Host–guest blue light-emitting electrochemical cells†
Antonio
Pertegás
a,
Nail M.
Shavaleev
*b,
Daniel
Tordera
a,
Enrique
Ortí
a,
Mohammad K.
Nazeeruddin
b and
Henk J.
Bolink
*a
aInstituto de Ciencia Molecular, Universidad de Valencia, C/Catedrático J. Beltrán 2, ES-46980 Paterna, Valencia, Spain. E-mail: henk.bolink@uv.es
bLaboratory of Photonics and Interfaces, Institute of Chemical Sciences and Engineering, École Polytechnique Fédérale de Lausanne, CH-1015 Lausanne, Switzerland. E-mail: shava@mail.ru; Fax: +41 21 693 4111; Tel: +41 21 693 6124
First published on 15th January 2014
Abstract
Carbazole, a commonly used hole-transporter for organic electronics, has been modified with an imidazolium cation and a hexafluorophosphate counter-anion to give an ionic hole-transporter. It has been applied as one of the hosts in a host–guest blue light-emitting electrochemical cell (LEC) with the neutral blue emitter FIrPic. We have obtained efficient and bright blue LECs with an electroluminescence maximum at 474 nm and efficacy of 5 cd A−1 at a luminance of 420 cd m−2, thereby demonstrating the potential of the ionic organic charge-transporters and of the host–guest architecture for LECs.
Introduction
Electroluminescent devices using organic semiconductors are becoming a serious alternative to conventional inorganic technology as their efficiencies and stabilities have improved significantly over the last few years.1 The most efficient and stable organic light-emitting devices (OLEDs) are based on a multi-stack of low molecular-weight components that use air-sensitive charge-injection layers.2 The multi-layer architecture is obtained by sequentially evaporating the active species under vacuum. OLEDs require rigorous encapsulation to prevent degradation of the charge-injection layers.3,4Another type of electroluminescent device, referred to as the light-emitting electrochemical cell (LEC), has a simpler architecture and does not rely on air-sensitive charge-injection layers,5–10 which simplifies its preparation and makes it more cost-efficient. In its simplest form, the LEC consists of a single emitting layer of either an ionic transition-metal complex (iTMC)9 or a neutral light-emitting material (usually a polymer) mixed with an ionic transporter and a salt.8,10 The presence of mobile ions facilitates the formation of ionic junctions that lower the barrier for charge injection and make the LEC independent of the work function of the electrode material.11–17
Despite these advances, the lack of efficient blue LECs remains a problem. Although several blue LECs have been reported, they exhibit low efficiency, luminance and lifetime.18–23 Efficacies of up to 18.3 cd A−1 at a luminance of 14.5 cd m−2 have been reported for blue-green LECs.22 For deeper-blue emission, the performances are worse: 2.6 cd A−1 at 5.3 cd m−2 for a sky-blue LEC23 and 0.65 cd A−1 at 39 cd m−2 for the bluest LEC reported so far.19 With polymers as the active material in a tri-layer structure, an efficient LEC was made (5.3 cd A−1).24 However, in general, the stability of blue LECs is low, from minutes to a few hours; moreover, their colour stability is rarely discussed.
In iTMC–LECs, the iTMC is the only component involved both in charge transport and in emission. With blue LECs, these processes are challenging, because they involve high-energy excitons (blue emitters have a large energy gap between the lowest unoccupied and the highest occupied molecular orbitals). The same problem occurs in OLEDs, which is a reason for the continued search for stable and efficient blue emitters. In OLEDs, charge transport is usually not performed by the emitter but by the specially designed electron- and hole-transporters.25 In contrast, in LECs, charge transport occurs via the emitter and involves its reduction and oxidation. Because charge transport in blue LECs forms high-energy species, efficient blue LECs are hard-to-make. Hence, in LECs, it is of interest to decouple the charge transport and the emission by using different molecules for these processes. LECs rely on ionic movement to reduce the charge injection barriers; therefore, in the LEC, the charge-transporter must be mixed with the emitter and, more importantly, the ionic movement must be maintained; for example, host–guest orange and red LECs have been made with moderate band-gap iTMCs as the host.26–29
Here, we report a wide band-gap ionic hole-transporter NMS25 suitable for blue light-emitting guests. NMS25 is an aryl-carbazole modified with an imidazolium cation and a hexafluorophosphate counter-anion (Scheme 1). NMS25 was mixed with a neutral polar electron-transporter SPPO13 and a neutral blue-phosphorescent iridium(III) emitter FIrPic (Scheme 1). We show that the combination of these low molecular weight compounds, when they are sandwiched between two air-stable electrodes, gives efficient blue-electroluminescent devices that function as LECs, with a slow turn-on determined by the movement of ions, and that reach an efficacy of 5 cd A−1 at a luminance of 420 cd m−2.
