
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Calcium phosphate increases the encapsulation efficiency of hydrophilic drugs (proteins, nucleic acids) into poly(D,L-lactide-co-glycolide acid) nanoparticles for intracellular delivery
Gregor
Dördelmann
a,
Diana
Kozlova
a,
Sarah
Karczewski
b,
Rosario
Lizio
c,
Shirley
Knauer
b and
Matthias
Epple
*a
aInorganic Chemistry and Center for Nanointegration Duisburg-Essen (CeNIDE), University of Duisburg-Essen, Universitaetsstr. 5-7, 45117 Essen, Germany. E-mail: matthias.epple@uni-due.de; Fax: +49 201 1832621; Tel: +49 201 1832402
bMolecular Biology, Center for Medical Biotechnology (ZMB), University of Duisburg-Essen, Universitaetsstr. 5 45117 Essen, Germany
cEvonik Industries AG, Rodenbacher Chaussee 4, 63457 Hanau, Germany
First published on 10th September 2014
Abstract
Calcium phosphate/poly(D,L-lactide-co-glycolide) (PLGA) nanoparticles with a diameter below 200 nm, loaded with either nucleic acids or proteins, were synthesized by a water-in-oil-in-water (W1/O/W2) emulsion solvent evaporation technique. The particles were stabilized by polyvinyl alcohol (PVA) and had a negative charge (zeta potential −26 mV). By the addition of calcium phosphate into the inner aqueous phase of the W1/O/W2-emulsion, the encapsulation efficiency of siRNA was increased to 37%, of DNA to 52%, and of bovine serum albumin to 78%, i.e. by a factor of 3 to 10 compared to PLGA nanoparticles without calcium phosphate. Total loadings of 8 μg siRNA, 5 μg DNA and 280 μg fluorescein isothiocyanate-labelled bovine serum albumin (FITC-BSA) per mg of PLGA nanoparticles were achieved by this method. The addition of an outer layer of either chitosan or polyethyleneimine (PEI) reversed the charge of the particles (zeta potential > +30 mV) and improved the cellular uptake as well as the endosomal escape of these particles as demonstrated by confocal laser scanning microscopy. Calcium phosphate-PLGA nanoparticles loaded with DNA encoding for enhanced green fluorescent protein (eGFP-DNA) showed a good transfection efficiency for epithelial cells (HeLa). Gene silencing with HeLa cells expressing eGFP gave knockdown efficiencies of 53% for anionic nanoparticles, of 68% for chitosan-coated cationic nanoparticles, and of 89% for polyethyleneimine-coated cationic nanoparticles.
Introduction
According to the biopharmaceutical classification system, it is a challenge in modern drug delivery to administrate water-soluble, especially negatively charged (bio-)molecules to cells because they are often unable to penetrate the cell membrane on their own and are also sensitive to enzymatic degradation, e.g. by nucleases or proteases.1–3 Classical examples for hydrophilic drugs are nucleic acids, i.e. DNA or siRNA, for transfection, gene silencing and gene therapy, and proteins. Typically, carriers like liposomes, viruses or nanoparticles are necessary to transport such biomolecules across the cell membrane.2,4–8Poly(D,L-lactide-co-glycolide) (PLGA) is a biocompatible and biodegradable polymer.9 It has been approved by the Food and Drug Administration (FDA) and is frequently used as a polymeric nanoparticulate carrier system for the delivery of biopharmaceuticals.10–13 In addition to its biocompatibility and biodegradability, PLGA possesses excellent controllable release properties. The drug release profile can be tuned by the molecular weight, the block copolymer composition, the size and the morphology of the nanoparticles.14 Although previous reports have shown that PLGA nanoparticles are promising carriers for plasmid DNA and siRNA, it is still challenging to encapsulate hydrophilic biomolecules (especially siRNA) into a PLGA matrix by classical emulsion techniques.15–17 Because of their high water solubility, nucleic acids and proteins tend to diffuse into the continuous aqueous phase during the emulsion process. Therefore, the encapsulation efficiency for such compounds is typically low.18 A well-established method to increase the encapsulation efficiency of nucleic acids into polymeric matrices is to prepare polyplexes of siRNA or DNA with cationic polymers like polyethyleneimine (PEI). Alternatively, the nucleic acid can be inserted into a liposome or lipid nanoparticles, formed by surfactants like 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP).19,20 However, in addition to an altered and retarded release profile, many of these compounds are cytotoxic.21–23
In contrast to polyplexes or liposomes, calcium phosphate as the inorganic part of hard tissue like bone, teeth, and tendons is known for its biocompatibility.24,25 In addition, calcium phosphate nanoparticles have a high affinity to nucleic acids and proteins and are efficiently taken up by cells and subsequently dissolved in lysosomes at a pH below 5.26–32 Thus, they can be used as carriers for biomolecules7 and synthetic molecules8,33–35 across the cell membrane.
The aim of this study was to synthesize a drug delivery system based on calcium phosphate and PLGA nanoparticles for sensitive hydrophilic biomolecules like siRNA, DNA, and proteins with increased encapsulation efficiencies compared to classical PLGA nanoparticles (Fig. 1). In addition, the improvement of the storability compared to “single shell” calcium phosphate nanoparticles by the incorporation into a PLGA matrix, followed by freeze-drying, was investigated.36–38
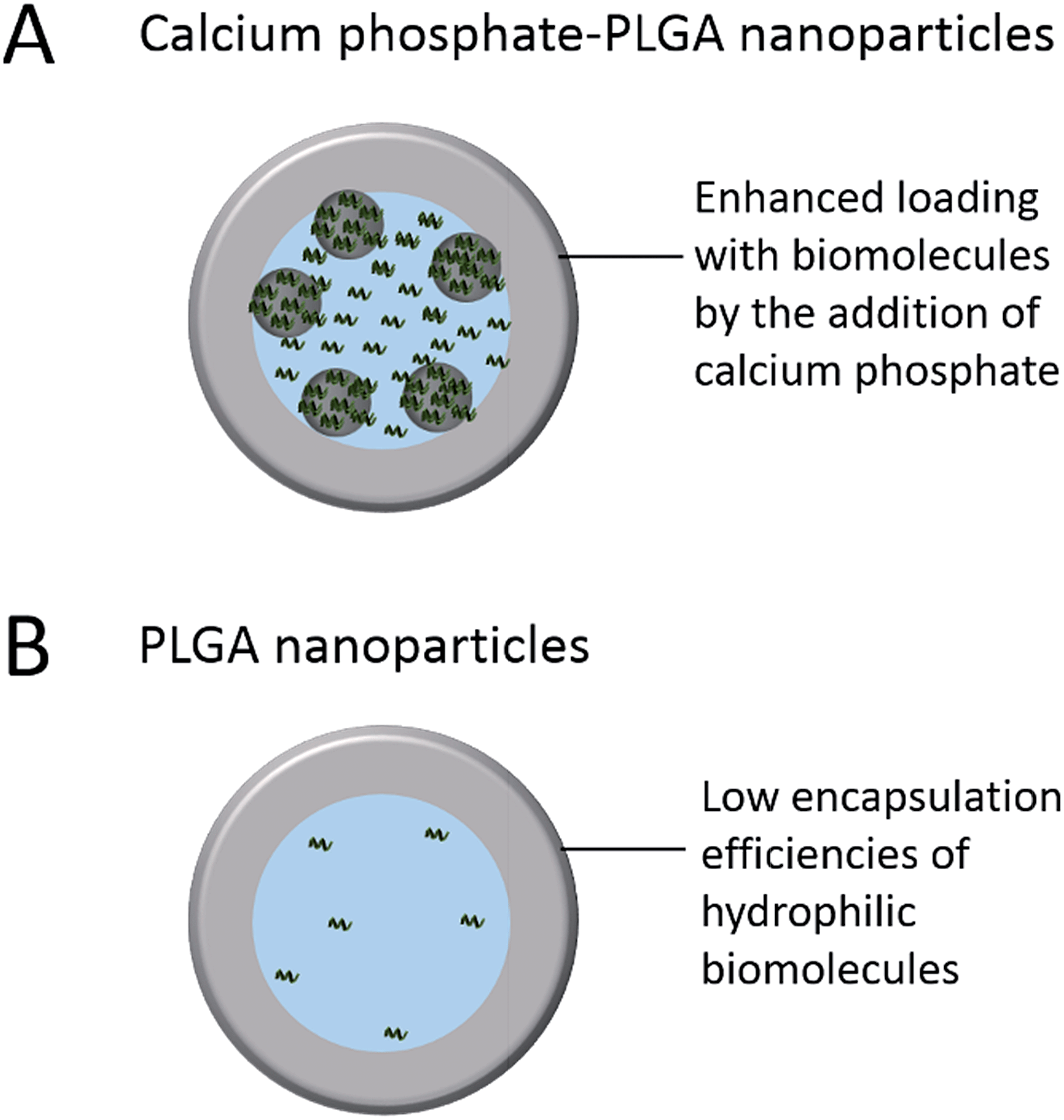 | ||
| Fig. 1 Schematic illustration of the improved loading efficiency of nucleic acids by the addition of calcium phosphate nanoparticles. | ||
Experimental
Materials
Poly(D,L-lactide-co-glycolide) 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (Resomer® RG 502 H, Mw = 7000–17000 g mol−1) was obtained from Evonik Industries (Darmstadt). Polyvinyl alcohol (PVA, Mw = 30000–70000 g mol−1, 87–90% hydrolysed), chitosan (Aldrich, low molecular weight, 75–85% deacetylated) and polyethyleneimine (PEI, Mw = 25000 g mol−1) were purchased from Sigma-Aldrich. For gene silencing experiments with anti-eGFP-siRNA, desalted, double-stranded siRNA from Invitrogen, Ambion® (Carlsbad, USA), sense, 5′-GCAAGCUGACCCUGAAGUUCAU-3′ and antisense, 5′-AUGAACUUCAGGGUCAGCUUGC-3′ (Mw = 14019.5 g mol−1) was used. For transfection experiments with plasmid DNA, pcDNA3-eGFP (Mw = 3621600 g mol−1, computed from the number of base pairs in the plasmid DNA) encoding for enhanced fluorescent protein (eGFP) was isolated from Escherichia coli using a Nucleobond endotoxin-free plasmid DNA kit (Macherey-Nagel, Dueren, Germany). All other chemicals were of analytical grade and used without further purification.
