
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Biomolecule detection in porous silicon based microcavities via europium luminescence enhancement†
S. N. Aisyiyah
Jenie
a,
Zhangli
Du
b,
Steven J. P.
McInnes
a,
Phuc
Ung
c,
Bim
Graham
c,
Sally E.
Plush
b and
Nicolas H.
Voelcker
*a
aARC Centre of Excellence in Convergent Bio-Nano Science and Technology, Mawson Institute, University of South Australia, GPO Box 2471, Mawson Lakes, Adelaide, SA 5095, Australia. E-mail: nico.voelcker@unisa.edu.au; Fax: +61 8 8302 5613; Tel: +61 8 8302 5508
bSchool of Pharmacy and Medical Sciences, University of South Australia, Adelaide, SA 5000, Australia
cFaculty of Pharmacy and Pharmaceutical Sciences, Monash University, VIC 3800, Australia
First published on 2nd October 2014
Abstract
In this paper, we demonstrate the detection of europium-complex-labeled streptavidin in a porous silicon microcavity (pSiMC) via luminescence enhancement. The pSiMC platform was modified for optimized luminescence enhancement which encompassed changing the pore size of the microcavity to ensure molecular infiltration and adjusting the optical quality of the microcavity. Characterization of the optimized surface was performed by infrared spectroscopy, interferometric reflectance spectroscopy and luminescence measurements. Luminescence enhancement of the bound Eu(III) complex by a factor of 3 was observed on the optimized pSiMC as compared to that on a single pSi layer. The ability of a pSiMC to act as a luminescence enhancing sensor was confirmed using streptavidin as a model analyte on a biotin-modified pSiMC. The sensor was able to detect Eu(III) complex labeled streptavidin with a concentration as low as 150 nM. Furthermore, streptavidin was selectively detected when spiked in human wound fluid. The concept of detecting Eu(III) labeled bioconjugates on pSiMC may be incorporated into the design of highly sensitive and specific point-of-care biosensors.
Introduction
Nanostructured porous materials have been successfully applied for the development of a plethora of biosensors due to their unique electrical, optical and magnetic properties.1–3 One porous material of particular importance in the context of biosensors is porous silicon (pSi) and this is due to a range of favourable material properties; (1) ease of fabrication, (2) high surface area, (3) tuneable configuration of alternating layers of different porosities, (4) ability to bind biomolecules covalently to the surface, (5) ability to reflect light, enabling detection of biomolecules via optical reflectivity, (6) photoluminescence properties and (7) biocompatibility and biodegradability.4–9Over the past decade, pSi platforms based on photonic structures have been intensively studied2,4,10–12 and often offered remarkable sensitivity in detecting the molecule of interest. pSi can be fabricated as photonic reflectors such as Bragg reflectors or rugate filters, which are composed of periodic layers of alternating high and low refractive indexes. This produces a photonic band gap in the reflectance spectrum.4 A resonant microcavity is generated when a spacer layer with certain porosity and thickness is located between the multilayers of high and low refractive indexes (i.e. porosity). This 1D photonic structure generates an allowed mode in the stop band of the reflectors. A spacer layer with an optical thickness of λ/2 will result in a symmetrical breaking of the stop band of the Bragg reflector and hence a dip in the reflectance peak.13,14
Previous reports have established that besides being sensitive to changes in the optical thickness of the spacer layer,14–16 pSi microcavities (pSiMC) are an excellent host-matrix for luminescent molecules since they both sharpen and amplify the emission.10,17–23 Biosensors can exploit this effect by monitoring target analytes or molecules either directly by their emission or via an additional luminescent label.8 Recent work investigating the performance of pSiMCs for fluorescence based detection include the ones conducted by Palestino, et al.18 The performance of the pSiMC structure for emission enhancement was performed by comparing the fluorescence intensities between the microcavity and other pSi structures (i.e. single layer and Bragg reflectors), infiltrated with emitting molecules.18,22 The emitting molecules incorporated were glucose oxidase (GOX showing natural green fluorescence) and fluorescein isothiocyanate (FITC) labeled streptavidin. pSi microcavities demonstrated excellent fluorescence enhancement for both proteins. The enhancement was attributed to the interaction between the molecules and the optical field of the spacer layer in the microcavity structure. The fluorescence enhancement feature has also been observed on a more complex structure namely a pSi-coupled microcavity. A coupled microcavity consists of two spacer layers, which are surrounded by adjacent distributed Bragg reflectors resulting in a double resonance in the reflectance spectrum. The double resonance causes an enhancement of both the excitation and the emission of the confined fluorescein-labelled protein as observed by Sciacca, et al.10 Using this double microcavity, a labeled protein with a concentration as low as 10−9 M was still detected. More recent study employing the use of a resonant microcavity has been conducted in detecting the Matrix Metalloproteinase enzyme. In this work, the limit of detection of the microcavity surface was observed at 7.5 × 10−19 M, confirming the sensitivity of the detection platform.24 Taken together, these studies show that the microcavity structure could be used for the detection of low concentrations of the confined emitting biomolecules.
Herein, we demonstrate the application of a modified pSi platform with the ability to detect analytes labeled with a luminescent lanthanide ion complex. The use of luminescent lanthanide ion complexes as optical sensors has gained much interest in recent years owing to their unique properties e.g. narrow line-like emission,25 large Stokes shift exceeding 200 nm26 reducing overlap between the excitation and emission, which is often observed in organic fluorescent dyes,25,27 and long lived emission lifetimes (∼ms). The latter characteristic allows time gating of the lanthanide emission, hence removal of background autofluorescence. The low extinction coefficient (due to the Laporte forbidden f–f transitions) of the lanthanide ions themselves can be easily overcome through the incorporation of a chromophore (antenna) capable of populating the excited state of the lanthanide ion. This process is commonly referred to as sensitization.28,29 Moreover, these complexes are popular as being sensitive labeling reagents that permit the simultaneous analysis of multiple targets at low concentrations due to their distinctive emission wavelengths and emission lifetimes, improving the current labeling/detection technologies.27,30,31 Recent studies have shown the application of visible emitting lanthanide complexes as labels in ultrasensitive sensor systems with limits of detection (LOD) as low as 10−11 M.30,32
The lanthanide complex used in this study is a Eu(III) ion complexed by a functionalized cyclen (1,4,7,10-tetraazacyclododecane) ligand. Functionalization includes a carbostyril antenna for efficient sensitization of the lanthanide ion emission and three carboxylic acid pendant arms to provide a strong chelating environment (Fig. 1, 1). The first step of the study was to investigate the luminescence enhancement properties of the complex in a pSiMC modified with the Eu(III) complex when compared to a pSi single layer. We further investigated the ability of the pSiMC as a selective sensing platform for biomolecules labeled with the Eu(III) complex. We used streptavidin–biotin as the model system due to the very high binding affinity (dissociation constant ∼1.3 × 10−15 M).33 Streptavidin was labeled with the Eu(III) complex and then exposed to the biotinylated surface. Again, the emission intensity was compared between the pSiMC and the single pSi layer. The selectivity of the pSiMC sensing platform was demonstrated by exposing the sensor to human wound fluid spiked with the labeled streptavidin. We further demonstrate the optimization of the biotin coverage on the pSiMC surface and the concentration of the Eu(III) complex labeled streptavidin, hence improving the sensitivity of the model sensing platform.