 | ||
| Scheme 1 Synthesis of NMS25: (a) AlCl3, tert-butyl chloride, CH2Cl2, under Ar, 0 °C to RT; (b) 3-iodoanisole, Cs2CO3, Cu2O, DMF, under Ar, 120 °C; (c) pyridine hydrochloride, under Ar, 200 °C; (d) 1, K2CO3, DMF, under Ar, 60 °C. Structures of SPPO13 and FIrPic. | ||
Results and discussion
Synthesis and characterization
NMS25 was prepared in a multi-step procedure, which was up-scaled to give up to 4 g of the product (Scheme 1). In NMS25, the tert-butyl groups were added to improve the solubility and to block electrochemically-reactive C3 and C6 positions of the carbazole; a long hexyloxy-chain was introduced to prevent interaction of the imidazolium cation with the carbazole. Imidazolium was chosen because it is optically-transparent and electrochemically-inert. NMS25 is a white solid that is soluble in polar organic solvents; it exhibits electronic absorption with a cut-off at 365 nm in dichloromethane solution (Fig. S1, ESI†). The redox potentials for NMS25 were measured by cyclic voltammetry. In acetonitrile, NMS25 undergoes reversible oxidation of the carbazole at 0.77 V (against ferrocene couple), confirming its hole-transport properties, but no reduction down to −2.7 V (Fig. S2, ESI†).Electroluminescence devices
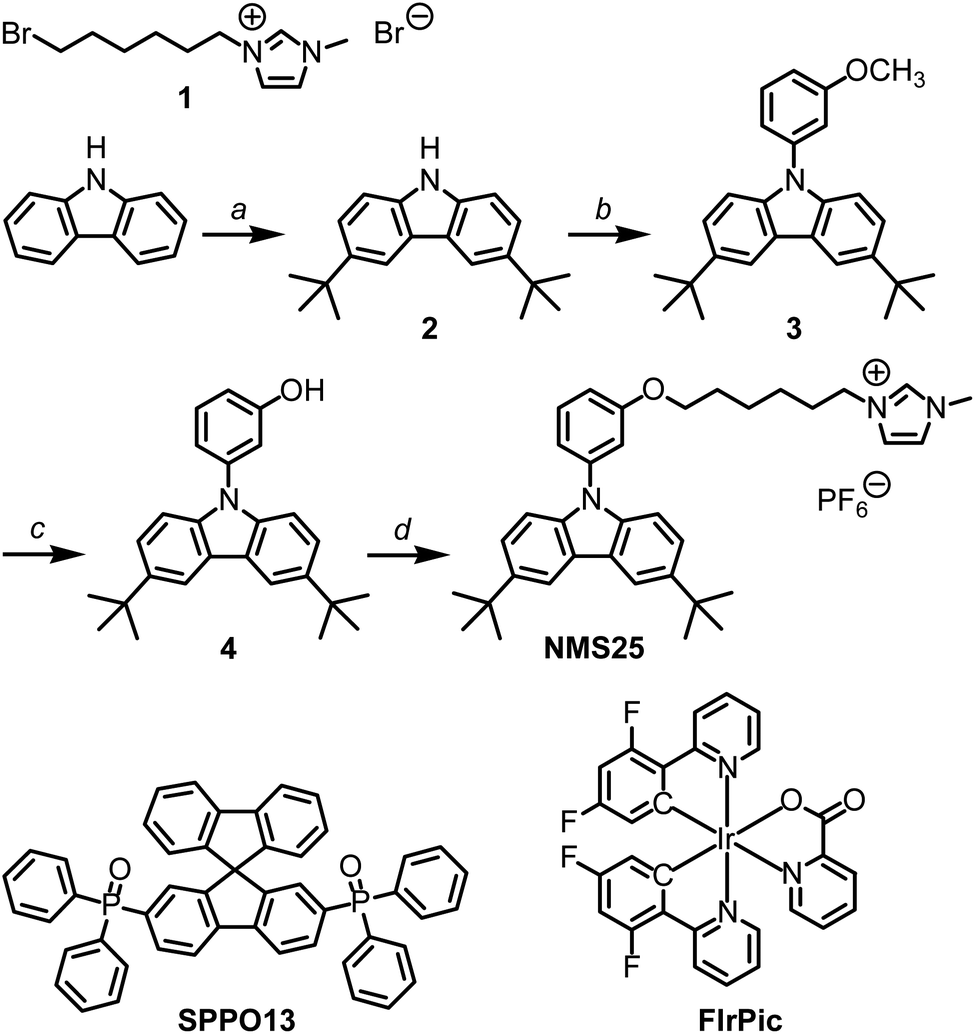
Two-layer LECs were prepared by spin-coating from solution. An 80 nm layer of PEDOT:PSS was spin-coated on ITO-glass to increase the reproducibility of the devices. Then, an 80 nm layer of NMS25, SPPO13 and FIrPic was deposited from an anisole solution. Aluminium was used as the top electrode.To find the optimum ratio of hole- and electron-transporters, we evaluated devices with various mass ratios of NMS25:SPPO13 (matrix; Fig. 1). The device configuration was: ITO/PEDOT:PSS (80 nm)/matrix:FIrPic (80 nm; with 10 wt% FIrPic) with the ionic liquid (IL) 1-butyl-3-methylimidazolium tetrafluoroborate added in a 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of matrix:IL/Al. The LECs were run with a block-wave pulsed current at a frequency of 1000 Hz and a duty cycle of 50% at an average density of 100 A m−2.
:1 molar ratio of matrix:IL/Al. The LECs were run with a block-wave pulsed current at a frequency of 1000 Hz and a duty cycle of 50% at an average density of 100 A m−2.
 | ||
| Fig. 1 Time-dependence of voltage and luminance for LECs with various mass ratios of NMS25:SPPO13 (matrix) with 10 wt% FIrPic and with a 4:1 molar ratio matrix:IL 80 nm emitting layer (block-wave pulsed current; 1000 Hz; 50% duty cycle; average density 100 A m−2). | ||
All of the devices exhibit blue electroluminescence from FIrPic with a maximum at 474 nm and with the typical characteristics of LECs (Fig. 1–4). In pulsed-current mode, the LECs exhibit a rapid decrease of the initially high driving voltage to a lower steady-state value (Fig. 1–4). This is caused by the decrease in injection barriers for electrons and holes as ions migrate to the electrodes. The maximum luminance (Lmax) and the time when it is reached (tmax) vary significantly for the different LECs, thereby complicating the lifetime comparison (as lifetime, for example, defined by the time it takes for the luminance to decrease to half of its maximum value, t1/2, counted from t = 0, depends on the luminance). Kalyuzhny et al. suggested30 to characterize the LEC by the total emitted energy (Etot) up to the time when the luminance decreases to the 1/5 of its maximum.