:50 (Resomer® RG 502 H, Mw = 7000–17000 g mol−1) was obtained from Evonik Industries (Darmstadt). Polyvinyl alcohol (PVA, Mw = 30000–70000 g mol−1, 87–90% hydrolysed), chitosan (Aldrich, low molecular weight, 75–85% deacetylated) and polyethyleneimine (PEI, Mw = 25000 g mol−1) were purchased from Sigma-Aldrich. For gene silencing experiments with anti-eGFP-siRNA, desalted, double-stranded siRNA from Invitrogen, Ambion® (Carlsbad, USA), sense, 5′-GCAAGCUGACCCUGAAGUUCAU-3′ and antisense, 5′-AUGAACUUCAGGGUCAGCUUGC-3′ (Mw = 14019.5 g mol−1) was used. For transfection experiments with plasmid DNA, pcDNA3-eGFP (Mw = 3621600 g mol−1, computed from the number of base pairs in the plasmid DNA) encoding for enhanced fluorescent protein (eGFP) was isolated from Escherichia coli using a Nucleobond endotoxin-free plasmid DNA kit (Macherey-Nagel, Dueren, Germany). All other chemicals were of analytical grade and used without further purification.
Instruments
For the formation of water-in-oil and water-in-oil-in-water emulsions, ultrasonication was carried out with a Hielscher UP50H instrument, sonotrode MS2, 70% amplitude, pulse 0.7, for 20 s. Scanning electron microscopy was performed with an ESEM Quanta 400 instrument with gold/palladium-sputtered samples. Dynamic light scattering and zeta potential determinations were performed with a Zetasizer nanoseries instrument (Malvern Nano-ZS, laser: λ = 532 nm) using the Smoluchowski approximation and taking the data from the Malvern software without further correction. The particle size data refer to scattering intensity distributions (z-average). Confocal laser scanning microscopy was performed with a confocal laser scanning microscope (SP5 LCSM, Leica) using a 63× water objective. The laser wavelength was 488 nm for FITC excitation (emission: 500–520 nm) and 561 nm for RFP excitation (emission: 580–600 nm). Centrifugation was performed at 4 °C with a Heraeus Fresco 21 instrument (Thermo Scientific). Transfection and gene silencing efficiencies were determined by transmission light and fluorescence spectroscopy with a Carl Zeiss Axiovert 40 CFL instrument. The viability of the cells was analysed by the MTT-Test by spectrophotometric analysis with a Multiscan FC instrument (ThermoFisher scientific, Vantaa, Finland) at λ = 570 nm. Freeze-drying was performed with a Christ, Alpha 2-4 LSC instrument. Statistical analysis was carried out using Student's t-test.Synthesis of calcium phosphate nanoparticles, loaded with nucleic acids
For the synthesis of calcium phosphate nanoparticles carrying either anti-eGFP-siRNA or eGFP-DNA, we modified the rapid precipitation method (Fig. 2A) that we had reported earlier.20,21 Aqueous solutions of calcium nitrate (6.25 mM, 105 μL) and di-ammonium hydrogen phosphate (3.74 mM, 105 μL) were mixed in a tube reactor with a syringe pump and pumped under continuous mixing (Vortex) either into an aqueous solution of eGFP-DNA (2.5 mg mL−1, 40 μL) or into a solution of anti-eGFP-siRNA (3.9 mg mL−1, 40 μL). The flow rate of both solutions was 16.6 μL s−1, and the residence (nucleation) time in the Y-adapter (7 mm length) was 1.3 s. After the completed precipitation, the dispersion of the nanoparticles (core: calcium phosphate; shell: nucleic acid) was cooled on ice and used after 5 min of incubation for the encapsulation into PLGA nanoparticles.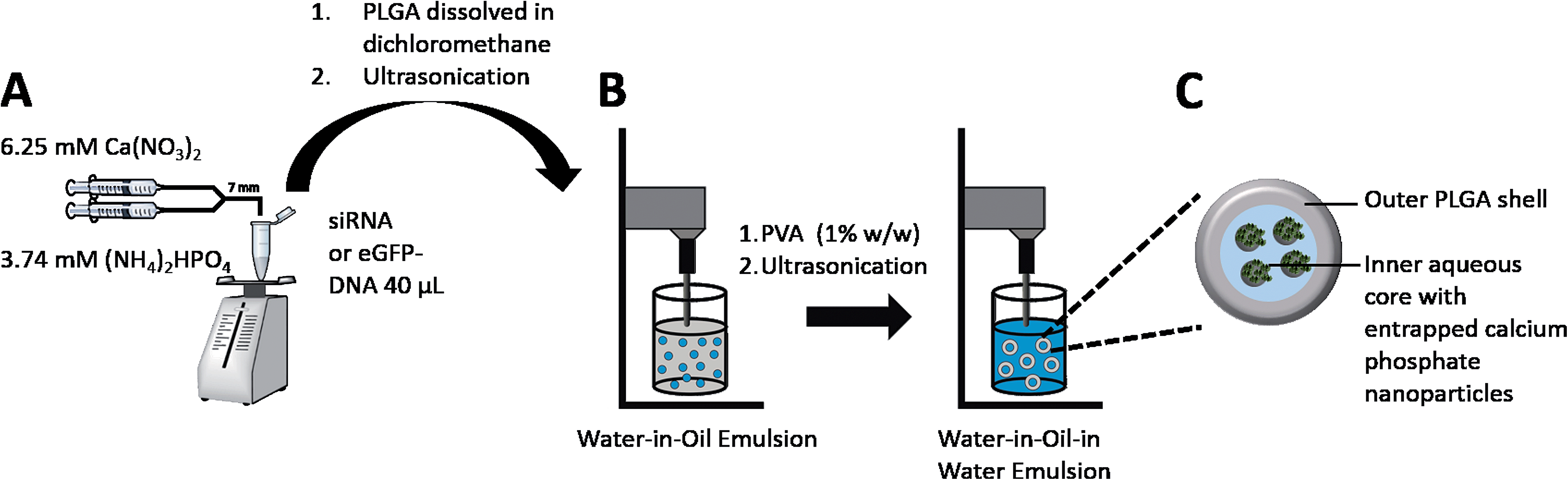 | ||
| Fig. 2 Synthesis of nucleic acid-loaded calcium phosphate nanoparticles (A) and their encapsulation into a PLGA matrix by the W1/O/W2-emulsion technique (B). Structure and composition of a PLGA particle which carries drug-loaded calcium phosphate nanoparticles (C). | ||
Synthesis of nucleic acid-loaded calcium phosphate PLGA nanoparticles
For the encapsulation of either eGFP-DNA- or siRNA-functionalized calcium phosphate nanoparticles into PLGA nanoparticles, a water-in-oil-in-water (W1/O/W2) double emulsion solvent evaporation method was applied (Fig. 2B). To a solution of 10 mg PLGA dissolved in 750 μL dichloromethane, 250 μL of the dispersion of calcium phosphate/nucleic acid nanoparticles was added. Then a solution of 200 μg RNase-free acetylated bovine serum albumin (BSA) in 40 μL water as dispersant was added. The mixture was ultrasonicated (sonotrode) to form the primary, milky white W1/O-emulsion. The W1/O-emulsion was then immediately poured into the continuous water phase (3 mL), containing 30 mg polyvinyl alcohol (PVA) as dispersant, and ultrasonicated again (sonotrode). Finally, the PLGA nanoparticles were precipitated after the dichloromethane was removed under reduced pressure (200–600 mbar) in a rotary evaporator. Thereby, the calcium phosphate nanoparticles carrying siRNA or DNA were incorporated into a nanoparticulate matrix of PLGA. The excess of PVA was removed by centrifugation (30 min at 14800 rpm), and redispersion of the particles in ultrapure water, repeated three times each. The resulting dispersion was shock-frozen in liquid nitrogen and finally lyophilized for 72 h at 0.31 mbar and −10 °C. The particles were easily redispersible in water by gentle shaking.
