
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Alternate layer-by-layer assembly of graphene oxide nanosheets and fibrinogen nanofibers on a silicon substrate for a biomimetic three-dimensional hydroxyapatite scaffold†
Jinhui
Wang‡
a,
Haixia
Wang‡
a,
Yizhu
Wang
a,
Jingfeng
Li
b,
Zhiqiang
Su
*a and
Gang
Wei
*b
aBeijing Key Laboratory on Preparation and Processing of Novel Polymeric Materials, Beijing University of Chemical Technology, 100029 Beijing, China. E-mail: suzq@mail.buct.edu.cn
bFaculty of Production Engineering, University of Bremen, D-28359 Bremen, Germany. E-mail: wei@uni-bremen.de
First published on 8th September 2014
Abstract
Layer-by-layer (LBL) assembly is a simple and effective method for the fabrication of a three-dimensional (3D) scaffold for nanotechnological and biomedical applications. Herein, a novel 3D scaffold based on an alternate LBL assembly of graphene oxide (GO) nanosheets and fibrinogen nanofibers (Fg NFs) on a silicon substrate was fabricated and utilized to create a 3D hydroxyapatite (HA) scaffold by biomimetic mineralization in 1.5× simulated body fluid for different nucleation periods. The obtained 3D (GO–NF)n–HA scaffold was characterized using atomic force microscopy, scanning electron microscopy, transmission electron microscopy, X-ray diffraction, Fourier transform infrared spectroscopy, and X-ray photoelectron spectroscopy. The results demonstrate that the Fg NFs could promote the nucleation and growth of HA crystals along the axis. The 3D (GO–NF)10–HA scaffold composed of 10 layers of GO alternating with 10 layers of NFs was successfully created by LBL assembly and subsequent biomimetic mineralization. The effects of the created 3D (GO–NF)10–HA scaffolds on the adhesion, morphology, and proliferation of L-929 cells were investigated. The in vitro cell culture indicates that the 3D (GO–NF)10–HA scaffold has a higher proliferation ability and better cytocompatibility than the other control samples.
1. Introduction
Bone is a hard tissue found in living systems, which serves as both a mechanical support and mineral reservoir in the body. The main source of complexity in bone arises from the process of mineralization, during which inorganic hydroxyapatite (HA, Ca10(PO4)6(OH)2) nanoplates are oriented and regularly dispersed within a type I collagen matrix.1 As a result of the increased understanding of how the mineralization process occurs in vivo, numerous in vitro studies have been devoted for the fabrication of materials that mimic the unique structure, strength, and bioactivity of bones.2,3 In the last two decades, HA has attracted considerable attention because of its biocompatibility, bioactivity, and nontoxicity, which makes it an excellent implant biomaterial that binds to the bone tissue and promotes bone repair.4,5In biomedical engineering, three-dimensional (3D) scaffolds have been designed to serve as a temporary artificial extracellular matrix (ECM) that can harness the natural progression of osseous growth by implantation to guide bone repair.6,7 In general, it should be noted that an ideal 3D scaffold can mimic the excellent characteristics of a natural ECM such as the cellular viability and some physical properties.8–10 In recent years, there has been an increasing interest in the preparation and application of 3D scaffolds as bone substitutes.11–14 For example, Liu et al. reported a 3D intrafibrillar mineralization with bone-like hierarchical nanostructures, which could improve the mechanical and biological properties of a collagen matrix.14
Collagen is the most abundant protein in bone and the formation of its nanoscale fibrillar structure in vivo has been proven to be important for the nucleation and growth of HA crystals.15 Inspired by the studies on collagen, many other protein and peptide nanofibers (NFs), such as silk fibers,16 fibrinogen NFs,17 and self-assembled peptide amphiphile NFs,18–20 have been created and utilized for the biomimetic synthesis of HA and apatite crystals. In one of our previous studies, we demonstrated that the self-assembled fibrinogen NFs possesses the ability to promote the nucleation and growth of HA crystals.17 It is evident that the self-assembled protein and peptide NFs are perfect candidates to replace collagen for the formation of HA.21 Firstly, the self-assembly and formation of these NFs are controllable in vitro to achieve different structures. Secondly, the created NFs contain rich and diverse functional groups on their surfaces, which provide the perfect nucleation sites for HA. Thirdly, these NFs exhibit excellent biocompatibility for the growth of bone.22
It is well known that the biomimetic mineralization of pure protein or peptide NFs is not capable of completely solving the problems such as poor tensile strength and fracture toughness associated with HA. To improve the mechanical properties of HA, the incorporation of HA into other materials, such as titanium alloy,23 TiO2,24,25 boron nitride,26 and carbon nanotubes, as a reinforcing phase27–30 has been explored. Graphene oxide (GO), an amphiphile building block with a largely hydrophobic basal plane and hydrophilic edges,31 could be an ideal and promising nanoscale reinforcement material for biomimetic mineralization due to its properties of high mechanical strength, large specific surface area (2600 m2 g−1), and good biocompatibility.32–36 GO nanosheets have been referred to as credible and excellent candidates to fabricate 3D scaffolds for bone regeneration.37–39 However, there are only limited studies on the fabrication of hierarchical 3D scaffolds by combining GO nanosheets with self-assembled protein and peptide NFs.
Fibrinogen (Fg) is a soluble plasma glycoprotein and it is an inactive precursor for fibrin, a protein that is involved in blood clotting.40 In addition, Fg has some unique properties, such as self-assembly,41 promotion of cell adhesion,42 and an ability to undergo biomimetic mineralization,43 which makes it a potential molecular template for the preparation of novel biomaterials. In this work, we first created the self-assembled Fg NFs, and then utilized an alternate layer-by-layer (LBL) assembly technique to fabricate a 3D scaffold containing GO and Fg NFs on a silicon substrate. We expected that the combination of GO nanosheets with Fg NFs will produce a novel scaffold with enhanced mechanical strength and bioactivity. To test this hypothesis, we synthesized 1-pyrenebutyric acid N-hydroxysuccinimide ester (PNHS)-functionalized GO nanosheets (GO–PNHS) and Fg NFs by a simple self-assembly method. Then, an alternate LBL assembly of GO nanosheets and Fg NFs was created on a silicon wafer to form a 3D scaffold with the desired layers. To investigate the formation of HA crystals, the obtained 3D scaffold was further soaked in 1.5× simulated body fluid (SBF) for different periods of time. In addition, the surface properties and the effects of the fabricated 3D scaffold on cell adhesion and growth were explored. To the best of our knowledge, this is the first report to create a hierarchical 3D GO- and NF-based HA scaffold with an alternate LBL assembly of GO nanosheets and Fg NFs on a silicon substrate.
