ylidene)malononitrile (2),29 2-(4-(diphenylamino)phenyl)buta-1,3-diene-1,1,4,4-tetracarbonitrile (3),30 and 2-(4-(3,3-dicyano-1-(4-(diphenylamino)phenyl)allylidene)cyclohexa-2,5-dien-1-ylidene)malononitrile (4)30 were synthesized according to the reported methods. Although the reaction yields were almost quantitative, 1–4 were carefully purified by column chromatography (SiO2, CH2Cl2) in order to avoid any contaminations. The spectroscopic characterization data of 1–4 agreed with the reported data. In addition, the purity was confirmed by elemental analysis: 1 calcd (%) for C16H11N5 (273.30): C 70.32, H 4.06, N 25.63; found: C 70.42, H 4.41, N 25.21; 2 calcd (%) for C22H15N5 (349.40): C 75.63, H 4.33, N 20.04; found: C 75.68, H 4.83, N 19.49; 3 calcd (%) for C26H15N5 (397.44): C 78.57, H 3.80, N 17.62; found: C 78.56, H 4.33, N 17.11; 4 calcd (%) for C32H19N5 (473.54): C 81.17, H 4.04, N 14.79; found: C 79.30, H 4.14, N 16.56%.
Cell fabrication and characterization
The DSSC devices were fabricated as follows. A main transparent layer (15 μm) with titania particles (about 20 nm) and a scattering layer (10 μm) with titania particles (about 400 nm) were screen-printed onto the fluorine-doped tin oxide (FTO)-conducting glass substrate. The films were then sintered at 500 °C for 1 h. The thicknesses of the films were measured by a Surfcorder ET 4000 (Kosaka Laboratory, Ltd). The films were treated with a 0.1 M aqueous solution of HCl before examination. Coating of the titania films was carried out by immersion in a 3 × 10−4 M solution of the sensitizers in CH3CN/tBuOH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) for 24 h under a nitrogen atmosphere. Deoxycholic acid (DCA, 20 mmol) was added to the solution of the dye as a coadsorbent. The dye-covered TiO2 electrode and the Pt counter electrode were assembled into a sandwich-type cell and sealed with a hot-melt gasket (60 μm thickness) that was made of the ionomer Surlyn1702 (DuPont). Finally, the electrolyte, which consisted of 2 M LiI and 0.025 M I2 in CH3CN, was injected into the cell and sealed with a cover glass. The current–voltage characteristics were measured using an Advantest R6243 current–voltage unit after a 10 min wait time in order to achieve thermal equilibrium under AM 1.5G simulated solar light of 100 mW cm−2, irradiated from a WACOM super solar simulator, with a 0.25 cm2 mask to define the cell area.
:1, v/v) for 24 h under a nitrogen atmosphere. Deoxycholic acid (DCA, 20 mmol) was added to the solution of the dye as a coadsorbent. The dye-covered TiO2 electrode and the Pt counter electrode were assembled into a sandwich-type cell and sealed with a hot-melt gasket (60 μm thickness) that was made of the ionomer Surlyn1702 (DuPont). Finally, the electrolyte, which consisted of 2 M LiI and 0.025 M I2 in CH3CN, was injected into the cell and sealed with a cover glass. The current–voltage characteristics were measured using an Advantest R6243 current–voltage unit after a 10 min wait time in order to achieve thermal equilibrium under AM 1.5G simulated solar light of 100 mW cm−2, irradiated from a WACOM super solar simulator, with a 0.25 cm2 mask to define the cell area.