The encapsulation efficiency (EE) of the nucleic acids was calculated by eqn (1). The content of nucleic acids in the supernatant was analysed by UV/Vis spectroscopy at 260 nm according to standard protocols after separation of the nanoparticles by centrifugation.
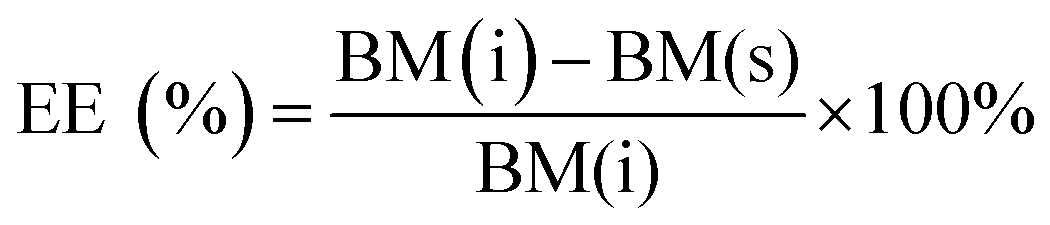 | (1) |
EE (%): encapsulation efficiency in percent; BM(i): total amount of biomolecule added initially; BM(s): amount of biomolecule in the supernatant measured by quantitative UV/Vis spectroscopy.
Synthesis of protein-loaded calcium phosphate-PLGA nanoparticles
Calcium phosphate-FITC-BSA-PLGA nanoparticles were synthesised by a W1/O/W2 emulsion solvent evaporation method (Fig. 3), by modification of a synthesis reported by Tang et al.39 First, two W/O emulsions (A and B) were prepared by ultrasonication. For emulsion A, 625 μg FITC-BSA was dissolved in 125 μL of a 10 mM solution of Na2HPO4. This was dispersed in a solution of PLGA in dichloromethane (13.3 mg mL−1, 375 μL). For emulsion B, 625 μg FITC-BSA was dissolved in 125 μL of a 1.25 M solution of CaCl2. This was dispersed in a solution of PLGA in dichloromethane (13.3 mg mL−1, 375 μL). Then, emulsions A and B were mixed by ultrasonication to form emulsion C. This W1/O-emulsion was then added dropwise to the continuous water phase (3 mL), containing 30 mg PVA as dispersant, and ultrasonicated again (sonotrode) to form a yellow, milky W1/O/W2-emulsion. After removal of dichloromethane under reduced pressure (200–600 mbar), the calcium phosphate-FITC-BSA nanoparticles were incorporated into the PLGA particle. Any excess of PVA and FITC-BSA was removed by centrifugation (30 min at 14800 rpm), and redispersion of the particles in ultrapure water by ultrasonication (sonotrode), repeated three times. The resulting dispersion was shock-frozen in liquid nitrogen and finally lyophilised for 72 h at 0.31 mbar and −10 °C. The particles were easily redispersible in water by gentle shaking.
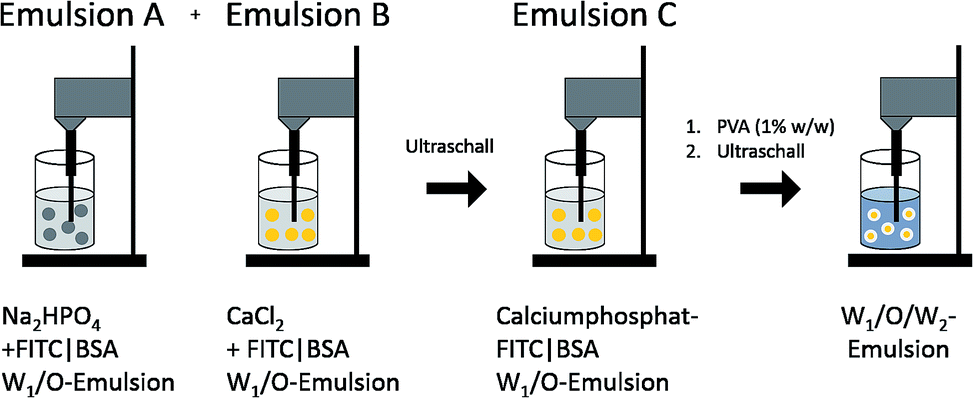 | ||
| Fig. 3 Schematic pathway of the synthesis of PLGA nanoparticles, loaded with calcium phosphate nanoparticles that carried FITC-labelled BSA (FITC-BSA). | ||
The encapsulation efficiency (EE) of FITC-BSA was calculated by eqn (1) as described above. The supernatants were analysed by UV/Vis spectroscopy at 460 nm after previous calibration with dissolved FITC-BSA.
Charge reversal of anionic PLGA nanoparticles by coating with either PEI or chitosan
To enhance the cellular uptake and the endosomal escape, the surface of the negatively charged PLGA nanoparticles was coated with a cationic polyelectrolyte, i.e. either PEI or chitosan. 1.5 mg of the freeze-dried particles were resuspended in 1 mL ultrapure water and added dropwise to either an aqueous PEI solution (2 mg in 1 mL) or an aqueous chitosan solution (5 mg in 1 mL, pH adjusted to 5 with acetic acid) under continuous stirring. After 30 min of continuous stirring at room temperature, the dispersion was purified three times by centrifugation (30 min at 14800 rpm), and redispersion (shaking, no ultrasonication necessary) in ultrapure water. For cell culture experiments, the particles were finally redispersed in the cell culture medium used.
Cell culture experiments
HeLa and HeLa-eGFP cells (genetically modified HeLa cells that express enhanced green fluorescent protein, eGFP) were cultivated in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS), 100 U mL−1 penicillin, and 100 U mL−1 streptomycin at 37 °C under 5% CO2 atmosphere. 12 h before the addition of the nanoparticles, the cells were trypsinized and seeded in 24-well plates with a density of either 2.5 × 104 cells per well (for transfection and gene silencing experiments) or 105 cells per well (for uptake studies).The cell viability was analysed by the MTT-assay 72 h after the transfection. MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; Sigma, Taufkirchen, Germany) was dissolved in PBS (5 mg mL−1) and then added to the required cell culture medium, giving a final MTT concentration of 1 mg mL−1. The cell culture medium of transfected cells was replaced by the MTT medium (300 μL) and incubated for 90 min at 37 °C under 5% CO2 in humidified atmosphere. Then, the MTT medium was removed and the blue precipitate was dissolved in DMSO (300 μL, each well) and incubated for 30 min. Finally, 100 μL of each well were taken for spectroscopic analysis at λ = 570 nm. The absorption of cells treated with nanoparticles was normalized to that of the control (untreated cells).