Table 1 summarizes the performance of LECs. The LEC with the longest lifetime and the highest total emitted energy had an emitting layer with a 1:1 mass ratio of NMS25:SPPO13 (Table 1); its steady-state driving voltage was the lowest, indicating optimized charge-injection/transport (Fig. 1). Hence, this LEC was chosen for optimization.
| NMS25:SPPO13 (mass) | t max (min) | L max (cd m−2) | t 1/2 (h) | E tot (J) | Effmax (cd A−1) | EQEmax (%) |
|---|---|---|---|---|---|---|
| 0:1 |
0.4 | 176 | 0.1 | 0.01 | 1.7 | 0.6 |
| 0.5:9.5 |
6.6 | 165 | 0.6 | 0.02 | 1.6 | 0.6 |
| 2.5:7.5 |
4.2 | 205 | 1.1 | 0.03 | 1.9 | 0.8 |
| 3.5:6.5 |
14 | 124 | 3.9 | 0.07 | 1.2 | 0.5 |
| 1:1 |
41 | 67 | 14 | 0.13 | 0.6 | 0.3 |
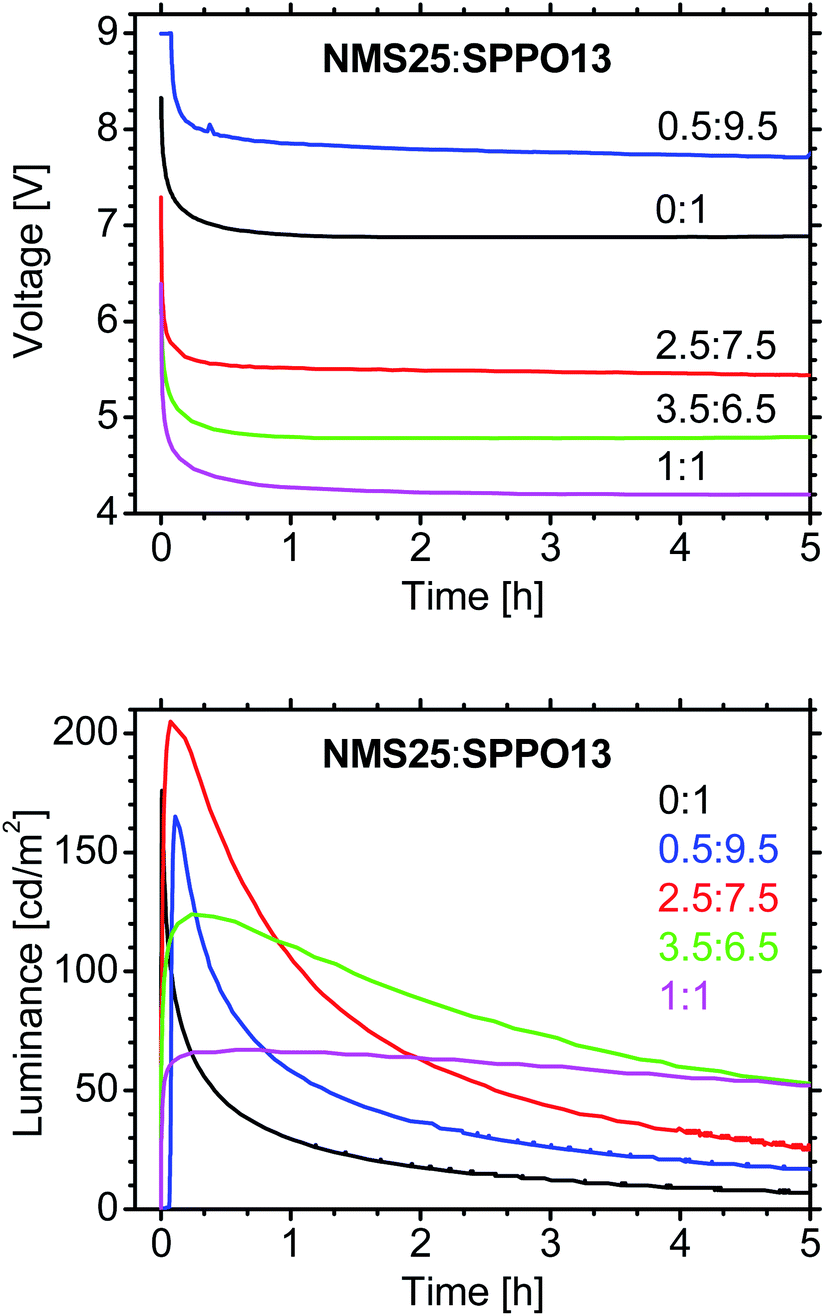
The thickness of the emitting layer changes the LEC performance;20 therefore, we varied it from 40 to 80 and to 100 nm (Fig. 2). Although the driving voltage increases for thicker layers, the highest luminance was achieved with an 80 nm layer (Fig. 2). The driving conditions were evaluated by changing the average current density31 from 25 to 100 A m−2, with the optimum determined to be 75 A m−2.