2. Experimental section
2.1 Reagents and materials
Natural graphite flake (99.8% purity), Type I human plasma Fg (>95% clottable protein), and (3-aminopropyl)triethoxysilane (APTES, ≥98.0% purity) were purchased from Sigma-Aldrich. Tris(hydroxymethyl)aminomethane (Tris, ≥98.0% purity) was obtained from J&K Scientific Ltd. (Beijing, China). 1-Pyrenebutyric acid N-hydroxysuccinimide ester (PNHS, ≥95.0%) was obtained from Santa Cruz Biotechnology. Silicon wafers were supplied by Beijing Zhongjing Technology Co., Ltd. (Beijing, China). N,N-Dimethylformamide (DMF), KMnO4, Na2HPO4, NaH2PO4, KCl, NaCl, CaCl2·2H2O, K2HPO4·3H2O, MgCl2·6H2O, NaHCO3, Na2SO4, absolute ethanol, and acetone were purchased from Beijing Chemicals Co., Ltd. (Beijing, China). Hydrogen peroxide (H2O2, 30% aqueous solution) and H2SO4 (analytical grade, 98% aqueous solution) were purchased from Tianjin Dongfang Chemical Plant (Tianjin, China). All chemicals used in this work were of analytical reagent grade and directly used without additional purification. The water used was purified through a Millipore system (∼18.2 MΩ cm).2.2 Synthesis of GO and GO–PNHS
Natural graphite powder was oxidized using a modified method to produce graphite oxide.44,45 In brief, a mixture of concentrated H2SO4/H3PO4 (v/v, 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was mixed with 3.0 g of graphite flakes under stirring at room temperature. Then, KMnO4 (18 g) and H2O2 (10%, 10 mL) were added. Finally, the mixture was centrifuged and the remaining solid material was washed in succession with deionized water, 30% HCl and ethanol. The remaining material was filtered and vacuum-dried overnight at room temperature. Finally, 6.0 g of product was obtained.
:1) was mixed with 3.0 g of graphite flakes under stirring at room temperature. Then, KMnO4 (18 g) and H2O2 (10%, 10 mL) were added. Finally, the mixture was centrifuged and the remaining solid material was washed in succession with deionized water, 30% HCl and ethanol. The remaining material was filtered and vacuum-dried overnight at room temperature. Finally, 6.0 g of product was obtained.
GO–PNHS was synthesized according to one of our previous studies.35,46 In brief, GO powder was diluted with DMF to 0.1 mg mL−1, and then sonicated using a bath sonicator (100 W, 40 kHz) for 1 h. After that, PNHS was added into the GO solution to keep the concentration of PNHS at 10 mM. Finally, the mixture was stirred for 24 h at room temperature, centrifuged (13k rpm) twice and diluted with water for further use.
2.3 Synthesis of Fg NFs
Human plasma Fg solution was diluted to 20 ng μL−1 with ultrapure water. The diluted Fg solution was then incubated in a water bath at 37 °C for 30 min. After that, the prepared Fg solution was mixed with ethanol in a ratio of 1:4. Amyloid-like Fg NFs were created after the mixed solution was incubated in a water bath at 37 °C for 1 h.41
2.4 Modification of silicon wafer
Silicon wafers (1 × 1 cm) were rinsed sequentially with ultrapure water, absolute ethanol, and acetone in a bath sonicator, immersed in “Piranha solution” (30% H2O2:H2SO4, 3:7),47 and heated to boil for 1 h to remove the organic covering on their surface. After being cleaned with ultrapure water, the silicon wafers were immersed in a solution of 15 mL absolute ethanol containing 1 mL APTES for 5 h to generate amine groups on their surfaces. Finally, the APTES-modified substrates were cleaned with ultrapure water and dried with nitrogen for further use.
2.5 Construction of the 3D (GO–NF)n scaffold on a silicon wafer
The 3D (GO–NF)n scaffold was constructed by an alternate LBL assembly technique. In detail, APTES-modified silicon wafers were first immersed in a GO–PNHS aqueous solution (0.1 mg mL−1) at room temperature for 1 h followed by washing with ultrapure water. Then, the covalently assembled GO–silicon wafers were immersed in a Fg NFs solution (20 ng μL−1) for 1 h followed by several washes with ultrapure water. 3D (GO–NF)n (where n is the number of layers of GO and NFs) scaffolds can be produced by repeating the above procedure n times. In this work, we created a (GO–NF)n scaffold with 10 layers, and the created 3D scaffold was named as (GO–NF)10.2.6 Biomimetic synthesis of the 3D (GO–NF)n–HA scaffold
Biomimetic mineralization of the 3D (GO–NF)n scaffold was performed according to one of our previous reports.13,41 Briefly, the multilayered (GO–NF)n scaffold on a silicon wafer was incubated in 10 mL of a 1.5× SBF solution (4.5 mM KCl, 205.2 mM NaCl, 3.8 mM CaCl2·2H2O, 1.5 mM K2PO4·3H2O, 2.3 mM MgCl2·6H2O, 6.3 mM NaHCO3, 0.75 mM Na2SO4, buffered at pH 7.4 with Tris–HCl) at 37 °C for the growth of bone-like HA. The samples were taken out after different time intervals, washed with ultrapure water to remove the adsorbed salts, and then dried in nitrogen for subsequent characterization.2.7 Cytotoxicity evaluation
L-929 cells were used to investigate the cytocompatibility of GO, (GO–NF)10, and the mineralized (GO–NF)10–HA scaffold. Samples were washed with ethanol (70%), rinsed in Dulbecco's PBS (DPBS, PAA Laboratories GmbH), and placed in a μ-slide (Ibidi GmbH). 300 μL of a cell suspension containing 50000 cells was seeded into each well and incubated at 37 °C under 5% CO2 atmosphere and 100% humidity. The viability of the cells in the samples was calculated after 24 h of incubation. After incubation, cells were stained with 100 μL of a vitality staining solution containing fluorescein diacetate (stock solution 0.5 mg mL−1 in acetone) and propidium iodide (stock solution 0.5 mg mL−1 in Ringer's solution, Fluka). The viable and dead cells were quantified by fluorescence microscopy.