Cell uptake studies
For co-localization experiments, HeLa cells were seeded in 8-well plates (Lab-Tek) and cultivated for 24 h. Then, HeLa cells were transfected with 50 ng of lysosomal-associated membrane protein 1 (Lamp1)-RFP plasmid-DNA and 0.3 μL Lipofectamine 2000 (Life technologies) according to the manufacturer's instructions. After 4 h, the cell culture medium was changed, and the cells were washed for several times with PBS. After additional 16 h, cells were treated with the nanoparticle dispersion (20 μL, 1 mg nanoparticles per mL) and examined with a confocal laser scanning microscope at different time points as described earlier.40Transfection of HeLa cells with either anti-eGFP-siRNA or eGFP-DNA functionalized calcium phosphate-PLGA nanoparticles
The transfection was carried out according to our protocols reported earlier.30,36,41 Before the transfection, the cell culture medium was replaced by the nanoparticles redispersed in fresh cell culture medium (0.1 mg nanoparticles in 0.5 mL corresponding to 0.29 μg DNA or 0.8 μg siRNA per well). After incubation for 7 h, the transfection medium was replaced by fresh cell culture medium. As control, the cells were transfected with Lipofectamine 2000 as recommended by the manufacturer. In brief, 50 μL of FCS-free DMEM were mixed with 1 μL Lipofectamine 2000 and incubated for 5 min at room temperature. Either eGFP-siRNA (20 pmol, 0.28 μg) or eGFP-DNA (1 μg) were added to 50 μL of FCS-free DMEM. Then, both solutions were mixed and incubated for 20 min before 100 μL of this solution and additionally 400 μL of DMEM were added to each well. After incubation for 7 h, the transfection medium was replaced by fresh cell culture medium. The transfection efficiency and the efficiency of the gene silencing were measured 72 h after the addition of the nanoparticles by light and fluorescence microscopy.The transfection efficiency of the experiments with eGFP-DNA-functionalized calcium phosphate-PLGA nanoparticles was calculated by the ratio of green fluorescing cells to the total number of cells. The efficiency of the gene silencing experiments with eGFP-siRNA-functionalized calcium phosphate-PLGA nanoparticles was calculated as reported earlier:38
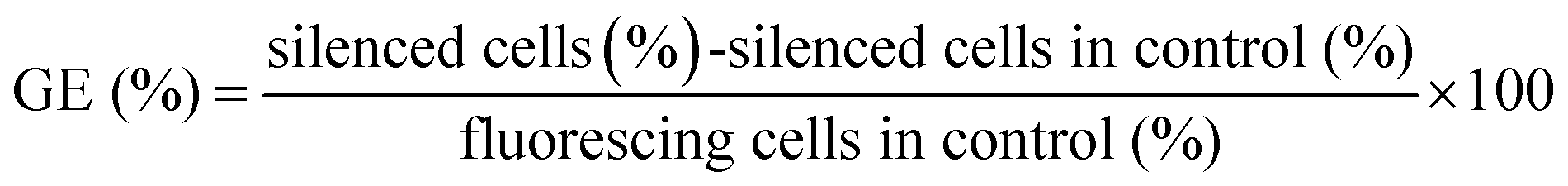 | (2) |
HeLa-eGFP cells cultivated under the same conditions but without any treatment were used as control.
Results and discussion
The size and surface charge of a drug carrier are crucial parameters for its ability to cross the cellular membrane by endocytosis.42–44 Thus, the aim of this study was to synthesize a carrier system in the size range below 300 nm with high encapsulation efficiencies for hydrophilic biologicals. With the classical W1/O/W2-emulsion solvent evaporation technique, anionic low molecular weight compounds (e.g., siRNA) easily escape from the inner aqueous phase (W1) into the outer water phase (W2) due to their hydrophilicity, low molecular weight, and to the electrostatic repulsion of the phosphate backbone and the carboxylic acid end groups of the polymer.18 Therefore, we modified the classical W1/O/W2-emulsion solvent evaporation technique and took advantage of the high adsorption capacity of calcium phosphate nanoparticles for anionic, hydrophilic compounds and encapsulated the biomolecule functionalized calcium phosphate nanoparticles into the polymeric PLGA matrix (Fig. 2).First of all, calcium phosphate nanoparticles were synthesized with a rapid precipitation method. As indicated by our previous work, the nucleic acids adsorb on the surface of the calcium phosphate nanoparticles. Hence, crystal growth is inhibited and the calcium phosphate nanoparticles are electrostatically stabilized. In this case, DNA or siRNA act both as pharmaceutical active ingredient and as stabilizing agent that protects the dispersion from aggregation.
To protect the outer shell of DNA or siRNA from degrading enzymes like DNases or RNases and to provide a sustained release profile to the particles, the calcium phosphate-DNA or calcium phosphate-siRNA nanoparticles were then encapsulated into a matrix of PLGA (Fig. 2). The aqueous calcium phosphate-DNA or calcium phosphate-siRNA dispersion was used as the inner aqueous phase (W1) in the emulsion process. The polymer was dissolved in dichloromethane (O). Sonication led to the primary W1/O emulsion with fine water droplets (containing the calcium phosphate nanoparticles) in the oil phase. Addition to the continuous water phase (PVA in water) and subsequent sonication led to a stable W1/O/W2-emulsion. Evaporation of the organic solvent under reduced pressure yielded an almost transparent dispersion. The resulting nanoparticles could be freeze-dried without the addition of any cryoprotectant. This is a major advantage compared to previously reported calcium phosphate nanoparticles, which lack of a good storability without the addition of high amounts of cryoprotectants.36,38,41,45–49
For the functionalization with biomolecules that adsorb to calcium phosphate nanoparticles but do not colloidally stabilize them (e.g., proteins like FITC-BSA), calcium phosphate was precipitated during the emulsion process in the primary W1/O-emulsion (Fig. 3).
According to Landfester et al., it is possible to precipitate inorganic salts in the inner aqueous droplets of the primary emulsion.50,51 Thus, we prepared two W1/O-emulsions in the first step (Fig. 3). Emulsion A contained the phosphate salt solution, and the biomolecules in the inner water droplets and PLGA were dissolved in the organic phase. Emulsion B contained the calcium salt solution in the inner aqueous phase and also PLGA dissolved in the organic phase. Mixing both emulsions under sonication led to the precipitation of calcium phosphate in the inner water droplets. Addition of the combined W1/O-emulsions into the continuous water phase (PVA in water) and sonication led to a stable W1/O/W2-emulsion. Evaporation of the organic solvent under reduced pressure gave an almost transparent, yellow dispersion. With this method, crystal growth was limited by the small volume of the water droplet serving as a microreactor in the organic solvent. We believe that BSA acts as a surfactant stabilizing the primary W1/O-emulsion and thus defining the volume of the microreactors. Without the addition of BSA, large aggregates of calcium phosphate precipitated during the emulsion process. Dynamic light scattering showed particles with an average size of 190 nm and a zeta potential of −24 mV. Scanning electron micrographs showed spherical particles with an average size of 105 ± 18 nm (Fig. 4A and C). Dynamic light scattering of the nucleic acid-loaded PLGA nanoparticles showed a dispersion with an average particle size of 200 nm and a zeta potential of −24 mV (Fig. 4B and D). The calcium phosphate-PLGA nanoparticles contained 5% calcium phosphate as determined by atomic absorption spectroscopy (computed from the content of calcium and assuming the stoichiometry of hydroxyapatite; Ca5(PO4)3OH).