 | ||
| Fig. 1 1,4,7-Tris(carbonylmethyl)-10-(4′-quinolineacetic acid, (7′-acetamide)-1′,2′-dihydro-2′-oxo)-1,4,7,10-tetraazacyclododecane.Eu, 1. | ||
Experimental
Materials and methods
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 48% (v/v) hydrofluoric acid (HF) (Scharlau, Australia) and ethanol (100%, ChemSupply, Australia) in a custom built Teflon cell at 25 °C using a source meter (Keithley, USA) as the current source. The cathode used was a platinum mesh and aluminum foil was used to contact the silicon anode. The area of the exposed region of the silicon wafer was 1.767 cm2. Prior to sample preparation, the parasitic layer of the samples was removed by etching the silicon wafers in a 1:1 HF/EtOH solution at 56.8 mA cm−2 for 30 s, thorough rinsing with ethanol, drying with N2 gas and immersing the layer in 1 M sodium hydroxide (NaOH, Merck, Australia) for 2 min. Finally, the wafer was cleaned with deionized water and ethanol consecutively before being dried under N2 gas.35
:1 mixture of 48% (v/v) hydrofluoric acid (HF) (Scharlau, Australia) and ethanol (100%, ChemSupply, Australia) in a custom built Teflon cell at 25 °C using a source meter (Keithley, USA) as the current source. The cathode used was a platinum mesh and aluminum foil was used to contact the silicon anode. The area of the exposed region of the silicon wafer was 1.767 cm2. Prior to sample preparation, the parasitic layer of the samples was removed by etching the silicon wafers in a 1:1 HF/EtOH solution at 56.8 mA cm−2 for 30 s, thorough rinsing with ethanol, drying with N2 gas and immersing the layer in 1 M sodium hydroxide (NaOH, Merck, Australia) for 2 min. Finally, the wafer was cleaned with deionized water and ethanol consecutively before being dried under N2 gas.35
The microcavity structures were fabricated by etching alternating layers of high and low refractive indices (Table 1). The corresponding current densities for each of the refractive indices are shown in Table 1. The configuration of the microcavities were designed through the commercial program SCOUT that is based on the transfer matrix method.36 This program allows a best-fit simulation and analysis between the theoretical and experimental reflectance spectrum. The software generates a theoretical spectrum of the pSi film, which is a function of the film's characteristics, i.e. dielectric function and thickness. A Bruggeman model was used to correlate the refractive index of each layer in the pSiMC to its porosity. The best fit between the theoretical and experimental spectra were obtained by adjusting the parameters of the simulated model (e.g. porosity and layer thickness), as previously reported.35 It should be noted that the angular dependence of the resonance wavelength was taken into consideration in designing the pSiMC.
| pSi Sample | Current density (mA cm−2) | Etching time (s) | Porosity (%) | Layer thickness (nm) | Refractive index | Pore size (nm) |
|---|---|---|---|---|---|---|
| MC57/14 | ||||||
| H layer | 56.8 | 5.6 | 83.8 | 137.7 | 1.25 | 65–100 |
| L layer | 14.2 | 18.2 | 70.8 | 117.7 | 1.60 | 22–35 |
|
||||||
| MC57/23 | ||||||
| H layer | 56.8 | 5.8 | 84.0 | 141.4 | 1.25 | 65–100 |
| L layer | 22.7 | 11.4 | 73.9 | 122.5 | 1.50 | 39–48 |
|
||||||
| Single layer | 56.8 | 81.3 | 84.0 | 1986.0 | 1.30 | 65–100 |
Two types of pSiMCs, labeled MC57/14 and MC47/23 were prepared. The numbers refer to the high and low current density applied during etching. For sample MC57/14, the first layer of the mirror was etched at 56.8 mA cm−2 (high porosity, H) while the second layer (low porosity, L) has a current density of 14.2 mA cm−2. Sample MC57/23 was etched at 56.8 mA cm−2 and 22.7 mA cm−2 for the high and low current density, respectively. The anodization time followed that listed in Table 1. From the SCOUT program, our optimum design resulted in a pSiMC configuration of (HL)3-HHHH-(LH)3, which consists of three periods of Bragg reflectors and four periods of H as the spacer layer. Freshly etched samples were washed thoroughly with ethanol and dried under N2 gas. In order to obtain the same film thickness as the pSiMC (∼2 μm), pSi single layers were prepared by etching the silicon wafers at 56.8 mA cm−2 for 81.3 s, then washed thoroughly with ethanol and dried under a stream of N2 gas.
The range of pore sizes for the H and L layers as well as the single layer were obtained through top view imaging of the samples by scanning electron microscopy. The pore sizes of the H and L layers were measured by preparing single layers of 56.8 mA cm−2 and 14.2 mA cm−2, respectively for sample MC57/14; and single layers of 56.8 mA cm−2 and 22.7 mA cm−2, respectively for sample MC57/23.