2.8 Experimental techniques
Atomic force microscopy (AFM) images were recorded using a NanoWizard 3 NanoScience atomic force microscope (JPK Instruments AG, Berlin, Germany) in tapping mode. Fourier transform infrared spectroscopy (FTIR, Nicolet 6700, Thermo-Fisher, USA), contact angle measurements (CA, JC2000C1, Zhongchen Digital tech. Ltd., China) and X-ray diffraction (XRD, Rigaku D/max-2500 VB+/PC) were used to compare the structures of GO, GO–NF, and (GO–NF)n–HA. Scanning electron microscopy (SEM) experiments were performed using a JSM-6700F scanning electron microscope (JEOL). Transmission electron microscopy (TEM) experiments were performed on a JEM-3010 transmission electron microscope with an accelerating voltage of 200 kV. The sample solution was transferred onto a copper grid and dried at room temperature. GO, (GO–NF)10, and (GO–NF)10–HA scaffolds modified with a fluorescence dye in water were characterized by confocal laser scanning microscopy (CLSM, Leica DM IRBE) with a 30 W UV lamp (λ = 350 nm) as the light source. Data was collected and analyzed utilizing the BioImage XD software.3. Results and discussion
3.1 Synthesis and characterization of the 3D (GO–NF)10–HA scaffold
Fig. 1 illustrates the construction of the 3D (GO–NF)n–HA architecture on silicon wafer by an alternate LBL assembly. First, APTES with –NH2 outer groups was covalently anchored onto the hydroxylated silicon wafer via Si–O–Si covalent bonding and the GO nanosheets were functionalized with PNHS via π–π stacking. Taking advantage of the chemical reactions between the succinimidyl ester group in PNHS and the amino group in APTES, the GO nanosheets were covalently attached to the APTES-functionalized silicon wafer. Because PNHS was anchored on both sides of GO surface, the other side of the GO nanosheets was still able to covalently react with the amino groups of the Fg NFs. We repeated this procedure 10 times such that the GO and Fg NFs were alternately attached onto the surface of the silicon wafer and a 3D scaffold was produced via covalent LBL assembly. After that, the fabricated 3D scaffold was immersed in 1.5× SBF to form HA crystals along the Fg NFs.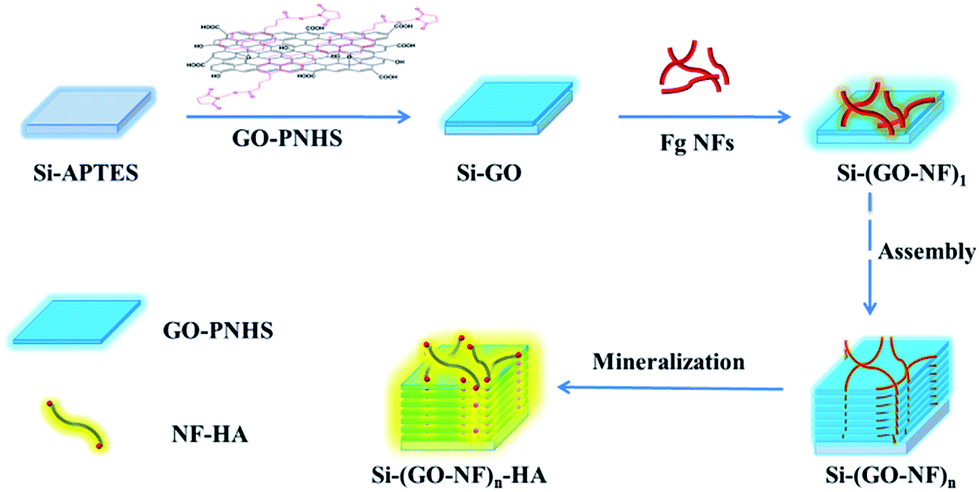 | ||
| Fig. 1 Schematic model for the construction of the 3D (GO–NF)n–HA hybrid material. | ||
The LBL assembly in this work is achieved by the covalent interactions between GO–PNHS and Fg NFs, which are very important for the enhancement of the chemical and structural stability of the fabricated 3D scaffold. AFM was used to confirm the immobilization of GO–PNHS and Fg NFs on the silicon wafer (Fig. 2). Fig. 2a shows the AFM height image of the silicon wafer after treatment with the Piranha solution. It is clear that the surface of the silicon wafer is smooth and that nothing else can be observed. In contrast, the surface topography of GO–PNHS is shown in Fig. 2b. After chemical interactions between GO–PNHS and Si–APTES, the silicon wafer was coated with GO nanosheets, which have wide size distribution from about 300 nm to 5 μm. Fig. 2c shows a typical AFM image of the self-assembled Fg NFs created by incubating 20 ng μL−1 Fg in 80% ethanol at 37 °C for 1 h. The created Fg NFs reveal a typical height of 2.2 ± 0.4 nm and lengths ranging from several hundred nm to a few μm, which agrees well with one of our previous report on the formation of Fg NFs.41 We suggest that the volume ratio of ethanol to Fg should be fine-tuned to promote the conformational transitions of Fg molecules and the self-assembly of Fg NFs.48,49 After modifying the Si–GO surface with the N-hydroxysuccinimide group, Fg NFs can react further with the GO surface. After chemical interactions between GO–PNHS and Fg NFs, Fg NFs were successfully immobilized on the surface of GO (Fig. 2d), which indicates that this preparation method is useful and feasible for the fabrication of the 3D (GO–NF)10 scaffold.