Results and discussion
Molecular design
Four different donor–acceptor dyes 1–4 were prepared by the formal [2 + 2] cycloaddition–retroelectrocyclisation reaction (Fig. 1). As previously reported, all compounds were obtained in quantitative yields at room temperature. The chemical structures were confirmed by NMR, IR, and MS spectrometry, which were consistent with the reported data. The longest wavelength absorption maxima (λmax) and redox potentials of 1–4 are summarized in Table S1.† Also, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels were estimated from the first oxidation potentials (Eox,1) and the first reduction potentials (Ered,1), respectively, under the assumption of Fc/Fc+ = −4.80 eV, and the values are shown in Fig. 1. The HOMO levels of the four dyes are in the range from −5.66 to −5.22 eV, which are sufficiently low as compared to the redox potential level of the iodide/triiodide electrolyte couple (−4.9 eV). This fact indicates the facilitated dye regeneration. On the other hand, the LUMO levels (−4.31 to −4.11 eV) are close to the conduction band of TiO2 (−4.3 eV), ensuring the formation of the surface complexes of the dyes and TiO2.26,27 A comparison of the acceptor moieties suggests that the LUMO levels of the TCNQ-adducts 2 and 4 are lower than those of the corresponding TCNE-adducts 1 and 3, respectively. This result reflects the strong electron-accepting power of TCNQ as compared to TCNE. The HOMO levels of 1 and 3 are almost the same, while 4 with the triphenylamine moiety shows a HOMO level lower than 2 with the dimethylaniline moiety. The lower HOMO or smaller ionization potential is a superior feature of the triphenylamine to dialkylaniline derivatives. Many triphenylamine frameworks have indeed been employed as hole-transporting materials in organic electroluminescence devices as well as stabilized cationic radicals of redox-active polymers.31–34
Dye adsorption onto TiO2
Since 1–4 do not possess any carboxylic acid anchoring groups, the adsorption behaviour on the TiO2 surface was first examined. The TiO2 powder does not show any visible absorption, but it became colourful when immersed into the dye solutions in CH3CN. It is known that both TCNE and TCNQ form surface complexes with TiO2, and this event newly produces a well-defined visible absorption ascribed to the charge-transfer from the TiO2 surface to TCNE or TCNQ.26,27 In contrast to these surface complexes, the donor–acceptor dyes 1–4 show visible absorption bands due to an intramolecular charge-transfer. Thus, the colours of TiO2 with organic dyes were similar to the original dye colours. However, the absorption bands became broadened and the end absorption bathochromically shifted when the dye adsorption onto TiO2 occurred (Fig. 2). The bathochromic shifts in the end absorption of the TCNQ-adducts were more significant than those of the corresponding TCNE-adducts, indicating the noticeable charge-transfer interactions due to the stronger electron-accepting characteristics. Moreover, 3 and 4 with the triphenylamine donor displayed a negligible bathochromic shift in λmax as compared to the corresponding dimethylaniline analogues 1 and 2, respectively. This might be due to the suppressed dye aggregation in terms of the propeller structure of the triphenylamine moiety. In addition to the absorption spectra, peak broadening was also observed in the IR spectra. For example, the cyano vibrational peak of 1 detected at 2212 cm−1 broadens on TiO2 (Fig. S1†). In the case of the surface complexes of highly symmetric and rigid dyes like TCNX (X = E or Q) on TiO2, the cyano vibrational peaks simply split due to the formation of coordinated and uncoordinated cyano groups.26,27 However, since the attached dimethylaniline and triphenylamine moieties break the symmetry, the cyano groups close to and far from the attached moieties give slightly different vibrational peaks and serve as the anchoring positions to provide the different chemical environments. Furthermore, it is possible to rotate the chemical structures around the single bond. Therefore, the dyes on TiO2 produce multiple peaks overlapping each other, resulting in the observed broad peak in the IR spectra.
 |
| | Fig. 2 Absorption spectra of organic dye (4 μm thickness)-adsorbed TiO2 films. | |
Photovoltaic performances of DSSCs
The chemically adsorbed dyes 1–4 on TiO2, as confirmed above, produce the photocurrent density–photovoltage (J–V) curves in the DSSCs (Fig. 3). The detailed photovoltaic data are listed in Table 1. The device sensitized by the triphenylamine-substituted TCBD 3 exhibited a current density (Jsc) of 0.65 mA cm−2, which is higher than that sensitized by the dimethylaniline-substituted TCBD 1 (0.12 mA cm−2). The TCNQ-adducts displayed a similar trend. The Jsc value of the device sensitized by the triphenylamine derivative 4 was 1.71 mA cm−2, while the device of the corresponding dimethylaniline analogue 2 displayed a Jsc of 1.41 mA cm−2. These results highlight the importance of the triphenylamine moiety as a donor unit. On the other hand, a comparison of the acceptor moieties clearly indicates the superiority of the TCNQ-adducts to the corresponding TCNE-adducts. In particular, the greater open-circuit voltages (Voc) of the TCNQ-adducts originate from the elongated molecular sizes and suitable energy levels.35,36 It is noteworthy that the Voc of the device sensitized by the TCNQ-adduct 2 is twice as high as that of the corresponding TCNE-adduct 1. Accordingly, the best photoconversion efficiency (PCE) of 0.25% was achieved for the device sensitized by the TCNQ-adduct 4 with the triphenylamine donor.