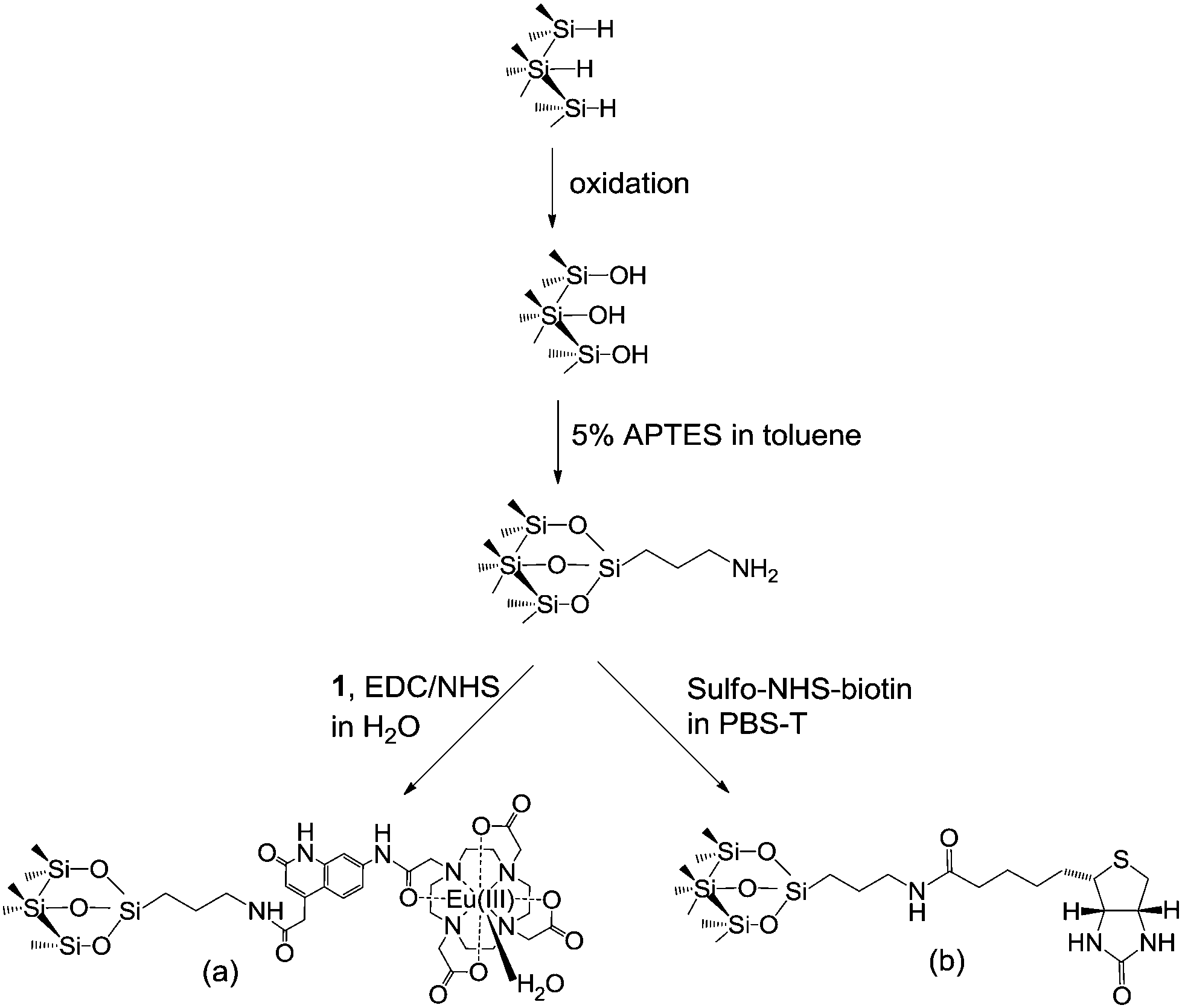 | ||
| Scheme 1 Surface modifications of the pSi resulting in (a) a surface with covalently attached complex 1 and (b) a surface functionalized with biotin for streptavidin detection. | ||
The attachment of Eu(III) complex to the pSi surfaces was carried out as follows. Complex 1 (1 mg) was dissolved in 250 μl of deionized water. The solution was then mixed with 5 mM of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC, Sigma Aldrich, Australia) and 5 mM of N-hydroxysuccinimide (NHS, Sigma Aldrich, Australia) in deionized water at room temperature for 15 min to form a succinimidyl ester group on compound 1.37 Amine-functionalized pSi samples were then exposed to 40 μl of the activated complex at room temperature, overnight and protected from light. Subsequently, samples were washed with deionized water and dried under N2 gas.
The biotin-functionalized surfaces were prepared as follows. Amine-functionalized pSi samples were exposed to 40 μl of 1 mM sulfo-NHS-biotin (Thermo Scientific, USA) in phosphate 0.1% Tween20 buffer (PBS-T) at room temperature for 1 h. Samples were washed with buffer and dried under N2 gas.

The biotinylated pSi surfaces were immersed in 40 μl of 5 μM Eu(III) complex labeled streptavidin at room temperature for 1 h. Subsequently, the pSi samples were rinsed with MES buffer, dried under N2 gas and stored in the dark for further interferometric and emission measurements.
The same procedure was applied for the optimization of biotin coverage and the determination of the limit of detection on the pSiMC surface. Optimization of biotin coverage on the surface was conducted by varying the biotin concentration of 0.006, 0.1, 1, 6.25, 12.5 and 25 mM exposed on the amine-terminated surfaces. Samples were then exposed to 40 μl of 5 μM Eu(III) complex labeled streptavidin and the emission at 614 nm was measured. To determine the limit of detection of the pSiMC surface, the amine-functionalized surface was exposed to 40 μl of 1 mM sulfo-NHS-biotin. The biotinylated surfaces was exposed to different concentrations of Eu(III) complex streptavidin, ranging from 0.15, 1, 2.5, 5, 10 and 20 μM. The emission at 614 nm was then measured.
The detection of Eu(III) complex labeled streptavidin on the pSi surfaces was also conducted in human wound fluid. The wound fluid was collected from six patients with chronic venous leg ulcers attending the multidisciplinary foot clinic at The Queen Elizabeth Hospital (Adelaide, Australia). Human wound fluid was diluted 10 times in 50 mM MES buffer and then incubated with 5 μM labeled streptavidin for 3 h at 37 °C. The biotinylated pSi surfaces were then exposed to spiked wound fluid for 1 h, rinsed with buffer and dried under N2 gas.
Streptavidin labeled with Cy5 dye was purchased from Invitrogen (Australia). The concentration of the stock solution was 1 mg ml−1. This was then diluted 3 times in 50 mM MES buffer and further used for the detection of streptavidin on the surface. The biotinylated pSi surfaces were exposed to the diluted Cy5 labeled streptavidin for 1 h, rinsed with buffer and dried under N2 gas.
Interferometric reflectance spectroscopy (IRS) measurements were conducted by applying white light from a tungsten lamp (Ocean Optics, USA), which was focused through a collimating lens onto the pSi surface at normal incidence. Light reflected from the surface was collected through the same optics and the distal end of the bifurcated fiber optic cable was connected to a CCD spectrometer (Ocean Optics S-2000). Reflectivity spectra were recorded over the wavelength range of 450–1000 nm.
Diffuse Reflectance Infrared Fourier Transform (DRIFT) spectra for p-type Si(100) wafers (0.0005–0.001 Ωcm) were obtained using a Thermo Nicolet Avatar 370MCT (Thermo Electron Corporation) instrument. A smart diffuse reflectance accessory was used, and spectra were recorded and analyzed using OMNIC version 7.3 software. Background spectra were taken from a clean unetched silicon wafer. Sample spectra were taken over the range of 800–3500 cm−1, accumulating 64 scans and selecting a 4 cm−1 resolution. All spectra were run in dry air to remove noise from CO2 and water vapor.