 | ||
| Fig. 2 AFM height images of (a) the silicon wafer, (b) GO–PNHS, (c) Fg NFs, and (d) one layer of the GO–NF hybrid. | ||
3.2 Surface morphology of the 3D (GO–NF)n–HA scaffold
Previous studies have indicated that the carboxylic groups on the surface of a matrix can absorb Ca2+ ions and facilitate the nucleation and growth of apatite.17,43 Therefore, Fg NFs can serve as a template for biomimetic mineralization because of the large amount of carboxylic groups on their surfaces. After the alternate LBL assembly of GO and Fg NFs, the created 3D (GO–NF)10 scaffold was incubated in 1.5× SBF to form HA crystals along the Fg NFs. AFM was utilized to observe the change in the apatite morphology on the GO–NF scaffold during the nucleation period. As is shown in Fig. 3a, the average height of the NFs is 3 ± 0.3 nm after exposing the 3D scaffold to a 1.5× SBF solution for 1 h. After 2 h of incubation, the average height of the NFs increases to 4 ± 0.5 nm, while the average height of the NFs further increases to 9.5 ± 2.5 nm after 7 h of incubation. Hence, we suggest that HA crystals nucleate and grow along the Fg NFs intercalated between the GO sheets. This structure closely mimics the lowest level of the hierarchical structure of natural bone in which HA crystals nucleate and grow along the collagen fibrils. In addition, the GO nanosheets support the mineralization of Fg NFs and keep the entire scaffold stable.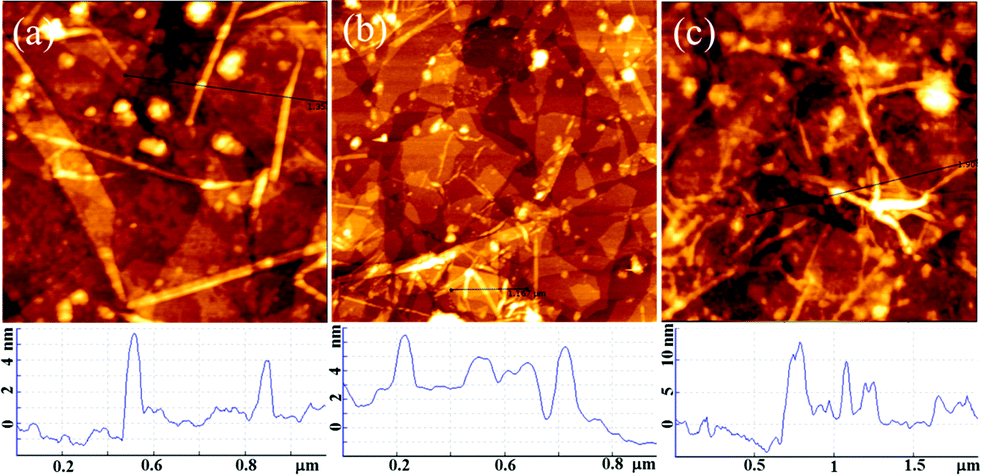 | ||
| Fig. 3 AFM height images of the 3D (GO–NF)10 scaffold at different times during mineralization: (a) 1 h; (b) 2 h; (c) 7 h. | ||
In addition, SEM was utilized to investigate the growth of HA crystals on the GO–NF scaffold. Fig. 4a shows a typical SEM image of the GO–NF scaffold after it was soaked in 1.5× SBF for 1 day. It is obvious that the surface of the scaffold becomes rough and that lots of branch-like minerals are formed on its surface. At a higher magnification, it can be seen that the branch-like minerals are composed of irregular aggregates (Fig. 4d). Furthermore, more minerals appear after 3 days of incubation (Fig. 4b), revealing that the apatite continues to grow. Interestingly, the higher magnification SEM image in Fig. 4e illustrates that the apatite grows regularly along the axis of the NFs with an average diameter of approximately 8 μm. For samples taken after 7 days of mineralization, the GO–NF surface is uniformly covered with branch-like apatite minerals (Fig. 4c and f). According to a previous report,38 no HA minerals were observed on a bare GO substrate after 14 days of mineralization. Therefore, we suggest that the Fg NFs play an important role in mediating the growth of apatite minerals on the 3D (GO–NF)10 scaffold. In addition, the cross-section analysis could display the multilayer structure (thickness of about 200 nm) of the fabricated 3D (GO–NF)10 scaffold, as shown in Fig. S1.†
 | ||
| Fig. 4 SEM images of the mineralized HA crystals on the 3D (GO–NF)10 scaffold after being soaked in 1.5× SBF for (a and d) 1 day, (b and e) 3 days and (c and f) 7 days. | ||
To further investigate the internal structure of 3D GO–NF–HA, a TEM measurement was carried out (Fig. 5). Fig. 5a shows the TEM image of the monolayer GO nanosheets. It can be seen that there is some corrugation and scrolling on the surface of GO. After being soaked in 1.5× SBF for 7 h, numerous fiber-like dark lines can be observed in the images of the 3D (GO–NF)10 (as indicated with arrows in Fig. 5c). Since the Fg NFs have not been stained, the contrast can only arise from the formation of HA crystals,50 which further confirm the nucleation and growth of HA along the Fg NFs. In addition, the lattice orientation of the HA crystals in the TEM image in Fig. 5d confirms the formation of HA on the 3D (GO–NF)10 scaffold with an interplanar distance of 0.28 nm that corresponds to the (211) planes of the HA hexagonal structure.
 | ||
| Fig. 5 TEM images of (a) GO nanosheets, and (b and c) 3D (GO–NF)10–HA after being soaked in 1.5× SBF for 7 h. (d) Corresponding HRTEM image. | ||
3.3 Structure and properties of 3D (GO–NF)10–HA
The chemical compositions of the products for each step were further confirmed by FTIR. Fig. 6 shows the FTIR spectra of Si–APTES, GO, 3D (GO–NF)10 and the 3D (GO–NF)10–HA hybrid after being soaked in 1.5× SBF for 1 day, 3 days, and 7 days. As shown in Fig. 6a, the APTES functionalized silicon wafer shows characteristic bands between 2850 and 2990 cm−1, corresponding to the CHx stretching of APTES.51 The bands between 950 and 1250 cm−1 correspond to the SiO-X region, and the band at 1570 cm−1 reveals the scissor vibration of the NH2 terminal group of APTES,51,52 indicating that the silicon wafer was successfully modified with APTES. The FTIR spectrum of GO clearly indicates the presence of oxygen-containing functional groups at 1100, 1594, and 1722 cm−1, which correspond to the C–O stretching vibration, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration, and CO stretching vibration of GO, respectively. Furthermore, the disappearance of the characteristic bands of Si–APTES can also be observed, verifying that the Si surface is covered with GO nanosheets. In addition, it can be observed that the bands at 1238 and 1455 cm−1, which correspond to C–N and C–H groups, respectively, appeared in the spectrum of GO–NF, showing the successful conjugation of Fg NFs with GO nanosheets through amidation. Fig. 6b shows the FTIR spectra of the 3D (GO–NF)10 scaffolds after being soaked in 1.5× SBF for different periods of time. The appearance of absorption bands at 1030 and 1375 cm−1 is ascribed to the symmetric stretching of PO43− and the stretching vibration of CO32−, respectively,53 which proves the successful formation of HA on the 3D GO–NF hybrid scaffold after being soaked in 1.5× SBF.