 | ||
| Fig. 2 Time-dependence of voltage and luminance for LECs with various thicknesses of the emitting layer: NMS25:SPPO13 (1:1 by mass) with 10 wt% FIrPic (without IL); pulsed current (1000 Hz; 50% duty cycle; average density 100 A m−2). | ||
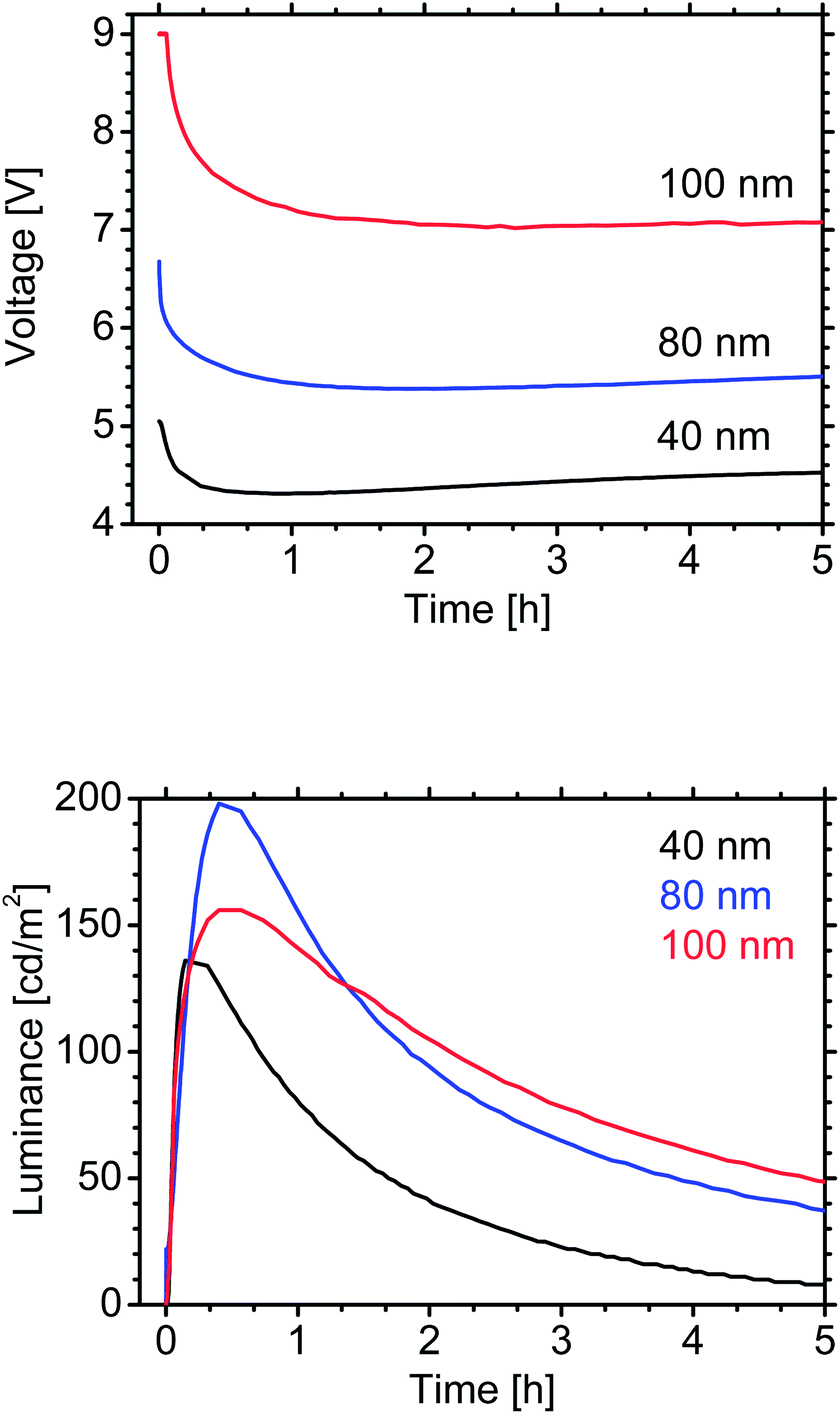
There are two salts in the optimized LEC: the ionic hole-transporter and the IL. A device was also prepared without IL to confirm that it can function as a LEC by using only the ions from NMS25 (Fig. 3). The LEC without the IL exhibits a higher maximum luminance and efficiency, but a higher driving voltage and a lower stability of the luminance than does the LEC with the IL (Fig. 3).
 | ||
| Fig. 3 Time-dependence of voltage and luminance for LECs with and without an ionic liquid (IL; in molar ratio): 80 nm emitting layer; NMS25:SPPO13 (matrix; 1:1 by mass) with 10 wt% FIrPic; pulsed current (1000 Hz; 50% duty cycle; average density 100 A m−2). | ||
Here, we demonstrate the potential of the host–guest LECs in terms of efficiency and luminance (in previous reports,19,23 the stability of blue LECs was low and the focus was on the efficiency). The LEC (without the IL) was optimized by annealing the emitting layer for 1 h at 100 °C prior to the evaporation of the top contact (the annealing changes the morphology and increases the photoluminescence quantum yield of the active layer32); the thus manufactured blue LEC exhibits a current efficacy of 5 cd A−1 at a luminance of 420 cd m−2 (Fig. 4). These values are significantly higher than are those previously reported for blue LECs.19,21,23 The lifetime of the optimized LEC (without the IL) is short (Fig. 4); however, it is possible to increase it by adding the IL (Fig. 3 and Table 1).