UV-Visible spectra for the Eu(III) complex labeled streptavidin were obtained using a Hewlett-Packard 8452 diode array spectrophotometer and analyzed using Agilent Technologies 8452 UV-vis Chemstation. The absorbance was recorded in the range of 190–1100 nm.
Luminescence intensities from the modified surfaces with the Eu(III) complex were measured on a LS55 fluorescence spectrometer (Perkin Elmer, USA) using excitation at 340 nm and recording the emission at the range of 550–650 nm. The measurement was conducted in phosphorescence mode with a cycle time of 20 ms, delay time of 0.1 ms and a photomultiplier voltage set to 900 V. Emission of the modified surfaces with Cy5 dye were measured using excitation at 640 nm and recording emission from 600 to 750 nm.
Results and discussions
Characterization of porous silicon samples
Mesoporous silicon samples were fabricated by electrochemical anodization on a heavily doped silicon wafer (p++) in ethanolic HF solution. The removal of the parasitic layer prior to sample preparation was conducted through short etching of the wafer followed by dissolution of the layer in NaOH. In order to construct a pSiMC, which is able to efficiently enhance the emission of a lanthanide complex, the infiltration of the desired emitting molecules through the microcavity layers and interaction between the infiltrated molecules and the electric field of the spacer layer of the pSiMC were optimized. This process involved the fine tuning of the thickness and refractive index of each layer of the microcavity, control over pore size and choice of the appropriate excitation configuration (i.e. laser wavelength and incidence angle). The pore size of the pSiMC configuration needs to be large enough to allow infiltration of the lanthanide complex or a lanthanide complex labeled protein, but still small enough to retain a high optical quality of the microcavity. The pore size in the pSiMC influences the porosity of the layers. This is critical for the design of pSiMC where the porosity of a layer defines its effective refractive index, which can be calculated from the Bruggeman model effective approximation. The refractive index contrast between alternating layers of H and L affects the photonic features of the microcavity.1 The energy stored within the pSiMC is measured by the quality factor (Q-factor) which is the ratio of the central resonance wavelength and the Full Width Half Maximum (FWHM) of the cavity resonance. A high Q-factor corresponds more efficient light confinement, translating into better luminescence enhancement.38Another important design criterion to develop an efficient luminescence enhancer, comes from the fact that complex 1 is excited at 340 nm. Since UV light is absorbed by pSi, a balance between high Q-factor which requires a large number repeats in the Bragg reflector and good excitation characteristics allowing the light to reach the spacer layer has to be found. Our simulation showed that a good trade-off between efficient excitation and efficient enhancement was obtained for a pSiMC configuration of (HL)3-HHHH-(LH)3, which consists of 3 periods of Bragg reflectors and 4 periods of H layer as the spacer layer. The porosities of each microcavity samples, its corresponding etching time and refractive indices are shown in Table 1.
Efficient luminescent enhancement of the Eu(III) complex within the pSiMC requires excellent spectral alignment between the resonance wavelength of the pSiMC and the emission maximum of the complex.10 It is also important to be mindful of the angular dependency of the pSiMC resonance. Here, the microcavity samples were designed for illumination at 45° angle of incidence to the surface since luminescence emission was recorded at this angle.
In Fig. 2, the experimental reflectance spectra under normal illumination (dashed curve) from two pSiMCs were compared with the corresponding best-fit simulations (solid curve) obtained by applying the transfer matrix method. Very good agreement between the experimentally observed and the simulated cavity modes was observed for both microcavities. Fig. 2(a) shows the reflectance spectrum of sample MC57/14. The spectrum showed a 235 nm wide reflectivity band between 653 and 935 nm, and the cavity mode of a freshly etched sample at normal incidence was positioned at 749 nm with an FWHM of ∼35 nm. The reflectance spectrum of sample MC57/23 is shown in Fig. 2(b). The experimental spectrum showed a reflectivity band between 633 and 917 nm with a cavity mode positioned at 735 nm with an FWHM of ∼38 nm. Similar Q values of 21.4 and 19.3 were obtained for MC57/14 and MC 57/23, respectively.
 | ||
| Fig. 2 Best-fit of the simulated and experimentally obtained reflectance spectra of freshly etched (a) MC57/14 and (b) MC57/23. The experimental spectra were recorded at normal incidence. | ||
Pore size, layer thickness and structure of the films were characterized by means of SEM measurements. Fig. 3 shows the SEM images of both top-view of the different layers of the pSiMCs. Fig. 3(a) and (b) shows the top view image of the H and L layers of MC57/14. The pore sizes for the H layers were in the range of 75–100 nm, while for L layers pore sizes ranging from 22 to 35 nm were determined. Both layers should therefore allow infiltration of the Eu(III) complex. Sample MC57/23 was designed with a higher porosity of L layer where the pore sizes were in the range of 39–45 nm (Fig. 3(c)). This range of pore size is around ten times larger than the diameter of streptavidin (∼4 nm) and should therefore allow protein infiltration.39Fig. 3(d) shows a cross sectional image of the ∼2 μm thick microcavity MC57/23, where the periods of the first and second Bragg reflectors and the spacer layer can be clearly observed. The measured thickness of the spacer layer (∼560 nm) was also in perfect agreement with that of the simulation (Table 1).