 |
| | Fig. 3 Photocurrent density–photovoltage (J–V) curves of DSSCs based on 1–4. | |
Table 1 Summary of DSSC performancesa
|
|
J
sc (mA cm−2) |
V
oc (V) |
FF |
PCE (%) |
|
0.25 cm2 TiO2 electrode composed of a transparent layer (15 μm) and scattering layer (10 μm) in CH3CN containing 2 M LiI and 0.025 M I2.
|
|
1
|
0.12 |
0.12 |
0.42 |
0.0058 |
|
2
|
1.41 |
0.24 |
0.61 |
0.20 |
|
3
|
0.65 |
0.19 |
0.52 |
0.063 |
|
4
|
1.71 |
0.24 |
0.61 |
0.25 |
Fig. 4 shows the action spectra of incident photon-to-current conversion efficiency (IPCE) as a function of the incident wavelength for the DSSCs. In good agreement with the absorption spectra shown in Fig. 2, the IPCE action spectra of the devices sensitized by the TCNQ-adducts are broader than those of the TCNE-adducts. This is due to the extended absorption range of the TCNQ-adducts. It should be noted that the IPCE of the TCNQ-adducts covers the entire visible region. In addition, the overall IPCE values of the TCNQ-adducts are significantly higher than those of the TCNE-adducts. Although the light absorption amount is not so different on the photoelectrode (Fig. 2) and the HOMO levels of all the dyes are low enough to accept electrons from the electrolyte, the TCNQ-adducts have more-expanded π electrons on the HOMO orbitals based on a density functional calculation.26 The expanded π orbitals would stabilize the oxidized states for the smooth electron relay to generate the photocurrent. Similarly, the triphenylamine moiety improves Jsc and IPCE due to the π-electron expansion on the HOMO orbitals and better redox properties. Note that reversible oxidation waves ascribed to the triphenylamine units of 3 and 4 are reported.30
 |
| | Fig. 4 IPCE spectra of DSSCs based on 1–4. | |
I–V parameter analysis
The J–V curves of the DSSCs based on 1–4 were analysed using the theoretical equation based on the equivalent-circuit model (eqn (1)):| | 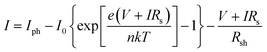 | (1) |
where I is the output electric current (I = 0.25J), V is the output electric voltage, e is the elementary charge, k is Boltzmann's constant, T is the absolute temperature, Iph is the photocurrent, I0 is the reverse saturation current of the diode, n is the ideality factor of the diode, Rs is the series resistance, and Rsh is the shunt resistance.37 By applying a fitting process to the J–V curves, the I–V parameters (Iph, I0, n, Rs, and Rsh) were obtained with a high accuracy. Assisted by some theoretical models or equations, e.g., Shockley equation for the p–n junction, the I–V parameters have been extensively used to analyse the physics in traditional semiconductor solar cells. Note that Jsc, Voc, and FF are convenient to characterise the J–V curve shapes and the PCE, but not appropriate to discuss the photovoltaic performances because the I–V parameters have a complex influence on the value of Jsc, Voc, and FF. To better understand the photovoltaic performances, the I–V parameter analysis is important in solar cell research. The validity of using the equivalent circuit to fit the J–V curves of organic solar cells and DSSCs was recently reported.38–40
Fig. 5 summarizes the I–V parameters obtained by fitting the J–V curves of the DSSCs based on 1–4. The Iph values increased when the dimethyaniline donor was replaced by the triphenylamine donor in both cases of the TCNE- (1 → 2) and TCNQ-adducts (3 → 4) (Fig. 5(a)). This increase in the Iph is due to the enhanced charge separation efficiencies at the interface between the dyes and TiO2 as already discussed. On the other hand, both of the diode parameter values, I0 and n, of the devices sensitized by the triphenylamine-based dyes were lower than those sensitized by the dimethylaniline-based dyes (Fig. 5(b) and (d)). Generally, it is difficult to determine whether or not the concept of suppressing the recombination current from TiO2 to the electrolyte functions from the values of Voc because Voc simultaneously correlates with Iph, I0, and n as follows:
