 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Incorporation of benzocarborane into conjugated polymer systems: synthesis, characterisation and optoelectronic properties†
Jonathan
Marshall
a,
Zhuping
Fei
a,
Chin Pang
Yau
a,
Nir
Yaacobi-Gross
b,
Stephan
Rossbauer
b,
Thomas D.
Anthopoulos
b,
Scott E.
Watkins
c,
Peter
Beavis
d and
Martin
Heeney
*a
aDepartment of Chemistry and Centre for Plastic Electronics, Imperial College London, London, SW7 2AZ, UK
bDepartment of Physics and Centre for Plastic Electronics, Imperial College London, London, SW7 2AZ, UK
cCSIRO Molecular and Health Technologies, VIC 3169, Australia
dAWE, Aldermaston, Reading, RG7 4PR, UK
First published on 12th November 2013
Abstract
We present the novel 1,4-difunctionalisation of benzocarborane with organometallic groups suitable for cross-coupling and its subsequent insertion for the first time into conjugated polymer backbones. Copolymers with solubilised cyclopentadithiophene and diketopyrrolopyrrole derivatives were prepared by Stille polymerisation in good molecular weight. The physical, material and optoelectronic properties of the resulting polymers were investigated, demonstrating that benzocarborane acts similarly to a stabilised, electron-deficient cis-diene linker. We also report the first polymer field effect transistors incorporating a benzocarborane in the backbone.
Introduction
The unusual properties of carboranes make them, in principle, highly attractive species to incorporate into optoelectronically active materials.1–6 Their unusual delocalized three-centre-two-electron bonding motif confers upon them extremely high temperature stability, pseudoaromaticity and a strong electron withdrawing nature, and has prompted much research into the properties of carborane containing materials.7,8 For example, carborane containing polysiloxanes have been demonstrated to show dramatically improved thermal and chemical stability over the parent polymers.9 In the area of conjugated, electroactive polymers it has been shown that the incorporation of carborane as a side-chain unit in polyfluorenes can increase the glass transition temperature and suppress aggregation in the solid state.1Carboranes exist in three isomers, differentiated by the relative position of carbon atoms in the cage structure. Analogous to benzene, carbon atoms occupy the 1 and 2 positions in ortho-carborane, the 1 and 7 positions in meta-carborane and the 1 and 12 position in para-carborane (Fig. 1). The polarity and electron withdrawing nature of the cage is determined by the position of these atoms, increasing from para < meta < ortho.7
 | ||
| Fig. 1 Structures of (left–right) ortho-carborane, meta-carborane, para-carborane and benzocarborane. Unlabelled vertices represent BH units. Protons omitted for clarity. | ||
Several approaches have been utilised to incorporate carboranes directly into conjugated polymer backbones. In 2007, Vicente and co-workers reported the synthesis and electrochemical polymerisation of the ortho, meta and para isomers of di(2-thiophenyl)carborane.10 They found that the isomer regiochemistry has a profound influence on polymer properties, with the para isomer exhibiting the highest ionisation potential, widest band gap and lowest conductivity while the ortho isomer had the lowest ionisation potential, smallest band gap and highest conductivity. These results suggested that effective delocalization through the meta and para isomers of carborane was poor. Further work upon incorporation of para and meta carborane into emissive conjugated backbones suggest that, whilst both systems serve to extend the conjugation length slightly, there was little through conjugation between the aromatic systems connected to the carborane ring.2,6,11,12
Several conjugated polymers containing ortho-carborane have been reported, typically showing enhanced delocalization between the aromatic systems compared to the meta and para analogues, as well as interesting aggregation induced emission (AIE) properties, where luminescence is enhanced and shifted under conditions of high concentration or in the solid state.12 Such ortho-substituted systems have shown interesting sensing behavior upon exposure to a variety or organic vapours.3 However, the incorporation of ortho-carborane results in a significant kink in the polymer backbone, as well as non-planar conformations of the aromatic groups linked to the carborane cages, typically resulting in the formation of amorphous polymers.13 We were interested to develop carborane containing conjugated polymers where the backbone was approximately linear, in the belief that this would induce aggregation in the solid state and benefit charge transport. In addition, the synthesis of carborane containing polymers displaying semiconducting behavior may be interesting for the future development of direct, electronic neutron detection due to the high neutron capture radius of 10B, a stable isotope of boron that makes up 20% of natural abundance.