C stretching vibration, and CO stretching vibration of GO, respectively. Furthermore, the disappearance of the characteristic bands of Si–APTES can also be observed, verifying that the Si surface is covered with GO nanosheets. In addition, it can be observed that the bands at 1238 and 1455 cm−1, which correspond to C–N and C–H groups, respectively, appeared in the spectrum of GO–NF, showing the successful conjugation of Fg NFs with GO nanosheets through amidation. Fig. 6b shows the FTIR spectra of the 3D (GO–NF)10 scaffolds after being soaked in 1.5× SBF for different periods of time. The appearance of absorption bands at 1030 and 1375 cm−1 is ascribed to the symmetric stretching of PO43− and the stretching vibration of CO32−, respectively,53 which proves the successful formation of HA on the 3D GO–NF hybrid scaffold after being soaked in 1.5× SBF.
 | ||
| Fig. 6 FTIR spectra of (a) Si–APTES, GO, and GO–NF, (b) 3D GO–NF–HA after incubation in 1.5× SBF for 1 day, 3 days and 7 days. | ||
The XRD patterns of the 3D (GO–NF)10 scaffold with different soaking times are displayed in Fig. 7. Compared to the XRD pattern of the 3D (GO–NF)10 scaffold (Fig. S2†), the XRD pattern of the 3D (GO–NF)10 scaffold after being soaked in 1.5× SBF for 1 h shows sharp peaks at 2θ = 31.8° and 45.6°, corresponding to the (211) and (222) planes, respectively. The XRD pattern of the mineralized scaffold is similar to the diffraction planes of octacalcium phosphate (OCP), which is a potential candidate that has already been suggested as a precursor for bone-like apatite in vivo.53–55 After its formation, OCP may be hydrolyzed to apatite by subsequent steps. When the soaking time is increased to 2 hours, the intensity of the peak at 2θ = 31.8° rises. In addition, a weak peak corresponding to the (210) plane at 2θ = 28.5° appears after 7 h of incubation. This result proves that HA forms on the 3D (GO–NF)10 scaffold and preferentially grows along the (211) plane. Furthermore, when the incubation period is prolonged to 7 days (Fig. 7b), it can be observed that more new peaks appear at 2θ = 25.9°, 27.5°, 32.9°, and 39.3°, which can be assigned to the (002), (102), (300), and (310) planes of HA crystals, respectively. These characteristic peaks are consistent with the standard diffraction pattern of the HA crystal (JCPDF 09-432),56,57 confirming the formation of HA on the 3D (GO–NF)10 scaffold.
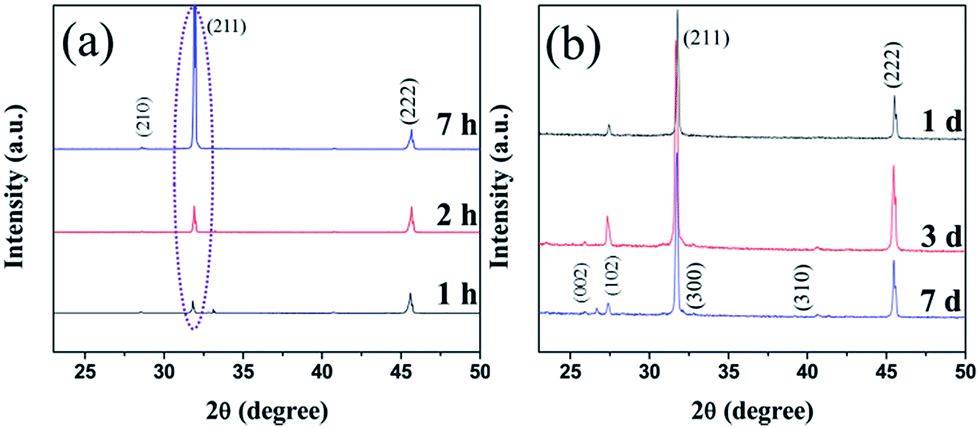 | ||
| Fig. 7 XRD patterns of the 3D (GO–NF)10 scaffold after being immersed in 1.5× SBF for (a) 1 h, 2 h, and 7 h, and (b) 1 day, 3 days, and 7 days. | ||
XPS was utilized to analyse GO and 3D (GO–NF)10–HA (Fig. 8). As shown in Fig. 8a, the spectrum of GO only exhibits the existence of C and O element. However, the signals of N 1s, Ca 2p, and P 2p emerge in the XPS spectrum of 3D (GO–NF)10–HA, indicating the covalent bonding of Fg NFs with GO nano-sheets and the formation of HA crystals. Fig. 8b shows the peak-differentiation-imitating analysis of the C 1s spectrum of 3D (GO–NF)10–HA. Five different types of carbon with peaks at 284.5, 285.5, 286.2, 286.8, and 288.4 eV are shown, corresponding to carbon atoms in CC, C–N, C–O, C–OH, and CO groups, respectively.48,58 The C–N peak at 285.5 eV further proves the formation of GO–NF. Fig. 8c provides the high-resolution XPS Ca 2p spectra of 3D (GO–NF)10–HA between 345 and 355 eV. A high-resolution P 2p spectrum centred at 132.8 eV is shown in Fig. 8d. These results are in agreement with the previously reported data.58
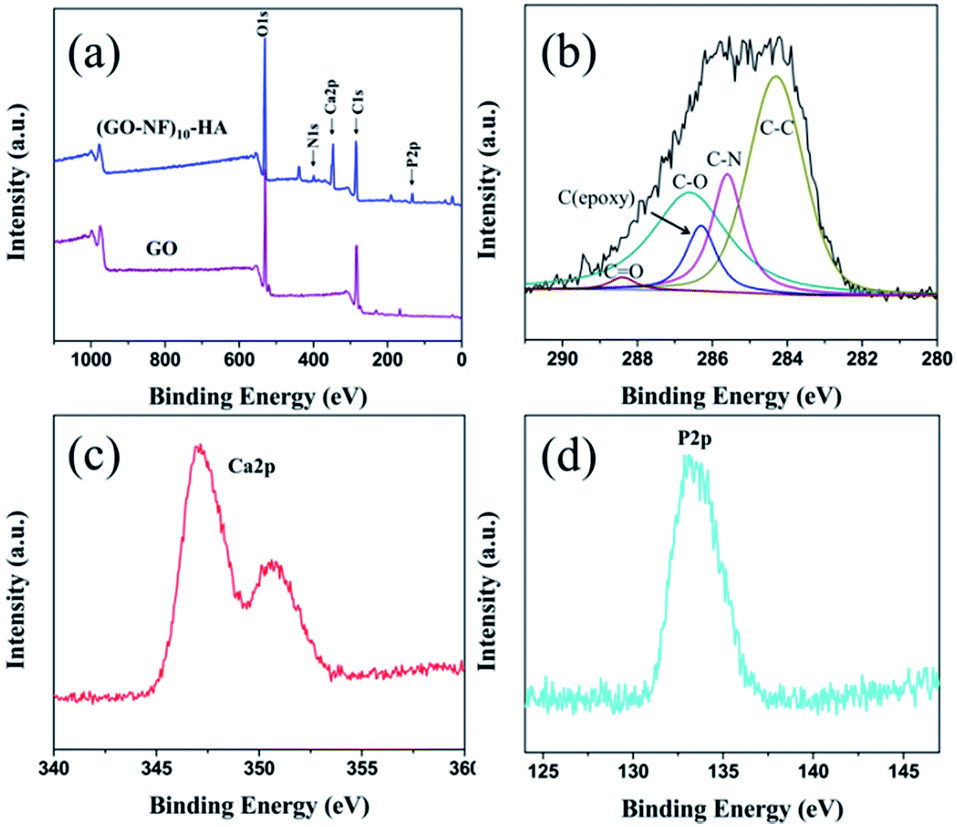 | ||
| Fig. 8 XPS spectra of (a) GO and 3D (GO–NF)10 after being soaked in 1.5× SBF for 7 days, (b) C 1s spectra, (c) Ca 2p spectrum, and (d) P 2p spectrum of 3D (GO–NF)10–HA. | ||
3.4 Possible growth mechanism of HA on the (GO–NF)10 scaffold