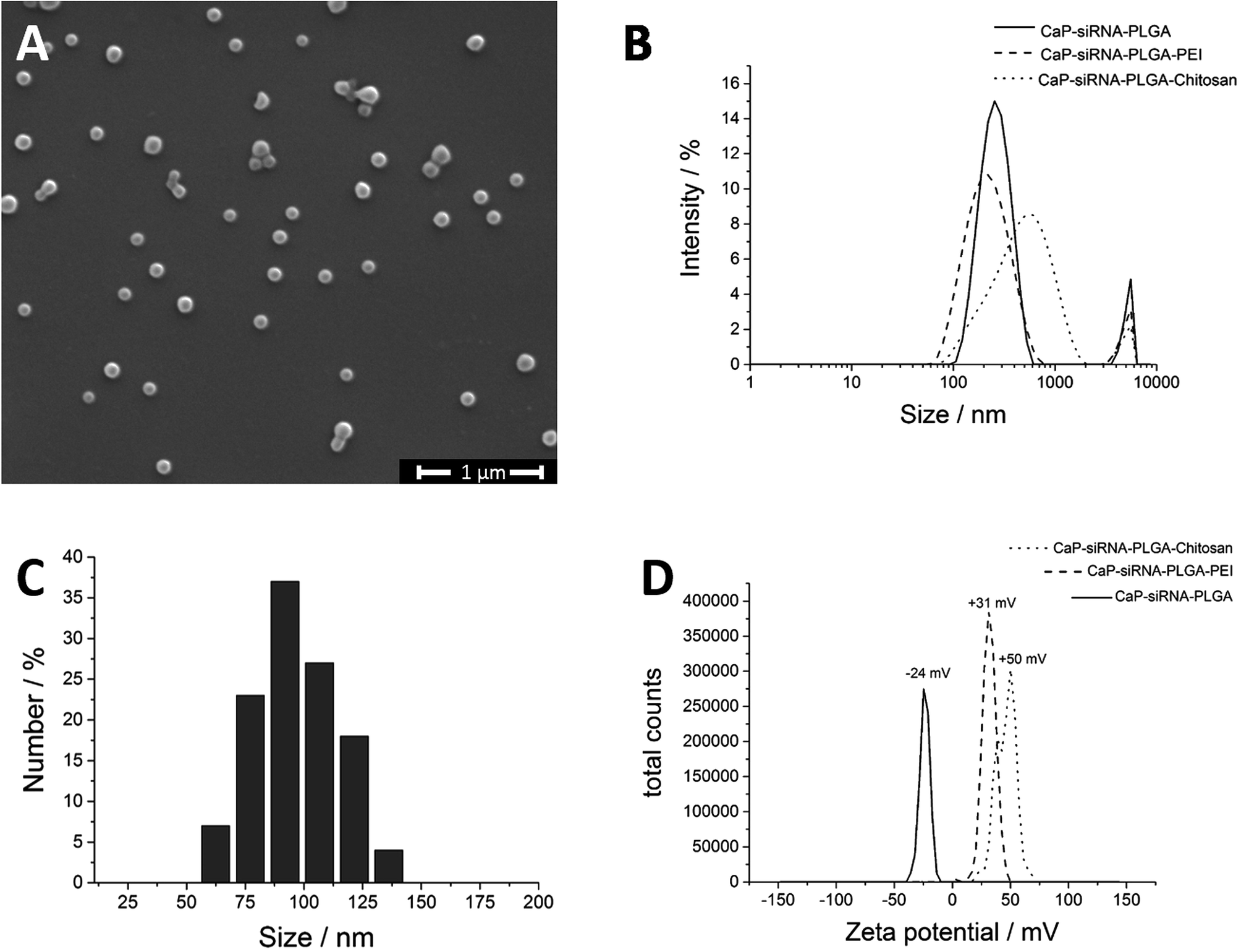 | ||
| Fig. 4 Scanning electron micrograph and particle size distribution of FITC-BSA-loaded PLGA nanoparticles. The average diameter of the spherical particles was 105 ± 18 nm (A, C). Dynamic light scattering data of anionic and cationic calcium phosphate/nucleic acid/PLGA nanoparticles (B, D). | ||
To enhance the cellular uptake of the calcium phosphate-PLGA nanoparticles, the surface charge can be reversed by layer-by-layer deposition of cationic polymers like chitosan or PEI. Furthermore, these polymers are capable to induce the proton sponge effect.52 This leads to an enhanced endosomal escape of the nanoparticles and an increased therapeutic efficiency. As shown by zeta potential measurements, the surface charge of the calcium phosphate-PLGA nanoparticles was easily reversed by layer-by-layer deposition of chitosan from about −24 mV to +50 mV (chitosan) or to +31 mV (PEI) (Fig. 4D).
Especially for expensive biopharmaceutical ingredients like antibodies, siRNA or DNA, the encapsulation efficiency of a drug carrier is an important factor in galenics. By the addition of calcium phosphate nanoparticles during the emulsion process, the encapsulation efficiency of the hydrophobic polymer PLGA could be optimized to 37% for siRNA, 52% for pDNA and 78% for FITC-BSA (Fig. 5) with total loadings of 8 μg siRNA, 5 μg pDNA and 280 μg FITC-BSA per mg of PLGA nanoparticles. These total loadings correspond to 1880 molecules FITC-BSA, 250 molecules siRNA, and 0.6 molecules pDNA per nanoparticle. Although these numbers seem to be low, especially for nucleic acids, the total loadings are high compared to previously reported PLGA nanoparticles with total loadings of 0.7 μg to 10 μg per mg of PLGA nanoparticles.10,18
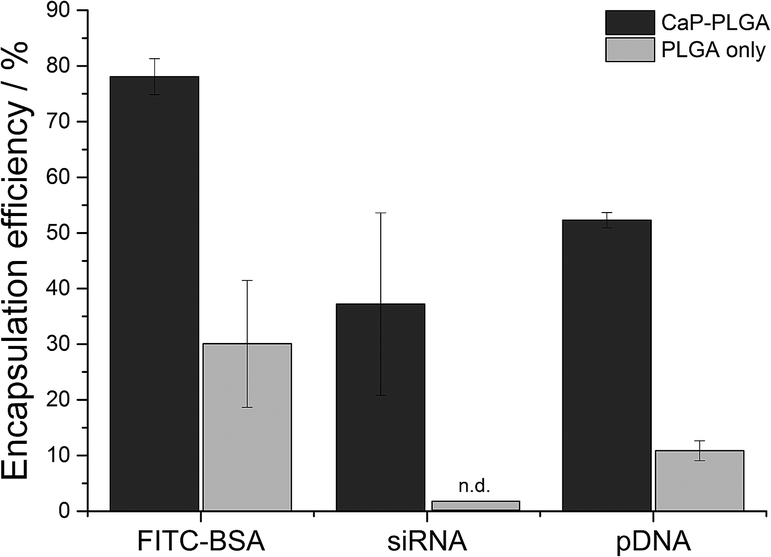 | ||
| Fig. 5 Comparison of the encapsulation efficiency of FITC-BSA, siRNA, and pDNA into PLGA nanoparticles with (black) and without (grey) the addition of calcium phosphate nanoparticles. Without the addition of calcium phosphate, siRNA was not detectable (n.d.) inside the PLGA nanoparticles. Values are given as the mean ± SD of triplicates. * indicates p < 0.05. | ||
The number of nanoparticles were calculated by taking the concentration of calcium phosphate-PLGA nanoparticles in the dispersion and assuming spherical nanoparticles with a diameter of 100 nm (eqn (3)). The number of biomolecules in the dispersion and per nanoparticle was determined by eqn (4) and (5), respectively.
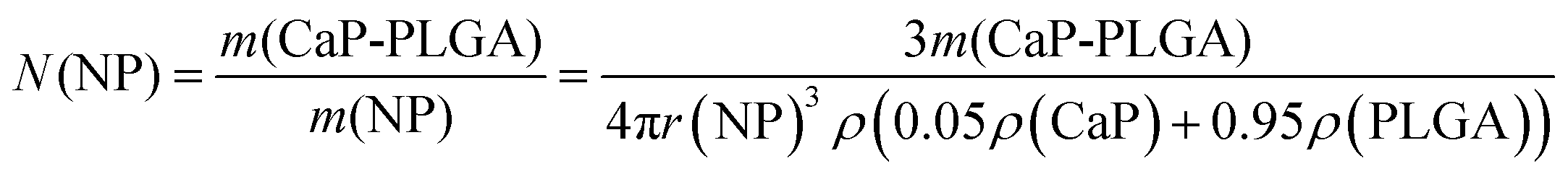 | (3) |
| N(biomolecule) = c(biomolecule) × NA | (4) |
:Avogadro constant: NA = 6.022 × 1023 mol−1.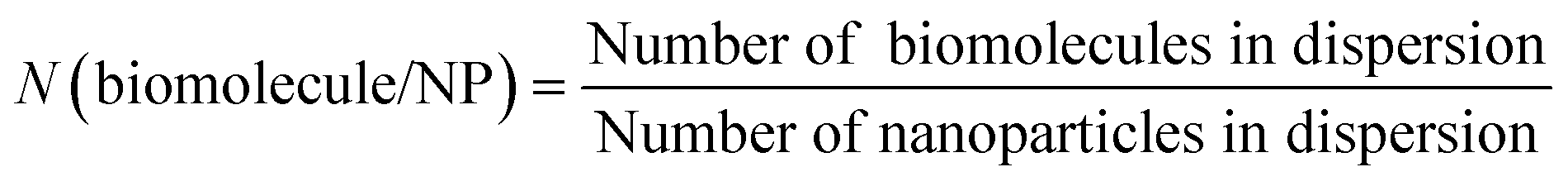 | (5) |
Incubation of HeLa cells expressing the lysosomal marker protein Lamp1-RFP with calcium phosphate-FITC-BSA-PLGA nanoparticles and co-localisation analyses demonstrated that the nanoparticles were efficiently taken up by the cells. As shown in Fig. 6, nanoparticles with a negative surface charge had a low affinity to the cell membrane and were taken up only moderately, while nanoparticles with a positive surface charge (chitosan- or PEI-functionalized nanoparticles) covered the cell membrane after 1 h of incubation due to electrostatic interactions of the negatively charged cell membrane and the cationic surface of the nanoparticles. In addition, the majority of the negatively charged calcium phosphate-FITC-BSA-PLGA nanoparticles ended up in the endo-/lysosome as shown by the co-localisation of Lamp1-RFP and the green fluorescence of FITC-BSA (yellow fluorescence by overlay of green and red). In contrast, chitosan- and PEI-functionalized calcium phosphate-FITC-BSA-PLGA nanoparticles (positive surface charge) induced the proton sponge effect and escaped the endosome as shown by the diffuse green fluorescence in the cytosol after 3 h of incubation (no co-localisation with Lamp1-RFP).