| |  | (2) |
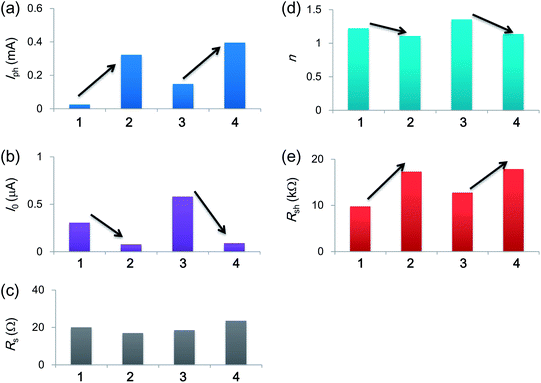 |
| | Fig. 5 Analysis results of the photocurrent density–photovoltage curves of DSSCs based on 1–4 using the equivalent-circuit model. | |
However, the decrease in I0 clearly suggests that the triphenylamine moiety suppresses the amount of recombination current to improve Voc as well as the enhanced Iph. The Rs values did not show any remarkable differences between the four devices, implying that the transparent electrode, counter electrode, and electrolyte solutions do not affect the photovoltaic parameters in the cell-assembling process (Fig. 5(c)). Although it is well known that the reduction of Rs improves FF,41 the triphenylamine moiety could realise the high FF without decreasing Rs. This improvement is explained by the increase in Voc based on the theoretical background of an ideal solar cell with no Rs and no Rsh; the relationship between Voc and FF is given as follows:42
| |  | (3) |
In contrast, a clear increase in the Rsh values occurred when the dimethylaniline donor was exchanged by the triphenylamine donor (Fig. 5(d)). This increase clearly reflects the decrease in the leak current, e.g., that from TiO2 to organic dyes. In short, the triphenylamine moiety regulates the current on the photovoltaic interface to enhance the photoinduced current and to suppress the recombination and leak currents. All these results again support the significant utility of the triphenylamine unit as a donor moiety of DSSC dyes even when using the new anchoring group.43–47
Conclusions
New types of organic donor–acceptor dyes for DSSCs were introduced. They can be synthesized by click chemistry reactions, enabling large scale production at a low cost. The tetracyanated acceptor moieties of the dyes were efficiently attached to the TiO2 surface without special treatments. In particular, the expanded tetracyanated acceptor, originating from TCNQ, showed better photovoltaic performances than the simple TCBD analogue. Moreover, it was demonstrated that the use of the triphenylamine donor offers significant advantages, such as enhanced charge separation efficiencies, a lower charge recombination, and suppressed dye aggregation. These are important design concepts of the donor and acceptor moieties. Further development of effective π-spacers that control the electronic communication between the donor and acceptor moieties will lead to high performance organic dyes.
Acknowledgements
This work is partially supported by a Grant-in-Aid for Scientific Research from MEXT, Japan (no. 22685023), the Konica Minolta Science and Technology Foundation, and the Murata Science Foundation (T. M.). We acknowledge R. Yoshida (NIMS) for his technical assistance and S. Genseki (Center for Advanced Materials Analysis, Tokyo Institute of Technology) for the elemental analyses.
Notes and references
- A. Hagfeldt, G. Boschloo, L. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595–6663 CrossRef CAS PubMed
 .
.
- A. Listorti, B. O'Regan and J. R. Durrant, Chem. Mater., 2011, 23, 3381–3399 CrossRef CAS .
- M. Grätzel, R. A. J. Janssen, D. B. Mitzi and E. H. Sargent, Nature, 2012, 488, 304–312 CrossRef PubMed .