Since earlier reports have suggested that the direct incorporation of para-carborane – which would be expected to afford the most linear conjugated backbone – into the conjugated backbone has a detrimental effect on performance and inhibits conjugation between the aromatics connected to the antipodal carbons,10 we therefore sought a suitable carborane containing comonomer which could effectively delocalize along the conjugated backbone. An attractive carborane containing moiety is 1,2(buta-1′-3′-diene-1′,4′-diyl)-1,2-dicarbadodecaborane (benzocarborane) in which a benzenoid ring is fused to the adjacent carbon atoms in an ortho-carborane cage (Fig. 1).4,12 The potential to introduce functionality on the six-membered ring opens the door to a new range of carborane-containing conjugated materials in which it may be possible to observe the effects of carborane incorporation – stability, band gap energy modification, altered solid-state morphologies – without hindering full delocalisation along the conjugated backbone as observed with para-carborane. In order to maintain our desired linear linker suitable 1,4-difunctionalised benzocarboranes were initially targeted.
Previously reported syntheses of diaryl-functionalized benzocarboranes typically employ a metal catalyzed [2 + 2 + 2] cycloaddition using carboryne – 1,2-dehydro-o-carborane – and two functionalized alkynes.15,16 Ren and co-workers developed a zirconium-mediated route where the position of substituents on the benzenoid ring can be readily controlled.17 However, this route is not suitable for the synthesis of our desired 1,4-diarylated species due to the substituent position being determined by the steric bulk of the species flanking the C–C triple bond, which would lead to 1,3-substitution when arylated species are present. This meta-substituted benzenoid system would not enable through conjugation, hindering electron delocalization along the polymer backbone and would additionally lead to the presence of a kink in the conjugated backbone. We note that polymers containing such 1,3-substituted benzocarboranes have recently been reported as interesting emissive materials.4
We were further interested to explore the potential electron accepting behavior of such benzocarboranes, since ortho-carboranes are known to have a strong electron accepting character. It is, however, unknown how benzocarborane acts upon inclusion in an extended aromatic system. We therefore decided to copolymerize 1,4-benzocarborane with two well investigated comonomers of different donor strength, namely cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) and bis(2-thien-5-yl)2,5-pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (DPP) to investigate if benzocarborane could act as the electron deficient component of donor–acceptor polymer systems.18–22 We hereby report a novel effective 1,4-difunctionalisation of benzocarborane with reactive tributylstannyl and boronic ester groups and subsequent polymerization with solubilized CDT and DPP derivatives.
Experimental
ortho-Carborane was purchased from Katchem and sublimed before use. All other reagents were purchased from Sigma-Aldrich, VWR, Alfa Aesar or Apollo scientific and were used without further purification. Dry solvents for anhydrous reactions were purchased from Sigma Aldrich. All reactions were carried out under an inert argon atmosphere unless otherwise stated. 1H {11B} NMR and 13C NMR spectra were recorded on a BRUKER 400 spectrometer in CDCl3 solution at 298 K unless otherwise stated. Proton NMRs were recorded after boron decoupling. Number-average (Mn) and weight-average (Mw) molecular weights were determined with an Agilent Technologies 1200 series GPC in chlorobenzene at 80 °C using two PL mixed B columns in series, and calibrated against narrow polydispersity polystyrene standards. A customer build Shimadzu recSEC system was used to purify the polymers. The system comprises a DGU-20A3 degasser, an LC-20A pump, a CTO-20A column oven, an Agilent PLgel 10 μm MIXED-D column and a SPD-20A UV detector. UV-Vis absorption spectra were recorded on a UV-1601 Shimadzu UV-Vis spectrometer. Solutions were filtered through a 0.45 μm PTFE filter at ca. 80 °C prior to measurement at room temperature. Column chromatography was carried out on silica gel (VWR) or on a Biotage Isolera One. Microwave reactions were performed in a Biotage initiator v.2.3. Photo Electron Spectroscopy in Air (PESA) measurements were recorded using a Riken Keiki AC-2 PESA spectrometer with a power setting of 5 nW and a power number of 0.5. Differential scanning calorimetry (DSC) measurements were made using a TA instruments DSC TZero Q20 instrument and analysed using TA universal analysis software. Thermal Gravimetric analysis measurements were recorded using a Perkin Elmer Pyris 1 TGA. X-ray Diffraction (XRD) measurements were carried out using a Panalytical X'pert-pro MRD diffractometer fitted with a nickel-filtered Cu Kα1 beam and an X'celerator detector using a current of 40 mA and an accelerating voltage of 40 kV.Bis(1,4-tributylstannyl)-benzocarborane