 | ||
| Fig. 6 Confocal laser scanning microscopic images of HeLa cells (life cell imaging) 1 h (A1–C1) and 3 h (A2-C2) after incubation with anionic calcium phosphate-FITC-BSA-PLGA nanoparticles (A1 and A2), cationic calcium phosphate-FITC-BSA-PLGA-chitosan nanoparticles (B1 and B2), and cationic calcium phosphate-FITC-BSA-PLGA-PEI nanoparticles (C1 and C2). The particles showed a green fluorescence in all cases, and the lysosomes were stained red by transfection of Lamp1-RFP. Note that due to the life cell imaging experiment, there are many fluorescing particles in the dispersion, unlike as in post-hoc experiments with fixed and washed cells. The scale bar corresponds to 20 μm. | ||
Calcium phosphate-PLGA nanoparticles loaded with eGFP-encoding plasmid DNA (eGFP-DNA) showed only a moderate transfection efficiency in HeLa cells (8 ± 2%). However, in comparison to the liposomal transfection agent Lipofectamine 2000 which showed a transfection efficiency of 57%, the viability of the cells after the transfection was very high (85 ± 4% in comparison to 26 ± 14%, Fig. 7).23
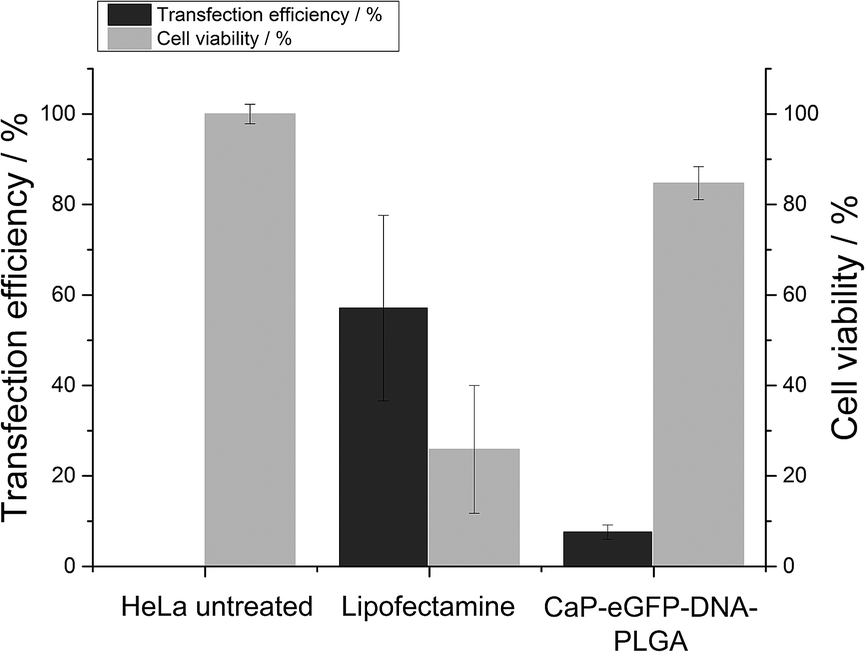 | ||
| Fig. 7 Transfection efficiency and viability of HeLa cells after the treatment with calcium phosphate-eGFP-DNA-PLGA nanoparticles in comparison to Lipofectamine. | ||
Calcium phosphate-PLGA nanoparticles functionalized with eGFP-siRNA efficiently silenced the eGFP-expression in eGFP-expressing HeLa cells. Anionic calcium phosphate-PLGA nanoparticles showed a knockdown efficiency of 53%, which is comparable to anionic triple shell nanoparticles (calcium phosphate/siRNA/calcium phosphate/siRNA) or anionic calcium phosphate/siRNA nanoparticles coated with a conjugate of 3,4-dihydroxy-L-phenylalanine (dopa) and hyaluronic acid described earlier.38,53 The cationic calcium phosphate-PLGA-siRNA nanoparticles coated with either chitosan or PEI showed knockdown efficiencies of 68% and 89%, respectively (Fig. 8 and 9). In accordance to the transfection experiments, the vitality of the cells after the treatment with calcium phosphate-PLGA nanoparticles was twice as high in comparison to liposomal transfection agents such as Lipofectamine. The cationic calcium phosphate-PLGA-siRNA nanoparticles coated with either chitosan or PEI showed only moderate cytotoxic effects, although PEI is known for its high cytotoxicity.21,54
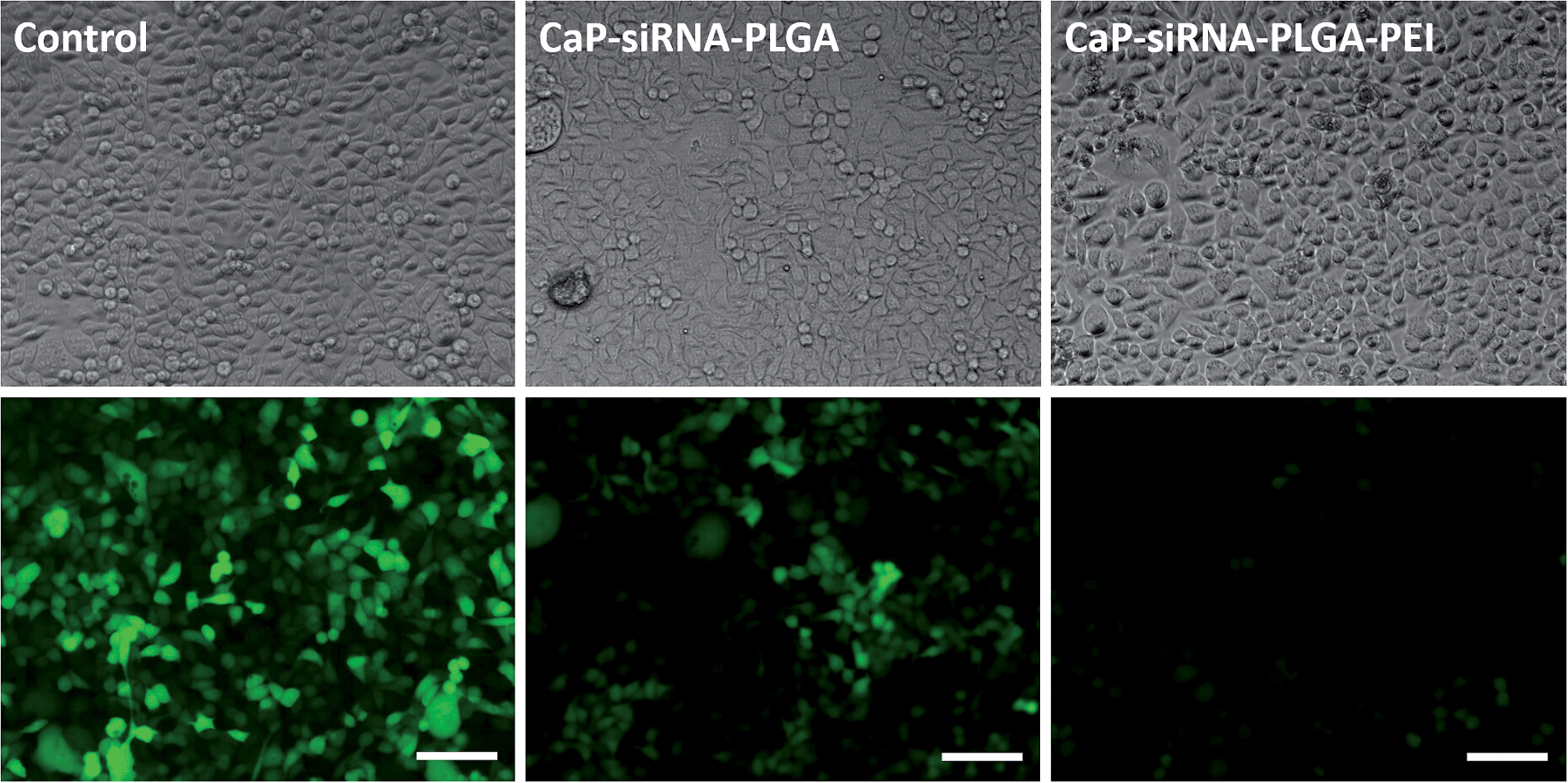 | ||
| Fig. 8 Light and fluorescence microscopic images of eGFP-expressing HeLa cells as control, calcium phosphate-siRNA-PLGA nanoparticles, and calcium phosphate-siRNA-PLGA-PEI nanoparticles. The scale bar corresponds to 100 μm. | ||
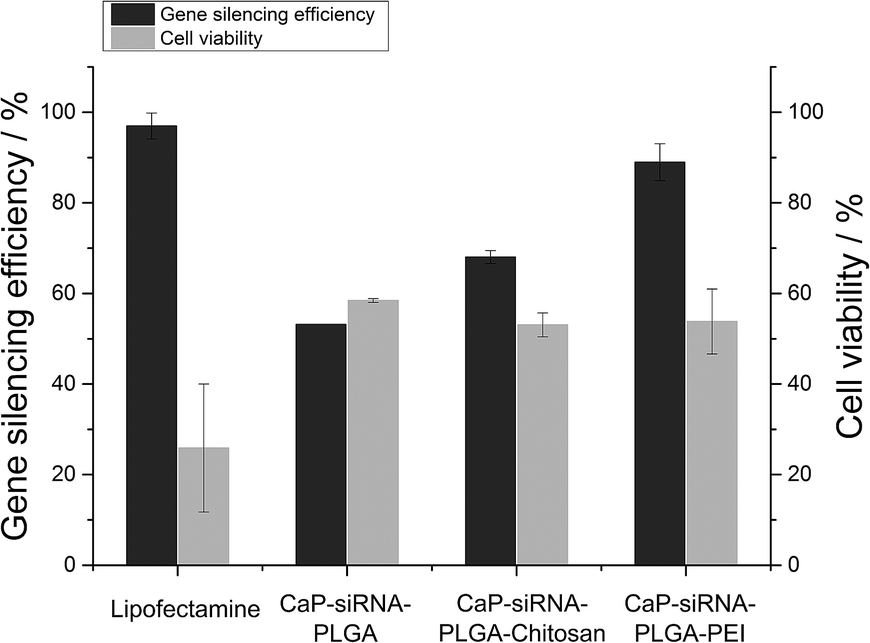 | ||
| Fig. 9 Gene silencing efficiency and cell viability of eGFP-expressing HeLa cells after the treatment with calcium phosphate-siRNA-PLGA nanoparticles, calcium phosphate-siRNA-PLGA-chitosan nanoparticles, and calcium phosphate-siRNA-PLGA-PEI nanoparticles in comparison to Lipofectamine. | ||
We have tried to determine the release rate of a labelled oligonucleotide from calcium phosphate-PLGA nanoparticles by UV spectroscopy. However, this turned out to be impossible. At the physiological pH 7.4 at 37 °C, there was no release of the oligonucleotide during one week, probably because the degradation of PLGA was very slow and the diffusion of the oligonucleotide was prevented. The calcium phosphate nanoparticles also do not dissolve at a pH 7.4. This is advantageous for a later in vivo drug release application because the drug does not leach out of the nanoparticles during circulation.
After crossing the cell membrane, the particles will end up first in an endosome and then in a lysosome at a pH around 4.31,55 Therefore, the delivery will occur after dissolution of the nanoparticle at a low pH. In this case, calcium phosphate will dissolve and PLGA will slowly degraded by acid, but also by nucleases.9
Under the given precipitation conditions, calcium phosphate nanoparticles are spherical with a diameter of about 50–100 nm. They are typically X-ray amorphous but show domains of hydroxyapatite in the transmission electron microscope, with indications for carbonate inclusion.56 As they are prepared around neutral pH, they probably consist of calcium-deficient hydroxyapatite (CDHA).24,57 In the case of nanoparticles, the dissolution rate at a pH of 4 as encountered in the lysosome should not be significantly affected by the stoichiometry.58,59