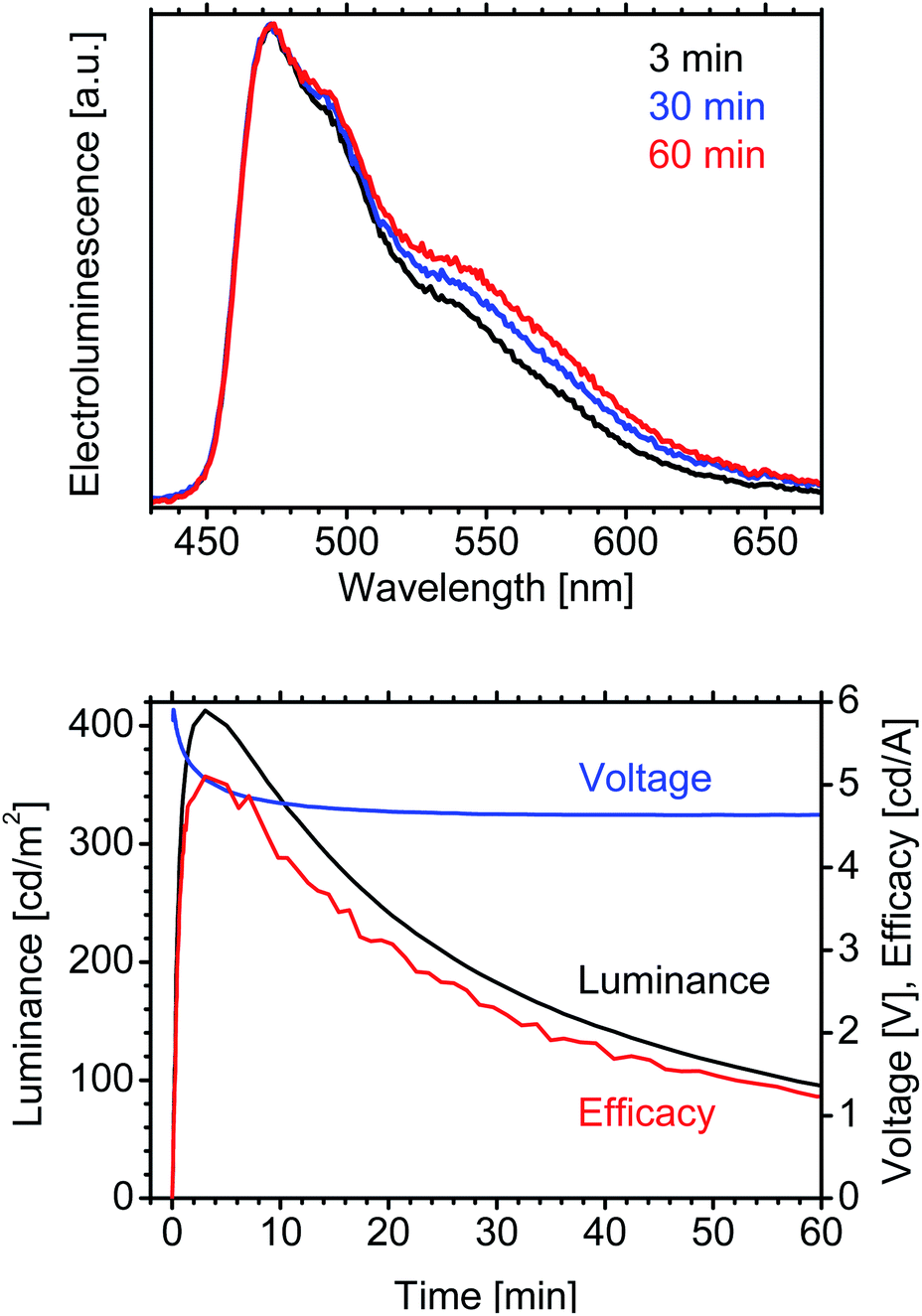 | ||
| Fig. 4 Time-dependence of electroluminescence spectrum, luminance, voltage and efficacy of optimized LEC (1:1 by mass of NMS25:SPPO13 with 10 wt% FIrPic; 80 nm emitting layer; annealed at 100 °C for 1 h; without IL; block-wave pulsed current; 1000 Hz; 50% duty cycle; average density 75 A m−2). | ||
In the optimized device, FIrPic exhibits only small red-shift of electroluminescence, that is, the host–guest LEC gives stable blue electroluminescence during the 60 min of work (Fig. 4 and Table 2). In contrast, in previous reports, the electroluminescence of blue iTMC–LECs underwent significant red-shift.33,34
| t (min) | CIE (x) | CIE (y) | λ max (nm) |
|---|---|---|---|
| 3 | 0.214 | 0.377 | 473 |
| 5 | 0.217 | 0.379 | 474 |
| 10 | 0.219 | 0.380 | 474 |
| 30 | 0.230 | 0.389 | 473 |
| 60 | 0.242 | 0.400 | 474 |
Conclusions
Ionic derivatives of charge-transport hosts are successfully applied in host–guest LECs. The performance of these LECs depends on the amount and type of the ions. For a blue LEC, we achieve high colour stability and a maximum efficacy of 5 cd A−1 at a luminance of 420 cd m−2. One can modify neutral charge-transporters with a suitable ionic group to optimize their performance in organic electronics.Experimental
Materials and methods
Purification and handling of all compounds were carried out under air. All products were stored in the dark. Chemicals from commercial suppliers were used without purification. Chromatography was performed on a column with an i.d. of 30 mm on silica gel 60 (Fluka, Nr 60752). The progress of reactions and the elution of products were followed on TLC plates (silica gel 60 F254 on aluminum sheets, Merck).Elemental analyses were performed by Dr E. Solari, Service for Elemental Analysis, Institute of Chemical Sciences and Engineering (ISIC EPFL). 1H and 13C NMR spectra were recorded with Bruker AV400 (400 MHz) and AVIII-400 (400 MHz) spectrometers. Mass spectra were recorded with Q-TOF Ultima (Waters) and TSQ7000 (Thermo Fisher) spectrometers (Mass-Spectroscopy Service, ISIC EPFL).