Benzocarborane (1 g, 5.2 mmol) and anhydrous N,N,N′,N′-tetramethylethane-1,2-diamine (1.7 g, 14.6 mmol) were dissolved in anhydrous THF (50 mL) and cooled to −78 °C. To this solution was added n-BuLi (1.6 M in hexanes, 7.1 mL, 11.3 mmol) slowly before the mixture was allowed to warm to room temperature and stirred for 30 minutes. Following re-cooling to −78 °C, tributyltin chloride (3.6 g, 11.1 mmol) was added and the solution was allowed to warm to room temperature overnight. The reaction mixture was diluted with water and extracted with diethyl ether. The organic layers were combined, washed with brine and dried over anhydrous sodium sulphate before being concentrated under reduced pressure. Column chromatography (eluent: hexane) afforded the desired product as colourless, viscous oil (1.83 g, 2.4 mmol, 46%). 1H NMR (400 MHz, CDCl3): δ 6.26 (s, 2H), 3.00–1.60 (m, 10H), 1.49 (m, 12H), 1.33 (m, 12H), 1.07 (m, 12H), 0.90 (t, 18H). 13C NMR (100 MHz, CDCl3): δ 128.8, 122.1, 119.5, 28.8, 27.3, 13.7, 11.9. 11B {1H} NMR (400 MHz, CDCl3): δ 29.55, −6.89, −9.41, −12.55. MS (EI): m/z = 772.45 (M+).Poly[(4,4-bis(dodecyl)-4H-cyclopenta-[2,1-b:3,4-b′]-dithiophene-2,6-diyl-alt-(benzocarborane)-1,4-diyl]] (pBZ-CDT)
Bis(1,4-tributylstannyl)-benzocarborane (121.2 mg, 0.16 mmol), 2,6-dibromo-4,4′-bis(2-dodecyl)-4H-cyclopenta[1,2-b:5,4-b]dithiophene (105.2 mg, 0.16 mmol), tris (dibenzylideneacetone)dipalladium(0) (2.9 mg, 0.003 mmol) and tri-o-tolylphosphine (3.8 mg, 0.012 mmol) were added to a dry microwave vial which was flushed thoroughly with argon. Anhydrous, degassed toluene (2 mL) was added and the resulting mixture was degassed for 45 minutes. The vial was sealed and the reaction mixture was heated at 140 °C for 3 days. After cooling to room temperature the reaction mixture was poured into vigorously stirring methanol and the resulting precipitate was filtered. The precipitate was purified by Soxhlet extraction first in methanol (24 h), acetone (24 h), n-hexane (24 h) and finally chloroform (24 h). The chloroform extract was stirred at 50 °C in the presence of an aqueous sodium diethyldithiocarbamate trihydrate for two hours. After cooling to room temperature the organic layer was separated and washed with water, dried and concentrated under reduced pressure. Final purification was by preparative size exclusion chromatography. The desired polymer was afforded following concentration under reduced pressure and precipitation in methanol (71 mg, 70%) as a dark purple solid. GPC (chlorobenzene): Mn = 14 kDa, Mw = 17 kDa, PDI = 1.2. 1H {11B} NMR (400 MHz, CDCl3): δ 7.34 (br, 2H), 6.98 (br, 2H), 3.20–0.85 (m, br, 60H).Poly[[2,5-bis(2-octyldodecyl)-2,3,5,6-tetrahydro-3,6-dioxopyrrolo-[3,4-c]pyrrole-1,4-diyl]]-alt-[[2,2′-(2,5-thiophene)bis-benzocarborane-1,4-diyl]]) (pBZ-DPP)
Bis(1,4-tributylstannyl)-benzocarborane (108.6 mg, 0.14 mmol), 3,6-bis(2-bromothien-5-yl)2,5-bis(2-octyldodecyl)pyrrolo[3,4-c]pyrrole-1,4(2H,5H)-dione (153.2 mg, 0.14 mmol), tris (dibenzylideneacetone)dipalladium(0) (2.54 mg, 0.0028 mmol) and tri-o-tolylphosphine (3.5 mg, 0.011 mmol) were added to a dry microwave vial which was flushed thoroughly with argon. Anhydrous, degassed toluene (2 mL) was added and the resulting mixture was degassed for 45 minutes. The vial was sealed and the reaction mixture was heated at 140 °C for 3 days. After cooling to room temperature the reaction mixture was poured into vigorously stirring methanol and the resulting precipitate was filtered. The precipitate was purified by Soxhlet extraction first in methanol (24 h), acetone (24 h), n-hexane (24 h) and finally chloroform (24 h). The chloroform extract was stirred at 50 °C in the presence of an aqueous sodium diethyldithiocarbamate trihydrate for two hours. After cooling to room temperature the organic layer was separated and washed with water, dried and concentrated under reduced pressure. Final purification was by preparative size exclusion chromatography. The desired polymer was afforded following concentration under reduced pressure and precipitation in methanol. (66 mg, 42%) as a dark blue solid. GPC (chlorobenzene): Mn = 12 kDa, Mw = 22 kDa, PDI = 1.8. 1H {11B} NMR (400 MHz, CDCl3): δ 8.93 (br, 2H), 7.60 (br, 2H), 7.40 (br, 2H) 4.03–0.83 (m, br, 50H).Results and discussion
The synthesis of benzocarborane followed previously reported conditions;14 thus, commercially available ortho-carborane (1) was dilithiated with n-BuLi before reaction with cis-1,4-dichloro-but-2-ene to afford 2 in 79% yield. Radical allylic bromination with NBS yielded a mixture of 2-bromo-1,2-dihydrobenzocarborane and 1-bromo-1,4-dihydrobenzocarborane in 62% yield (3). Dehydrobromination of the mixture of products was achieved by refluxing in DMF to afford benzocarborane (4) in 51% yield (Scheme 1). | ||