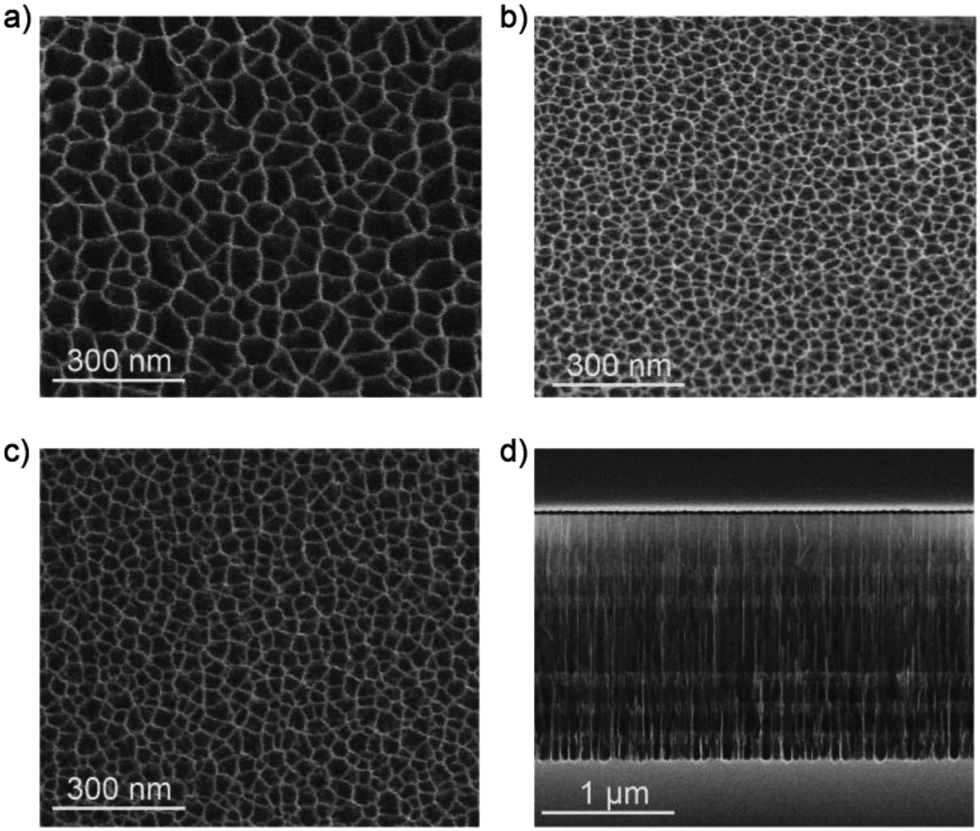 | ||
| Fig. 3 SEM images of (a) top-view H surface, (b) top-view L surface of sample MC57/14, (c) top-view L surface of sample MC57/23 and (d) cross-sectional view of the MC57/23. | ||
Infrared characterization of modified surfaces
The pSi films (pSiMC and single layer) were functionalized as shown in Scheme 1. Both experimental sets required an amine-terminated surface to covalently attach complex 1 and to generate a biotinylated surface. The freshly etched pSi samples were oxidized and subsequently silanized with APTES. The APTES modified samples were either exposed to the Eu(III) complex 1 to evaluate the luminescence properties of the resulting Eu(III) complex functionalized pSi or reacted with sulfo-NHS-biotin to facilitate detection of Eu(III) complex labeled streptavidin.Diffuse reflectance infrared Fourier transform (DRIFT) spectra were acquired for the pSi samples after each functionalization step. Fig. 4 shows the DRIFT spectra for (A) the pSi film after thermal and ozone oxidation, (B) after the reaction with APTES, (C) after the immobilization of Eu(III) complex and (D) after the reaction with sulfo-NHS-biotin. Spectrum (A) features prominent peaks located between 950 and 1200 cm−1. Peaks at 1024 and 856 cm−1 can be assigned to the Si–O–Si and Si–O vibrational modes, respectively, as previously reported.40 The absence of the specific band of the Si-Hx stretching modes at 2100 cm−1 confirms that all the hydrides on the surface had been oxidized.
 | ||
| Fig. 4 DRIFT spectra of modified pSi after (a) oxidation, (b) APTES modification, (c) modified with complex 1 and (d) modified with NHS-biotin. | ||
Spectrum (B) corresponding to the amine-functionalized surface shows peaks at 2840 and 2902 cm−1 attributed to the –CH2 asymmetric and symmetric stretching modes of the propyl chain. The evidence of an amine terminus on the surface after APTES modification is confirmed by the NH2 scissor vibration at 1577 cm−1. In addition to this mode, features at 1644 and 1488 cm−1 are seen that are attributed to the asymmetric and symmetric –NH3+ deformation modes respectively, suggesting partial protonation of the amine group.41–43
The surface modified with Eu(III) complex 1 in spectrum (C) shows spectral features confirming attachment of the NHS activated Eu(III) complex. The clear peak at 1654 and 1540 cm−1 correspond to the amide I and amide II vibrational modes, respectively, indicating the successful formation of an amide bond between the succinimidyl ester group and the amine group on the surface. The shoulder at 1616 cm−1 was assigned to the –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O– stretching vibrations of the carbonyl groups in the cyclen ligand.34 The aromatic –C–C– stretching vibrations the Eu(III) complex were observed at 1463 cm−1. The broad peak at ∼3250 cm−1 suggest the presence of hydroxyl groups from the coordinated water of the Eu(III) complex.
O– stretching vibrations of the carbonyl groups in the cyclen ligand.34 The aromatic –C–C– stretching vibrations the Eu(III) complex were observed at 1463 cm−1. The broad peak at ∼3250 cm−1 suggest the presence of hydroxyl groups from the coordinated water of the Eu(III) complex.
The optical shifts in the resonance of the two microcavities after different surface modifications were studied. The results summarized in Table 2 clearly show that modification of the surface had occurred supporting the DRIFT results. In the case of MC57/14, both thermal and ozone oxidation of the freshly etched samples resulted in a combined blue shift of the resonance wavelength by ∼25 nm. In turn, silanization produced a red shift of ∼20 nm. Subsequent reaction of the silanized surface with the NHS activated Eu(III) complex resulted in a further red shift of ∼3 nm.
