
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and characterization of a biocompatible monotyrosine-based polymer and its interaction with DNA†
Radhika
Mehta
a,
Rina
Kumari
a,
Prolay
Das
a and
Anil K.
Bhowmick
*ab
aDepartment of Chemistry, Indian Institute of Technology, Patna, India-800013. E-mail: director@iitp.ac.in; anilkb@rtc.iitkgp.ernet.in; Fax: +91-6122277384; Tel: +91-6122277380
bDepartment of Materials Science, Indian Institute of Technology, Patna, India-800013
First published on 24th July 2014
Abstract
A novel tyrosine-based copolymer containing L-tyrosine (Tyr) and diglycidylether of bisphenol A(DGEBA) was synthesized and studied for its interaction with DNA for potential applications in biological systems. The synthesis of the polymer was optimized by varying monomer ratios using 4-(dimethylamino)pyridine (DMAP) as a catalyst to yield polymers with a Mw of 7500–8000. Further characterization by FTIR, NMR and thermal analysis supported the formation of the monotyrosine–DGEBA polymer. The interaction of the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 DGEBA–tyrosine copolymer with DNA was investigated by gel electrophoresis, thermal melting, and fluorescence spectroscopy in ratios ranging from 0.5:1 to 12:1 polymer–DNA (w/w). The copolymer was seen to lend stability to the DNA without damaging it and demonstrated endonuclease resistivity that is conducive for biological applications. Scanning electron microscopy, dynamic light scattering and zeta potential studies of the polymer–DNA complex also established that the polymer is capable of encapsulating DNA leading to the formation of the DNA–polymer polyplex nano-assembly. The potential of the polymer for biological applications was further reinstated by its non-cytotoxicity.
:1 DGEBA–tyrosine copolymer with DNA was investigated by gel electrophoresis, thermal melting, and fluorescence spectroscopy in ratios ranging from 0.5:1 to 12:1 polymer–DNA (w/w). The copolymer was seen to lend stability to the DNA without damaging it and demonstrated endonuclease resistivity that is conducive for biological applications. Scanning electron microscopy, dynamic light scattering and zeta potential studies of the polymer–DNA complex also established that the polymer is capable of encapsulating DNA leading to the formation of the DNA–polymer polyplex nano-assembly. The potential of the polymer for biological applications was further reinstated by its non-cytotoxicity.
Introduction
Tyrosine, while classified as a non-essential amino acid, is used as a building block for several important neurotransmitters, including norepinephrine,1–3 epinephrine3 and dopamine,4 as well as plays an important role in photosynthesis.5 Apart from its biological roles, researchers have used tyrosine as an amino acid of choice to create unique polymers and supramolecular structures.6–8 Such polymers have been researched for applications in various biomedical areas including tissue engineering and drug and gene delivery amongst others.9–12 The choice of tyrosine for creating novel polymers stems from the unique structure of the neutral amino acid that harbours an aromatic ring and three functional groups that can be made to react with various reagents after careful manipulation of the reaction conditions.Herein, we report the synthesis and characterization of a copolymer that incorporates tyrosine, with DGEBA as a linker through generation of ester and amine functionalities at linkages. The use of L-tyrosine as one of the main constituents of a copolymer with DGEBA has not been reported before to the best of our knowledge. Our synthetic methodology leads to the formation of a polymer that incorporates the less reactive, poorly soluble L-tyrosine with the highly hydrolysable DGEBA. The incorporation of DGEBA in the polymer is due to its ability to polymerize efficiently through its highly reactive epoxide moiety that can be easily broken under mild acidic or basic conditions. The π–π interaction between the aromatic rings of the constituents has been found to facilitate the formation of nanostructures through the self-assembly of such polymers13 which also prompted us to investigate tyrosine in the context of polymerization with DGEBA. DGEBA has been extensively polymerized to generate copolymers with lactones and amines.14,15 Synthesis of the DGEBA homopolymer is also reported16 by the anionic polymerization mechanism. The structure and properties of several copolymers have been discussed previously by one of the authors.17,18
Peripheral coupling of tyrosine to polycationic polymers has been achieved that displayed reduced cytotoxicity and stability for in vitro and in vivo applications.8,13 Tyrosine polymerization resulting in the generation of dityrosine units in the polymer matrix has been reported for potential bio-applications.9,10,19 As such, many tyrosine polymers have been found to be biocompatible by common cell cytotoxicity assays and many researchers have pointed out their potential bioapplications including drug delivery, gene delivery and tissue engineering.9,11 However, low cytotoxicity essentially does not mean that the concerned polymer does not inflict any damage to biomolecules like DNA and scores of proteins inside the cell at the molecular level. The interactions of the polymers with important cellular contents need to be evaluated in detail to ensure the safety of the cellular contents for the desired in vitro or in vivo applications. In this context, the interaction of tyrosine-based polymers with biomolecules like DNA and proteins at the molecular level is still largely unknown. Thus, apart from the synthesis and the meticulous characterization of the novel tyrosine–DGEBA polymer herein, we also report a detailed study of the interaction of the synthesized tyrosine–DGEBA with various forms of DNA viz. calf thymus DNA (ctDNA) and plasmid DNA. Our study also discusses the non-cytotoxicity and the ability of the polymer to resist the endonuclease activity on the DNA.
Experimental section
Materials
Diglycidyl ether of bisphenol A (DGEBA), 4-(dimethylamino) pyridine (DMAP), L-tyrosine (Tyr), calf thymus DNA (ctDNA), agarose, ethidium bromide (EtBr) and chemicals for buffer preparation were purchased either from Sigma or from Merck and used as received. The pUC19 plasmid and Hind III restriction enzyme were purchased from New England Biolabs, USA. All other materials and solvents were used as received without further purification. All experiments of polymer–DNA interactions were done in triplicate and error bars were generated after calculating the mean SD from the average value wherever applicable.Synthesis of the DGEBA–tyrosine polymer
The optimized synthetic route is reported herewith (Scheme 1). | ||
| Scheme 1 Scheme showing the formation of the polymer by the reaction of DGEBA with tyrosine through (a) ester and (b) amine linkages via ring opening. | ||
Variations in solvent, catalyst, time, temperature and concentration of the catalyst and monomers were also employed. Copolymers were synthesized by adding DGEBA to L-tyrosine in varying monomer ratios of 1:0.5 (Bp-Ty 1), 1:1 (Bp-Ty 2) and 1:1.5 (Bp-Ty 3).
The synthetic route mechanistically involves ring opening of the DGEBA epoxide moiety with the activation of amine and carboxyl groups on tyrosine (Scheme 1). The synthesis of copolymer Bp-Ty 2 employed for DNA interaction studies is as follows.
L-Tyrosine (2 mmol, 0.3622 g) and DMAP (0.4 mmol, 0.0488 mg) were dissolved in 4-N,N-dimethyl formamide (DMF) (5 mL) in a 25 mL round bottom flask and heated to 70 °C, to which a solution of DGEBA (2 mmol, 0.6808 mg) in DMF (5 mL) was added dropwise with constant stirring. The reaction mixture was monitored by silica gel TLC in a 60:40 ethyl acetate–hexane solvent system (by volume) that shows complete disappearance of DGEBA spots after 48 hours. The product was precipitated in water after solvent evaporation. A sticky brown solid separated out of the solution and was washed with water (5 × 10 mL) and ether (5 × 5 mL) to remove DMF, DMAP and unreacted monomers. The crude product thus obtained was further purified by dissolving in DMF and precipitating with water in an ice bath (0 °C), following water and ether washes and drying under vacuum for 24 hours. The method for re-precipitation was repeated twice to obtain an ultrapure compound for polymer characterization and further downstream studies with DNA. Alternatively, the reaction mixture in DMF was concentrated under vacuum to a volume of 5 mL. To this, 20 mL cold methanol was added in an ice bath with stirring to precipitate out unreacted tyrosine. The extract was filtered and solvent evaporated to remove methanol and DMF. The polymeric product was then precipitated out in water as discussed above. 1H-NMR (400 MHz, DMSO-d6) δ: 1.5 (s, 6H), 2.2–3.0 (m, 5H), 3.1–4.5 (m, 12H), 6.2–8.0 (m, 12H). 13C-NMR (100 MHz, DMSO-d6) δ: 30.72, 40.52, 41.11, 62.74, 67.29, 69.40, 69.95, 113.78, 115.03, 127.36, 129.94, 142.45, 142.82, 156.45, 162.30. ESI-MS m/z: 495.53 (Tyr + DGEBA − CO), 521.53 (Tyr + DGEBA − H), 227.40 (bisphenol A fragment of DGEBA).
Variations in the above synthetic route were the (a) use of tetrahydrofuran (THF) as a solvent, (b) use of triphenyl phosphine (TPP) and NaOH as catalysts at 10 mole percent, (c) variation of DMAP concentration at 5 and 10 mole percent, (d) variation of temperature from 25 to 100 °C and (e) DGEBA–Tyr molar ratios of 1:0.5 (Bp-Ty 1) and 1:1.5 (Bp-Ty 3).
Characterization of polymers
Infrared spectroscopy was performed on a Perkin-Elmer Spectrum-400 FTIR Spectrometer using KBr pellets in transmission mode in the spectral range of 4000–650 cm−1 with a total of 16 scans per sample. 1H-NMR (400 MHz) and 13C-NMR (100 MHz) were performed on a Bruker Avance II FT-NMR Spectrometer using DMSO-d6 as a solvent and TMS as an internal standard and chemical shifts are reported in the δ-ppm scale. Molecular weight determination was carried out using an Agilent PLGPC 50 Integrated Gel Permeation Chromatograph (GPC) using a PL gel 5 μm Mixed-D column equipped with a refractive index detector. The sample was prepared in HPLC-grade DMF and calibrated with polystyrene standards. Thermogravimetric analysis (TGA) was performed on a TA SDT-Q600 under a nitrogen atmosphere. Powdered samples were heated from 25 to 800 °C at 10 °C min−1. The polymer and polymer–DNA complexes were imaged using a Field Emission Scanning Electron Microscope (FE-SEM), Hitachi-S4800 (Tokyo, Japan). An accelerating voltage of 5 kV and a working distance of 8 mm were used to image the samples. Electron spray ionisation (ESI)-mass spectrometry was performed on a Thermo LCQ Deca XP MAX in the negative mode with a m/z range of 100–1000.Thermal melting studies
The stability of calf thymus DNA (ctDNA) in the presence of Bp-Ty 2 was assessed by measuring the absorbance (A) at 260 nm (average time 1 °C min−1, ramp rate 0.5 °C min−1, from 90 °C to 25 °C and in reverse order) as a function of temperature (T) (dA/dT) on a Peltier controlled Bio Quest CE2501 spectrophotometer (Cecil, UK). The absorbance was measured with 50 μL aliquots containing 7.5 μg of ctDNA mixed with the polymer in concentration ratios of 0.25:1, 0.5:1 and 1:1 in 10 mM sodium phosphate (NaPi) buffer at pH 7.4 in 5% DMSO.