Synthesis of NMS25
Scaled-up synthesis. 1-Methylimidazole (2 mL, 2.06 g, 25 mmol) and 1,6-dibromohexane (15 mL, 24 g, 97 mmol, excess) in acetone (25 mL) gave 6 g (18.4 mmol, 74%) of the product. Chromatography was performed on silica (30 g) with 0–1% CH3OH in CH2Cl2 to remove the starting material and with 7.5% CH3OH in CH2Cl2 to recover 1.
Scaled-up synthesis. 3-Iodoanisole (1.8 mL, 3.53 g, 15.1 mmol), di-tert-butyl carbazole 2 (4 g, 14.3 mmol), Cs2CO3 (17 g, 52 mmol), Cu2O powder (296 mg, 1.55 mmol) in DMF (12 mL; 99.8%, extra dry over molecular sieve, AcroSeal, Acros) were stirred at 120 °C for 24 h. Chromatography on silica (20 g) and re-crystallization from CH2Cl2 and ethanol (20 mL) gave 4.6 g (11.9 mmol, 83%) of 3.
Scaled-up synthesis. Aryl carbazole 3 (2.12 g, 5.49 mmol) and Py·HCl (40 g, 0.35 mol) were stirred at 200 °C for 7.5 h. Chromatography on silica (30 g) gave 1.25 g (3.36 mmol, 61%) of 4.
Scaled-up synthesis. Aryl carbazole 3 (4.6 g, 11.9 mmol) and Py·HCl (80 g, 0.69 mol) were stirred at 200 °C for 8 h. Chromatography on silica (30 g) gave 3.18 g (8.56 mmol, 72%) of 4.
Batch 1. The reaction was performed under argon in dry solvents. Phenol-carbazole 4 (241 mg, 0.65 mmol), imidazolium salt 1 (212 mg, 0.65 mmol), and K2CO3 (90 mg, 0.65 mmol) were stirred in DMF (4 mL; 99.8%, extra dry over molecular sieve, AcroSeal, Acros) at 60 °C for 24 h to give a white suspension. It was diluted with water (100 mL) containing KPF6 (for anion exchange; 0.55 g, 2.99 mmol, excess). The resulting suspension was stirred for 30 min and filtered. The solid was washed with water and hexane. It was extracted with water–CH2Cl2. The organic layer was washed with water and evaporated. Chromatography was performed on silica (15 g) with 0.5% CH3OH in CH2Cl2 to remove impurities and with 2% CH3OH in CH2Cl2 to recover the pure product. The product may separate as thick and easily foaming oil; in this case, mixing with ether and sonication followed by evaporation of the solvent converts the oil to a solid. White solid: 300 mg (0.44 mmol, 68%). Anal. calcd for C36H46F6N3OP (MW 681.73): C, 63.42; H, 6.80; N, 6.16. Found: C, 63.66; H, 6.84; N, 6.24. 1H NMR (400 MHz, CD2Cl2): δ = 8.55 (s, 1H), 8.18 (d, J = 2.0 Hz, 2H), 7.55–7.47 (m, 3H), 7.40 (d, J = 8.4 Hz, 2H), 7.25 (s, br, 1H), 7.22 (s, br, 1H), 7.17 (dd, J = 7.6, 1.2 Hz, 1H), 7.09 (t, J = 2.4 Hz, 1H), 7.00 (dd, J = 8.0, 2.0 Hz, 1H), 4.20 (t, J = 7.2 Hz, 2H), 4.04 (t, J = 6.0 Hz, 2H), 3.94 (s, 3H), 2.01–1.91 (m, 2H), 1.90–1.80 (m, 2H), 1.65–1.40 (m, 4H, obscured by signal of water protons), 1.48 (s, 18H) ppm. 1H NMR (400 MHz, DMSO-d6): δ = 9.08 (s, 1H), 8.28 (d, J = 2.0 Hz, 2H), 7.76 (t, J = 1.6 Hz, 1H), 7.67 (t, J = 1.6 Hz, 1H), 7.54 (t, J = 8.0 Hz, 1H), 7.47 (dd, J = 8.4, 1.6 Hz, 2H), 7.32 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 7.6 Hz, 1H), 7.10 (t, J = 2.0 Hz, 1H), 7.04 (dd, J = 8.4, 2.4 Hz, 1H), 4.15 (t, J = 6.8 Hz, 2H), 4.04 (t, J = 6.4 Hz, 2H), 3.82 (s, 3H), 1.86–1.69 (m, 4H), 1.41 (s, 18H), 1.52–1.20 (m, 2H), 1.37–1.26 (m, 2H) ppm. 13C NMR (100 MHz, DMSO-d6): δ = 160.6, 143.2, 139.2, 139.2, 137.2, 131.5, 124.3, 124.3, 123.6, 122.9, 118.9, 117.3, 114.3, 112.7, 109.8, 68.3, 49.4, 36.4, 35.1, 32.5, 30.0, 29.1, 26.0, 25.6 ppm. ESI+ TOF MS: m/z 536.36 ({M − PF6}+, 100%).