| pSi surface | Δλoxidized (nm) | ΔλAPTES (nm) | Δλcomplex1 (nm) | ΔλSulfo-NHS-biotin (nm) |
|---|---|---|---|---|
| a (−) denotes a blue shift, (+) denotes a red shift. All shifts are compared to the peak wavelength of the samples in the previous step. | ||||
| MC57/14 | −30 | +20 | +3 | n.a. |
| MC57/23 | −25 | +25 | n.a. | +14 |
Luminescence enhancement
The position of the resonance wavelength for the modified surface MC57/14 of 749 nm determined in the previous section was measured at normal incidence. At 45° incidence, the resonance wavelength blueshifts and overlaps with the narrow emission of complex 1 at 614 nm as per our initial design requirement (Fig. 5(a)). The luminescence intensity of the tuned pSiMC functionalized with the Eu(III) complex was then compared to that of a functionalized single pSi layer of identical thickness. Fig. 5(c) shows the emission spectra of the Eu(III) complex on the microcavity surface (spectrum A) and the single layer (spectrum B). The luminescence of the Eu(III) complex on the microcavity was enhanced threefold compared to the single layer. This result demonstrates that the microcavity indeed generates a luminescence enhancement due to light confinement within the spacer resulting in higher emission intensity. Spectral alignment of the wavelength further boosts the emission, allowing coupling between the resonance wavelength and the light generated from the Eu(III) complex. This was confirmed by comparing the emission intensities of the aligned microcavity with that of a non-aligned microcavity of identical thickness (Fig. 5(b)). The comparison between spectra A and C of Fig. 5(c) show four-fold increase of intensity was observed on the microcavity with aligned resonance wavelength compared to that of non-aligned microcavity. | ||
| Fig. 5 Luminescence measurement of Eu(III) complex 1 on pSi surfaces (a) pSiMC with aligned resonance wavelength, (b) pSiMC with non-aligned resonance wavelength and (c) luminescence of Eu(III) complex 1 on a aligned microcavity (A), a single layer (B) and a non-aligned pSiMC (C). The dashed line show the simulated reflectance spectra of MC57/14 modified with complex 1 at 45° incidence. | ||
The emission intensities of the covalently bound Eu(III) complex 1 on MC57/14 was further compared to the luminescence of complex 1 in solution. Eu(III) complex 1 in water generally exhibits 5 emission bands (5D0 → 7FJ, J = 0–4) (ESI, Fig. S1†). The most prominent emission bands that contribute to the emission of the Eu(III) complex, are the ones originating from the 5D0 → 7F1 (λ = 585 nm) and 5D0 → 7F2 (λ = 614 nm) transitions. As shown in Fig. 5(a), the luminescence of immobilized Eu(III) complex on the modified microcavity surface caused the intensity of the 5D0 → 7F2 transition to increase twofold relative to the 5D0 → 7F1 transition as compared to that in solution phase. The electrons of the 5D0 → 7F2 energy transition are profoundly dependent upon the local coordination of the Eu(III) complex, i.e. host material, while the 5D0 → 7F1 transition is more susceptible to magnetic dipole transitions.44,45 This implies that the pSi matrix with a microcavity configuration is a suitable host for the Eu(III) complex and that the covalent attachment of complex 1 to the amine-functionalized pSi surface strengthens the 5D0 → 7F2 transition. The 5D0 → 7F2 to 5D0 → 7F1 ratio has been reported to be indicative of the quality of the luminescence of Eu(III) complex, with higher ratios showing increased color purity.44
Detection of Eu(III) complex labeled streptavidin
The use of the Eu(III) complex 1 as a signal amplifier was demonstrated next, following the observation of luminescence enhancement of the complex inside a pSi microcavity structure. The well-known noncovalent high affinity interaction between biotin and streptavidin was chosen as the model detection system.33 Surface MC57/23 was employed for the capturing of labeled streptavidin, owing to its larger pore size of the L layer in comparison to MC57/14. As a consequence, the porosity contrast between the H and L layers is decreased and the Q-value of the surface is slightly lower than that of MC57/14. The capturing of a Eu(III) complex-labeled streptavidin (Scheme 2) was conducted on a biotinylated pSiMC surface. Initially, the freshly etched surface was stabilized by thermal and ozone oxidation followed by silanization with APTES. The resulting amine-functionalized surface was stable in ethanol over a 2 h period (ESI, Fig. S2†) demonstrating that no silane was removed from the surface during that time. NHS-activated biotin was then reacted with the surface. In Fig. 4, spectrum (D), the formation of biotinylated surface was confirmed through the appearance of peaks at 1544 cm−1 and 1660 cm−1 assigned to amide II (N–H bend and C–N stretch) and amide I (CO stretch) vibrational modes, respectively. The C–H stretching vibrations at ∼2840 and 2902 cm−1 were also observed as expected. Reaction of the amine-functionalized surface MC57/23 with sulfo-NHS-biotin also resulted in a ∼14 nm red shift in the microcavity resonance, confirming the formation of a biotin layer on the silanized surface (Table 2 and Fig. S3†). For the same biotin concentration, the observed red shift is higher than in a previous report where the authors measured 4–5 nm of a red shift.15 This is expected due to increased thickness of the spacer layer causing a larger surface area for molecule binding and a higher shift of wavelength in the resonance cavity.18
 | ||
| Scheme 2 Schematic of the luminescence-based detection of Eu(III) complex-labeled streptavidin on a biotinylated pSi surface. | ||
Detection of labeled streptavidin (Scheme 2) on the biotinylated surface resulted in a small but measurable shift of ∼2 nm of the microcavity resonance. The Q-value of the pSi detection platform remained constant after the different surface modifications, indicating that the microcavity retained its structure and the porosity contrast remained the same (Fig. S3†). The preserved spectrum features, e.g. shape and width of the stopping band, indicated uniform modification throughout the layers of the microcavity.12
Luminescence and reflectance measurements were conducted to demonstrate the detection of Eu(III) complex labeled streptavidin on the pSi surfaces. Fig. 6(a) shows the luminescence spectrum of the biotinylated MC57/23 after capturing the Eu(III) complex labeled streptavidin. The concentration of the labeled streptavidin was 5 μM. In Fig. 6(b), the emission spectra of labeled streptavidin are compared between the biotinylated microcavity MC57/23 (spectrum A) and a single pSi layer of identical thickness functionalized in the same way (spectrum B). It can be clearly observed that the emission originated from the MC57/23 surface was increased three-fold compared to that from a single layer, showing that the microcavity structure is able to amplify the detection signal of the Eu(III) labeled biomolecule.
 | ||
| Fig. 6 (a) Luminescence spectra of Eu(III) complex labeled streptavidin captured on a biotin-functionalized MC57/23 (solid line) overlaid with the corresponding reflectance spectrum (dashed line) of the microcavity and (b) luminescence spectra of Eu(III) complex labeled streptavidin on (A) MC57/23 in MES buffer, (B) a single pSi layer in MES buffer, and (C) biotin-functionalized surface without labeled streptavidin. | ||
To further establish the application of the microcavity platform as a biosensing device in a complex biological system, we applied the biotinylated surface to detect the Eu(III) complex labeled streptavidin spiked at 5 μM concentration in human wound fluid at 37 °C. When the biotin-functionalized MC57/23 was exposed to spiked wound fluid, luminescence could be detected from the microcavity (ESI, Fig. S4†). The observed luminescence in this case was lower than in buffer but could be easily observed. The reason for the reduced emission compared to buffer could possibly be caused by the higher pH of the wound fluid, affecting the emission of the Eu(III) complex. The wound fluid has a basic pH ∼ 8.346 while the MES buffer was slightly acidic at pH 6.5. The luminescence of the Eu(III) complex, 1 decreases as the pH increases as shown in Fig. S5 (ESI†).
Control experiments were performed in buffer on samples without biotin on the surface to ensure that no labeled streptavidin would attach or adsorbed onto the surface alone. These control experiments showed no changes in the position of the resonance wavelength and no emission on the luminescence measurement (Fig. 6(b), spectrum C).
We also compared the performance of the sensor with a Eu(III) complex labeled streptavidin and a conventional fluorophore labeled streptavidin at the same concentration in buffer. The Cy5 dye has a maximum excitation of 640 nm which induces a fluorescence emission maximum of 670 nm. Due to the Stokes shift, the scattered light signal around 640 nm can be easily confused with the much smaller emission 670 nm as shown in Fig. S6 (ESI†). On the other hand, the large Stokes shift (∼270 nm) and long emission lifetime of the Eu(III) complex allow the emission to be easily distinguished from background fluorescence and scattering.