- B. E. Hardin, H. J. Snaith and M. D. McGehee, Nat. Photonics, 2012, 6, 162–169 CrossRef CAS .
- C. Qin, W.-Y. Wong and L. Han, Chem. – Asian J., 2013, 8, 1706–1719 CrossRef CAS PubMed .
- S. Zhang, Y. Xudong, Y. Numata and L. Han, Energy Environ. Sci., 2013, l6, 1443–1464 Search PubMed .
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629–634 CrossRef CAS PubMed .
- T. Kinoshita, J. T. Dy, S. Uchida, T. Kubo and H. Segawa, Nat. Photonics, 2012, 7, 535–539 CrossRef .
- L. Han, A. Islam, H. Chen, C. Malapaka, B. Chiranjeevi, S. Zhang, X. Yang and M. Yanagida, Energy Environ. Sci., 2012, 5, 6057–6060 CAS .
- J. N. Clifford, E. Martínez-Ferrero, A. Viterisi and E. Palomares, Chem. Soc. Rev., 2011, 40, 1635–1646 RSC .
- Y. Ooyama, S. Inoue, T. Nagano, K. Kushimoto, J. Ohshita, I. Imae, K. Komaguchi and Y. Harima, Angew. Chem., Int. Ed., 2011, 50, 7429–7433 CrossRef CAS PubMed .
- Y. Ooyama, T. Nagano, S. Inoue, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. – Eur. J., 2011, 17, 14837–14843 CrossRef CAS PubMed .
- M. Katono, T. Bessho, S. Meng, R. Humphry-Baker, G. Rothenberger, S. M. Zakeeruddin, E. Kaxiras and M. Grätzel, Langmuir, 2011, 27, 14248–14252 CrossRef CAS PubMed .
- J. Mao, N. He, Z. Ning, Q. Zhang, F. Guo, L. Chen, W. Wu, J. Hua and H. Tian, Angew. Chem., Int. Ed., 2012, 51, 9873–9876 CrossRef CAS PubMed .
- L. Macor, M. Gervaldo, F. Fungo, L. Otero, T. Dittrich, C.-Y. Lin, L.-C. Chi, F.-C. Fang, S.-W. Lii, K.-T. Wong, C.-H. Tsai and C.-C. Wu, RSC Adv., 2012, 2, 4869–4878 RSC .
- J. Zhao, X. Yang, M. Cheng, S. Li and L. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 5227–5231 CAS .
- Y. Ooyama, N. Yamaguchi, I. Imae, K. Komaguchi, J. Ohshita and Y. Harima, Chem. Commun., 2013, 49, 2548–2550 RSC .
- Y. Ooyama, Y. Hagiwara, T. Mizumo, Y. Harima and J. Ohshita, New J. Chem., 2013, 37, 2479–2485 RSC .
- M. Kivala and F. Diederich, Acc. Chem. Res., 2009, 42, 235–248 CrossRef CAS PubMed .
- S.-i. Kato and F. Diederich, Chem. Commun., 2010, 46, 1994–2006 RSC .
- T. Michinobu, Chem. Soc. Rev., 2011, 40, 2306–2316 RSC .
- T. Michinobu, C. Boudon, J.-P. Gisselbrecht, P. Seiler, B. Frank, N. N. P. Moonen, M. Gross and F. Diederich, Chem. – Eur. J., 2006, 12, 1889–1905 CrossRef CAS PubMed .
- M. Morimoto, K. Murata and T. Michinobu, Chem. Commun., 2011, 47, 9819–9821 RSC .
- Y.-L. Wu, M. C. Stuparu, C. Boudon, J.-P. Gisselbrecht, W. B. Schweizer, K. K. Baldridge, J. S. Siegel and F. Diederich, J. Org. Chem., 2012, 77, 11014–11026 CrossRef CAS PubMed .
- M. Štefko, M. D. Tzirakis, B. Breiten, M.-O. Ebert, O. Dumele, W. B. Schweizer, J.-P. Gisselbrecht, C. Boudon, M. T. Beels, I. Biaggio and F. Diederich, Chem. – Eur. J., 2013, 19, 12693–12704 CrossRef PubMed .