Batch 2, scaled-up synthesis. Phenol-carbazole 4 (1.25 g, 3.36 mmol), imidazolium salt 1 (1.10 g, 3.37 mmol), and K2CO3 (0.47 g, 3.40 mmol) in DMF (8 mL; 99.8%, extra dry over molecular sieve, AcroSeal, Acros) at 60 °C for 24 h gave a pale orange suspension. Work-up with KPF6 (for anion exchange; 4 g, 22 mmol) and chromatography on silica (35 g) with 0.5–2.0% CH3OH in CH2Cl2 (pale green impurity precedes the product) gave 1.45 g of the product that contained a small amount of impurity. The product was dissolved in CH2Cl2 (5 mL) and poured into ether (>200 mL). A solution formed at first, but a fine dense precipitate appeared after 1 min of stirring. It was filtered and washed with ether to provide the pure product: 1.38 g (2.03 mmol, 60%).
Batch 3, scaled-up synthesis. Phenol-carbazole 4 (3.18 g, 8.56 mmol), imidazolium salt 1 (2.79 g, 8.56 mmol), and K2CO3 (1.20 g, 8.68 mmol) in DMF (9 mL; 99.8%, Extra Dry over Molecular Sieve, AcroSeal, Acros) at 60 °C for 24 h gave a pale orange suspension. Work-up was performed with KPF6 (for anion exchange; 13 g, 71 mmol) and by chromatography on silica (30 g) with 0.5–2.0% CH3OH in CH2Cl2. The product was dissolved in CH2Cl2 (15 mL) and poured into ether (>350 mL). A solution formed at first, but a fine dense precipitate appeared after 1 min of stirring. It was filtered and washed with ether to provide the pure product: 4.17 g (6.12 mmol, 71%).
The LEC devices were made as follows. Indium tin oxide ITO-coated glass plates were patterned by conventional photolithography (Naranjo Substrates). The substrates were cleaned ultrasonically in water-soap, water and 2-propanol baths. After drying, the substrates were placed in a UV–ozone cleaner (Jelight 42-220) for 20 min. An 80 nm layer of PEDOT:PSS was spin-coated on the ITO-glass substrate. For the emitting layer preparation, the devices were spin-coated at 1000 rpm for 30 s from an anisole solution containing hosts (with 10% wt of FIrPic). To find the optimum ratio of NMS25:SPPO13 (matrix), we employed solutions with mass ratios of 1:0, 0.5:9.5, 2.5:7.5, 3.5:6.5, 1:1 (matrix + FIrPic; 30 mg mL−1) for each device and we added [Bmim][BF4] to give a 4:1 molar ratio of matrix:IL. The effect of the IL was checked by preparing the emitting layer from a solution with a 1:1 mass ratio NMS25:SPPO13 (matrix + FIrPic; 30 mg mL−1), without adding any IL. To test the effect of thickness, the solutions of 20, 30 and 40 mg mL−1 with a 1:1 mass ratio NMS25:SPPO13 without IL were spin-coated to give a film thickness of 40, 80 and 100 nm, respectively. After deposition of the emitting layer, the devices were transferred into an inert atmosphere glovebox, where the aluminium electrode was thermally evaporated using a shadow mask. The size of the LEC was 6.5 mm2.
The thickness of the films was determined with an Ambios XP-1 profilometer. Thin film photoluminescence spectra and quantum yields were measured with a Hamamatsu C9920-02 Absolute PL Quantum Yield Measurement System. It consists of an excitation light source (a xenon lamp linked to a monochromator), an integration sphere and a multi-channel spectrometer. Time dependence of luminance and voltage was measured by applying pulsed current and by monitoring the voltage and the luminance simultaneously by a True Colour Sensor MAZeT (MTCSICT Sensor) using a Lifetime Test System designed by BoTEST (Botest OLT OLED Lifetime-Test System). Electroluminescence spectra were recorded with an Avantes fiber-optics photo-spectrometer. The devices were not encapsulated and were characterized inside the glovebox.
Acknowledgements
This work is supported by the European Union (CELLO, STRP 248043; http://www.cello-project.eu/) and the Spanish Ministry of Economy and Competitiveness (MINECO) (MAT2011-24594, CSD2007-00010, and CTQ2009-08790). A.P. and D.T. acknowledge the support of a FPI and FPU grant of the MINECO and MECD, respectively.References
- S. Reineke, F. Lindner, G. Schwartz, N. Seidler, K. Walzer, B. Lüssem and K. Leo, Nature, 2009, 459, 234 CrossRef CAS PubMed.
- K. Walzer, B. Maennig, M. Pfeiffer and K. Leo, Chem. Rev., 2007, 107, 1233 CrossRef CAS PubMed.
- L. Xiao, S.-J. Su, Y. Agata, H. Lan and J. Kido, Adv. Mater., 2009, 21, 1271 CrossRef CAS.
- C. Rothe, C.-J. Chiang, V. Jankus, K. Abdullah, X. Zeng, R. Jitchati, A. S. Batsanov, M. R. Bryce and A. P. Monkman, Adv. Funct. Mater., 2009, 19, 2038 CrossRef CAS.
- Q. B. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef CAS PubMed.
- K. M. Maness, R. H. Terrill, T. J. Meyer, R. W. Murray and R. M. Wightman, J. Am. Chem. Soc., 1996, 118, 10609 CrossRef CAS.