| Scheme 1 Synthesis of benzocarborane monomers. (a) n-BuLi then 1,4-dichlorobut-2-ene (b) NBS, benzoylperoxide (c) DMF, reflux (d) n-BuLi, TMEDA then Bu3SnCl (e) n-BuLi, TMEDA then iPrO-B(pin). | ||
Initially, dibromination was investigated as a route to achieve 1,4-difunctionalisation. However, attempted reaction with electrophilic sources of bromine – NBS or molecular bromine – resulted in the addition of two equivalents of bromine across the diene double bonds to afford the dibrominated alkene as a mixture of isomers. This behaviour, rather than the desired electrophilic substitution, is more typical of dienes than aromatics and was an indication that the electronic structure of benzocarborane was significantly different to that of a traditional aromatic system.
Therefore deprotonation with n-BuLi and trapping of the resulting anion was investigated as an alternative. Gratifyingly, we found that treatment with 2.5 equivalents of n-BuLi in the presence of TMEDA resulted in exclusive deprotonation of the 1,4-positions. Initially we trapped the resulting anion with isopropylboronic acid pinacol ester to afford the bis(boronic ester) (6) in 71% yield. However our attempts to polymerise 6 with 2,6-dibromo-4,4-bis(dodecyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) resulted in low molecular weights under a variety of conditions, due to competing deboronation reactions, so focus was shifted to Stille polymerisation. Therefore quenching of the dilithiated benzocarborane with tributylstannyl chloride afforded the difunctionalised monomer in 46% yield. It is worth noting that, unlike the majority of organostannyl compounds, the C–Sn bond was stable to the acidic conditions associated with silica gel chromatography, affording a monomer that could be easily purified by conventional means.
Stille copolymerizations with 2,6-dibromo-4,4-bis(dodecyl)-4H-cyclopenta[2,1-b:3,4-b′]dithiophene (CDT) and 3,6-bis(2-bromothien-5-yl)2,5-bis(2-octyldodecyl)pyrrolo[3,4-c]pyrole-1,4(2H,5H)-dione (DPP) were performed by heating in toluene in sealed microwave tubes at 140 °C for 3 days (Scheme 2).23 Following precipitation the materials were purified by sequential soxhlet extractions using methanol, acetone, n-hexane and chloroform, the latter of which was retained. Potential catalyst impurities were removed by heating a solution of the polymer in the presence of sodium diethyldithiocarbamate trihydrate followed by preparative size exclusion chromatography (SEC).24,25 Following concentration and precipitation, pBZ-CDT was afforded as a dark purple solid in 70% yield and pBZ-DPP as a dark blue solid in 42% yield. Molecular weights and polydispersities were determined by SEC in hot (80 °C) chlorobenzene (Table 1), demonstrating reasonable degrees of polymerisation.
| Polymer | M n/Mwa (kDa) | λ max (nm) | E g (eV) | HOMOc (eV) | LUMOd (eV) | |
|---|---|---|---|---|---|---|
| PhCl | Film | |||||
| a Determined by SEC and reported as their poly(styrene) equivalents. b Determined by onset of optical absorption. c Determined as a thin film by UV-PESA. d Estimated by the subtraction of the optical band gap from the HOMO. e Following annealing at 160 °C under an argon atmosphere. | ||||||
| pBZ-CDT | 14/17 | 571 | 584 | 1.88 | −5.1 ± 0.05 | −3.2 ± 0.05 |
| pBZ-DPP | 12/22 | 728 | 742e | 1.23 | −5.2 ± 0.05 | −4.0 ± 0.05 |
 | ||
| Scheme 2 Structures of polymers. Both polymers were synthesised by reaction of the appropriate dibromo-monomer with Pd2(dba)3/P(o-tol)3 in toluene. | ||
Both polymers were soluble in chlorobenzene at room temperature and in chloroform upon heating. The optical absorption of pBZ-CDT and pBZ-DPP in chlorobenzene solution and as a thin film are shown in Fig. 2. The solution spectrum of pBZ-CDT displays a maximum at 571 nm, changing to 584 nm upon film formation. This red shift, along with the formation of a long-wavelength shoulder around 650 nm, is indicative of backbone planarization compared to solution and suggests some degree of ordering in the solid state. Upon annealing the film at 160 °C, no further changes were observed. From the onset absorption in the solid state, the optical band gap was calculated as 1.88 eV. Compared to CDT copolymers with strong acceptors like benzothiadiazole (which absorbs at 775 nm in the solid state)26 or relatively weak acceptor comonomers such as 5,8-quinoxaline which absorbs around 625 nm in the solid state,19 it appears that benzocarborane is not acting as a strong electron acceptor. However we do observe a small red-shift of approximately 50 nm compared to a CDT–benzene copolymer synthesized for comparative purposes (see Fig. S11†).