Towards an optical biosensor
To improve the sensitivity of the Eu(III) complex-streptavidin detection platform, the biotin coverage on the surface and the concentration of Eu(III) labeled streptavidin were optimized. The concentration of biotin applied on the APTES-modified surface were observed in order to avoid saturation of the surface with sulfo-NHS-biotin therefore decreasing the possibility of the surface capturing the labeled streptavidin. APTES-modified MC57/23 surfaces were incubated with different concentrations of reactive sulfo-NHS-biotin of 0.005, 0.1, 1, 6.25, 12.5 and 25 mM. Fig. 7(a) shows the effect of varying the concentration of sulfo-NHS-biotin during functionalization on the shift of the resonance wavelength of the microcavity after incubation. The graph shows an increase of resonance wavelength with sulfo-NHS-biotin concentration up to 12.5 mM consistent. Following the biotin functionalization, these surfaces were then exposed to the same concentration of Eu(III) complex labeled streptavidin of 5 μM. Fig. 7(b) depicts that the luminescence intensity of the Eu(III) complex labeled streptavidin captured on the surface which reached an optimum value at a 1 mM sulfo-NHS-biotin concentration or around 440 μg ml−1 of sulfo-NHS-biotin. The drop in luminescence intensity at higher concentrations was unexpected and is most likely due to the high biotin density which could reduce the likelihood of binding with the streptavidin, hence leading to a decrease in luminescence emission.15 Alternatively, self-quenching of the Eu(III) complex could occur at too high surface densities resulting in low luminescence intensity. The sulfo-NHS-biotin concentration of 1 mM was hence used for further experiments. | ||
| Fig. 7 (a) Shifts in resonance wavelength of the amine-functionalized MC57/23 microcavity after reaction with sulfo-NHS-biotin at various concentrations and (b) measured luminescence intensity of the biotin-functionalized microcavities (using different sulfo-NHS-biotin concentrations during functionalization) at 614 nm upon incubation with labeled streptavidin (5 μM). | ||
In order to determine the sensitivity of sensing platform, the sulfo-NHS-biotin functionalized MC57/23 (using a 1 mM concentration of the sulfo-NHS-biotin, as previously optimized) were exposed to six different concentrations of Eu(III) complex labeled streptavidin ranging from 150 nM to 20 μM in MES buffer for 1 h. In Fig. 8, the luminescence intensity of the captured labeled streptavidin on the pSiMC surface was plotted as a function of streptavidin concentration. The luminescence intensity was the intensity corresponding to the 5D0 → 7F2 transition of the Eu(III) complex which occurs at 614 nm. An increase of intensity was observed as the concentration of the labeled streptavidin increased up to 20 μM. A concentration as low as 150 nM was detected on the biotinylated MC57/23 surface by a luminescence signal six fold of that observed from pure buffer.
 | ||
| Fig. 8 Luminescence intensity of Eu(III) complex labeled streptavidin captured on MC57/23 as a function of its concentration. n = 5. | ||
The results provide proof-of-concept of Eu(III) complex labeled biomolecule detection on pSi microcavities through luminescence enhancement. The detection was conducted not only in buffer solutions, but also in a complex matrix, human wound fluid. The long luminescence lifetime of the Eu(III) complex and the large Stokes shift (∼270 nm) coupled with the ability of the pSiMC as an emission enhancer show advantages over the existing detection systems employing fluorescent dyes or pSi single layers.
Conclusions
In conclusion, immobilization of a covalently bound Eu(III) complex on a pSiMC and the resulting luminescence enhancement has been successfully demonstrated. The design of the pSiMC involves several parameters that need to be taken into account in order to obtain an optimum configuration. These parameters include pore size and the optical properties of the pSiMC, i.e. porosity contrast and thickness. A trade-off between the mentioned parameters was required to achieve homogenous infiltration of the Eu(III) complex molecules and ensure coupling between the emitting molecules and the electric field of the pSiMC layers. Luminescence measurements on an aligned pSiMC demonstrated an enhancement effect compared to the luminescence from a single pSi layer and a non-aligned microcavity of identical thickness.Proof-of-concept of detection of Eu(III) complex labeled streptavidin was achieved on the pSiMC both in buffer and human wound fluid. We were able to detect concentrations of streptavidin as low as 150 nM on the microcavity with an optimal biotin density. The approach of detecting Eu(III) complex labeled bioconjugates on pSi microcavities via luminescence enhancement opens up the possibilities of employing such system in the future as a biosensor platform in a range of applications including monitoring of biomarkers in chronic wounds.
Acknowledgements
SNAJ would like to thank the Australian Government for the Australia Award Scholarship and acknowledge funding from the Wound Management Innovation CRC (Australia). The authors would also like to thank Prof. Allison Cowin (Mawson Institute, University of South Australia) for providing human wound fluid samples. This research was conducted and funded by the Australian Research Council Centre of Excellence in Convergent Bio-Nano Science and Technology (project number CE140100036).Notes and references
- S. Chan, P. M. Fauchet, Y. Li, L. J. Rothberg and B. L. Miller, Phys. Status Solidi A, 2000, 182, 541–546 CrossRef CAS.
- F. Cunin, T. A. Schmedake, J. R. Link, Y. Y. Li, J. Koh, S. N. Bhatia and M. J. Sailor, Nat. Mater., 2002, 1, 39–41 CrossRef CAS PubMed.
- J. Wang, Analyst, 2005, 130, 421–426 RSC.
- Y. Zhao, X. Zhao and Z. Gu, Adv. Funct. Mater., 2010, 20, 2970–2988 CrossRef CAS.
- A. Jane, R. Dronov, A. Hodges and N. H. Voelcker, Trends Biotechnol., 2009, 27, 230–239 CrossRef CAS PubMed.
- M. J. Sailor and E. C. Wu, Adv. Funct. Mater., 2009, 19, 3195–3208 CrossRef CAS.
- V. S.-Y. Lin, K. Motesharei, K.-P. S. Dancil, M. J. Sailor and M. R. Ghadiri, Science, 1997, 278, 840–843 CrossRef CAS.
- M. Bosch, A. Sánchez, F. Rojas and C. Ojeda, Sensors, 2007, 7, 797–859 CrossRef CAS PubMed.
- A. Janshoff, K.-P. S. Dancil, C. Steinem, D. P. Greiner, V. S. Y. Lin, C. Gurtner, K. Motesharei, M. J. Sailor and M. R. Ghadiri, J. Am. Chem. Soc., 1998, 120, 12108–12116 CrossRef CAS.