Agarose gel assay
Polymer solutions at the desired concentrations were mixed with 250 ng of pUC19 plasmid DNA and incubated at 37 °C for 24 hours in 10 mM NaPi buffer (pH 7.4) with 5% DMSO (v/v). These samples were then loaded into wells of 1% agarose gel (TAE buffer pH 8, 100 V, 1.5 h) and run with control samples as the plasmid DNA only. The DNA bands were visualised with ethidium bromide staining on a UVP GelDoc-It 300 gel documentation system (UK). The quantitative estimation of the presence of supercoiled (SC), open circular (OC) or the linear (L), if any, was done using Vision Work Ls Image Acquisition and Analysis software from UVP (UK). A nuclease resistance assay was also carried out to evaluate the stability of the DNA in the polymer–DNA complex against restriction enzyme Hind III. The polymer–DNA complex and free DNA were separately incubated with Hind III buffer including Hind III (2 units per 250 ng DNA) at 37 °C for 1 h. The results were analyzed by 1% agarose gel electrophoresis.EtBr displacement assay
Fluorescence studies involving EtBr displacement from DNA in the presence of the polymer were recorded on a Horiba Jobin Yuon Fluoromax-4 spectrofluorometer at excitation and emission wavelengths of 540 nm and 600 nm respectively. Each aliquot (50 μL) containing 30 μM pUC19 DNA and 5 μM EtBr was incubated for 12 hours with Bp-Ty 2 in different ratios in 10 mM NaPi buffer at pH 7.4 with 5% DMSO (v/v). Fluorescence of pure EtBr and DNA–EtBr was also recorded under the same conditions. The percentage of relative fluorescence upon polymer binding was calculated using the following equation:20 | (1) |
Particle size and zeta potential determination
The hydrodynamic diameter and zeta potential of the polymer–DNA complexes were measured by Dynamic Light Scattering (DLS) experiments on a Delsa NanoC Particle Analyser (Beckman-Coulter). Individual samples of ctDNA (15 μg mL−1) in 1 mM NaPi at pH 7.0 (2 mL) filtered through a membrane filter (PVDF, 0.2 μm) were prepared and aliquots of polymer solutions in DMSO were added to achieve different polymer–DNA (w/w) ratios with overall 5% DMSO (v/v) in 1 mM NaPi buffer at pH 7.0. The DLS measurements were performed after 30 minutes as well as after 12 hours incubation in duplicate at 25 °C and a scattering angle of 165°. The average particle size of each sample was obtained by using CONTIN analysis as the mean hydrodynamic diameter (standard deviation of five determinations including polydispersity). For zeta potential measurements, polymer–DNA complexes at ratios of 1:1, 1:5, 1:10 and 1:20 containing 2.5 μg mL−1 ctDNA were prepared in distilled water with 5% DMSO (v/v). These were incubated for 12 hours and the zeta potential was measured across an electric field of 16.3 V cm−1, scattering angle of 15° and cell positions of 0 mm, ±0.35 mm and ±0.7 mm.
MTT assay for cell activity
The MTT assay was performed to measure the metabolic activity of cells. 1 mg of Bp-Ty 2 was dissolved in 0.5 mL of 5% DMSO to make 2 mg mL−1 stock solution. The stock solution was diluted to 100 μg mL−1, 75 μg mL−1, 50 μg mL−1, 25 μg mL−1 and 10 μg mL−1 concentration solutions in the culture medium with serum. Cells cultured in normal medium were considered as cell control and 5% DMSO in culture medium as reagent control (0.5% DMSO). An equal volume (100 μL) of various dilutions of test samples, extract of negative control UHMWPE (ultra high molecular weight polyethylene), cell control, reagent control and positive control (dilute phenol) were placed on subconfluent monolayer of L-929 cells (mouse fibroblast). After incubation of the cells with various concentrations of the test samples and controls at 37 ± 1 °C for 24 hours, extract and control medium was replaced with 50 μL MTT solution (1 mg mL−1 in medium without supplements), wrapped with aluminium foil and incubated at 37 ± 1 °C for 2 hours. After discarding the MTT solution, 100 μL of isopropanol was added to all the wells and subsequently the color developed was quantified by absorbance measurements at 570 nm. The data obtained for test sample, reagent control negative control (UHMWPE) and positive control (dilute phenol) were compared with cell control.Results and discussion
Synthesis of tyrosine–DGEBA polymers
Polymerization of tyrosine and DGEBA is hypothesized to proceed via nucleophilic attack of DMAP on tyrosine to facilitate its activation and the ring opening of the DGEBA epoxide moiety (Scheme 1). Tyrosine, having low solubility in most organic solvents, is appreciably soluble in aqueous solvents depending on pH. It exists primarily in zwitterionic form, and hence is not readily reactive. The reaction between tyrosine and DGEBA was carried out in an organic medium as the epoxide moiety of DGEBA gets hydrolyzed in aqueous solution. Initial reactions were performed using tetrahydrofuran as a solvent. However, the low solubility of tyrosine in THF led us to employ DMF as the solvent.The reaction in DMF with a 1:1 monomer ratio (Bp-Ty 2) was monitored until the consumption of at least one of the monomers. The reaction was followed for 48 hours at 70 °C when the disappearance of DGEBA spots on TLC and no sedimentation of unreacted tyrosine were observed. The reaction at 16 hours and 24 hours at 10% DMAP concentration showed DGEBA spots on TLC and sedimentation of excess tyrosine indicative of incomplete reaction. It is apparent from Fig. 1A that the tyrosine consumed in the reaction is linearly proportional with time up to 50 hours. The higher the catalyst concentration, the higher is the slope or higher is the reaction rate (Fig. 1A and B). 5 mole percent DMAP concentration gave only 15% yield compared to 58% with 10 mole percent DMAP at 48 hours and 70 °C. DMAP was not used at concentrations above 10% to avoid its embedding in the polymer matrix. With 5% catalyst, excess tyrosine sediments out of the reaction and TLC confirmed the presence of excess DGEBA in the solution, indicating that 10 mole percent DMAP was required for complete consumption of the reagents (Fig. 1A).
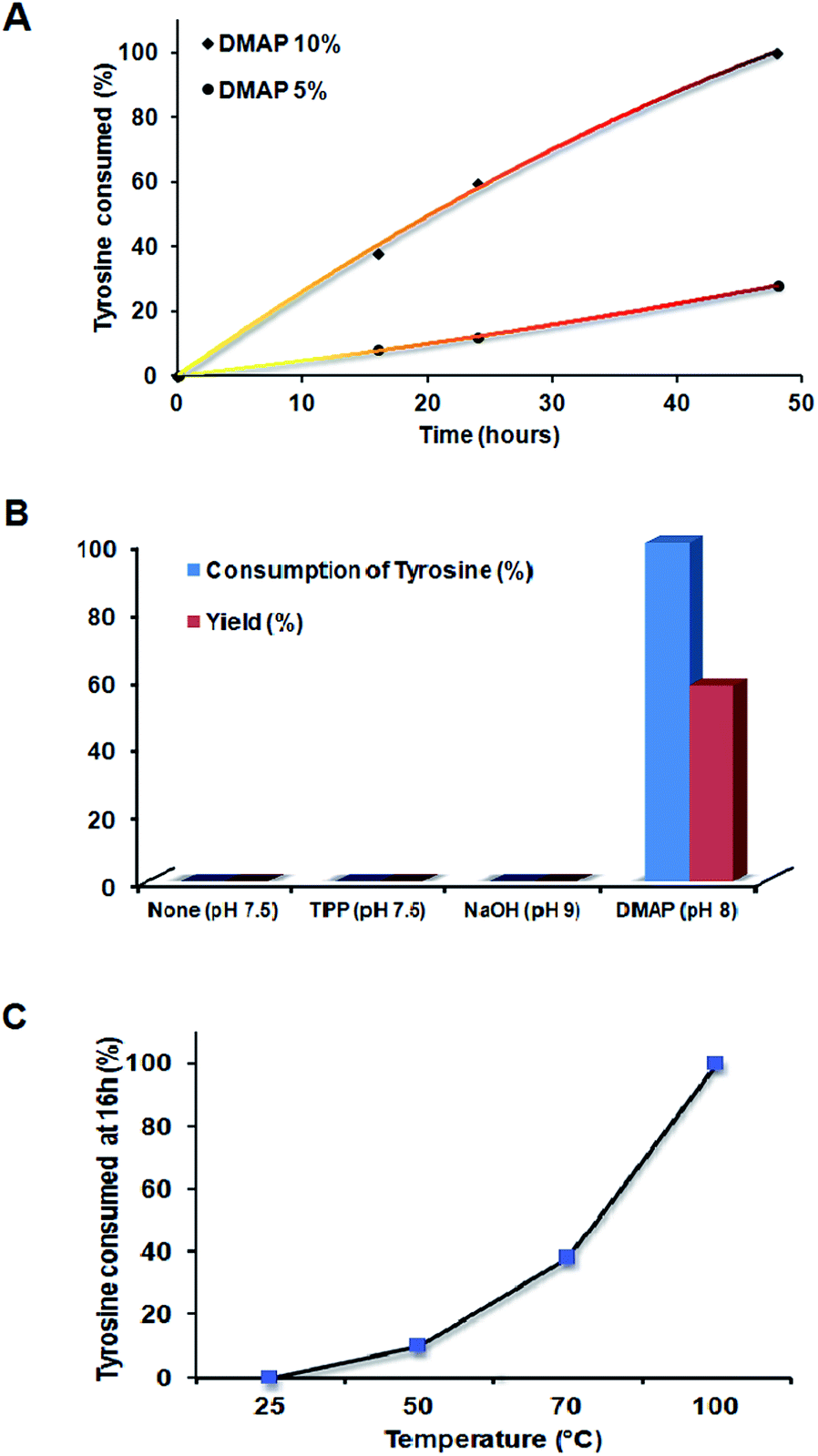 | ||
| Fig. 1 (A) Plot showing percentage consumption of tyrosine from the reaction mixture with time for the 1:1 molar ratio of monomers at varying DMAP concentrations. The colour gradient of the curve resembles the reaction mixture at the respective time intervals. (B) Progress of the reaction between DGEBA and tyrosine with the choice of catalyst and pH as a function of tyrosine consumption and yield. (C) Plot showing consumption of tyrosine as a function of temperature. | ||
The reaction between tyrosine and DGEBA was initially performed without any catalyst, with variations in temperature from 25, 40 to 70 °C without any yield. Excess tyrosine was seen to sediment in the flask at the end of each reaction. Thus, an initiator/catalyst was employed such that it may catalyze the reaction. NaOH and triphenyl phosphine (TPP) were also used, at 10 mole percent in the basic pH range. Both failed to digest tyrosine in the reaction (Fig. 1B) unlike DMAP, which demonstrated consumption of tyrosine and change in colour of solution from a white colloidal mixture to dark brown clear solution indicating the reaction between tyrosine and DGEBA.
Temperature variation was carried out to ascertain the dependence of the reaction on temperature. As seen from Fig. 1C, the reaction proceeds faster on increasing the temperature. At room temperature, the reaction failed to proceed, indicating the need to heat to activate the reaction. On going from 70 to 100 °C, complete consumption of tyrosine was observed at the end of 16 hours.
The reaction with lower ratios of tyrosine–DGEBA (Bp-Ty 1) showed the presence of residual DGEBA as confirmed by TLC. However, no sedimentation of tyrosine was observed indicating complete addition of tyrosine to the polymer matrix. For Bp-Ty 3, i.e. at a higher ratio of tyrosine–DGEBA, sedimentation of excess tyrosine at the end of 48 hours was observed. Thus, this reaction occurs primarily in 1:1 molar ratios of DGEBA–Tyr which has been the focus of our study with DNA.
The polymerization method employed also generates the DGEBA homopolymer but failed to homopolymerize tyrosine. The DGEBA homopolymer was light yellow in colour and insoluble in common organic solvents including MeOH, DCM, THF, hexane, CHCl3, DMF and DMSO. In contrast, the synthesized copolymers Bp-Ty 1, 2 and 3 were brown in colour and soluble in organic solvents including DMF, DMSO and a mixture of MeOH–DCM, which supports the fact that the synthesized polymer is a copolymer comprising both DGEBA and tyrosine. Further characterization of the synthesized copolymers via GPC, FTIR, NMR and TGA is discussed below.
Molecular weight determination
:1 DGEBA–Tyr (Bp-Ty 2) copolymer had a Mw of 7800 and a polydispersity index (PDI) of 1.21. The GPC data for polymers Bp-Ty 1 and Bp-Ty 3 indicate a similar molecular weight range for higher and lower tyrosine ratios. With a low PDI of 1.2, there is an indication that the synthesized polymers are not branched. Bp-Ty 2 is hypothesized to consist of 12–15 units each of tyrosine and DGEBA as an AB type random copolymer, which is further supported by NMR, ESI-MS and thermal studies.
| Sample | M n | M w | PDI |
|---|---|---|---|
| Bp-Ty 1, 48 h, 70 °C | 6450 | 7800 | 1.21 |
| Bp-Ty 2, 48 h, 70 °C | 6230 | 7560 | 1.21 |
| Bp-Ty 3, 48 h, 70 °C | 6410 | 7780 | 1.21 |
Spectroscopic analysis of Tyr–DGEBA copolymers
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1580 cm−1 (medium, aromatic CC and N–H bend from tyrosine), 1505 cm−1 (strong, aromatic CC), 1243 cm−1 (strong, broad, C–N stretch for tyrosine) and 1235 cm−1 (C–O–C bend for DGEBA). The disappearance of the characteristic peak of DGEBA at 914 cm−1 for epoxide bending indicates ring opening of the DGEBA moiety. The strong peak appearing at 1653 cm−1 in the product is an indication of the formation of a carbonyl group in the copolymer.
C), 1580 cm−1 (medium, aromatic CC and N–H bend from tyrosine), 1505 cm−1 (strong, aromatic CC), 1243 cm−1 (strong, broad, C–N stretch for tyrosine) and 1235 cm−1 (C–O–C bend for DGEBA). The disappearance of the characteristic peak of DGEBA at 914 cm−1 for epoxide bending indicates ring opening of the DGEBA moiety. The strong peak appearing at 1653 cm−1 in the product is an indication of the formation of a carbonyl group in the copolymer.
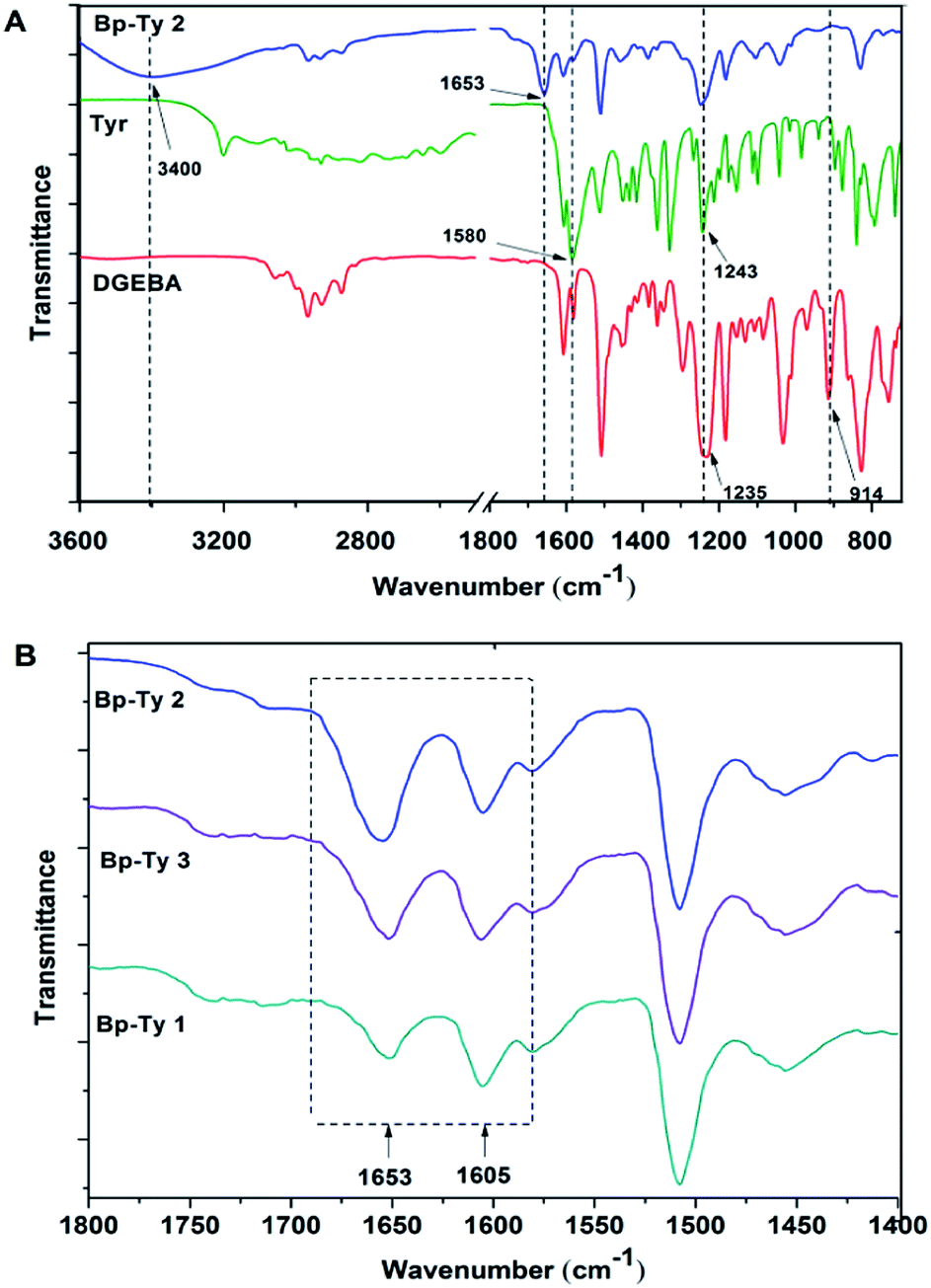 | ||
| Fig. 2 (A) FTIR spectra of pristine DGEBA, tyrosine and Bp-Ty 2 copolymer. (B) FTIR spectra of Bp-Ty 1, 2 and 3 with reference to the peak at 1653 cm−1. | ||
The peak at 1653 cm−1 was normalized with respect to the 1605 cm−1 peak (CC stretching in aromatic compounds) for the synthesized Bp-Ty copolymers. As depicted in Fig. 2B, this peak had high transmittance for Bp-Ty 2 and Bp-Ty 3. As expected, the transmittance decreased for the 1:0.5 DGEBA–Tyr (Bp-Ty 1) copolymer due to the low amount of tyrosine incorporated into the polymer.
 | ||
| Fig. 3 NMR spectra of the synthesized polymer Bp-Ty 2: (A) 13C NMR spectra and (B) 1H NMR spectra. | ||
In the 1H-NMR, the protons are accounted for in the 4–9 ppm range. For amide functionality, a peak between 4 and 6 ppm is expected21 that is absent here, indicative of ester bonding between tyrosine and DGEBA, which correlates with the mass spectrometry result discussed previously. The monomeric ratios in the synthesized copolymers were calculated from the NMR spectra by taking a ratio of the number of aliphatic methyl protons of DGEBA to aromatic protons of DGEBA and tyrosine. With a substrate ratio of DGEBA–Tyr as 1:1, the ratio of aliphatic methyl protons to aromatic protons was calculated to be 6:12 or 1:2. The NMR spectrum for the synthesized polymer Bp-Ty 2 (Fig. 3B) shows that this aliphatic to aromatic proton ratio is close to 1:2.
Thermal analysis of tyrosine–DGEBA copolymers
Thermogravimetric analysis (TGA) was performed on the synthesized copolymers to study the effect of temperature on degradation. TGA curves of Bp-Ty 1, 2 and 3 are shown in Fig. 4. The derivative curves of the copolymers (ESI, Fig. S2†) were analyzed22 to determine ratios of tyrosine (320 °C, 50% degradation) and DGEBA (370 °C, 95% degradation) in the sample with reference to the monomers. The calculated Tyr:DGEBA ratios of Bp-Ty 1, Bp-Ty 2 and Bp-Ty 3 are 0.49:1, 1:1 and 1.1:1 respectively. These data indicate that even with a higher ratio of tyrosine as a substrate, the amount of tyrosine remains equimolar to DGEBA. This was evident from the sedimentation of excess tyrosine at the end of the synthesis route for Bp-Ty 3. At a 0.5 :1 tyrosine–DGEBA ratio (Bp-Ty 1), the synthesized copolymer contains monomers in the same ratio, which is supported by the fact that DGEBA undergoes homopolymerization in the above method. This also indicates the presence of copolymer units and DGEBA homopolymer within the polymer matrix of Bp-Ty 1. The copolymer Bp-Ty 2 has a Tg centred at 77 °C after which the copolymer starts to melt beyond 120 °C (ESI, Fig. S3†).
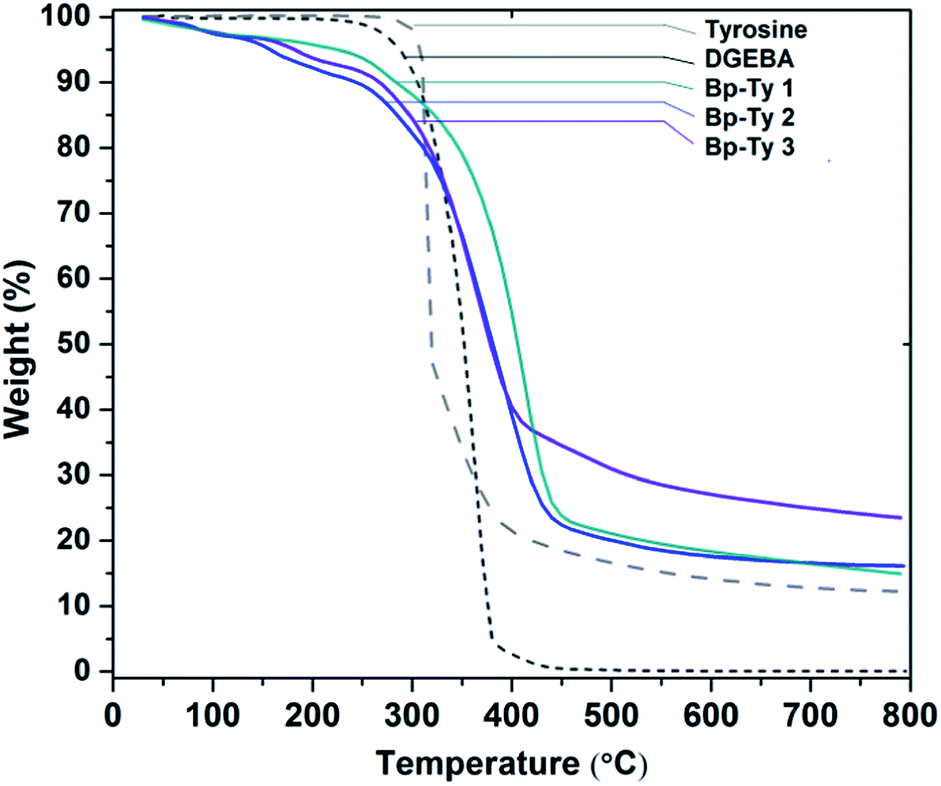 | ||
| Fig. 4 TGA curve showing thermal degradation of Bp-Ty 1, 2 and 3 with respect to the monomers DGEBA and tyrosine. | ||
DGEBA–tyrosine polymers do not damage DNA
Agarose gel assay was conducted to determine the effect of the copolymer on the DNA structure.The supercoiled form of pUC19 plasmid DNA after incubation with the polymer does not show the presence of any linear or nicked form even after incubation for 24 hours (Fig. 5A). This result corroborates the findings of Tseng et al. where poly(esterurethane) and poly(amino ester glycol urethane) polymers showed a similar effect.23 However, in the present study with an increase in polymer concentration, the intensity of the supercoiled plasmid band gradually decreased in the gel (Fig. 5B). For a polymer to DNA ratio of 1:1, the decrease in the intensity of the band was ca. 10% which further decreased by ca. 63% when compared to the plasmid only control band in the agarose gel with an increase in polymer concentration.
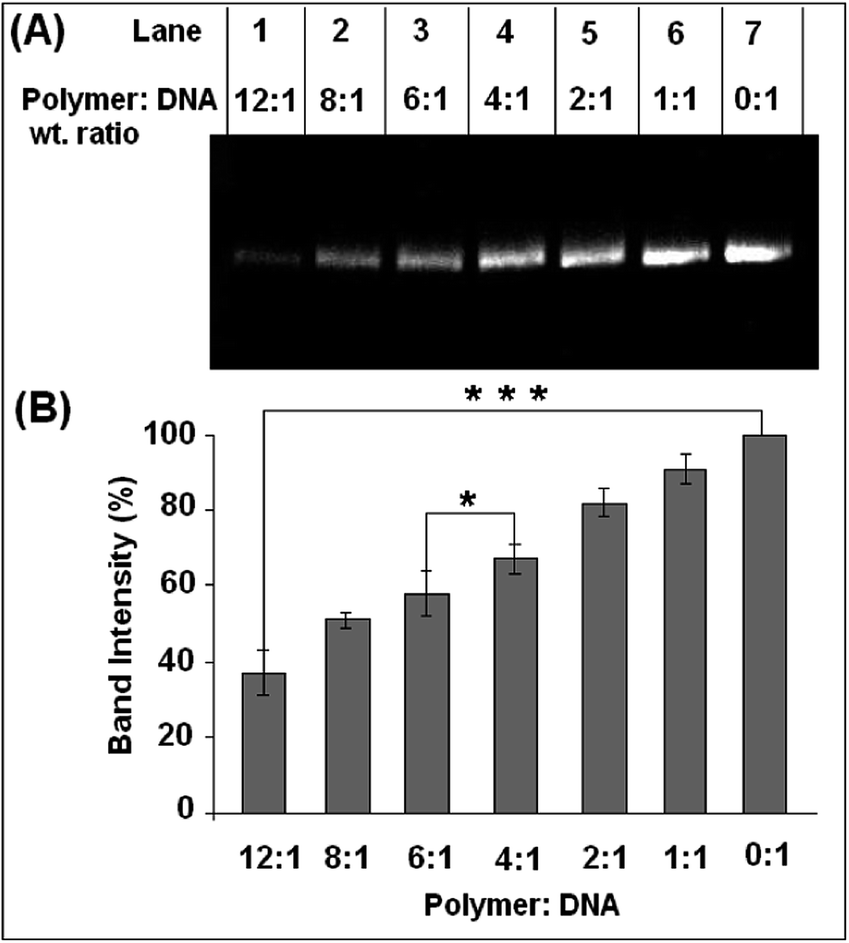 | ||
| Fig. 5 Effect of the polymer on the supercoiled form of pUC19 plasmid DNA after incubation for 24 hours. (A) 1% agarose gel showing results of incubation of plasmid with Bp-Ty 2 in various weight ratios. No relaxed or linear form was detected. (B) Quantitative estimation of the decrease of the intensity of the plasmid band in the agarose gel due to groove binding of the polymer with DNA. Each value represents mean ± SD. An * denotes a statistical significance of p < 0.005 and *** denotes a statistical significance of p < 0.00001 between samples as measured by the unpaired t-test. | ||
Further, we compared the band intensity and mobility pattern of DNA in agarose gel preincubated with the Bp-Ty polymer (neutral polymer) and poly(ethyleneimine) (PEI) (Mn 10000), a highly researched polycationic polymer for delivery applications. The results are similar where the disappearance of DNA bands in agarose gels was observed, albeit at lower concentrations and incubation time in the case of PEI (Fig. S4, ESI†). Additionally, mobility shift of the DNA bands incubated with Bp-Ty was not observed whereas DNA incubated with PEI shows mobility shift at a concentration ratio of 0.75:1 polymer–DNA (w/w). This suggests that PEI tends to neutralise the peripheral charge of DNA at lower concentrations due to its polycationic nature. Since the agarose gels were stained with EtBr after completion of the gel run, the decrease in the intensity of the plasmid bands is attributed to insufficient intercalation of the EtBr into DNA due to the shielding effect of the polymer on DNA at higher concentrations. The band for the supercoiled plasmid does not show any mobility shift, indicative of the absence of any unwinding in the DNA brought about by the polymer. Thus, conformational destabilization in DNA is not being introduced by the Bp-Ty polymer.
The absence of any nuclease activity and unwinding of the DNA in the presence of the polymer is a good indication that the polymer does not inflict any damage or modification to the DNA, which is desired for any biological application. The effect of the restriction enzyme, Hind III, on the polymer–DNA complex to determine the endonuclease activity is shown in Fig. 6. Plasmid DNA, after incubation with the polymer at higher concentrations, resists the endonuclease activity of the enzyme Hind III to an appreciable degree (Fig. 6A). As can be seen from Fig. 6A, the free DNA (Lane 7) is completely linearized in the presence of Hind III. While up to the polymer to DNA ratio of 2:1, the nuclease activity of Hind III remained unhindered, thereafter a gradual decrease in the nuclease activity of Hind III with an increase in polymer concentration was observed. The nuclease activity of Hind III was reduced to ca. 50% at a polymer concentration 8 times more than the plasmid concentration (Fig. 6B).
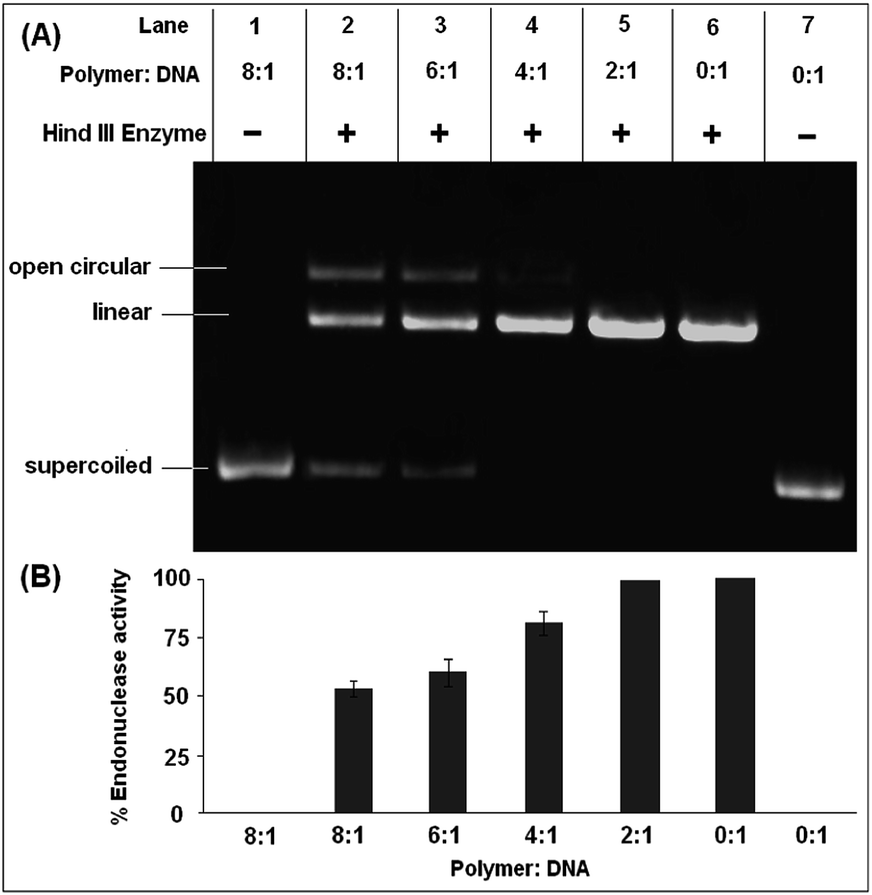 | ||
| Fig. 6 Cleavage efficiency of restriction endonuclease Hind III on pUC19 plasmid DNA (A) 1% agarose gel (pH 7.5) showing the effect of different concentrations (w/w) of Bp-Ty 2 on the endonuclease activity of Hind III following 24 hour incubation with pUC19. (B) Quantitative estimation of Bp-Ty 2 induced resistance to the endonuclease activity of Hind III on pUC19. | ||
The decrease in nuclease activity suggests that at higher polymer concentrations, the DNA strands are not exposed to the surrounding due to the shielding and encapsulation effect of the polymer that prevents recognition of DNA bases by the endonucleases giving rise to an insufficient amount of the linear form of the plasmid.23 To check whether the polymer has any effect on Hind III enzyme, the enzyme was co-incubated with the polymer at higher concentration followed by dialysis of the solution and incubation of the purified enzyme with DNA. No effect on the endonuclease activity of Hind III was observed, which further confirms the fact that interaction of the polymer with DNA is responsible for inhibition of the endonuclease activity. This result is noteworthy since in the light of the possible use of any DNA cargo to be transported to a particular cellular location, the presence of numerous endogenous nucleases can degrade the DNA even before being released from the polyplex, thus rendering the delivery ineffective. In this case, the polymer can offer some resistance to the endonuclease activity to the DNA cargo, albeit at higher concentrations.
DGEBA–tyrosine copolymer lends stability to the DNA
The increase in DNA melting temperature (Tm) followed by addition of a compound or polymer is indicative of the increase in the thermal stability of the DNA because of intercalation, groove binding, etc. The melting temperatures of the complexes formed between ctDNA and the polymer at different ratios are shown in Table 2. The Tm of native ctDNA was observed at 66 °C, and a gradual increase in the Tm was documented, up to 71 °C with a 1:1 weight ratio of polymer to DNA. Reference thermal melting data obtained to determine the effect of DMSO on Tm (not shown) showed a decrease in Tm to 65.5 °C. However, the highest increase in melting temperature on complexation was found with the DNA–polymer ratio of 1:1 that confirmed the stabilisation of the DNA in the presence of the copolymer (Table 2). However, sharp Tm was unavailable at higher polymer concentrations, possibly due to formation of higher order structures and aggregates.
| DNA–polymer ratio (w/w) |
1:0
|
1:0.25
|
1:0.5
|
1:1
|
| T m (°C) | 66.0 | 66.9 | 68.5 | 71.0 |
Partial intercalation of polymers into DNA replaces EtBr
The EtBr replacement assay was performed to measure the degree of association of the polymer with DNA (Fig. 7). The decrease in fluorescence of EtBr intercalated into DNA results from the quenching of EtBr after it comes out from the DNA into the solvent.24 A gradual decrease in the fluorescence of EtBr was observed upon the addition of the polymer.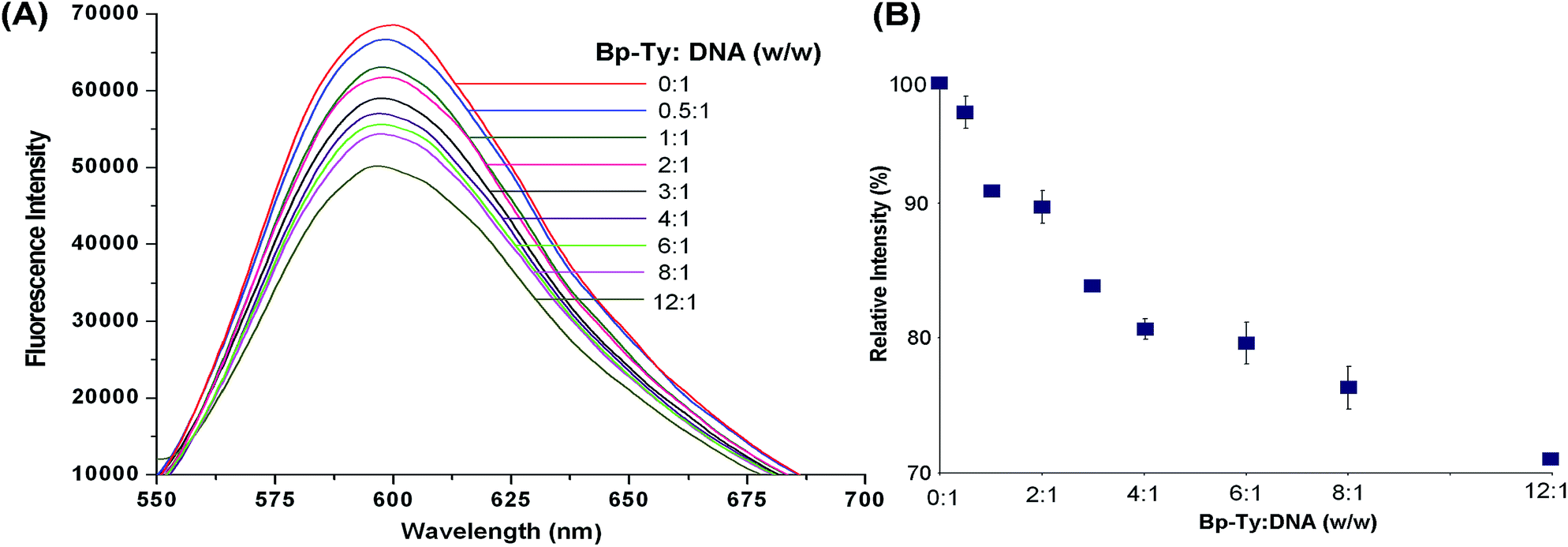 | ||
| Fig. 7 EtBr displacement assay. (A) Decrease in fluorescence intensity upon addition of By-Ty 2 to DNA observed at λex = 540 nm and λem = 600 nm. (B) Plot of relative fluorescence intensity with DNA–EtBr at different polymer concentrations. | ||
As observed from Fig. 7, EtBr fluorescence is seen to decrease by nearly 30% only when the DNA and the polymer ratio is 12:1. This decrease in fluorescence is attributed to partial intercalation, thereby releasing bound EtBr in solution. The presence of other non-covalent interactions between the polymer and DNA may also introduce changes in DNA conformations, thereby releasing bound EtBr. Insertion of aromatic residues of the polymer between base pairs of DNA may lead to bending of the helix at the point of intercalation, essentially releasing EtBr and hence the decrease in fluorescence.25 Most notably, the replacement of intercalated EtBr is not substantial at comparable DNA and polymer ratios. This observation points to the fact that at lower concentrations of the polymer, its intercalation into DNA is negligible, which means that the conformational distortion leading to DNA damage is minimal under such conditions.
The data correlate well with the agarose gel assay data (Fig. 5) wherein a gradual decrease in band intensity is observed on increasing the amount of the polymer. Thus, the DNA–copolymer complex formed is shown to be stable, with efficient binding and ability to restrict endonuclease degradation as desirable for a biocompatible material.
Size and surface charge determination of polymers in the presence of DNA
The size and the surface charge of the polymer in the presence of the DNA were investigated by DLS. As depicted in Fig. 8A, good correlation was not obtained with only DNA or the polymer. However, upon addition of the polymer to DNA, the correlation function gets stabilized that indicate the formation of stable particles of finite size. The same trend was observed with the intensity distribution, where only the DNA or the polymer had broad distributions that consequently narrowed down when co-incubated at respective concentrations (Fig. 8B).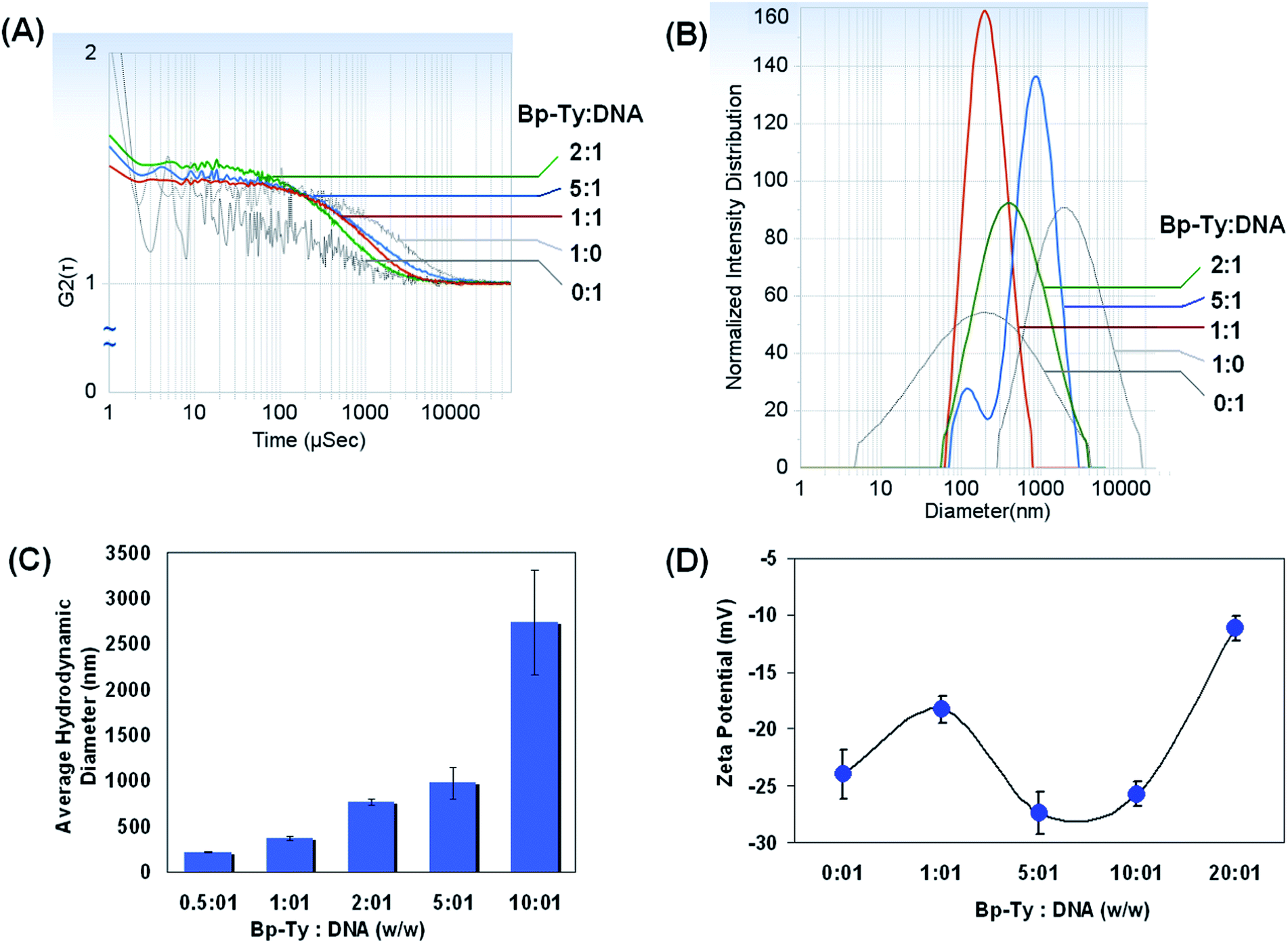 | ||
| Fig. 8 Dynamic light scattering experiments. (A) Correlation factor for measurement of hydrodynamic diameter by incubating different concentrations of Bp-Ty 2 to a definite concentration of DNA. (B) Plot of intensity distribution of DNA–polymer complexes with their respective hydrodynamic diameter. (C) Plot of average hydrodynamic diameters with different polymer to DNA ratios after 12 hour incubation. (D) Measurement of zeta potential at different polymer to DNA ratios after 12 hour incubation. | ||
The hydrodynamic diameter of DGEBA–Tyr copolymer–DNA complexes at various copolymer to DNA w/w ratios was measured using DLS after incubation for 30 min (ESI, Table S1†) as well as after 12 hours (Fig. 8C). Our results indicate possible formation of a polyplex between the polymer and the DNA. A gradual increase in the hydrodynamic diameter of the polyplex was observed until the polymer concentration was 5–6 times that of DNA. Our study shows that even after incubation of the DNA with the polymer for 12 hours, the polyplexes are stable as is evident from the correlation and the particle size distribution. This is in contrast to PEI incubated DNA where aggregation is more prominent after 12 hours of incubation that can deter data acquisitions. In the present case of the Bp-Ty polymer, there is a gradual increase in the average hydrodynamic radii whereas with PEI the hydrodynamic radii increase with increase or decrease in polymer concentration after the 2:1 PEI–DNA ratio (w/w).26 It is to be noted that DLS data with PEI and DNA could only be obtained after addition of relevant concentration of NaCl unlike the Bp-Ty polymer. Nevertheless, the average hydrodynamic radii of DNA condensates obtained by incubating with the Bp-Ty polymer and PEI (Mn 10000) were found to be similar (∼300 nm in the concentration range 5:1 w/w).
The best results in terms of correlation and intensity distribution were obtained when the concentration of the polymer is between 1 and 5 times greater than the DNA. The sudden increase of the diameter at a polymer concentration 10 times more than the DNA suggests the formation of aggregates at such concentrations. This is hypothesised to be due to the condensation of polymeric sheets with DNA leading to a more linearized complex. At a 0.5:1 polymer to DNA ratio, the hydrodynamic diameter is around 219 nm with a standard deviation and polydispersity of 3 nm and 0.2 respectively indicating a uniform entity. With increasing polymer ratio to 1:1, the polydispersity and scattering intensity remain the same, but the diameter increases, which is in agreement with the literature.20 At higher ratios of polymer to DNA such as 10:1, the polydispersity is seen to increase significantly indicating a higher degree of aggregation. Overall, stable particle formation was observed within a certain range of polymer concentrations with DNA that shows appreciable interactions between the polymer and DNA. Comparison of the DLS data obtained after incubation of the DNA with the polymer for 30 minutes and 12 hours revealed some interesting differences (ESI, Table S1†). On incubation for 30 minutes, the hydrodynamic radii of the particles formed with a certain ratio of polymer to DNA are smaller as compared to those incubated for 12 hours, indicating slow aggregation with time. It is also to be noted that the diameter observed in DLS is the average hydrodynamic diameter and not the actual diameter of the polyplex, which is proven to be much lesser than that of the hydrated form.27
The zeta potential was recorded for copolymer–DNA complexes to gain insight into the complexes' surface charge and their potential stability. The effective charge density is considered as a crucial parameter in determining the structure and morphology of DNA in the condensed state. DNA condensation occurs when ∼90% of the surface negative charge of DNA is neutralized in an aqueous solution and that is irrespective of the side chain functionality of the condensing agent.28 The observed pKa of the polymer at 7.6 and 11.3 (ESI, Table S2†) confirms 80–100% ionization of the polymer according to the Henderson–Hasselbalch equation, thereby supporting DNA condensation leading to the formation of the polyplex. The zeta potential results show that the free polymer has a zeta potential of 17 mV, possibly due to the basicity of the amine groups of tyrosine. The positive zeta potential for the polymer is also indicative of few free hydroxyl and carboxyl moieties in the polymer matrix. The surface charge for free DNA was observed to be −23.7 mV which is expected due to the phosphate groups on DNA. The zeta potential for the polymer–DNA complexes became negative after incubation indicating that the phosphate groups on DNA interact with the free amine groups thereby making the whole entity negative. It has also been reported previously that the condensed DNA may well exist as a partially dissociated polyanion, effectively bringing down the zeta potential.29
As is evident from Fig. 8D, the zeta potential stabilized for 5:1 and 10:1 polymer to DNA ratios at a negative value, indicating the presence of free phosphate groups for binding. With further increase in polymer concentration, a decrease in zeta potential was observed due to the availability of fewer free phosphate groups coupled with the fact of higher degree of aggregation and low stabilisation of the polyplex. The 20:1 complex was observed to be least stable as expected.
Imaging studies with DNA via SEM
The morphology of the copolymer–DNA complexes formed at a ratio of 0.5:1 was examined using FE-SEM. The images of the pure polymer were also obtained for reference (Fig. 9A and B).
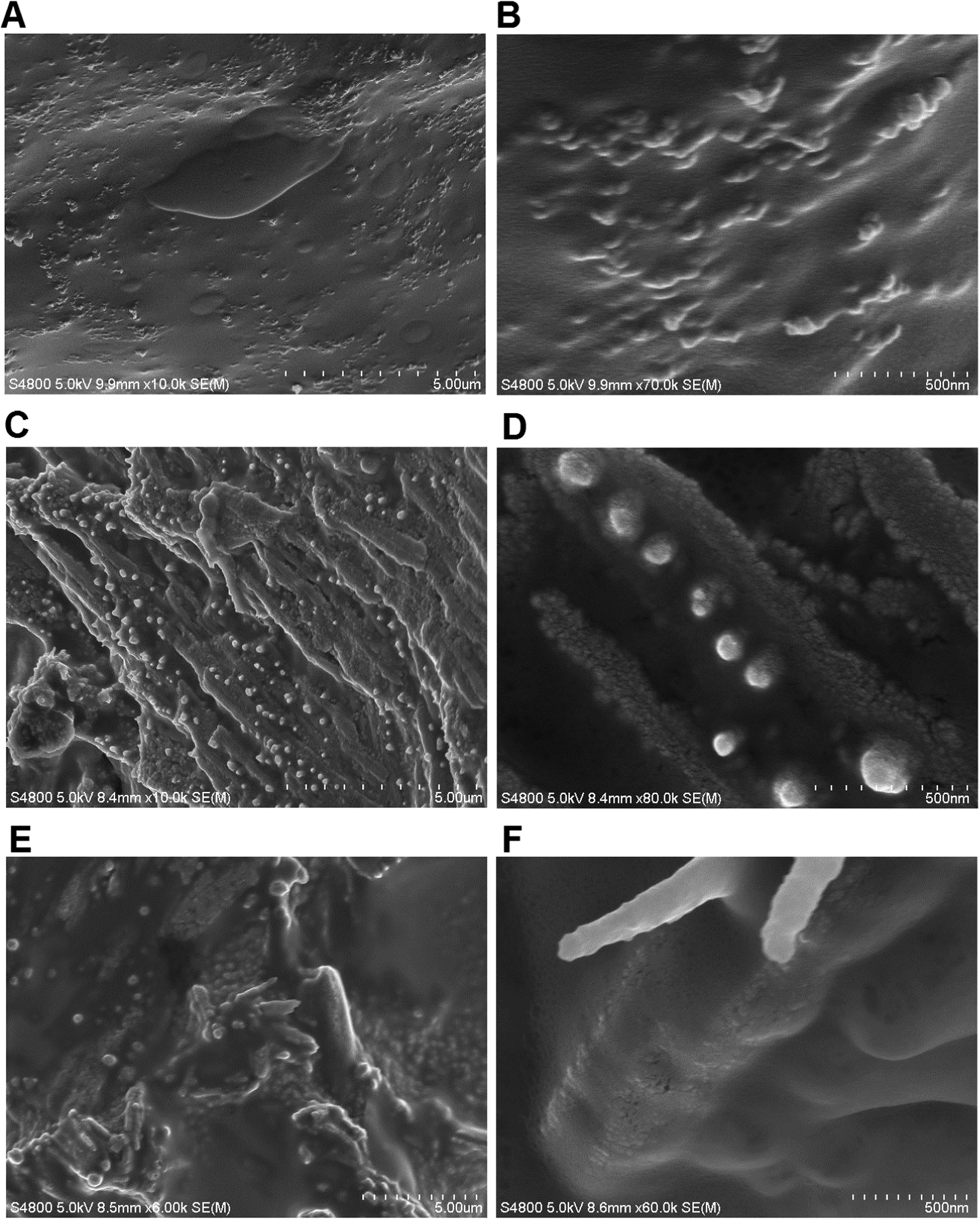 | ||
| Fig. 9 SEM images of the Bp-Ty 2 polymer (A and B) and the polyplex with the 0.5:1 polymer–DNA ratio (C and D) and 1:1 polymer–DNA ratio (E and F). | ||
The Bp-Ty 2 polymer had a sheet-like appearance with protrusions (50–100 nm) to the single-layered epoxy sheets. After incubation with DNA, the polymer sheets start aggregating around the DNA. In the SEM image for the 0.5:1 polymer–DNA complex (Fig. 9C and D), dense globular DNA fragments are observed. The morphology of the polymer sheet also changes after the DNA interaction, showing smooth and rough patches. The smooth patches are attributed to the free polymer. The rough patches as seen in Fig. 9D are clusters of DNA spread throughout the polymer. The SEM images for the 0.5:1 polymer–DNA polyplex display DNA on the surface of the polymer as well. For the 1:1 polyplex (Fig. 9E and F), the DNA could hardly be seen on the surface of the polymer sheet possibly due to its encapsulation by the polymer. As seen in Fig. 9F, the DNA is much more discreetly spread out than in 9D. Thus, the SEM images of copolymer–DNA show DNA clusters and free polymer indicating that there are portions on the polymer that are not engaged in the DNA interaction, which increase with the polymer concentration. This also supports the agarose gel and EtBr experiments portraying the entrapment of DNA within the polyplex with increasing polymer concentration.
The interaction of DNA with the polymer at the interface and as a whole entity is pictorially depicted in Scheme 2. In summary, the polymer does not show any noteworthy nuclease activity and even protects plasmid DNA from the endonuclease activity. Partial displacement of EtBr from DNA and elevated Tm were observed following incubation of the polymer with ctDNA. As deduced from SEM, the DNA is assembled in clusters and entrapped within the polymer matrix. DLS studies showed concentration dependent stable particle formation with ctDNA in the presence of the copolymer.
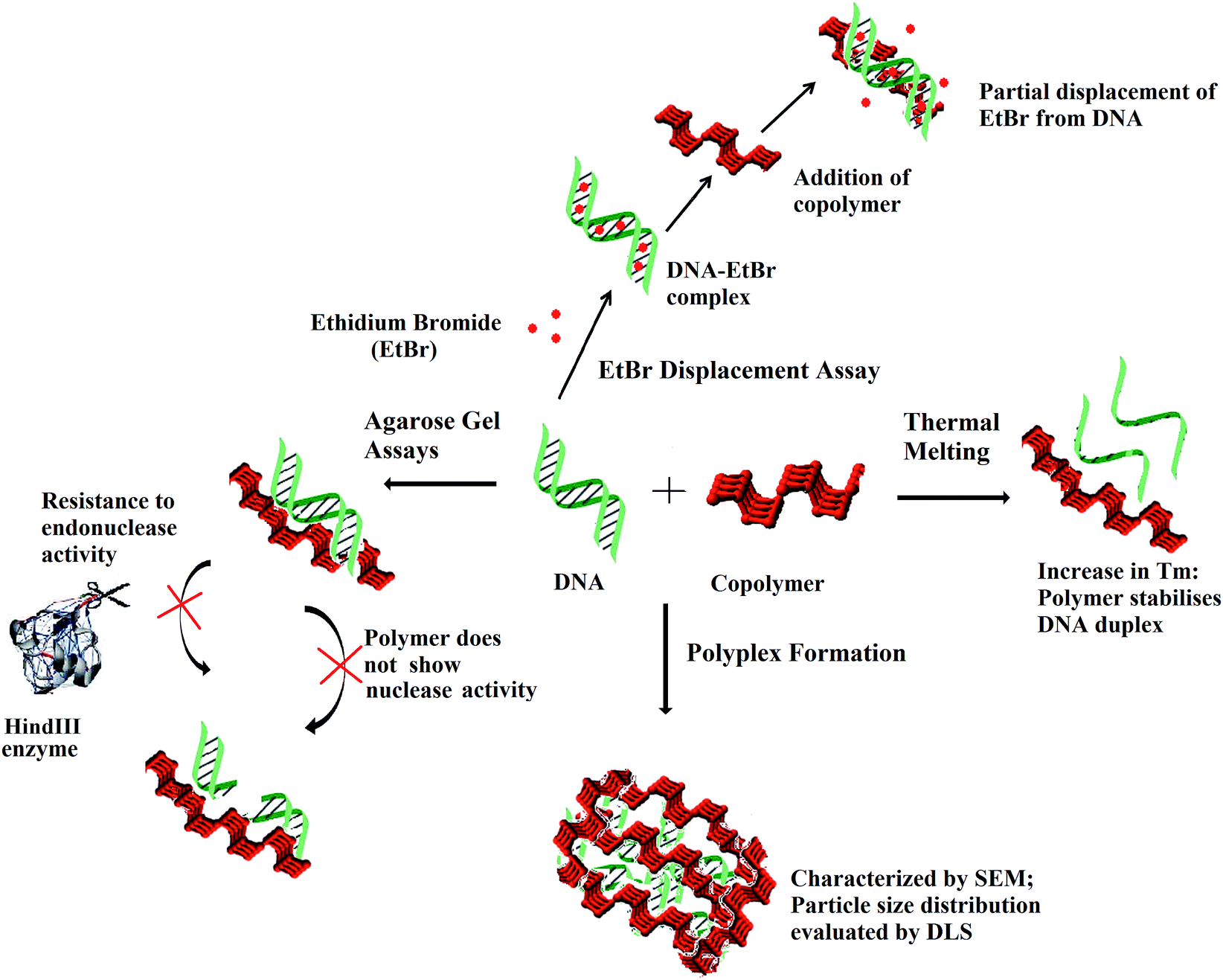 | ||
| Scheme 2 Scheme visualising the interaction of the copolymer with DNA via EtBr displacement assay and thermal melting at the interface and DNA–polymer complex formation as indicated by DLS and SEM. | ||
Cell viability
Tyrosine-based polymers have been previously reported to be non-cytotoxic.9 In the present study, the cell viability of L929 mammalian cells (mouse fibroblast) incubated with the copolymer Bp-Ty 2 was evaluated by assessment of the cell metabolic activity (MTT assay). The metabolic activity of the cells in the presence of Bp-Ty 2 in the concentration range of 10–100 μg mL−1 is seen to remain high (Fig. 10).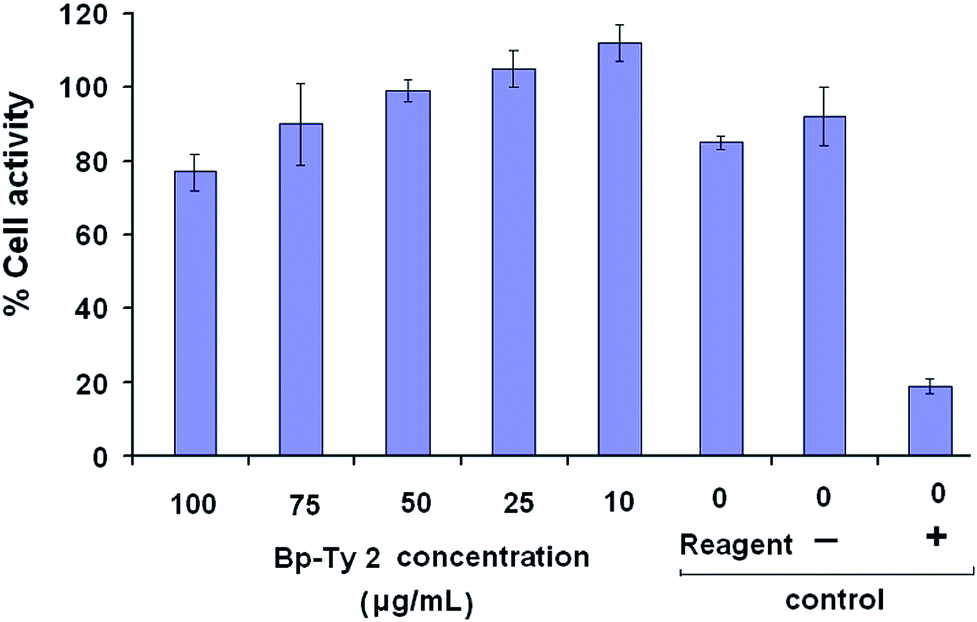 | ||
| Fig. 10 Percentage cellular metabolic activity of the polymer Bp-Ty 2 with L929 mouse fibroblast cells at different concentrations of Bp-Ty 2 after 24 hours of incubation. The negative control contains UHMWPE and the positive control contains dilute phenol. | ||
At 100 μg mL−1 concentration of Bp-Ty 2, the metabolic activity is close to the reagent control and significantly higher than the positive control phenol, which is indicative of low cytotoxicity of the polymer at this concentration. At lower concentrations of 10–75 μg mL−1, the metabolic activity is observed to be greater, even higher than the reagent control that does not contain any polymer. This signifies that in the presence of the polymer at relevant concentrations, the normal functioning of the cells is not interrupted. Overall, the synthesized polymer is observed to be non-cytotoxic at reasonable concentrations. The biocompatibility of the polymer is noticeable even in the presence of DGEBA moieties, probably owing to the occurrence of the tyrosine units. In case the polymer finds applications in vector delivery in future, its non-cytotoxicity may be an advantageous property since commonly used few PEIs have been reportedly found to be cytotoxic.26,30
Conclusions
DGEBA and tyrosine derived Bp-Ty copolymers with different monomer ratios were synthesized successfully using DMAP as a catalyst. Characterization of the polymer was done by FTIR spectroscopy that indicates the presence of characteristic wavelengths for both the starting materials as well as the formation of carbonyl bonds. 1H and 13C NMR indicated the presence of carbonyl linkages and the former also displayed the aliphatic to aromatic proton ratio being close to 1:2. Thermal degradation studies confirmed the monomer ratios as 0.49:1, 1:1 and 1.1:1 in the synthesised polymers Bp-Ty 1, 2 and 3 with an equimolar content of tyrosine with DGEBA as the maximum incorporated into the polymers. Cell viability studies performed using the MTT assay confirmed the non-cytotoxic nature of the polymer.
The Bp-Ty 2 polymer was thus studied with DNA to ascertain its employability in biological applications. Our experiments indicate possible formation of a polyplex at relevant concentration of the DNA and the polymer. The polymer was found to increase the melting temperature by 4 °C, thereby lending stability to the DNA after polyplex formation. Agarose gel assays proved that the polymer does not damage the DNA and even offer resistance to the endonuclease activity, a fact promising for its potential bioapplications. Fluorescence assay conducted with the intercalated EtBr suggested partial intercalation or groove binding of the polymer with the DNA helix at higher polymer concentrations. DLS experiments showed stable particle formation at polymer–DNA ratios of 1:1 up to 5:1 and a decrease in zeta potential of the polymer upon complexation with DNA. The polymer–DNA conjugate was imaged on a SEM that showed that clusters of DNA spread discreetly on the polymer sheet. Overall, a novel and biocompatible polymer with DGEBA and tyrosine was created and characterized. The interaction of the polymer with DNA was evaluated that gave important insights into its potential for use in biological applications. In particular, our studies indicate that the polymer has potential biological applications including but not limited to scaffolding and vector delivery.
Acknowledgements
RM is thankful to Bridgestone Corporation (Japan) for fellowship and RK is thankful for fellowship to IIT Patna. The authors are thankful to SAIF Panjab University for NMR spectroscopy and IISc Bangalore for ESI-mass spectrometry. The authors are also thankful to SCTIMST, Trivandrum for conducting MTT Assay to determine the cell viability. Financial assistance from Dept. of Science and Technology, Govt. of India (grant no. SR/FT/LS/36/2010) to PD is gratefully acknowledged.Notes and references
- M. Levitt, S. Spector, A. Sjoerdsma and S. Udenfriend, J. Pharmacol. Exp. Ther., 1965, 148, 1 CAS.
- S. Meyers, Altern. Med. Rev., 2000, 5, 64 CAS.
- S. Udenfriend and J. B. Wyngaarden, Biochim. Biophys. Acta, 1956, 20, 48 CrossRef CAS.
- J. D. Elsworth and R. H. Roth, Exp. Neurol., 1997, 144, 4 CrossRef CAS PubMed.
- B. A. Barry and G. T. Babcock, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7099 CrossRef CAS.
- (a) V. Tangpasuthadol, A. Shefer, K. A. Hooper and J. Kohn, Biomaterials, 1996, 17, 463 CrossRef CAS; (b) M. H. R. Magno, J. Kim, A. Srinivasan, S. McBride, D. Bolikal, A. Darr, J. Hollinger and J. Kohn, J. Mater. Chem., 2010, 20, 888 RSC; (c) J. Rickerby, R. Prabhakar, M. Ali, J. Knowles and S. Brocchini, J. Mater. Chem., 2005, 15, 1849 RSC.
- A. S. Gupta and S. T. Lopina, J. Biomater. Sci., Polym. Ed., 2002, 13, 1093 CrossRef CAS PubMed.
- G. Creusat, A. S. Rinaldi, E. Weiss, R. Elbaghdadi, J. S. Remy, R. Mulherka and G. Zuber, Bioconjugate Chem., 2010, 21, 994 CrossRef CAS PubMed.
- S. L. Bourke and J. Kohn, Adv. Drug Delivery Rev., 2003, 55, 447 CrossRef CAS.
- A. S. Gupta and S. T. Lopina, Polymer, 2005, 46, 2133 CrossRef PubMed.
- D. Y. Sarkar, J.-C. Yang, N. Klettlinger and S. T. Lopina, eXPRESS Polym. Lett., 2007, 1, 724 CrossRef CAS.
- F. E. Ali, K. J. Barnham, C. J. Barrow and F. Separovic, J. Inorg. Biochem., 2004, 98, 173 CrossRef CAS PubMed.
- C. Troiber, D. Edinger, P. Kos, L. Schreiner, R. Kläger, A. Herrmann and E. Wagner, Biomaterials, 2013, 34, 1624 CrossRef CAS PubMed.
- L. González, X. Ramis, J. M. Salla, A. Mantecón and A. Serra, J. Appl. Polym. Sci., 2009, 111, 1805 CrossRef PubMed.
- M. Arasa, X. Ramis, J. M. Salla, A. Mantecón and A. Serra, Polymer, 2009, 50, 2228 CrossRef CAS PubMed.
- I. E. Dell'Erba and R. J. J. Williams, Polym. Eng. Sci., 2006, 46, 351 Search PubMed.
- N. Roy and A. K. Bhowmick, Polymer, 2010, 51, 5172 CrossRef CAS PubMed.
- H. Datta, A. K. Bhowmick and N. K. Singha, Polymer, 2009, 50, 3259 CrossRef CAS PubMed.
- (a) T. Fukuoka, Y. Tachibana, H. Tonami, H. Uyama and S. Kobayashi, Biomacromolecules, 2002, 3, 768 CrossRef CAS PubMed; (b) L. H. Jones, A. Narayanan and E. C. Hett, Mol. BioSyst., 2014, 10, 952 RSC.
- U. Rungsardthong, T. Ehtezazi, L. Bailey, S. P. Armes, M. C. Garnett and S. Stolnik, Biomacromolecules, 2003, 4, 683 CrossRef CAS PubMed.
- C. Yu and J. Kohn, Biomaterials, 1999, 20, 253 CrossRef CAS.
- A. Choudhury, A. K. Bhowmick and C. Ong, J. Appl. Polym. Sci., 2010, 116, 1428 CAS.
- S. J. Tseng and S. C. Tang, Biomacromolecules, 2007, 8, 50 CrossRef CAS PubMed.
- A. Schallon, C. V. Synatschke, D. V. Pergushov, V. Jérôme, A. H. Müller and R. Freitag, Langmuir, 2011, 27, 12042 CrossRef CAS PubMed.
- V. H. Mulimani and R. A. Day, Indian J. Biochem. Biophys., 1982, 19, 292 CAS.
- X. Yan, Y. Zhang, H. Zhang, P. G. Wang, X. Chu and X. Wang, Org. Biomol. Chem., 2014, 12, 1975 CAS.
- S. J. Hwang, N. C. Bellocq and M. E. Davis, Bioconjugate Chem., 2001, 12, 280 CrossRef CAS PubMed.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269 CrossRef CAS.
- F. Ke, Y. K. Luu, M. Hadjiargyrou and D. Liang, PLoS One, 2010, 5, 13308 Search PubMed.
- (a) M. Thomas and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14640 CrossRef CAS PubMed; (b) S. M. Moghimi, P. Symonds, J. C. Murray, A. C. Hunter, G. Debska and A. Szewczyk, Mol. Ther., 2005, 11, 990 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Mass spectrometry figure, TGA derivative curves, differential scanning calorimetry data, agarose assay with PEI, DLS data for 30 minutes and pKa and ionization of the polymer. See DOI: 10.1039/c4tb00854e |
| This journal is © The Royal Society of Chemistry 2014 |