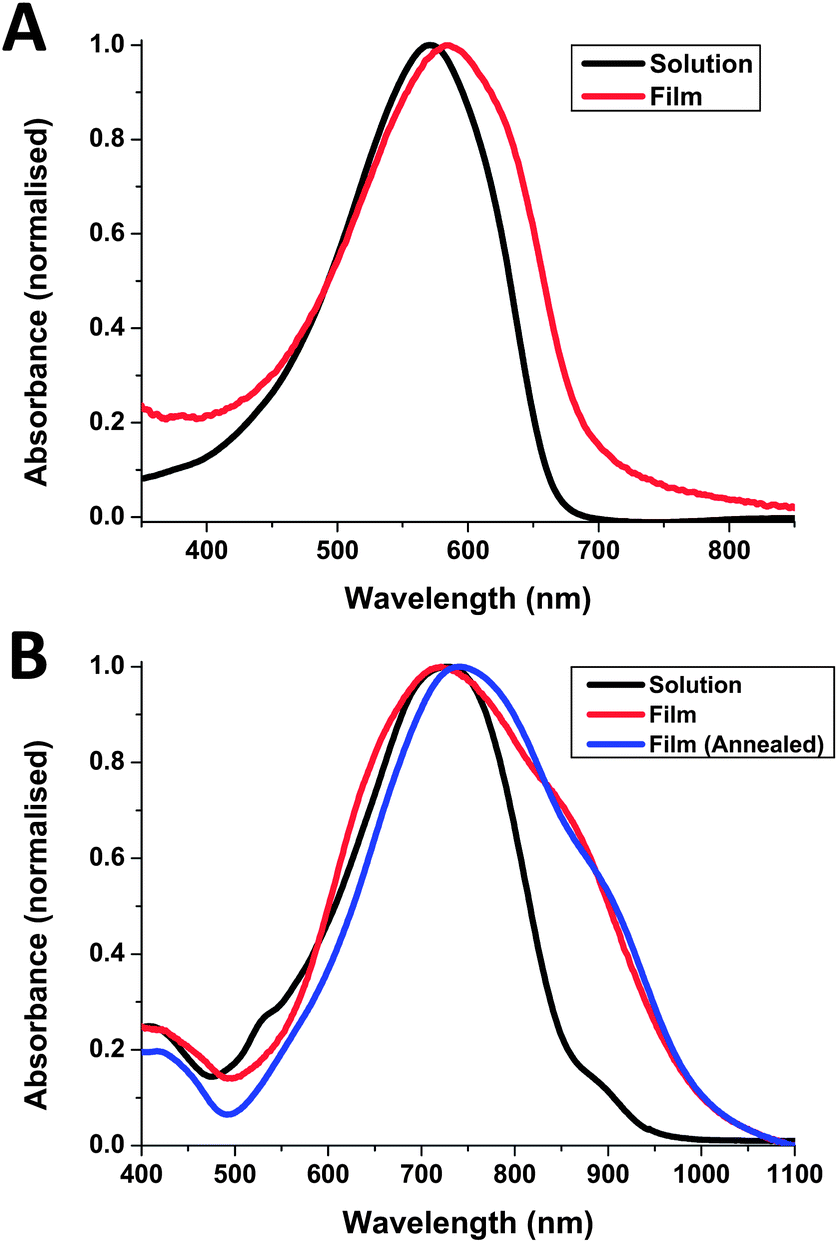 | ||
| Fig. 2 UV-Vis absorption spectra of pBZ-CDT (A) and pBZ-DPP (B) in solution (chlorobenzene) and in thin film (as-spun from chlorobenzene). | ||
The λmax of pBZ-DPP in dilute chlorobenzene solution was significantly red-shifted compared to pBZ-CDT at 728 nm, reflecting the fact that, unlike CDT, DPP is an electron acceptor. A small shoulder was also observed around 900 nm, suggesting some degree of aggregation in solution. Upon film formation a significant broadening is observed in the absorption profile along with a slight blue shift of λmax to 722 nm and the strengthening of the shoulder absorption around 900 nm. This long wavelength shoulder is commonly attributed to aggregation in the solid state. When compared to a previously reported benzene copolymer, which exhibits a dual peak absorption with peaks around 680 nm and a longer wavelength aggregation band at 750 nm, it is noticed that the aggregation-induced shoulder for pBZ-DPP at ca. 900 nm is comparatively much less intense in solution than for the benzene analogue.27 Similarly the absorption of the long wavelength shoulder in the solid state is much less pronounced for pBZ-DPP than for the benzene analogue, or for other DPP polymers.28 This suggests that aggregation in both solution and in the solid state is much weaker for pBZ-DPP compared to other DPP copolymers, possibly due to the steric demands of the cage-like carborane system. Upon annealing of the thin film at 160 °C, the λmax is red-shifted slightly to 742 nm, implying that it is possible to improve ordering in the solid state by thermal treatments. The broad absorption of pBZ-DPP results in a small optical band gap of 1.23 eV, which is 0.3 eV smaller than the benzene analogue. The reduced band gap for both benzocarborane polymers compared to their benzene analogues suggests that electron delocalization along the backbone is facilitated, which we attribute to the reduced resonance stabilization energy of benzocarborane relative to benzene. In agreement with our attempts at electronic substitution reactions, it appears benzocarborane, rather than acting as an aromatic linker, acts rather similarly to a cis-diene linker.
The HOMO levels of both polymers in thin films were determined by ambient photoelectron spectroscopy (PESA), as shown in Table 1. For pBZ-CDT the ionization potential was significantly smaller (0.3 eV) than measured for the CDT–benzene copolymer by the same technique, in agreement with the enhanced backbone delocalization of benzocarborane versus benzene. Both values are similar to the work function of gold, suggesting that charge injection should be facilitated in an OFET with gold electrodes.
Differential scanning calorimetry (DSC) of pBZ-CDT showed no obvious thermal transitions between 0 °C and 250 °C (Fig. S5†). Additional signs of an absence of crystallinity were inferred from wide-angle XRD where no diffraction peaks were observed (Fig. S6†). This was unchanged upon annealing at 160 °C, suggesting an absence of long-range order despite the bathochromic shift in absorption upon moving from solution to the solid state. The comparable benzothiadiazole polymer also shows amorphous character,29 so it is not surprising to see an absence of crystallinity in this system.
In contrast, the DSC of pBZ-DPP showed two clear endotherms on heating, at 120 °C and 215 °C, with a corresponding exotherm at 192 °C upon cooling (Fig. 3). We believe that the transitions around 200 °C correspond to backbone melting and recrystallisation respectively, whereas the lower endotherm may be related to sidechain melting. The transitions were fully reversible upon repeated cycling indicating good thermal stability of the system. This was further confirmed by thermogravimetric analysis (TGA) which indicated that the onset of degradation for both polymers was above 380 °C (Fig. S7 and S8†) X-ray diffraction measurements on drop-cast films exhibited a clear diffraction peak at 2θ = 4.0°, corresponding to a d-spacing of 22.1 nm (Fig. 3). Upon annealing at 160 °C the spacing does not change but the diffraction peak increases in intensity and a second order peak at 2θ = 8.0° is observed (Fig. 3), suggesting an increase in film crystallinity. The aggregating properties of DPP are well known and it is encouraging to see that the presence of bulky, three-dimensional carborane cages does not impede this.
 | ||
| Fig. 3 (top) Second heating–cooling DSC cycle of pBZ-DPP at 10 °C min (bottom) X-ray diffraction spectra of pBZ-DPP as cast and annealed at 160 °C under argon. | ||
Theoretical density functional theory (DFT) calculations were performed using the B3LYP/6-31G* basis set to calculate the minimum energy conformation of both polymers. In both instances a clear twist can be seen along the conjugated backbone, due to interactions between each benzocarborane unit and the adjacent α-hydrogen of the neighbouring CDT or thiophene unit (in the case of DPP). This dihedral angle is calculated as 34° for pBZ-CDT and 37° for the DPP copolymer. For pBZ-DPP it is comparable to that seen in the benzene analogue.27 This conformation is in agreement with the lack of crystallinity noted in XRD experiments for pBZ-CDT, since a twisted backbone is more likely to suppress intermolecular interactions. In the case of pBZ-DPP, we believe the well-known tendency for the DPP cores to aggregate leads to some order being observed despite the backbone torsion.