- B. Sciacca, F. Frascella, A. Venturello, P. Rivolo, E. Descrovi, F. Giorgis and F. Geobaldo, Sens. Actuators, B, 2009, 137, 467–470 CrossRef CAS PubMed.
- K. A. Kilian, T. Böcking, K. Gaus, M. Gal and J. J. Gooding, ACS Nano, 2007, 1, 355–361 CrossRef CAS PubMed.
- G. Priano, L. N. Acquaroli, L. C. Lasave, F. Battaglini, R. D. Arce and R. R. Koropecki, Thin Solid Films, 2012, 520, 6434–6439 CrossRef CAS PubMed.
- L. Pavesi and V. Mulloni, J. Lumin., 1998, 80, 43–52 CrossRef CAS.
- S. M. Weiss and P. M. Fauchet, Phys. Status Solidi A, 2003, 197, 556–560 CrossRef CAS.
- H. Ouyang, M. Christophersen, R. Viard, B. L. Miller and P. M. Fauchet, Adv. Funct. Mater., 2005, 15, 1851–1859 CrossRef CAS.
- L. De Stefano, L. Moretti, I. Rendina and A. M. Rossi, Sens. Actuators, A, 2003, 104, 179–182 CrossRef CAS.
- S. Setzu, P. Ferrand and R. Romestain, Mater. Sci. Eng., B, 2000, 69–70, 34–42 CrossRef.
- G. Palestino, V. Agarwal, R. Aulombard, E. a. Pérez and C. Gergely, Langmuir, 2008, 24, 13765–13771 CrossRef CAS PubMed.
- S. Setzu, S. Létant, P. Solsona, R. Romestain and J. C. Vial, J. Lumin., 1998, 80, 129–132 CrossRef CAS.
- H. Qiao, B. Guan, T. Bocking, M. Gal, J. J. Gooding and P. J. Reece, Appl. Phys. Lett., 2010, 96, 161106 CrossRef PubMed.
- C. B. Poitras, M. Lipson, H. Du, M. A. Hahn and T. D. Krauss, Appl. Phys. Lett., 2003, 82, 4032–4034 CrossRef CAS PubMed.
- G. Palestino, M. Martin, V. Agarwal, R. Legros, T. Cloitre, L. Zimányi and C. Gergely, Phys. Status Solidi C, 2009, 6, 1624–1628 CrossRef CAS.
- L. A. DeLouise and H. Ouyang, Phys. Status Solidi C, 2009, 6, 1729–1735 CrossRef CAS.
- F. S. H. Krismastuti, S. Pace and N. H. Voelcker, Adv. Funct. Mater., 2014, 24, 3639–3650 CrossRef CAS.
- P. Huhtinen, M. Kivelä, O. Kuronen, V. Hagren, H. Takalo, H. Tenhu, T. Lövgren and H. Härmä, Anal. Chem., 2005, 77, 2643–2648 CrossRef CAS PubMed.
- H. L. Handl and R. J. Gillies, Life Sci., 2005, 77, 361–371 CrossRef CAS PubMed.
- K. Matsumoto and J. Yuan, in Metal Ions in Biological Systems: The Lanthanides and Their Interrelations with Biosystems, ed. H. Sigel, CRC Press, 2003 Search PubMed.
- E. G. Moore, A. P. S. Samuel and K. N. Raymond, Acc. Chem. Res., 2009, 42, 542–552 CrossRef CAS PubMed.
- L. D. Carlos, R. A. S. Ferreira, V. d. Z. Bermudez and S. J. L. Ribeiro, Adv. Mater., 2009, 21, 509–534 CrossRef CAS PubMed.
- H. Harma, T. Soukka and T. Lovgren, Clin. Chem., 2001, 47, 561–568 CAS.
- I. Hemmilä, J. Alloys Compd., 1995, 225, 480–485 CrossRef.
- K. Ai, B. Zhang and L. Lu, Angew. Chem., 2009, 121, 310–314 CrossRef.
- G. T. Hermanson, in Bioconjugate Techniques, Academic Press, New York, 2nd edn, 2008, pp. 900–923 Search PubMed.
- Z. Du, Synthesis and Characterization of Folate and Methotrexate Labelled Luminescent Lanthanide Complexes, Ph.D. Thesis, University of South Australia, 2013.
- S. Pace, R. B. Vasani, F. Cunin and N. H. Voelcker, New J. Chem., 2013, 37, 228–235 RSC.
- SCOUT, M., Theiss Hard- and Software, http://www.wtheiss.com.
- S. Sam, L. Touahir, J. Salvador Andresa, P. Allongue, J. N. Chazalviel, A. C. Gouget-Laemmel, C. Henry de Villeneuve, A. Moraillon, F. Ozanam, N. Gabouze and S. Djebbar, Langmuir, 2009, 26, 809–814 CrossRef PubMed.
- H. Ouyang and P. M. Fauchet, Proc. SPIE, 2005, 6005, 600508–600515 CrossRef PubMed.
- H. Li, S. H. Park, J. H. Reif, T. H. LaBean and H. Yan, J. Am. Chem. Soc., 2003, 126, 418–419 CrossRef PubMed.
- N. A. Lapin and Y. J. Chabal, J. Phys. Chem. B, 2009, 113, 8776–8783 CrossRef CAS PubMed.
- N. Aissaoui, L. Bergaoui, J. Landoulsi, J.-F. Lambert and S. Boujday, Langmuir, 2011, 28, 656–665 CrossRef PubMed.
- J. Kim, P. Seidler, L. S. Wan and C. Fill, J. Colloid Interface Sci., 2009, 329, 114–119 CrossRef CAS PubMed.
- C.-H. Chiang, H. Ishida and J. L. Koenig, J. Colloid Interface Sci., 1980, 74, 396–404 CrossRef CAS.
- Q. Xu, L. Li, B. Li, J. Yu and R. Xu, Microporous Mesoporous Mater., 2000, 38, 351–358 CrossRef CAS.
- K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed.
- F. S. H. Krismastuti, S. Pace, E. Melville, A. Cowin, T. R. Dargaville and N. H. Voelcker, Aust. J. Chem., 2013, 66, 1428–1434 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis of Eu(III) complex (1), luminescence spectrum of Eu(III) complex (1) in aqueous solution, stability test of APTES-modified surface in ethanol, optical response of MC57/23 after each surface modification, luminescence spectrum of Eu(III) complex labeled streptavidin on MC57/23 in wound fluid, luminescence spectrum of Eu(III) complex (1) in H2O at different pH and fluorescence spectrum of Cy5 labeled streptavidin on the biotin modified surface. See DOI: 10.1039/c4tb01409j |
| This journal is © The Royal Society of Chemistry 2014 |