- J. D. Slinker, A. A. Gorodetsky, M. S. Lowry, J. J. Wang, S. Parker, R. Rohl, S. Bernhard and G. G. Malliaras, J. Am. Chem. Soc., 2004, 126, 2763 CrossRef CAS PubMed.
- Q. J. Sun, Y. F. Li and Q. B. Pei, J. Disp. Technol., 2007, 3, 211 CrossRef CAS.
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- S. Tang, W.-Y. Tan, X.-H. Zhu and L. Edman, Chem. Commun., 2013, 49, 4926 RSC.
- D. J. Dick, A. J. Heeger, Y. Yang and Q. B. Pei, Adv. Mater., 1996, 8, 985 CrossRef CAS.
- J. D. Slinker, J. A. DeFranco, M. J. Jaquith, W. R. Silveira, Y. W. Zhong, J. M. Moran-Mirabal, H. G. Craighead, H. D. Abruña, J. A. Marohn and G. G. Malliaras, Nat. Mater., 2007, 6, 894 CrossRef CAS PubMed.
- P. Matyba, K. Maturova, M. Kemerink, N. D. Robinson and L. Edman, Nat. Mater., 2009, 8, 672 CrossRef CAS PubMed.
- S. van Reenen, P. Matyba, A. Dzwilewski, R. A. J. Janssen, L. Edman and M. Kemerink, J. Am. Chem. Soc., 2010, 132, 13776 CrossRef CAS PubMed.
- M. Lenes, G. García-Belmonte, D. Tordera, A. Pertegás, J. Bisquert and H. J. Bolink, Adv. Funct. Mater., 2011, 21, 1581 CrossRef CAS.
- P. Zalar, Z. B. Henson, G. C. Welch, G. C. Bazan and T. Q. Nguyen, Angew. Chem., Int. Ed., 2012, 51, 7495 CrossRef CAS PubMed.
- E. Zysman-Colman, J. D. Slinker, J. B. Parker, G. G. Malliaras and S. Bernhard, Chem. Mater., 2008, 20, 388 CrossRef CAS.
- A. B. Tamayo, S. Garon, T. Sajoto, P. I. Djurovich, I. M. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 8723 CrossRef CAS PubMed.
- L. He, L. Duan, J. Qiao, R. J. Wang, P. Wei, L. D. Wang and Y. Qiu, Adv. Funct. Mater., 2008, 18, 2123 CrossRef CAS.
- M. K. Nazeeruddin, R. T. Wegh, Z. Zhou, C. Klein, Q. Wang, F. De Angelis, S. Fantacci and M. Grätzel, Inorg. Chem., 2006, 45, 9245 CrossRef CAS PubMed.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS.
- L. He, L. A. Duan, J. A. Qiao, G. F. Dong, L. D. Wang and Y. Qiu, Chem. Mater., 2010, 22, 3535 CrossRef CAS.
- B. Chen, Y. H. Li, W. Yang, W. Luo and H. B. Wu, Org. Electron., 2011, 12, 766 CrossRef CAS PubMed.
- S. Tang, A. Sandström, J. F. Fang and L. Edman, J. Am. Chem. Soc., 2012, 134, 14050 CrossRef CAS PubMed.
- A. Chaskar, H.-F. Chen and K.-T. Wong, Adv. Mater., 2011, 23, 3876 CrossRef CAS PubMed.
- A. R. Hosseini, C. Y. Koh, J. D. Slinker, S. Flores-Torres, H. D. Abruña and G. G. Malliaras, Chem. Mater., 2005, 17, 6114 CrossRef CAS.
- H. C. Su, C. C. Wu, F. C. Fang and K. T. Wong, Appl. Phys. Lett., 2006, 89, 26118 Search PubMed.
- H. C. Su, H. F. Chen, Y. C. Shen, C. T. Liao and K. T. Wong, J. Mater. Chem., 2011, 21, 9653 RSC.
- C.-T. Liao, H.-F. Chen, H.-C. Su and K.-T. Wong, Phys. Chem. Chem. Phys., 2012, 14, 1262 RSC.
- G. Kalyuzhny, M. Buda, J. McNeill, P. Barbara and A. J. Bard, J. Am. Chem. Soc., 2003, 125, 6272 CrossRef CAS PubMed.
- D. Tordera, J. Frey, D. Vonlanthen, E. C. Constable, A. Pertegás, E. Ortí, H. J. Bolink, E. Baranoff and M. K. Nazeeruddin, Adv. Energy Mater., 2013, 3, 1338 CrossRef CAS.
- C. Y. Liu and A. J. Bard, Appl. Phys. Lett., 2003, 83, 5431 CrossRef CAS PubMed.
- H. J. Bolink, L. Cappelli, S. Cheylan, E. Coronado, R. D. Costa, N. Lardies, M. K. Nazeeruddin and E. Orti, J. Mater. Chem., 2007, 17, 5032 RSC.
- S. B. Meier, W. Sarfert, J. M. Junquera-Hernandez, M. Delgado, D. Tordera, E. Orti, H. J. Bolink, F. Kessler, R. Scopelliti, M. Gratzel, M. K. Nazeeruddin and E. Baranoff, J. Mater. Chem. C, 2013, 1, 58 RSC.
- A. Correa and C. Bolm, Adv. Synth. Catal., 2007, 349, 2673 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Spectroscopy and electrochemistry of NMS25. See DOI: 10.1039/c3tc31983k |
| This journal is © The Royal Society of Chemistry 2014 |