Visualisation of the frontier molecular orbitals (HOMO and LUMO) show that for both polymers there is no electronic communication between the carborane cage and polymer backbone and therefore the main influence of carborane inclusion is an inductive electron withdrawing effect. This supports the conclusions that benzocarborane does not act as a strong acceptor, a hypothesis supported by the optical properties of pBZ-CDT. In both instances it can be seen that the benzocarborane unit is more akin to an electron deficient diene than a benzene ring, a theory supported by the preference towards electrophilic addition rather than substitution, vide supra. It can also be observed that both wave functions are delocalized along the backbone in spite of the torsional twist (Fig. 4), suggesting that the non-planarity does not impede electronic delocalization.
 | ||
| Fig. 4 Left: minimum energy conformation of pBZ-CDT trimer, face-on (top) and side-on (second from top). Visualised frontier molecular orbitals of pBZ-CDT trimer, LUMO (second from bottom) and HOMO (bottom). Right: minimum energy conformation of pBZ-DPP trimer, face-on (top) and side-on (second from top). Visualised frontier molecular orbitals of pBZ-DPP trimer, LUMO (second from bottom) and HOMO (bottom). Calculated in Gaussian with B3LYP/6-31G* basis set. | ||
Finally the performance of the two polymers in field effect transistors was investigated in bottom gate-bottom contact (BGBC) FET devices, using HMDS treated SiO2 dielectric and gold electrodes. pBZ-CDT exhibited p-type semiconductor behaviour, albeit with rather low saturation mobilities of 1.7 × 10−5 cm2 V−2 s−1, a threshold of 21 V and an on/off ratio of 103 (Fig. 5). Despite showing ambipolar behaviour, the results for pBZ-DPP were less encouraging. The absence of a saturation regime for either hole or electron transport meant that performance characteristics could not be reliably calculated (Fig. S10†). A similar lack of saturation was also observed for bottom contact, top gate devices. The relatively poor performance of the DPP copolymer is somewhat surprising, given the evidence of solid state ordering. We note however that other DPP-T copolymers with comonomers containing bulky sidechains like 9,9-dioctylfluorene30 or 9,9-dioctylgermafluorene31 exhibit semicrystalline order in the solid state but have similarly low charge carrier mobilities. Thus we believe the relatively high steric demand of the benzocarborane unit, coupled with the backbone torsional twist prevents close overlap of the conjugated chains and therefore suppresses charge carrier mobility. Nevertheless this is the first time benzocarborane containing conjugated polymers have been shown to operate in FET devices.
 | ||
| Fig. 5 Representative output (a) and transfer (b) characteristics of bottom-gate, bottom contact organic field-effect transistor (OFET) of pBZ-CDT (transistor channel = 7.5 μm, transistor width = 1000 μm). | ||
Conclusions
In conclusion, we report the synthesis of two novel derivatives of benzocarborane incorporating organoboron or organostannane functional groups. These are able to participate in standard cross-coupling chemistries, and Stille copolymerization afforded polymers with 1,4-benzocarborane incorporated in the conjugated backbone. Preliminary investigation of optoelectronic properties showed potential for the use of carborane containing conjugated polymers in organic field effect transistors, although significant device optimization may be required. We also note that this synthetic route provides scope for further arylation of benzocarborane to afford molecules useful in fields beyond organic electronics.Acknowledgements
We thank AWE for financial support of an EPSRC CASE award and for their input into this project. We also thank Ester Buchaca-Domingo at Imperial College London for XRD measurements.Notes and references
- (a) Y. C. Simon, J. J. Peterson, C. Mangold, K. R. Carter and E. B. Coughlin, Macromolecules, 2009, 42, 512–516 CrossRef CAS; (b) A. R. Davis, J. J. Peterson and K. R. Carter, ACS Macro Lett., 2012, 1, 469–472 CrossRef CAS.
- J. J. Peterson, M. Werre, Y. C. Simon, E. B. Coughlin and K. R. Carter, Macromolecules, 2009, 42, 8594–8598 CrossRef CAS.
- J. J. Peterson, A. R. Davis, M. Werre, E. B. Coughlin and K. R. Carter, ACS Appl. Mater. Interfaces, 2011, 3, 1796–1799 CAS.
- K. Kokado, M. Tominaga and Y. Chujo, Macromol. Rapid Commun., 2010, 31, 1389–1394 CrossRef CAS PubMed.
- Y. Morisaki, M. Tominaga and Y. Chujo, Chem.–Eur. J., 2012, 18, 11251–11257 CrossRef CAS PubMed.
- (a) K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 2925–2930 CrossRef CAS; (b) J. J. Peterson, Y. C. Simon, E. B. Coughlin and K. R. Carter, Chem. Commun., 2009, 4950–4952 RSC.
- R. N. Grimes, Carboranes, Academic Press, 2nd edn, 2011 Search PubMed.
- E. D. Jemmis, J. Am. Chem. Soc., 1982, 35, 7017–7020 CrossRef.
- E. Peters, Ind. Eng. Chem. Prod. Res. Dev., 1984, 2, 28–32 CrossRef.