Based on the AFM results shown in Fig. 3 and the SEM results in Fig. 4, we suggest that biomineralization in short term is totally different from than in long term. The formation of apatite nanocrystals along the axis of the Fg NFs can be clearly observed when the incubation time is of several hours (Fig. 3), but the surface of the 3D (GO–NF)10 scaffold is coated by HA microcrystals when the incubation time is increased to several days (Fig. 4). The nucleation and growth of HA crystals in this work is similar to our observations in previous studies with Fg NFs, polymer NFs, and fibronectin–CNT hybrids.13,17,27Based on these results, we propose a possible mechanism for the formation of the 3D (GO–NF)10–HA scaffold, as shown in Fig. 9. The functionalization of GO by Fg NFs is achieved through amidation. This chemical reaction ensures that Fg NFs are firmly adsorbed on the GO nanosheets. The Fg NFs possess many amino, hydroxyl, and carboxyl groups, and they can ionize and mediate the mineralization of HA in 1.5 × SBF. Therefore, when 3D (GO–NF)10 is immersed in 1.5× SBF, the negatively charged functional groups of the Fg NFs provide pinning forces for Ca2+ and react with Ca2+ through electrostatic attractions. After the nucleation sites are created, PO43− and OH− ions will be absorbed around the sites to produce the HA crystal. In a short-term mineralization process, only apatite nanocrystals can grow along the axis of the Fg NFs. With increase in incubation time to several days, HA nanocrystals connect with each other and grow along the vertical direction of a NF axis. Finally, tabular HA microcrystals are created to cover the entire surface and interlayers of the 3D (GO–NF)10 scaffold. We suggest that GO nanosheets are important because they can support the formation of tabular HA microcrystals on their surfaces.
 | ||
| Fig. 9 Schematic presentation of the formation mechanism of the 3D (GO–NF)10–HA scaffold. | ||
3.5 Wettability test
Contact angle (CA) analysis provided an important strategy to evaluate surface chemistry properties. Fig. 10a shows the CA images and values for the Si substrate modified with Fg NFs, GO nanosheets, a GO–NF monolayer, GO–NF–GO, a (GO–NF)2 bilayer, and the mineralized (GO–NF)10–HA scaffold. It is evident that these modified silicon wafers show different CA values (Fig. 10b), indicating differences in the surface compositions of the silicon wafers after the alternate assembly of GO nanosheets and Fg NFs. The silicon wafer modified with Fg NFs shows a CA of 50°, while the silicon wafer modified with GO nanosheets exhibits more of a hydrophilic property with a CA of 36.5° due to the multiple oxygen-containing groups on the surfaces of GO nanosheets. In addition, the silicon wafer modified with a GO–NF monolayer has approximately the same CA value as the wafer modified with Fg NFs, which confirms that the GO nanosheets are almost completely covered by the Fg NFs on the GO–NF substrate. However, a lower CA value was observed after a second alternating GO and NF coating on the surface of Si–GO–NF, which reveals that the wettability of the 3D scaffold is sensitive to the number of GO–NF layers. We suggest that the surface roughness may change with the LBL assembly, thus affecting the CA value of the obtained surface.59 In addition, we investigated the wettability of the mineralized 3D (GO–NFs)10–HA scaffold, and it shows the best hydrophilicity out of all six surfaces.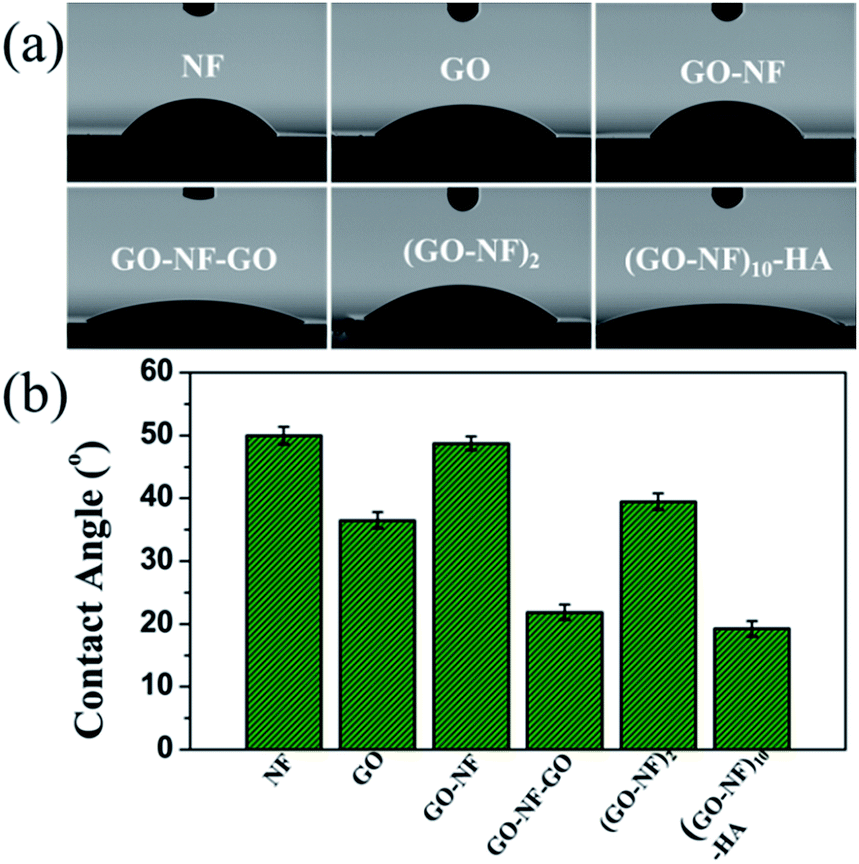 | ||
| Fig. 10 (a) The water droplet photographs and (b) corresponding air/water contact angle values for NF, GO, GO–NF, GO–NF–GO, and (GO–NF)2 modified silicon wafers and the mineralized (GO–NF)10–HA scaffold. | ||
3.6 In vitro cytotoxicity evaluation
To evaluate the potential applications of the created 3D (GO–NF)10–HA scaffold for biomedical engineering, its cytocompatibility was tested. We performed a live-dead assay via direct contact of cells (fibroblast cell line L-929) with GO, (GO–NF)10, and the 3D (GO–NF)10–HA scaffold after 24 h of incubation (with TCPS as a control), and the result is shown in Fig. 11a–d. In this test, the dead cells appear in red and the living cells appear in green when observed with a CLSM. It is clear that the nuclear fluorescence intensities of the cells on GO, (GO–NF)10, and the 3D (GO–NF)10–HA scaffold are distinctively higher than the intensity of the cells cultured on the TCPS control. In addition, nearly no dead cells are found in any of the samples, suggesting that all the materials (GO, (GO–NF)10, and (GO–NF)10–HA) possess effective cytocompatibility. It can also be observed that the L-929 cells cultured on the 3D (GO–NF)10–HA scaffold exhibit the highest viability and proliferation compared to other samples (Fig. 11e). Based on the cell culture result, we suggest that this novel 3D (GO–NF)10–HA scaffold has no obvious inhibitory effects on the in vitro cell proliferation of L-929 cells, and exhibits excellent in vitro cytocompatibility. | ||
| Fig. 11 (a–d) CLSM images of L-929 cells (green) on (a) the TCPS control, (b) GO, (c) (GO–NF)10, and (d) the 3D (GO–NF)10–HA scaffold after 24 h of incubation. (e) The number of live cells cultured in the corresponding samples. | ||
Conclusions
In summary, we demonstrated a feasible method to fabricate a hierarchical 3D (GO–NF)10–HA scaffold with an alternate LBL assembly of GO nanosheets and Fg NFs for further biomimetic mineralization. Fg NFs and GO nanosheets can be successfully assembled via covalent interactions onto a silicon substrate. The Fg NFs layers between the GO nanosheets can successfully mediate the nucleation and growth of HA crystals on the fabricated 3D (GO–NF)10 scaffold, forming a novel hierarchical 3D (GO–NF)10–HA hybrid scaffold. After 24 h of incubation with the created 3D (GO–NF)10–HA scaffold, the L-929 cells reveal a high proliferation ability and excellent cytocompatibility, which proves the excellent cytocompatibility of this GO-, NF-, and HA-based scaffold. We expect that this novel biocompatible scaffold will show great potential in biomedical applications like tissue engineering and drug delivery.Acknowledgements
The authors gratefully acknowledge the financial supports from the Fundamental Research Funds for the Central Universities (project no. ZZ1307). We would like to thank the financial support of the China Scholarship Council (CSC) for a PhD scholarship in University of Bremen.Notes and references
- K. A. Mosiewicz, K. Johnsson and M. P. Lutolf, J. Am. Chem. Soc., 2010, 132, 5972–5974 CrossRef CAS PubMed.
- B. Balakrishnan and R. Banerjee, Chem. Rev., 2011, 111, 4453–4474 CrossRef CAS PubMed.
- C. P. Barnes, S. A. Sell, E. D. Boland, D. G. Simpson and G. L. Bowlin, Adv. Drug Delivery Rev., 2007, 59, 1413–1433 CrossRef CAS PubMed.
- S. Weiner and H. D. Wagner, Annu. Rev. Mater. Sci., 1988, 28, 271–298 CrossRef.
- T. A. Taton, Nature, 2001, 412, 491–492 CrossRef CAS PubMed.
- T. G. Kim, H. Shin and D. W. Lim, Adv. Funct. Mater., 2012, 22, 2446–2468 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- P. X. Ma, Adv. Drug Delivery Rev., 2008, 60, 184–198 CrossRef CAS PubMed.
- H. Shin, S. Jo and A. G. Mikos, Biomaterials, 2003, 24, 4353–4364 CrossRef CAS.
- K. Anselme, Biomaterials, 2000, 21, 667–681 CrossRef CAS.
- X. Liu, L. A. Smith, J. Hu and P. X. Ma, Biomaterials, 2009, 30, 2252–2258 CrossRef CAS PubMed.
- A. L. Oliveira, S. A. Costa, R. A. Sousa and R. L. Reis, Acta Biomater., 2009, 5, 1626–1638 CrossRef CAS PubMed.
- Z. Su, J. Li, Z. Ouyang, M. M. Arras, G. Wei and K. D. Jandt, RSC Adv., 2014, 4, 14833–14839 RSC.
- Y. Liu, D. Luo, X. X. Kou, X. D. Wang, F. R. Tay, Y. L. Sha, Y. H. Gan and Y. H. Zhou, Adv. Funct. Mater., 2013, 23, 1404–1411 CrossRef CAS.
- Y. Wang, T. Azaïs, M. Robin, A. Vallée, C. Catania, P. Legriel, G. Pehau-Arnaudet, F. Babonneau, M. Giraud-Guille and N. Nassif, Nat. Mater., 2012, 11, 724–733 CrossRef CAS PubMed.
- A. Takeuchi, C. Ohtsuki, T. Miyazaki, H. Tanaka, M. Yamazaki and M. Tanihara, J. Biomed. Mater. Res., Part A, 2003, 65, 283–289 CrossRef PubMed.
- G. Wei, J. Reichert, J. Bossert and K. D. Jandt, Biomacromolecules, 2008, 9, 3258–3267 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- T. D. Sargeant, C. Aparicio, J. E. Goldberger, H. Cui and S. I. Stupp, Acta Biomater., 2012, 8, 2456–2465 CrossRef CAS PubMed.
- H. Ceylan, S. Kocabey, H. Unal Gulsuner, O. S. Balcik, M. O. Guler and A. B. Tekinay, Biomacromolecules, 2014, 15, 2407–2418 CAS.
- C. Li, A. K. Born, T. Schweizer, M. Zenobi-Wong, M. Cerruti and R. Mezzenga, Adv. Mater., 2014, 26, 3207–3212 CrossRef CAS PubMed.
- N. P. Reynolds, K. E. Styan, C. D. Easton, Y. Li, L. Waddington, C. Lara, J. S. Forsythe, R. Mezzenga, P. G. Hartley and B. W. Muir, Biomacromolecules, 2013, 14, 2305–2316 CrossRef CAS PubMed.
- A. Carradó, ACS Appl. Mater. Interfaces, 2010, 2, 561–565 Search PubMed.
- A. Pittrof, S. Bauer and P. Schmuki, Acta Biomater., 2011, 7, 424–431 CrossRef CAS PubMed.
- H. Li, K. A. Khor and P. Cheang, Biomaterials, 2002, 23, 85–91 CrossRef CAS.
- D. Lahiri, V. Singh, A. P. Benaduce, S. Seal, L. Kos and A. Agarwal, J. Mech. Behav. Biomed. Mater., 2011, 4, 44–56 CrossRef CAS PubMed.
- G. Wei, J. T. Zhang, L. Xie and K. D. Jandt, Carbon, 2011, 49, 2216–2226 CrossRef CAS PubMed.
- Q. Tan, K. Zhang, S. Gu and J. Ren, Appl. Surf. Sci., 2009, 255, 7036–7039 CrossRef CAS PubMed.
- D. Lahiri, V. Singh, A. Keshri, S. Seal and A. Agarwal, Carbon, 2010, 48, 3103–3120 CrossRef CAS PubMed.
- L. Debrupa, P. B. Ana, K. Lidia and A. Arvind, Nanotechnology, 2011, 22, 355703–355712 CrossRef PubMed.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS PubMed.
- X. Zhao, Y. Li, J. Wang, Z. Ouyang, J. Li, G. Wei and Z. Su, ACS Appl. Mater. Interfaces, 2014, 6, 4254–4263 CAS.
- Y. Chang, S. T. Yang, J. H. Liu, E. Dong, Y. Wang, A. Cao, Y. Liu and H. Wang, Toxicol. Lett., 2011, 200, 201–210 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. T. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- H. Wang, D. Sun, N. Zhao, X. Yang, Y. Shi, J. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2014, 2, 1362–1370 RSC.
- P. Zhang, Y. Huang, X. Lu, S. Zhang, J. Li, G. Wei and Z. Su, Langmuir, 2014, 30, 8980–8989 CrossRef CAS PubMed.
- M. Li, Y. Wang, Q. Liu, Q. Li, Y. Cheng, Y. Zheng, T. Xi and S. Wei, J. Mater. Chem. B, 2013, 1, 475–484 RSC.
- H. Liu, J. Cheng, F. Chen, F. Hou, D. Bai, P. Xi and Z. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 3132–3140 CAS.
- H. Liu, J. Cheng, F. Chen, D. Bai, C. Shao, J. Wang, P. Xi and Z. Zeng, Nanoscale, 2014, 6, 5315–5322 RSC.
- T. Lisman, C. Weeterings and P. G. de Groot, Front. Biosci., Landmark Ed., 2005, 10, 2504–2517 CrossRef CAS PubMed.
- G. Wei, J. Reichert and K. D. Jandt, Chem. Commun., 2008, 3903–3905 RSC.
- J. Koo, D. Galanakis, Y. Liu, A. Ramek, A. Fields, X. Ba, M. Simon and M. H. Rafailovich, Biomacromolecules, 2012, 13, 1259–1268 CrossRef CAS PubMed.
- S. Areva, T. Peltola, E. Säilynoja, K. Laajalehto, M. Linden and J. B. Rosenholm, Chem. Mater., 2002, 14, 1614–1621 CrossRef CAS.
- S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. B. T. Nguyen and R. S. Ruoff, Carbon, 2007, 45, 1558–1565 CrossRef CAS PubMed.
- P. Zhang, X. Zhang, S. Zhang, X. Lu, Q. Li, Z. Su and G. Wei, J. Mater. Chem. B, 2013, 1, 6525–6531 RSC.
- G. Wei, Y. Zhang, S. Steckbeck, Z. Su and Z. Li, J. Mater. Chem., 2012, 22, 17190–17195 RSC.
- H. Li, J. Zhang, X. Zhou, G. Lu, Z. Yin, G. Li, T. Wu, F. Boey, S. S. Venkatraman and H. Zhang, Langmuir, 2009, 26, 5603–5609 CrossRef PubMed.
- J. Wang, X. Zhao, J. Li, X. Kuang, Y. Fan, G. Wei and Z. Su, ACS Macro Lett., 2014, 3, 529–533 CrossRef CAS.
- S. Goda, K. Takano, K. Yutani, Y. Yamagata, R. Nagata, H. Akutsu, S. Maki, K. Namba and K. Yutani, Protein Sci., 2000, 9, 369–375 CrossRef CAS PubMed.
- Y. Wang, L. Cao, S. Guan, G. Shi, Q. Luo, L. Miao, L. Thistlethwaite, Z. Huang, J. Xu and J. Liu, J. Mater. Chem., 2012, 22, 2575–2581 RSC.
- R. Pena-Alonso, F. Rubio, J. Rubio and J. L. Oteo, J. Mater. Sci., 2007, 42, 595–603 CrossRef CAS PubMed.
- R. M. Pasternack, S. Rivillon Amy and Y. J. Chabal, Langmuir, 2008, 24, 12963–12971 CrossRef CAS PubMed.
- M. C. Hsiao, S. H. Liao, M. Y. Yen, P. I. Liu, N. W. Pu, C. A. Wang and C. C. M. Ma, ACS Appl. Mater. Interfaces, 2010, 2, 3092–3099 CAS.
- C. Drouet, BioMed Res. Int., 2013, 2013, 490946 Search PubMed.
- W. E. Brown, L. W. Schroeder and J. S. Ferris, J. Phys. Chem., 1979, 83, 1385–1388 CrossRef CAS.
- K. Lin, J. Chang, Y. Zhu, W. Wu, G. Cheng, Y. Zeng and M. Ruan, Cryst. Growth Des., 2008, 9, 177–181 Search PubMed.
- K. Lin, Y. Zhou, Y. Zhou, H. Qu, F. Chen, Y. Zhu and J. Chang, J. Mater. Chem., 2011, 21, 16558–16565 RSC.
- Z. Fan, J. Wang, Z. Wang, Z. Li, Y. Qiu, H. Wang, Y. Xu, L. Niu, P. Gong and S. Yang, J. Phys. Chem. C, 2013, 117, 10375–10382 CAS.
- Y. J. Shin, Y. Wang, H. Huang, G. Kalon, A. T. S. Wee, Z. Shen, C. S. Bhatia and H. Yang, Langmuir, 2010, 26, 3798–3802 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb01324g |
| ‡ These authors contributed to this work equally. |
| This journal is © The Royal Society of Chemistry 2014 |