The lysosomal degradation was impossible to replicate in this case. The immersion of the calcium phosphate-PLGA nanoparticles in buffer at pH4 led to agglomeration because the colloidal stability was lost and the particles agglomerated. This will not occur in the lysosome, as the particles will be taken up individually, but it constrains the experimental release study. However, the fact that we observe a successful transfection and gene silencing proves that the nucleic acids are released into the cytoplasm after cellular uptake without harmful degradation.
Conclusions
We have shown that the encapsulation efficiency of hydrophilic and anionic biomolecules such as siRNA, plasmid DNA, and FITC-BSA into PLGA nanoparticles can be strongly increased by the addition of calcium phosphate nanoparticles into the inner aqueous phase (W1) during a classic W1/O/W2 emulsion solvent evaporation synthesis. DNA or siRNA interact with the surface of the calcium phosphate nanoparticles and act both as pharmaceutical active ingredient and as a stabilizing agent. The additional PLGA matrix protects the outer shell of DNA or siRNA from degrading enzymes like nucleases and proteases and provides the particles with a good storability in freeze-dried form, without the necessity of a cryoprotectant. A special challenge during this synthesis route was to obtain nanoparticles with a diameter below 200 nm. Therefore, it was necessary to add acetylated BSA into the inner aqueous phase (W1) to stabilize the primary W1/O emulsion. To enhance the cellular uptake of anionic calcium phosphate-PLGA nanoparticles, the surface of these particles was coated with cationic polymers such as chitosan and PEI.As shown by confocal laser scanning microscopy and co-expression of the endosomal marker protein Lamp1-RFP, the nanoparticles are very efficiently taken up by endocytosis. Chitosan- and PEI-coated nanoparticles have a strong affinity to the anionic cellular membrane, induce the proton sponge effect and escape the endosome. In addition, transfection and gene silencing experiments showed that sensitive biomolecules like siRNA and DNA can be encapsulated inside PLGA nanoparticles by the addition of calcium phosphate, and efficiently delivered across the cell membrane. Furthermore, the cytotoxicity of calcium phosphate-PLGA nanoparticles is low in comparison to liposomal transfection agents. Thus, calcium phosphate-PLGA nanoparticles represent a very promising delivery system for hydrophilic anionic drugs, e.g. for nucleic acids in gene therapy, because of their good biocompatibility and high encapsulation efficiency.
Acknowledgements
This work was supported by Evonik Industries, Darmstadt and by the Deutsche Forschungsgemeinschaft (DFG) in the framework of the Collaborative Research Center “Supramolecular Chemistry on Proteins” (SFB 1093).We thank Dr J. Mueller-Albers and Dr S. Grimm for helpful discussions.References
- G. L. Amidon, H. Lennernaes, V. P. Shah and J. R. Crison, Pharm. Res., 1995, 12, 413–420 CrossRef CAS.
- C. X. Li, A. Parker, E. Menocal, S. Xiang, L. Borodyansky and J. H. Fruehauf, Cell Cycle, 2006, 5, 2103–2109 CrossRef CAS.
- G. L. Verdine and L. D. Walensky, Clin. Cancer Res., 2007, 13, 7264–7270 CrossRef CAS PubMed.
- Z. P. Xu, Q. H. Zeng, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS PubMed.
- J. Kurreck, Angew. Chem., Int. Ed., 2009, 48, 1378–1398 CrossRef CAS PubMed.
- V. Sokolova and M. Epple, Angew. Chem., Int. Ed., 2008, 47, 1382–1395 CrossRef CAS PubMed.
- V. Sokolova, O. Rotan, J. Klesing, P. Nalbant, J. Buer, T. Knuschke, A. M. Westendorf and M. Epple, J. Nanopart. Res., 2012, 14, 910 CrossRef.
- O. Rotan, V. Sokolova, P. Gilles, W. Hu, S. Dutt, T. Schrader and M. Epple, Materialwiss. Werkstofftech., 2013, 44, 176–182 CrossRef CAS.
- B. D. Ulery, L. S. Nair and C. T. Laurencin, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 832–864 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, B. A. Le and V. Preat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- A. Kumari, S. K. Yadav and S. C. Yadav, Colloids Surf., B, 2010, 75, 1–18 CrossRef CAS PubMed.
- J. Panyam and V. Labhasetwar, Adv. Drug Delivery Rev., 2003, 55, 329–347 CrossRef CAS.
- J. K. Vasir and V. Labhasetwar, Adv. Drug Delivery Rev., 2007, 59, 718–728 CrossRef CAS PubMed.
- A. Giteau, M. C. Venier-Julienne, A. Aubert-Pouessel and J. P. Benoit, Int. J. Pharm., 2008, 350, 14–26 CrossRef CAS PubMed.