- E. Hao, B. Fabre, F. R. Fronczek and M. G. H. Vicente, Chem. Commun., 2007, 4387–4389 RSC.
- M. Fox and J. MacBride, J. Chem. Soc., Dalton Trans., 1998, 401–412 RSC.
- (a) K. Kokado and Y. Chujo, Macromolecules, 2009, 42, 1418–1420 CrossRef CAS; (b) K. Kokado, Y. Tokoro and Y. Chujo, Macromolecules, 2009, 42, 9238–9242 CrossRef CAS.
- N. I. Bekasova and N. G. Komarova, Russ. Chem. Rev., 1992, 61, 647–667 CrossRef CAS PubMed.
- D. S. Matteson and N. K. Hota, J. Am. Chem. Soc., 1971, 93, 2893–2897 CrossRef CAS.
- Z. Qiu, Z. Xie and H. Kong, J. Am. Chem. Soc., 2009, 2084–2085 CrossRef CAS PubMed.
- L. Deng, H. Chan and Z. Xie, J. Am. Chem. Soc., 2006, 128, 7728–7729 CrossRef CAS PubMed.
- S. Ren, Z. Qiu and Z. Xie, J. Am. Chem. Soc., 2012, 134, 3242–3254 CrossRef CAS PubMed.
- K. Li, J. Huang, Y. Hsu and P. Huang, Macromolecules, 2009, 42, 3681–3693 CrossRef CAS.
- J. C. Bijleveld, M. Shahid, J. Gilot, M. M. Wienk and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 3262–3270 CrossRef CAS.
- M. Horie, L. A. Majewski, M. J. Fearn, C.-Y. Yu, Y. Luo, A. Song, B. R. Saunders and M. L. Turner, J. Mater. Chem., 2010, 20, 4347 RSC.
- H. Bronstein, Z. Chen, R. S. Ashraf, W. Zhang, J. Du, J. R. Durrant, P. S. Tuladhar, K. Song, S. E. Watkins, Y. Geerts, M. M. Wienk, R. A. J. Janssen, T. Anthopoulos, H. Sirringhaus, M. Heeney and I. McCulloch, J. Am. Chem. Soc., 2011, 133, 3272–3275 CrossRef CAS PubMed.
- I. Meager, R. S. Ashraf, S. Mollinger, B. C. Schroeder, H. Bronstein, D. Beatrup, M. S. Vezie, T. Kirchartz, A. Salleo, J. Nelson and I. McCulloch, J. Am. Chem. Soc., 2013, 135, 11537–11540 CrossRef CAS PubMed.
- S. Tierney, M. Heeney and I. McCulloch, Synth. Met., 2005, 148, 195–198 CrossRef CAS PubMed.
- K. T. Nielsen, H. Spanggaard and F. C. Krebs, Macromolecules, 2005, 38, 1180–1189 CrossRef CAS.
- R. S. Ashraf, B. C. Schroeder, H. Bronstein, Z. Huang, S. Thomas, R. J. Kline, C. J. Brabec, P. Rannou, T. D. Anthopoulos, J. R. Durrant and I. McCulloch, Adv. Mater., 2013, 25, 2029–2034 CrossRef CAS PubMed.
- D. Mühlbacher, M. Scharber, M. Morana, Z. Zhu, D. Waller, R. Gaudiana and C. Brabec, Adv. Mater., 2006, 18, 2884–2889 CrossRef.
- J. C. Bijleveld, V. S. Gevaerts, D. Di Nuzzo, M. Turbiez, S. G. J. Mathijssen, D. M. de Leeuw, M. M. Wienk and R. A. J. Janssen, Adv. Mater., 2010, 22, E242–E246 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859–1880 CrossRef CAS PubMed.
- M. Horie, J. Kettle, C.-Y. Yu, L. a. Majewski, S.-W. Chang, J. Kirkpatrick, S. M. Tuladhar, J. Nelson, B. R. Saunders and M. L. Turner, J. Mater. Chem., 2012, 22, 381 RSC.
- A. P. Zoombelt, S. G. J. Mathijssen, M. G. R. Turbiez, M. M. Wienk and R. A. J. Janssen, J. Mater. Chem., 2010, 20, 2240 RSC.
- N. Allard, R. B. Aich, D. Gendron, P.-L. T. Boudreault, C. Tessier, S. Alem, S.-C. Tse, Y. Tao and M. Leclerc, Macromolecules, 2010, 43, 2328 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, figures showing synthetic schemes for the preparation of compounds reported in this study, 1H and 13C NMR spectra of novel materials, TGA data, OFET graphs, XRD data. See DOI: 10.1039/c3tc31663g |
| This journal is © The Royal Society of Chemistry 2014 |