- R. Jono, J.-i. Fujisawa, H. Segawa and K. Yamashita, J. Phys. Chem. Lett., 2011, 2, 1167–1170 CrossRef CAS .
- S. Manzhos, R. Jono, K. Yamashita, J.-i. Fujisawa, M. Nagata and H. Segawa, J. Phys. Chem. C, 2011, 115, 21487–21493 CAS .
- T. Michinobu, J. C. May, J. H. Lim, C. Boudon, J.-P. Gisselbrecht, P. Seiler, M. Gross, I. Biaggio and F. Diederich, Chem. Commun., 2005, 737–739 RSC .
- M. Kivala, C. Boudon, J.-P. Gisselbrecht, P. Seiler and F. Diederich, Chem. Commun., 2007, 4731–4733 RSC .
- X. Tang, W. Liu, J. Wu, C.-S. Lee, J. You and P. Wang, J. Org. Chem., 2010, 75, 7273–7278 CrossRef CAS PubMed .
- Y. Shirota and H. Kageyama, Chem. Rev., 2007, 107, 953–1010 CrossRef CAS PubMed .
- T. Michinobu, H. Kumazawa, E. Otsuki, H. Usui and K. Shigehara, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 3880–3891 CrossRef CAS .
- H.-J. Yen and G.-S. Liou, Polym. Chem., 2012, 3, 255–264 RSC .
- Y. Li and T. Michinobu, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 2111–2120 CrossRef CAS .
- A. Reynal, A. Forneli, E. Martinez-Ferrero, A. Sánchez-Díaz, A. Vidal-Ferran, B. C. O'Regan and E. Palomares, J. Am. Chem. Soc., 2008, 130, 13558–13567 CrossRef CAS PubMed .
- M. Miyashita, K. Sunahara, T. Nishikawa, Y. Uemura, N. Koumura, K. Hara, A. Mori, T. Abe, E. Suzuki and S. Mori, J. Am. Chem. Soc., 2008, 130, 17874–17881 CrossRef CAS PubMed .
- J. H. Cai, N. Satoh, M. Yanagida and L. Y. Han, J. Nonlinear Opt. Phys. Mater., 2010, 19, 637–643 CrossRef CAS .
- J. Cai, N. Satoh, M. Yanagida and L. Han, Rev. Sci. Instrum., 2009, 80, 115111 CrossRef PubMed .
- J. Cai, N. Satoh and L. Han, J. Phys. Chem. C, 2011, 115, 6033–6039 CAS .
- N. Satoh and L. Han, Phys. Chem. Chem. Phys., 2012, 14, 16014–16022 RSC .
- L. Han, N. Koide, Y. Chiba, A. Islam, R. Komiya, N. Fuke, A. Fukui and R. Yamanaka, Appl. Phys. Lett., 2005, 86, 213501 CrossRef PubMed .
-
P. Würfel, in Physics of Solar Cells: From Basic Principles to Advanced Concepts, Wiley-VCH, Weinheim, 2007, pp. 155–175 Search PubMed .
- U. Bach, D. Lupo, P. Comte, J. E. Moser, F. Weissörtel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef CAS PubMed .
- N. Hirata, J.-J. Lagref, E. J. Palomares, J. R. Durrant, M. K. Nazeeruddin, M. Gratzel and D. D. Censo, Chem. – Eur. J., 2004, 10, 595–602 CrossRef CAS PubMed .
- N. Satoh, T. Nakashima and K. Yamamoto, J. Am. Chem. Soc., 2005, 127, 13030–13038 CrossRef CAS PubMed .
- H. J. Snaith, S. M. Zakeeruddin, Q. Wang, P. Péchy and M. Grätzel, Nano Lett., 2006, 6, 2000–2003 CrossRef CAS PubMed .
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453–3488 RSC .
Footnotes |
| † Electronic supplementary information (ESI) available: Optical and electrochemical data of 1–4 and IR spectra. See DOI: 10.1039/c3tc32165g |
| ‡ Current address: Suzhou Institute of Nano-tech and Nano-Bionics, Chinese Academy of Science, no. 398 Ruoshui Road, SEID, Suzhou Industrial Park, Suzhou, Jiangsu Province, 215125, China. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a