- D. Luo, K. Woodrow-Mumford, N. Belcheva and W. M. Saltzman, Pharm. Res., 1999, 16, 1300–1308 CrossRef CAS.
- C. Perez, A. Sanchez, D. Putnam, D. Ting, R. Langer and M. J. Alonso, J. Controlled Release, 2001, 75, 211–224 CrossRef CAS.
- K. A. Woodrow, Y. Cu, C. J. Booth, J. K. Saucier-Sawyer, M. J. Wood and W. M. Saltzman, Nat. Mater., 2009, 8, 526–533 CrossRef CAS PubMed.
- D. Cun, D. K. Jensen, M. J. Maltesen, M. Bunker, P. Whiteside, D. Scurr, C. Foged and H. M. Nielsen, Eur. J. Pharm. Biopharm., 2011, 77, 26–35 CrossRef CAS PubMed.
- C. G. Oster, N. Kim, L. Grode, L. Barbu-Tudoran, A. K. Schaper, S. H. E. Kaufmann and T. Kissel, J. Controlled Release, 2005, 104, 359–377 CrossRef CAS PubMed.
- Y. C. Tseng, Z. Xu, K. Guley, H. Yuan and L. Huang, Biomaterials, 2014, 35, 4688–4698 CrossRef CAS PubMed.
- C. Brunot, L. Ponsonnet, C. Lagneau, P. Farge, C. Picart and B. Grosgogeat, Biomaterials, 2007, 28, 632–640 CrossRef CAS PubMed.
- W. T. Godbey, M. A. Barry, P. Saggau, K. K. Wu and A. G. Mikos, J. Biomed. Mater. Res., 2000, 51, 321–328 CrossRef CAS.
- H. T. Lv, S. B. Zhang, B. Wang, S. H. Cui and J. Yan, J. Controlled Release, 2006, 114, 100–109 CrossRef CAS PubMed.
- S. V. Dorozhkin and M. Epple, Angew. Chem., Int. Ed., 2002, 41, 3130–3146 CrossRef CAS.
- L. Treccani, T. Y. Klein, F. Meder, K. Pardun and K. Rezwan, Acta Biomater., 2013, 9, 7115–7150 CrossRef CAS PubMed.
- V. Uskokovic and D. P. Uskokovic, J. Biomed. Mater. Res., Part B, 2011, 96, 152–191 CrossRef PubMed.
- M. Epple, K. Ganesan, R. Heumann, J. Klesing, A. Kovtun, S. Neumann and V. Sokolova, J. Mater. Chem., 2010, 20, 18–23 RSC.
- Y. Cai and R. Tang, J. Mater. Chem., 2008, 18, 3775–3787 RSC.
- A. E. Ewence, M. Bootman, H. L. Roderick, J. N. Skepper, G. McCarthy, M. Epple, M. Neumann, C. M. Shanahan and D. Proudfoot, Circ. Res., 2008, 103, e28–e32 CrossRef CAS PubMed.
- S. Neumann, A. Kovtun, I. D. Dietzel, M. Epple and R. Heumann, Biomaterials, 2009, 30, 6794–6802 CrossRef CAS PubMed.
- V. Sokolova, D. Kozlova, T. Knuschke, J. Buer, A. M. Westendorf and M. Epple, Acta Biomater., 2013, 9, 7527–7535 CrossRef CAS PubMed.
- D. Zhao, C. Q. Wang, R. X. Zhuo and S. X. Cheng, Colloids Surf., B, 2014, 118, 111–116 CrossRef CAS PubMed.
- V. Uskokovic, C. Hoover, M. Vukomanovic, D. P. Uskokovic and T. A. Desai, Mater. Sci. Eng., C, 2013, 33, 3362–3373 CrossRef CAS PubMed.
- N. L. Ignjatovic, P. Ninkov, R. Sabetrasekh and D. P. Uskokovic, J. Mater. Sci.: Mater. Med., 2010, 21, 231–239 CrossRef CAS PubMed.
- N. L. Ignjatovic, C. Z. Liu, J. T. Czernuszka and D. P. Uskokovic, Acta Biomater., 2007, 3, 927–935 CrossRef CAS PubMed.
- J. Klesing, S. Chernousova and M. Epple, J. Mater. Chem., 2012, 22, 199–204 RSC.
- V. Sokolova, Synthesis, characterization and application of calcium phosphate nanoparticles for the transfection of cells, PhD Thesis, University of Duisburg-Essen, Germany, 2006.
- V. Sokolova, A. Kovtun, O. Prymak, W. Meyer-Zaika, E. A. Kubareva, E. A. Romanova, T. S. Oretskaya, R. Heumann and M. Epple, J. Mater. Chem., 2007, 17, 721–727 RSC.
- J. Tang, J. Y. Chen, J. Liu, M. Luo, Y. J. Wang, X. W. Wei, X. Gao, B. L. Wang, Y. B. Liu, T. Yi, A. P. Tong, X. R. Song, Y. M. Xie, Y. Zhao, M. Xiang, Y. Huang and Y. Zheng, Int. J. Pharm., 2012, 431, 210–221 CrossRef CAS PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi, C. Reinhardt, K. Landfester, H. Schild, M. Maskos, S. K. Knauer and R. H. Stauber, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef CAS PubMed.
- V. V. Sokolova, I. Radtke, R. Heumann and M. Epple, Biomaterials, 2006, 27, 3147–3153 CrossRef CAS PubMed.
- I. Canton and G. Battaglia, Chem. Soc. Rev., 2012, 41, 2718–2739 RSC.
- T. G. Iversen, T. Skotland and K. Sandvig, Nano Today, 2011, 6, 176–185 CrossRef CAS PubMed.
- G. Sahay, D. Y. Alakhova and A. V. Kabanov, J. Controlled Release, 2010, 145, 182–195 CrossRef CAS PubMed.
- V. Sokolova, S. Neumann, A. Kovtun, S. Chernousova, R. Heumann and M. Epple, J. Mater. Sci., 2010, 45, 4952–4957 CrossRef CAS.
- Y. Xie, H. Qiao, Z. Su, M. Chen, Q. Ping and M. Sun, Biomaterials, 2014, 35, 7978–7991 CrossRef CAS PubMed.
- J. Wang, B. Chen, D. Zhao, Y. Peng, R. X. Zhuo and S. X. Cheng, Int. J. Pharm., 2013, 446, 205–210 CrossRef CAS PubMed.
- K. Lee, M. H. Oh, M. S. Lee, Y. S. Nam, T. G. Park and J. H. Jeong, Int. J. Pharm., 2013, 445, 196–202 CrossRef CAS PubMed.
- T. Ito, Y. Koyama and M. Otsuka, J. Pharm. Sci., 2014, 103, 179–184 CrossRef CAS PubMed.
- A. Hamberger, U. Ziener and K. Landfester, Macromol. Chem. Phys., 2013, 214, 691–699 CrossRef CAS.
- N. Nabih, K. Landfester and A. Taden, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 5019–5029 CrossRef CAS.
- O. Boussif, F. Lezoualch, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Calif. Acad. Sci., 1995, 92, 7297–7301 CrossRef CAS.
- M. S. Lee, J. E. Lee, E. Byun, N. W. Kim, K. Lee, H. Lee, S. J. Sim, D. S. Lee and J. H. Jeong, J. Controlled Release, 2014, 192, 122–130 CrossRef CAS PubMed.
- P. Dubruel, B. Christiaens, B. Vanloo, K. Bracke, M. Rosseneu, J. Vandekerckhove and E. Schacht, Eur. J. Pharm. Sci., 2003, 18, 211–220 CrossRef CAS.
- J. Dausend, A. Musyanovych, M. Dass, P. Walther, H. Schrezenmeier, K. Landfester and V. Mailänder, Macromol. Biosci., 2008, 8, 1135–1143 CrossRef CAS PubMed.
- H. Urch, M. Vallet-Regi, L. Ruiz, J. M. Gonzalez-Calbet and M. Epple, J. Mater. Chem., 2009, 19, 2166–2171 RSC.
- R. Z. LeGeros, Calcium phosphates in oral biology and medicine, Karger, Basel, 1991 Search PubMed.
- D. Tang, L. Wang, C. A. Orme, T. Bonstein, P. J. Bush and G. H. Nancollas, Angew. Chem., 2004, 116, 2751–2755 CrossRef.
- R. Detsch, D. Hagmeyer, M. Neumann, S. Schaefer, A. Vortkamp, M. Wuelling, G. Ziegler and M. Epple, Acta Biomater., 2010, 6, 3223–3233 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |