 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid hydrogels based on keratin and alginate for tissue engineering
Raquel
Silva
a,
Raminder
Singh
be,
Bapi
Sarker
a,
Dimitrios G.
Papageorgiou
c,
Judith A.
Juhasz
a,
Judith A.
Roether
d,
Iwona
Cicha
e,
Joachim
Kaschta
d,
Dirk W.
Schubert
d,
Konstantinos
Chrissafis
c,
Rainer
Detsch
a and
Aldo R.
Boccaccini
*a
aInstitute of Biomaterials, Department of Materials Science and Engineering, University of Erlangen-Nuremberg, 91058 Erlangen, Germany. E-mail: aldo.boccaccini@ww.uni-erlangen.de
bLaboratory of Molecular Cardiology, Medical Clinic 2, University Hospital Erlangen, 91054 Erlangen, Germany
cSolid State Physics Section, Physics Department, Aristotle University of Thessaloniki, 541 24 Thessaloniki, Greece
dInstitute for Polymer Materials, Department of Materials Science and Engineering, University of Erlangen-Nuremberg, 91058 Erlangen, Germany
eCardiovascular Nanomedicine Unit, Section of Experimental Oncology and Nanomedicine, ENT Department, University Hospital Erlangen, 91054 Erlangen, Germany
First published on 20th June 2014
Abstract
Novel hybrid hydrogels based on alginate and keratin were successfully produced for the first time. The self-assembly properties of keratin, and its ability to mimic the extracellular matrix were combined with the excellent chemical and mechanical stability and biocompatibility of alginate to produce 2D and 3D hybrid hydrogels. These hybrid hydrogels were prepared using two different approaches: sonication, to obtain 2D hydrogels, and a pressure-driven extrusion technique to produce 3D hydrogels. All results indicated that the composition of the hydrogels had a significant effect on their physical properties, and that they can easily be tuned to obtain materials suitable for biological applications. The cell–material interaction was assessed through the use of human umbilical vein endothelial cells, and the results demonstrated that the alginate/keratin hybrid biomaterials supported cell attachment, spreading and proliferation. The results proved that such novel hybrid hydrogels might find applications as scaffolds for soft tissue regeneration.
Introduction
Advanced synthesis techniques are leading to the development of innovative biomaterials that can better reproduce the mechanical nature of biological tissues, exhibiting higher potential in tissue engineering applications. Hydrogels from naturally occurring polymers are widely used due to their unique properties, especially regarding the cell environment and morphology similar to the extracellular matrix of native tissues.1–3Some recent studies have indicated that hydrogels, especially those derived from proteins and polysaccharides, are attractive matrices for tissue engineering, since they provide a suitable three-dimensional (3D) setting that mimic the extracellular environment of native tissues.4–8 Furthermore, their aqueous-milieu allows transport of substances such as nutrients and bioproducts of the cell metabolism. One specific material that has been extensively studied for tissue engineering applications due to its particular ability to form hydrogel is alginate. Hydrogel formation can be achieved through various approaches, such as ionic and covalent crosslinking.9,10 In addition, alginate is a suitable material for novel tissue engineering application concepts including cell encapsulation and biofabrication.11,12 However, alginate does not promote cell attachment, causing poor cell–material interactions, followed by slow degradation and unfavourable degradation kinetics.13–15 To overcome these drawbacks of alginate, several approaches are being considered. For example, the use of specific proteins is being investigated due to their similarity with the extracellular matrix to enhance cellular interaction and to tailor the degradability, biocompatibility and availability of the hydrogel.6,16,17 Among different proteins, those based on the structure of extra cellular matrix, such as collagen, elastin, fibrin, and keratin are of interest due to their potentially high bioactivity. In this context, keratin is a promising protein since it is the major component of hair, feathers, nails and horns of mammals, reptiles and birds and is being widely tested for biomedical applications.18,19 At the molecular level, this protein can be distinguished from other fibrous proteins due to its high concentration of cysteine residues (7–20% of total amino acid residues).20,21 The keratin proteins can be classified as alpha-, gamma-, and beta-keratins.22 The alpha-keratins, which reside in the hair fibre cortex, are characterized by the low sulphur content and have an alpha-helical tertiary structure, and an average molar mass in the range of 60–80 kDa.23 The beta-keratins are primarily protective and form the majority of the cuticle, thus being difficult to extract. Moreover, they do not form especially useful reconstituted structures.22 Gamma-keratins, on the other hand, are globular, high in sulphur content, and lower in molar mass (approximately 15 kDa) compared to the other types of keratins. Due to their high sulphur content they behave as disulphide crosslinkers, holding the cortical superstructure together.24–26 Importantly, keratins extracted from wool and human hair contain cellular binding motifs such as, RGD (arginine-glycine-aspartic acid), and LDV (leucine-aspartic acid-valine), which mimic the sites of cellular attachment found in the extra cellular matrix.27–31 Several methodologies have been presented in the literature regarding the chemical extraction of keratin from wool and hair for further development of various biomaterials with different geometries, such as hydrogels matrices for tissue engineering applications.32–36
In this context, it was our goal to prepare a novel type of hybrid hydrogel based on alginate and keratin protein extracted from wool to develop and study the effect of two different hydrogel geometries; films (2D) and hydrogel microcapsules (3D) regarding cell–material interaction. The synergistic effects of each blend component were analysed using different techniques. Furthermore, the cell–material interaction was assessed using primary human endothelial cells.
Experimental section
Materials
Sodium alginate (sodium salt of alginic acid from brown algae, suitable for immobilization of micro-organisms, MW 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–200000 g mol−1, guluronic acid content 65–70%) was acquired from Sigma-Aldrich, Germany. Calcium chloride di-hydrate (CaCl2·2H2O) was purchased from VWR International, Belgium. All other reagents were analytical grade and purchased from Sigma-Aldrich, Germany.
000–200000 g mol−1, guluronic acid content 65–70%) was acquired from Sigma-Aldrich, Germany. Calcium chloride di-hydrate (CaCl2·2H2O) was purchased from VWR International, Belgium. All other reagents were analytical grade and purchased from Sigma-Aldrich, Germany.
Keratin extraction from wool
The keratin (K) solution of 1% (w/v) was prepared by immersing 1 g of delipidated wool fabric, provided by Albano Antunes Morgado Lda (Portugal), in 10 mL of a solution containing 8 M urea, 0.2 M sodium dodecyl sulphate (SDS), and 0.5 M Na2S2O5. The mixture was heated to 60 °C for 12 h. The solution was then diluted with 90 mL of distilled water, filtered, and dialyzed against distilled water using cellulose tubing (molecular-weight cut off of 12000–14000 Da) over approximately 3 days.
Determination of free sulfhydryl (–SH) groups
The amount of free sulfhydryl groups in keratin extracted from wool was determined spectrophotometrically via Ellman's reagent (5,5′-dithiobis(2-nitrobenzoic acid)), as described previously.37 Briefly, 250 μL of keratin (1%) was added to 2.5 mL of 0.5 M phosphate buffer (pH 8.0), containing 50 μL of Ellman's reagent solution (4 mg mL−1). The samples were mixed, incubated for 30 min at room temperature in the dark and the absorbance was measured at a wavelength of 412 nm. The amount of sulfhydryl groups was determined from the calibration curve obtained using L-cysteine as a standard.Sonochemical preparation of 2D hydrogels
The experimental set-up used was composed of a probe type ultrasound source (20 kHz Sonics & Materials Vibracell CV 33) fitted with a 3 mm diameter titanium micro-tip and the power delivery was controlled as percentage amplitude. The reaction vessel was a similar device used in a previous investigation (open glass cell with 19 mm of diameter and 75 mm of height), containing 14 mL of sample solution.38The alginate solution (2%; w/v), previously heated to 37 °C to improve handling by pipetting, was added to the prepared protein solution (1%; w/v). The blend solution of alginate/keratin (Alg/K) and only alginate solution were further submitted to an ultrasound treatment. The sonication treatment was carried out for 3 min and monitored in 58 s increments. A pulsed duty cycle of “8 s on, 2 s off” was used. 14 mL of blend solution was transferred into a 10 cm glass Petri dish (VWR, Germany) and left at 37 °C for approximately 30 min to dry. 0.1 M of calcium chloride solution was poured on the formed hydrogel-films and left for 15 min to allow the ionic gelation. Afterwards, films were washed three times with ultrapure water (Direct-Q®, Merck Milli-Pore, Germany) or with cell culture medium and discs of 13.5 mm in diameter were punched out using a stainless steel cutter.
Pressure driven extrusion preparation of 3D hydrogels
Microcapsules were produced according to a previously published method17 by applying a pressure-driven extrusion technique. In brief, alginate solution (2%; w/v) and the blend solution of Alg/K (2% and 1% w/v, respectively) were transferred into an extrusion cartridge (Nordson EFD, USA) connected to a high precision fluid dispenser (Ultimus V, Nordson EFD, USA). Microcapsules obtained by applying different air pressure (0.55 bar to 2.5 bars) were collected in a beaker containing calcium chloride solution (0.1 M) and kept for 10 min to allow ionic gelation. A further washing procedure was performed three times with water or cell-culture medium (in the case of cell encapsulation) in order to remove adherent calcium chloride, and to form microcapsules in the size range of 700–800 μm.Physico-chemical characterization of 2D and 3D hydrogels
The water uptake ability of the hydrogels with known weight was determined by soaking them in two different media: Dulbecco's modified eagle medium (DMEM, Gibco, Germany) and Hanks' balanced salt solution (HBSS, Sigma-Aldrich, Germany) at pH = 7.4 up to 3 days at 37 °C. The swollen hydrogels were removed at predetermined time points. After removing the excess water using a filter paper (Whatman Pergamyn Paper), the hydrogels were weighed with an analytical balance (Scaltec, Germany). The water uptake was calculated, where Ww and Wd are the weights of swollen and dried hydrogels, respectively, according to the eqn (1).
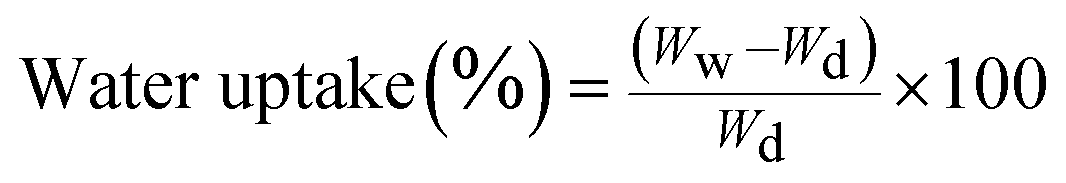 | (1) |
Each experiment was repeated 4 times and the average was considered to be the water uptake value (±standard deviation).
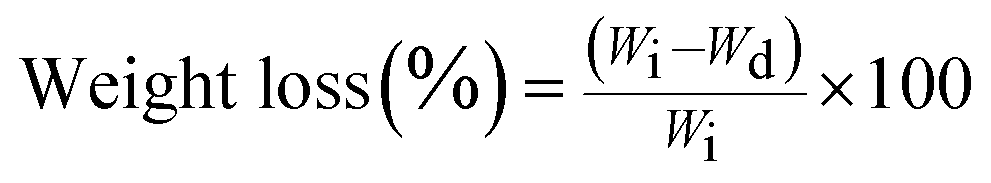 | (2) |
Each experiment was repeated four times, and the average value was considered to be the weight loss value (±standard deviation).
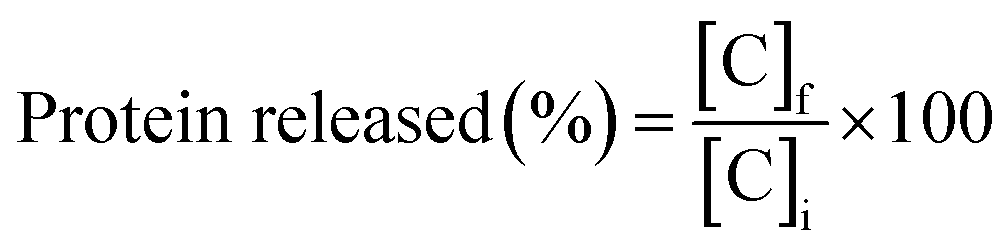 | (3) |
Measurements were recorded in triplicate and the results were expressed as mean value ± standard deviation.
Cell–material interaction using human umbilical vein endothelial cells (HUVEC)
Statistical analyses
Statistical analyses were performed by one-way analysis of variance (ANOVA) on the 2D and 3D hydrogels. The pairwise comparison of the means was performed with the Bonferroni's test (post hoc comparison), where p-values < 0.05 were considered statistically significant.Results and discussion
Quantification of sulfhydryl groups
Keratin was extracted from wool by sulphitolysis,40 where the cysteine disulphide bonds are cleaved by sulphite to give cysteine thiol (reduced keratin) and Bunte salts residues.40–42 Therefore, in order to analyse the presence of the sulfhydryl groups in the extracted keratin, the Ellman's reagent was used. The results showed that the extraction of keratin from the wool (1% w/v) leads to 0.13 mM of sulfhydryl groups. These groups, present in cysteine residues, are a common site for free radical reactions and hence the small amount of residues present in keratin can also contribute toward the gelation of keratin. Upon sonication, cysteine residues can be oxidized and establish inter-protein disulphide bonds that crosslink the proteins resulting in gelation and more organized structure, which was further confirmed by FTIR.Physico-chemical characterization of 2D and 3D hydrogels
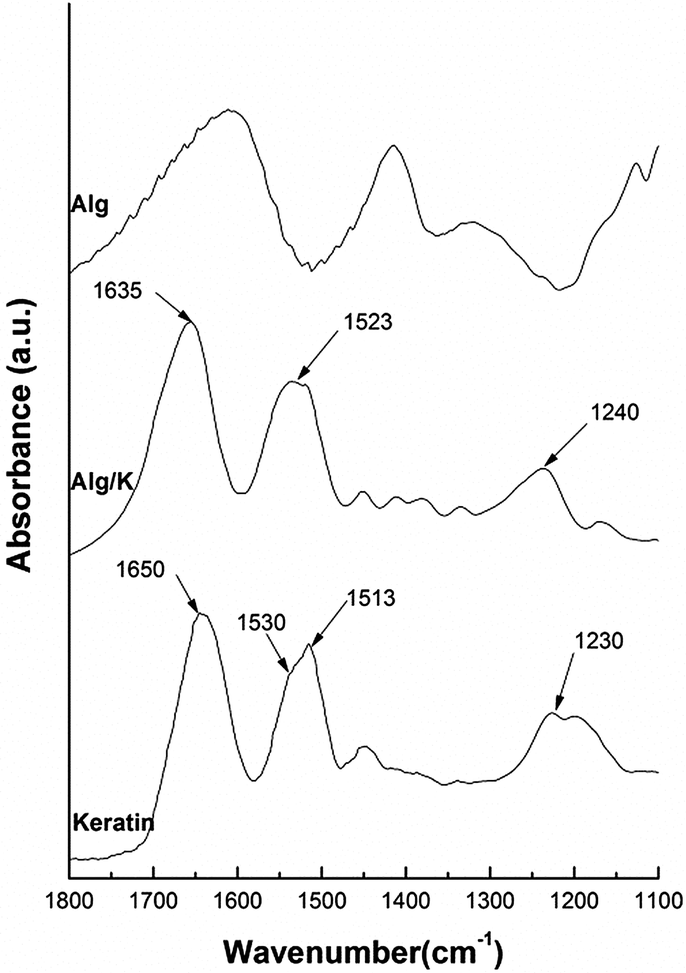 | ||
| Fig. 1 FTIR spectra intervals (1800–1100 cm−1) from the pure keratin without ultrasound treatment and 2D-hydrogels of Alg and Alg/K. | ||
The FTIR spectrum of Alg is widely reported in the literature and is characterized by the peaks which fall around 3447, 1615, 1421, and 1035 cm−1 and are attributed to the stretching of O–H, –COO (asymmetric), –COO− (symmetric), and C–O–C, respectively.43 The FTIR spectra of pure keratin and hydrogel films of Alg/K show characteristic absorption bands assigned to the peptide bonds (–CONH–) characterized with the bands known as amide I (1600–1700 cm−1, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), amide II (1540–1520 cm−1, NH bending and C–H stretching vibration) and amide III (1220–1300 cm−1 combination of C–N stretching and CO bending vibration).44,45 Concerning the literature, it has been shown that the spectral peaks from amide band I, which are mainly related to the CO stretching are commonly used to identify the protein secondary structure. The positions of these bands indicate the conformations of the protein materials: 1650 cm−1 (random coil) and 1630 cm−1 (β-sheet) for amide I; 1540 cm−1 (random coil) and 1520 cm−1 (β-sheet) for amide II; 1230 cm−1 (random coil) and 1270 cm−1 (β-sheet) for amide III.46,47 The spectrum from pure keratin (before ultrasound treatment) shows a peak at 1650 cm−1 from amide I, 1530 cm−1 with a shoulder at 1513 cm−1 amide II and 1230 cm−1 amide III. The absorption value of amide I band indicates that the secondary structure of keratin consist of α-helix and random coil conformation. The spectrum of Alg/K after ultrasound treatment presents a peak of amide I around 1626 cm−1 followed by a small shoulder at around 1650 cm−1, and amide II showed a broad peak around 1522 cm−1. The presence of these bands can indicate that after hydrogel formation, keratin protein assumes mainly a β-sheet conformation. However, the peak from amide II is not well defined, giving an indication that some disorder structures are also present. This occurs because ultrasound treatment leads to a change in the protein conformation, enhancing the electrostatic interactions and changes in the hydrophobic hydration, which can result in the accelerated formation of physical cross-links, such as initial chain interactions related to more organized structures. It is well known that the sonication treatment due to mechanical vibration causes the formation and collapse of bubbles, leading to the phenomena of cavitation, where the media may experience extreme local effects; including heating (10000 K), high pressure (200 bar), high strain rates (107 s−1)48–50 and production of hydroxyl (˙OH) and hydrogen (H˙) radicals, and in the presence of oxygen (O2), superoxide (O2˙−) and hydroperoxyl radicals (HO2˙).51–53 Moreover, these radicals (˙OH, O2˙−, and HO2˙) are potential protein crosslinking agents. Thus, keratin can be crosslinked through disulphide bonds resulting from cysteine oxidation, since keratin contains cysteine residues in its constitution, which are in the form of sulfhydryl groups after the reduction treatment. As a consequence, the molecular chains become closer and this organization can stabilize the supramolecular β-sheet structure of keratin.
O stretching), amide II (1540–1520 cm−1, NH bending and C–H stretching vibration) and amide III (1220–1300 cm−1 combination of C–N stretching and CO bending vibration).44,45 Concerning the literature, it has been shown that the spectral peaks from amide band I, which are mainly related to the CO stretching are commonly used to identify the protein secondary structure. The positions of these bands indicate the conformations of the protein materials: 1650 cm−1 (random coil) and 1630 cm−1 (β-sheet) for amide I; 1540 cm−1 (random coil) and 1520 cm−1 (β-sheet) for amide II; 1230 cm−1 (random coil) and 1270 cm−1 (β-sheet) for amide III.46,47 The spectrum from pure keratin (before ultrasound treatment) shows a peak at 1650 cm−1 from amide I, 1530 cm−1 with a shoulder at 1513 cm−1 amide II and 1230 cm−1 amide III. The absorption value of amide I band indicates that the secondary structure of keratin consist of α-helix and random coil conformation. The spectrum of Alg/K after ultrasound treatment presents a peak of amide I around 1626 cm−1 followed by a small shoulder at around 1650 cm−1, and amide II showed a broad peak around 1522 cm−1. The presence of these bands can indicate that after hydrogel formation, keratin protein assumes mainly a β-sheet conformation. However, the peak from amide II is not well defined, giving an indication that some disorder structures are also present. This occurs because ultrasound treatment leads to a change in the protein conformation, enhancing the electrostatic interactions and changes in the hydrophobic hydration, which can result in the accelerated formation of physical cross-links, such as initial chain interactions related to more organized structures. It is well known that the sonication treatment due to mechanical vibration causes the formation and collapse of bubbles, leading to the phenomena of cavitation, where the media may experience extreme local effects; including heating (10000 K), high pressure (200 bar), high strain rates (107 s−1)48–50 and production of hydroxyl (˙OH) and hydrogen (H˙) radicals, and in the presence of oxygen (O2), superoxide (O2˙−) and hydroperoxyl radicals (HO2˙).51–53 Moreover, these radicals (˙OH, O2˙−, and HO2˙) are potential protein crosslinking agents. Thus, keratin can be crosslinked through disulphide bonds resulting from cysteine oxidation, since keratin contains cysteine residues in its constitution, which are in the form of sulfhydryl groups after the reduction treatment. As a consequence, the molecular chains become closer and this organization can stabilize the supramolecular β-sheet structure of keratin.
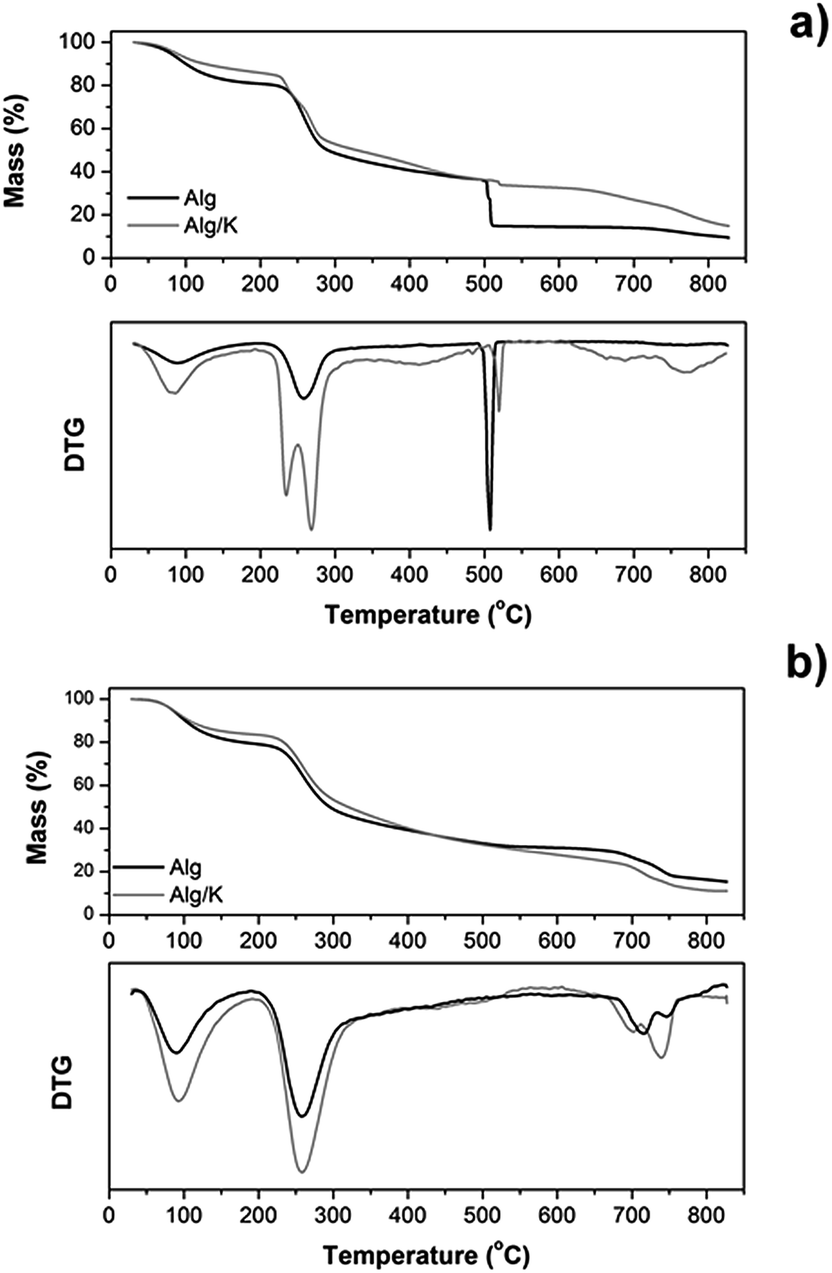 | ||
| Fig. 2 Mass loss (%) and derivative thermogravimetric analysis (DTG) curves of 2D (a) and 3D-hydrogels (b) obtained from Alg, and Alg/K. | ||
Generally, the thermogravimetric curves follow the same trend for both samples, indicating that the decomposition steps were practically the same. However, for 2D hydrogels the thermal stability of the samples was affected by the presence of keratin protein (Fig. 2a). The first decomposition stage starts almost immediately (after 30 °C), ranges up to 220 °C and corresponds to the dehydration process and water desorption of the samples. The temperature range between 220 and 470 °C corresponds to the gradual degradation of both samples and represents two different degradation steps. The first step for Alg/K begins at 225 °C presenting also a peak at 277 °C and for Alg starts at 240 °C. In the case of Alg/K hydrogels the peak at 225 °C is related to the melting/degradation of α-helices of the intermediate filaments proteins and the peak at 277 °C corresponds to the degradation of keratin associated proteins that comprise the high-sulfur (highly cross-linked) keratins of the intermacrofibrillar matrix. For Alg films the peak around 240 °C indicates the decomposition of alginate by the dehydration of the saccharide rings, breaking up of C–H bonds and breaking of the C–O–C glycoside bonds in the main polysaccharide chain. In the second stage, the blend with keratin presents higher thermal stability while the alginate sample exhibits lower thermal stability. In both degradation stages, dehydration and CO and C–C bond cleavage processes occur accompanied by the formation of carbon structures as previously determined by gas chromatography-mass spectrometry.54 The decomposition step, which occurs at the temperature range 475–530 °C, is also present in both samples, and represents a very fast and abrupt process, as indicated in derivative thermogravimetric analysis (DTG). This specific phenomenon is a consequence of a violent combustion and oxidation of the sample. The 2D hydrogel of Alg loses the highest percentage of its mass during this step (20 wt%), which starts at 510 °C. However, the blend with keratin protein presents the higher thermal stability up to that point, and starts this second degradation step at 513 °C loosing just 3% of its initial mass. This fact can be associated with the ability of keratin to form the disulphide bond during ultrasound treatment, which increases the thermal stability of the hybrid hydrogel, as previously mentioned. At the temperature range from 530 to 730 °C almost a plateau is formed in the thermograms of both hydrogel films, while from 730 to 800 °C the final degradation step of all samples is obvious, as indicated also in the DTG graphs. The decomposition of calcium carbonate, which had been formed during the heating process at low temperatures, is taking place at that temperature range. Regarding the remaining mass loss percentages, the Alg sample degraded almost completely and the mass residue was about 9%, while Alg/K the mass residue was approximately 14%. In the subsequent analyses, the thermal stability of the samples in microcapsule form was also studied under the same conditions as used for 2D hydrogels (Fig. 2b). However, the results from the thermogravimetric experiments performed on the microcapsule samples did not reveal significant differences in the thermal degradation between Alg and Alg/K 3D hydrogels. The trend observed during the degradation process was almost identical for both samples. A small difference was detectable at the turning point of 440 °C. Up to that temperature, the sample with keratin exhibited higher thermal stability than the Alg microcapsules demonstrating once again that keratin protein increases the thermal stability of the blend. From that point, Alg decomposed at slower rates than the hybrid samples, a fact which resulted in higher mass residue of Alg. Compared to the films, microcapsules underwent the same degradation steps at slightly elevated temperatures, but the most significant difference was that the fast decomposition, which was seen in the films at 475–530 °C, was not observed in microcapsules. This is an indication that this geometry contains a large amount of water (almost 94–98% w/w), which is evaporated at around 100 °C in addition to a small amount of materials which degrade after certain time. This conclusion was further confirmed by the derivative of the mass loss signal.
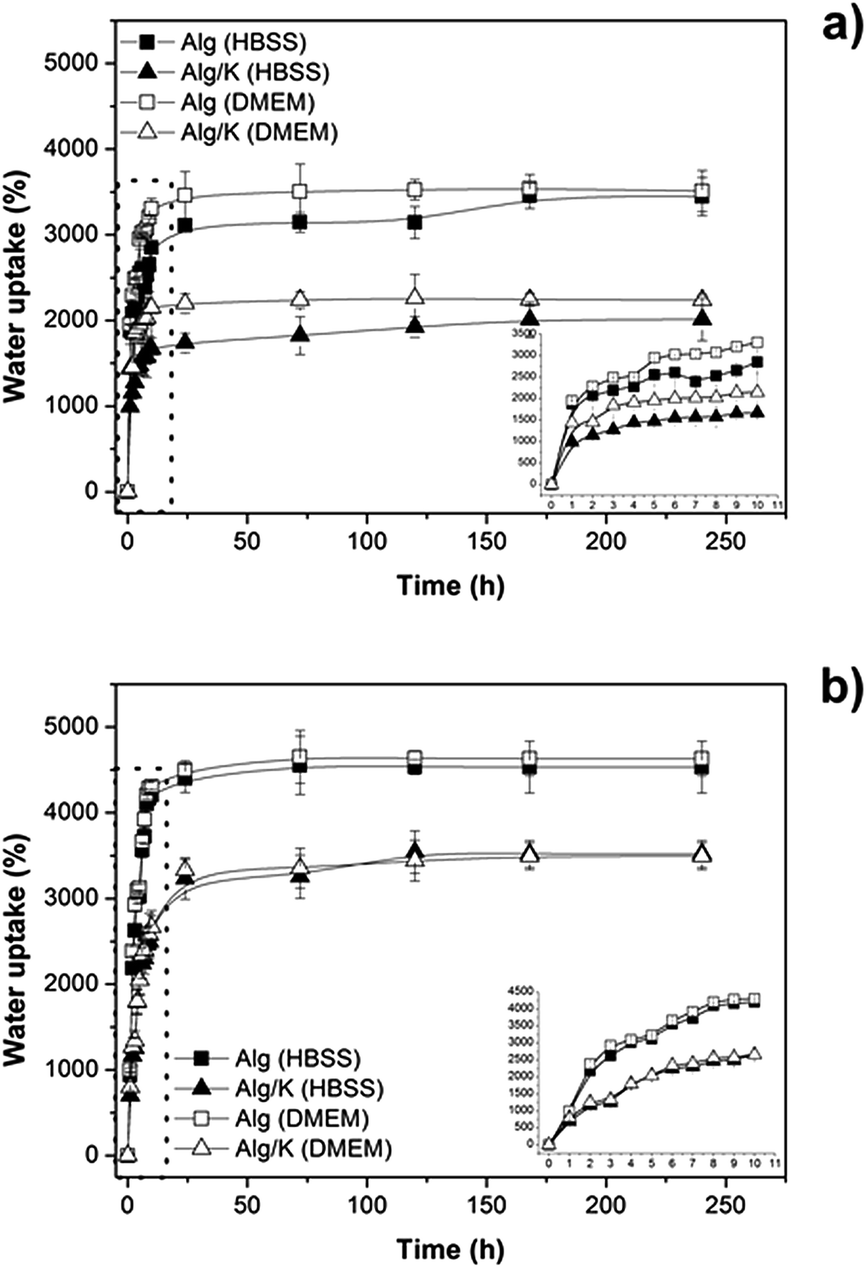 | ||
| Fig. 3 Water uptake studies of hydrogels in two different media (DMEM and HBSS): (a) 2D hydrogels and (b) 3D hydrogels. | ||
The results demonstrate that in both geometries produced the addition of keratin protein leads to a decrease of the amount of water uptake in the presence of either HBSS or DMEM medium (pH 7.4). This fact can be attributed to the diffusion of water inside the network, which decreases due to the electrostatic and hydrophobic interactions that occur between the protein chains and also with alginate molecules. However, comparing the 2D and 3D-hydrogels, the water uptake is higher for the 3D-hydrogels corroborating the results from thermogravimetric analysis, which verified the high content of water in microcapsules. Moreover, no obvious differences were observed for the water uptake analyses in different media. This result indicates the ability of these matrices to transport nutrients inside of the matrix which is extremely important for cell encapsulation purposes. However, it should be taken into account that the observed effect is not a linear relation since an increase of water uptake can also affects negatively the properties of a biomaterial, as previously mentioned, and therefore a balance should be found.55
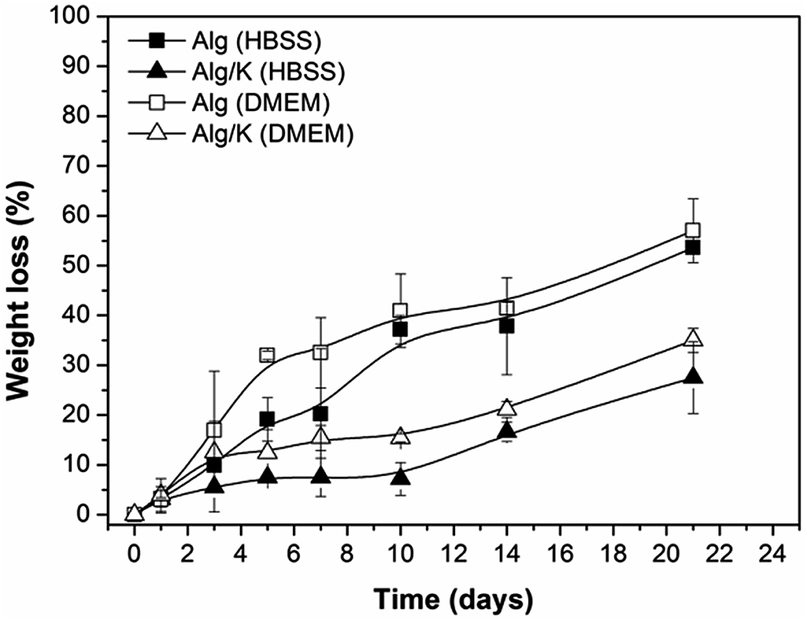 | ||
| Fig. 4 Weight loss measurements of 2D hydrogels (Alg and Alg/K) after 21 days of incubation in HBSS and DMEM. | ||
For a better understanding of the degradation processes of the investigated 2D and 3D hydrogels, the protein release was monitored over time. For that purpose, the analysis of the effect of different solutions (HBSS and DMEM solution at pH 7.4, 37 °C) on the behaviour of keratin release from 2D and 3D-hybrid hydrogels was performed (Fig. 5).
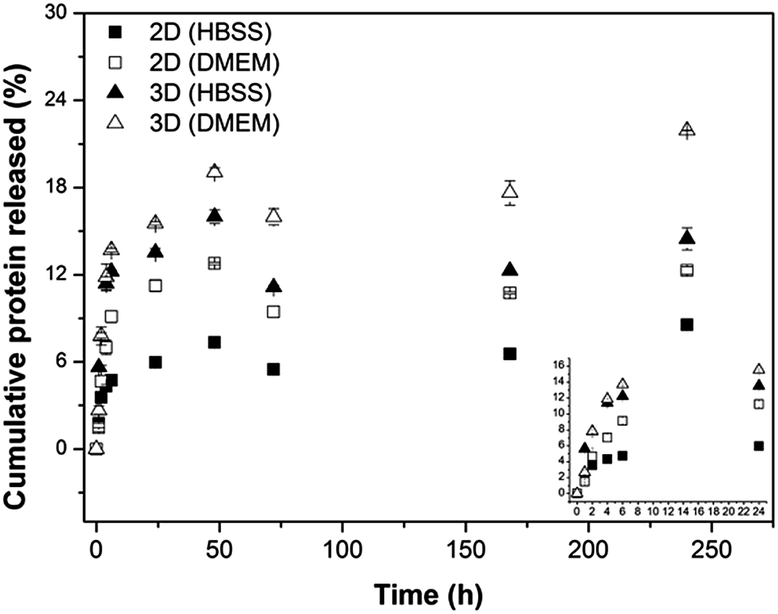 | ||
| Fig. 5 Cumulative keratin release (%) as a function of incubation time in HBSS and in DMEM of the different hydrogels investigated: 2D hydrogels and 3D hydrogels. | ||
For both geometries, the release of protein started immediately after immersion in the medium, with the protein release rate being slowed down with prolonged incubation time. This result can be related to the fact that a crucial molecular force involved in the formation of Alg/K hybrid hydrogels is a hydrophobic interaction, and the high initial protein release may be due to the slight instability of the protein network immediately upon rehydration. Comparing both geometries, the 2D hybrid hydrogels present lower amount of protein released, probably due to the fact that the sonication treatment can promote the stability of the network. Furthermore, the surface area of 3D hydrogels is higher and therefore a higher amount of proteins is released over time. The amount of keratin released from 2D geometries was found to be lower than from 3D hydrogels, indicating that a significant quantity of keratin remained in the material that could enhance cell–material interactions due to the presence of the cell adhesive peptide sequences.
 | ||
| Fig. 6 SEM cross-section images of Alg (a) and Alg/K (b) 2D hydrogels showing the network of 2D hydrogels. | ||
 | ||
| Fig. 7 SEM images of 3D hydrogels: Alg (a), whole and surface of microcapsule and Alg/K (b) whole and surface microcapsule. | ||
Regarding the morphology analysis of the 3D-hydrogels, it was possible to verify that alginate microcapsules exhibit a folded surface structure in regular configurations (Fig. 7). The blend with keratin protein leads to a less folded, smoother surface, indicating a more homogeneous material.
Mechanical properties
DMTA has been shown to be an adequate technique for characterizing the mechanical features of biomaterials under physiological-like conditions. In this study, DMTA was carried out to monitor the variations of viscoelastic properties of the 2D hydrogel upon immersion in DMEM in order to mimic the cell environment. Fig. 8 shows the results obtained by frequency scans from hydrogel samples that were immersed in DMEM for 21 days. | ||
| Fig. 8 DMTA scans of Alg and Alg/K hydrogels immediately after preparation and after 21 days of incubation in DMEM: (a) storage modulus (kPa); (b) tanδ. | ||
A small increase in storage modulus (E′) with the increase of frequency is verified for all samples. The data from Fig. 8 show that the blend with keratin promotes an increase of E′ from ≈100 to ≈400 kPa (evaluated to 10 Hz), indicating that the addition of protein leads to a stiffer material when compared with Alg hydrogels. The damping properties of the materials were also analysed and these data were obtained by monitoring the variation of tanδ with frequency as it is depicted in Fig. 8. tanδ is defined as the loss factor, is an important parameter due to the fact that it can give information about the energy that is being dissipated by viscous mechanisms relative to the energy stored in the elastic component.57 From our data, it is possible to observe that tanδ tends to decrease with the increase in frequency, indicating that the materials became less viscous and more elastic. Furthermore, within the uncertainty tanδ is constant for Alg which implies the formation of a real gel. For Alg/K tanδ is constant at high frequencies (higher gel behaviour) but increases at lower frequencies. This gives some indication of an additional relaxation process due to the presence of the keratin. Moreover, as shown in Fig. 8, tanδ exhibits higher values for Alg hydrogel, indicating that this material possesses higher capacity to dissipate energy in comparison to Alg/K. Additionally, the tanδ values of the Alg hydrogels do not exhibit significant variation over time, suggesting that the damping properties are not dependent on time. However, the presence of keratin results in a decrease of tanδ values after 21 days, which indicates that the hybrid hydrogels dissipate less energy over the storage time.
Cell–material interactions using human endothelial cells
The biocompatibility of tissue-engineered scaffolds is a decisive factor that must be considered prior to any in vivo application. It is therefore critically important to test the cell–material interactions in vitro, in order to verify the suitability of biomatrices for their potential use in humans. Alginate constitutes a matrix with a relatively poor cell compatibility profile.17 To investigate whether the cell–material interactions are improved by the presence of keratin in the hydrogel, the attachment and growth, as well as cell morphology and mitochondrial activity of primary HUVECs were analysed and compared between pure alginate and alginate/keratin blend, in two different geometries.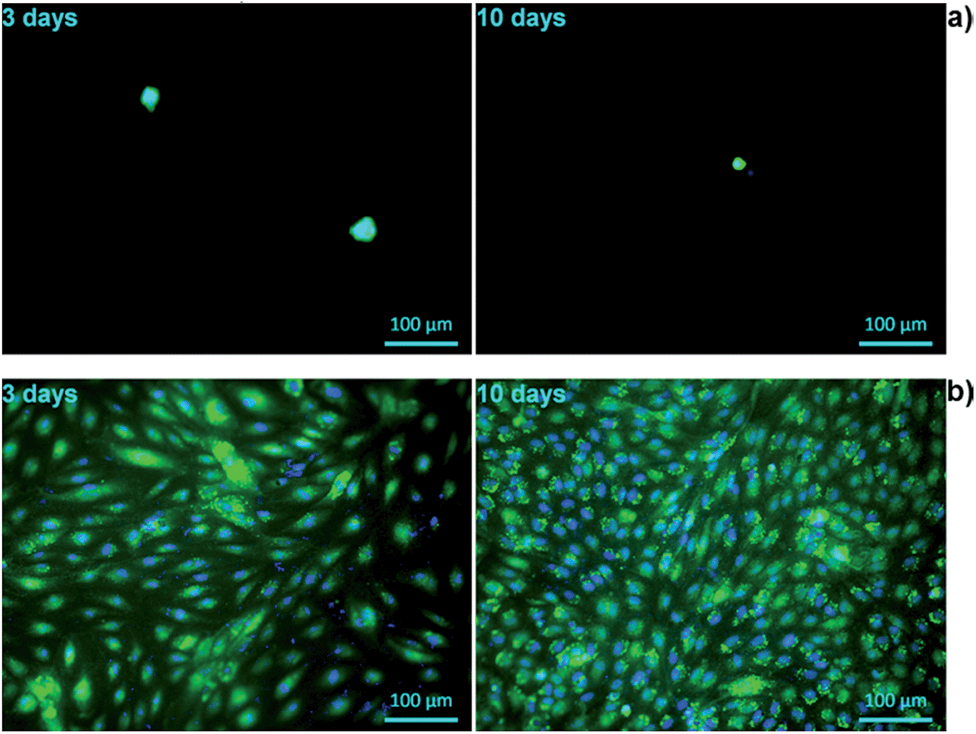 | ||
| Fig. 9 Fluorescent staining with calcein/DAPI of HUVECS on 2D hydrogels after 3 days and after 10 days of cultivation: (a) Alg and (b) Alg/K. The cells were stained for living cells (green) and nuclei (blue). | ||
The viability and morphology of living cells was evaluated using the calcein AM, which stains the cytoplasm of living cells.58 Esterases present in the cytoplasm of the living cells break down calcein, resulting in a florescent green product which is impermeable to cell membranes. Moreover, calcein staining further gives information about the cell shape and membrane integrity which are the hallmarks of the normal cell equilibrium. Furthermore, the cells were also stained with DAPI, a nucleic acid stain, to study the integrity of the nucleus.59 In Fig. 9, a strong reduction in viable cell numbers on alginate hydrogel after 3 days of incubation is shown. The results of morphological evaluation furthermore demonstrated that on hybrid Alg/K hydrogels, significantly more viable cells with intact nuclei and cell membranes were found as compared with pure alginate hydrogels.
After 3 days post-seeding on Alg/K films, the cells were attached and spread showing a typical endothelial morphology with an increase in the number of adherent cells being observed with time. At day 10, the films were completely covered with a dense monolayer of HUVECs. These results suggest that keratin-containing hydrogels are able to provide a suitable anchorage and a growth-supporting environment for endothelial cells. Initial attachment of cells promotes cell survival and plays a role in regulation of cell function and signalling.60,61 Once the endothelial cells are firmly attached, they can spread and proliferate, thus forming a monolayer. This step is critically important for the physiologic function of these cells, as endothelial responsiveness, vitality and barrier-function depend on the cell–cell contacts.62 Moreover, endothelial proliferation is essential for angiogenesis, which is necessary for efficient tissue/organ regeneration in vivo.61
The cell proliferation on 2D hydrogels was analysed by counting of fluorescently stained cell nuclei with Image J software after specific cultivation periods (Fig. 10). However, this analysis was performed only for alginate/keratin 2D hydrogels since it was not possible to count cells on pure alginate due to aggregation, which is shown in the fluorescence images of Fig. 9. The data showed that the cell number increased with cultivation time where a higher number of cells per unit area of hydrogel (cells per mm2) was found.
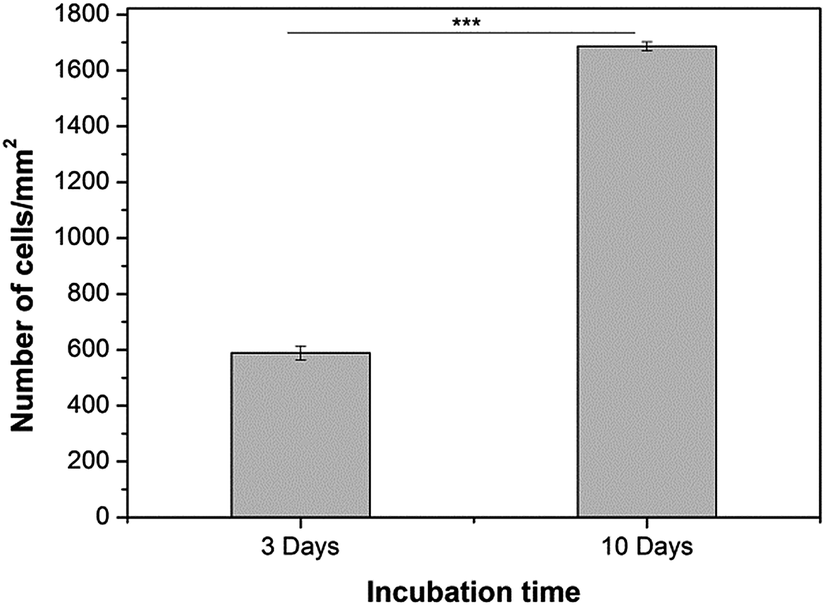 | ||
| Fig. 10 HUVECs proliferation on Alg/K 2D hydrogels after 3 days and 10 days of cultivation. A statistically significant difference is indicated: ***p < 0.001 (Bonferroni's post-hoc). | ||
Furthermore, to visualize cell morphology and cell spreading on to the hydrogels, actin cytoskeleton staining of the cells was performed using rhodamine phalloidin. Cell morphology plays an important role in the general cell hemostasis, and can be an early indicator of apoptotic responsiveness,63,64 or growth arrest.58 The F-actin cytoskeleton staining showed that cells were having typical cobblestone-like endothelial morphology on Alg/K and, with increase in time, an increase in cell number was observed as shown in the Fig. 11. As expected, on the Alg hydrogel, there were fewer cells as compared to the Alg/K hydrogels at day 3 and day 10. Moreover, cellular morphology was disturbed and typical endothelial morphology was not observed (Fig. 11), which is in accordance with the results observed by calcein staining.
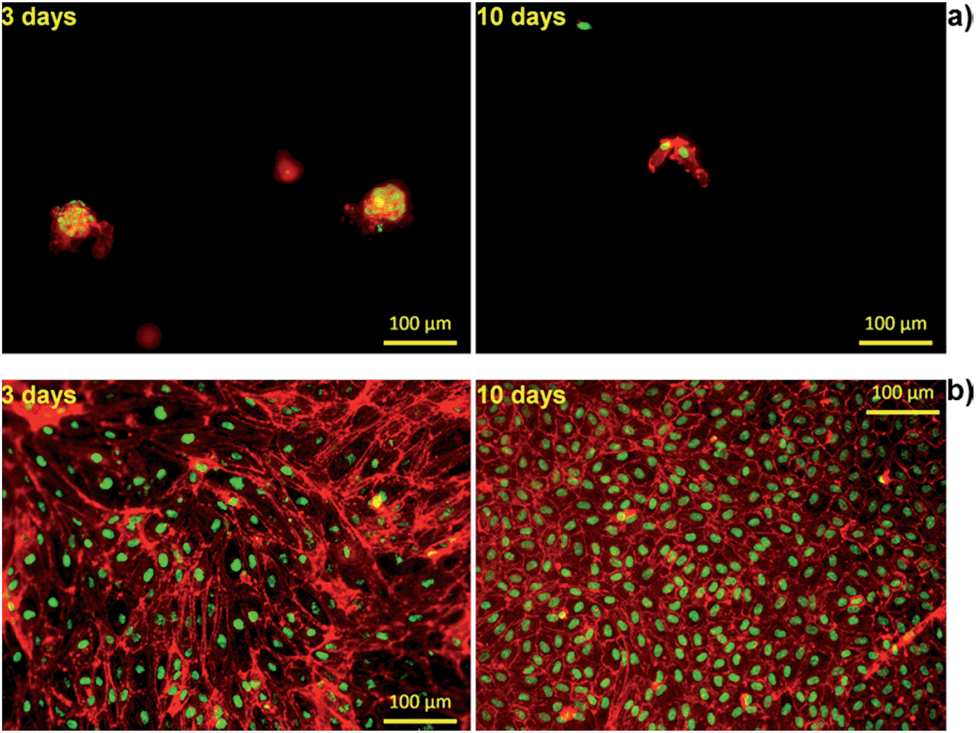 | ||
| Fig. 11 Fluorescent staining with Phalloidin/Sytox of HUVECSs on 2D hydrogels after 3 days and after 10 days of cultivation: (a) Alg and (b) Alg/K. The cells were stained for F-actin (red) and nuclei (green). | ||
In the case of the cell-encapsulation into the Alg/K microcapsules, fluorescent analyses demonstrated that the cells were able to grow and proliferate after four weeks of incubation (Fig. 12). This fact was not observed when the cells were encapsulated in alginate, proving the fact that the use of keratin, which provides cell adhesion motifs, enhances cell–material interaction.
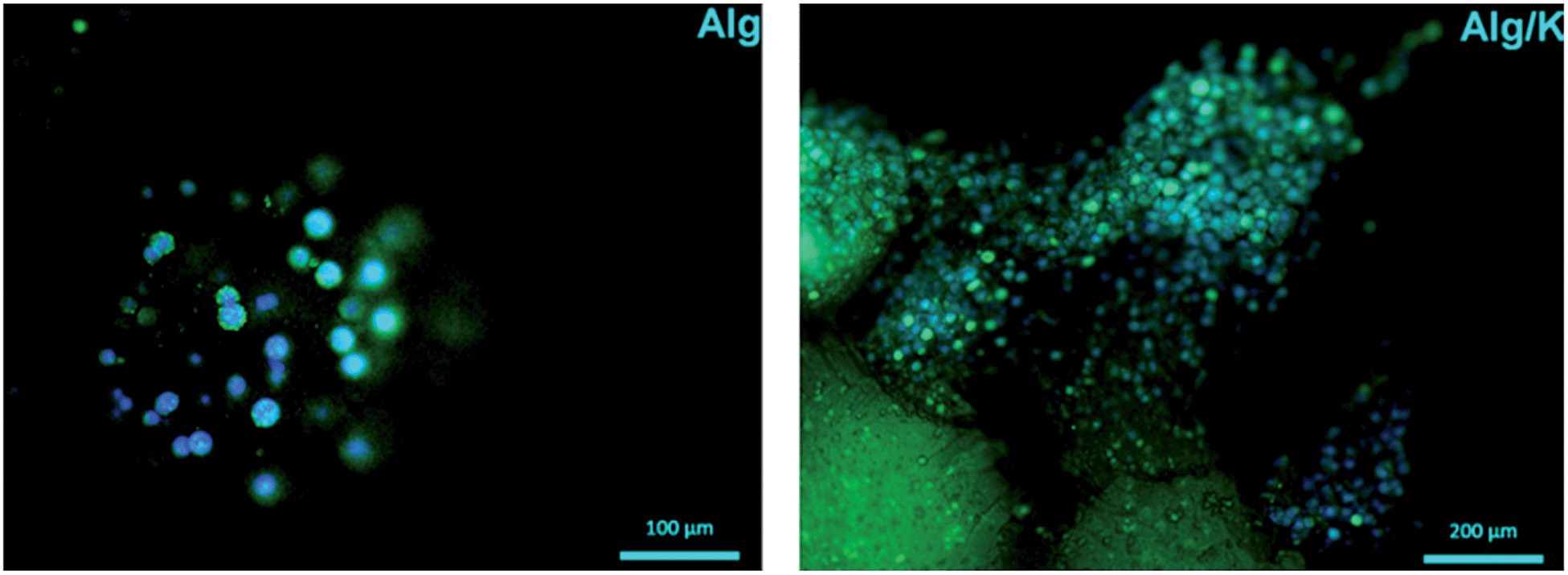 | ||
| Fig. 12 Fluorescent staining with calcein/DAPI of HUVECs into 3D hydrogels after 4 weeks of cultivation. The cells were stained for live cells (green) and nucleus (blue). | ||
Compared with the 2D geometry, where the typical HUVEC morphology was observed, HUVECs grown in the microcapsules showed more rounded cell bodies, with less spread phenotype. This may suggest that by encapsulation, an atypical mechanical load acting on the cells is created, limiting their full spreading ability and migration out of the microcapsules.
Gathering the results achieved, it is possible to conclude that the geometry of the biomaterial is an important parameter to take in consideration with relation to the type of application and the target cells.
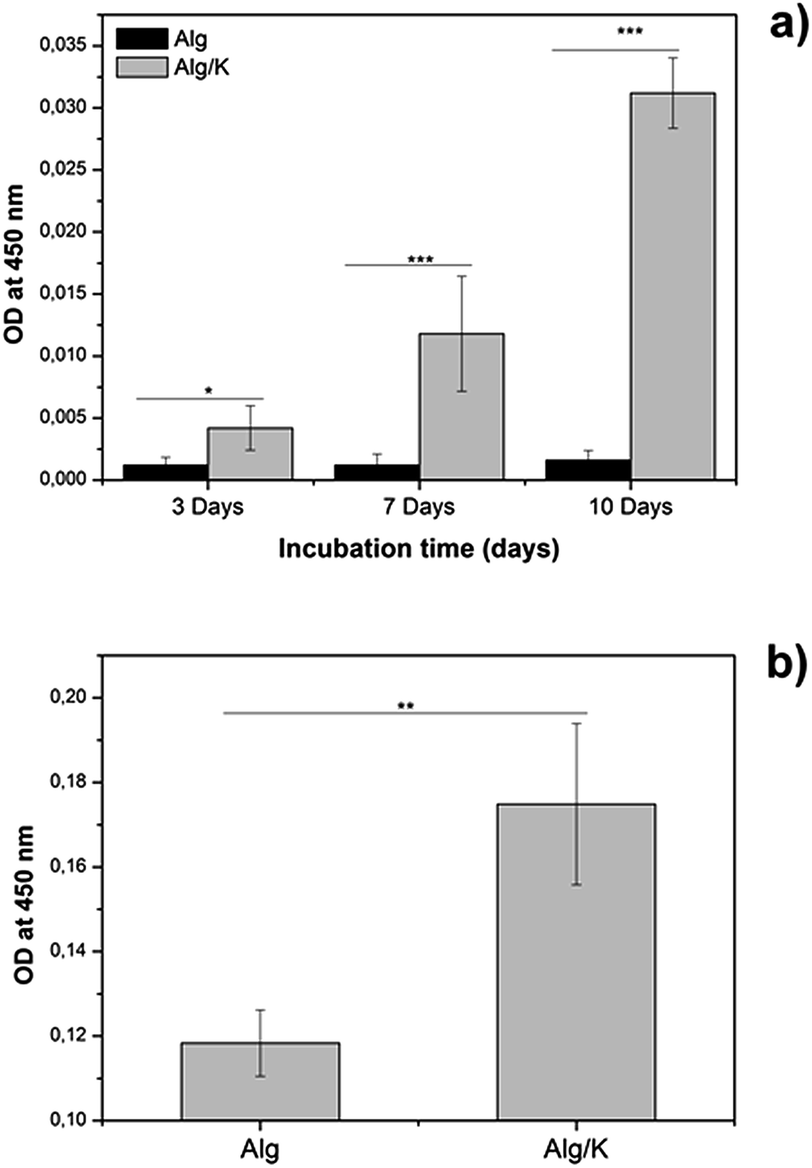 | ||
| Fig. 13 Mitochondrial activity of HUVECs on 2D (a) at different times of cultivation, and 3D (b) after 4 weeks of cultivation. Statistically significant differences are indicated comparing with Alg hydrogels in each time point: *p < 0.05, **p < 0.01 and ***p < 0.001 (Bonferroni's post-hoc test). | ||
In the metabolically active cells, tetrazolium compound is metabolized in the mitochondria and the product, formazan, is secreted into the medium, resulting in change of the colour which is measured spectrophotometrically. In the case of 2D hydrogels, after 3 days of incubation, the mitochondrial activity of HUVECs grown on hybrid hydrogels was significantly higher compared to those grown on alginate. Similar results were obtained for the HUVECs encapsulated into hybrid hydrogels, where a significant increase in the mitochondrial activity of the cells was observed. These results are in accordance with the results of the fluorescence staining of HUVECs which are presented in Fig. 9, 11 and 12. Our findings indicate that hydrogels composed of Alg/K blend possess superior endothelial compatibility and constitute a suitable matrix for example for applications in vascular tissue regeneration.
Conclusions
This study focused on the development and production of two different geometries of hybrid hydrogels based on a novel blend between polysaccharide (alginate) and protein (keratin). The hybrid hydrogel was successfully produced in 2D and 3D forms and an extensive physico-chemical characterization was carried out. Furthermore, cell studies performed with primary HUVECs demonstrate the ability of these hybrid hydrogels to promote cell attachment, proliferation, spreading and viability. The cells seeded on the surface of 2D hydrogels remained viable over 10 days of culture, forming a monolayer and demonstrating the typical cobblestone-like endothelial morphology. The encapsulated cells were viable over 4 weeks of cultivation. Within this culture time the number and mitochondrial activity of cells were increased and cells began to spread.Thus, the developed alginate/keratin hybrid hydrogels represent a promising candidate biomaterial for tissue engineering and regenerative medicine applications.
Acknowledgements
This study was supported by the Emerging Fields project “TOPbiomat” of the University of Erlangen-Nuremberg (Germany). The authors thank Nina Jaziri for help with HUVEC isolation and Dr. Menti Goudouri for FTIR measurements.Notes and references
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- B. V. Slaughter, S. S. Khurshid, O. Z. Fisher, A. Khademhosseini and N. A. Peppas, Adv. Mater., 2009, 21, 3307–3329 CrossRef CAS PubMed.
- S. Varghese and J. Elisseeff, Polym. Regen. Med., 2006, 95–144 CAS.
- N. C. Hunt and L. M. Grover, Biotechnol. Lett., 2010, 32, 733–742 CrossRef CAS PubMed.
- B. Balakrishnan and R. Banerjee, Tissue Engineering, Chem. Rev., 2011, 111, 4453–4474 CrossRef CAS PubMed.
- B. Balakrishnan, M. Mohanty, P. R. Umashankar and A. Jayakrishnan, Biomaterials, 2005, 26, 6335–6342 CrossRef CAS PubMed.
- J. Park, Med. Oral Patol. Oral Cir. Bucal., 2011, 16, e115–e118 CrossRef.
- J. Elisseeff, C. Puleo, F. Yang and B. Sharma, Orthod. Craniofac. Res., 2005, 8, 150–161 CrossRef CAS PubMed.
- Q. Zeng, Y. Han, H. Li and J. Chang, J. Biomed. Mater. Res., Part B, 2013, 1–10 Search PubMed.
- A. D. Augst, H. J. Kong and D. J. Mooney, Macromol. Biosci., 2006, 6, 623–633 CrossRef CAS PubMed.
- H.-B. Li, H. Jiang, C.-Y. Wang, C. M. Duan, Y. Ye, X.-P. Su, Q.-X. Kong, J.-F. Wu and X.-M. Guo, Biomed. Mater., 2006, 1, 42–47 CrossRef CAS PubMed.
- S.-J. Song, J. Choi, Y.-D. Park, S. Hong, J. J. Lee, C. B. Ahn, H. Choi and K. Sun, Artif. Organs, 2011, 35, 1132–1136 CrossRef PubMed.
- C. Gao, M. Liu, J. Chen and X. Zhang, Polym. Degrad. Stab., 2009, 94, 1405–1410 CrossRef CAS PubMed.
- L. Wang and J. Shansky, Tissue Eng., Part A, 2012, 18, 2000–2007 CrossRef CAS PubMed.
- A. Leal-Egaña, U.-D. Braumann, A. Díaz-Cuenca, M. Nowicki and A. Bader, J. Nanobiotechnol., 2011, 9, 24 CrossRef PubMed.
- F. P. W. Melchels, M. A. N. Domingos, T. J. Klein, J. Malda, P. J. Bartolo and D. W. Hutmacher, Prog. Polym. Sci., 2012, 37, 1079–1104 CrossRef CAS PubMed.
- B. Sarker, D. G. Papageorgiou, R. Silva, T. Zehnder, F. Gul-E-Noor, M. Bertmer, J. Kaschta, K. Chrissafis, R. Detsch and A. R. Boccaccini, J. Mater. Chem. B, 2014, 2, 1470–1482 RSC.
- A. Aluigi, M. Zoccola, C. Vineis, C. Tonin, F. Ferrero and M. Canetti, Biol. Macromol., 2007, 41, 266–273 CrossRef CAS PubMed.
- T. Aboushwareb, D. Eberli, C. Ward, C. Broda, J. Holcomb, A. Atala and M. Van Dyke, J. Biomed. Mater. Res., Part B, 2009, 90, 45–54 Search PubMed.
- B. Srinivasan, R. Kumar, K. Shanmugam, U. T. Sivagnam, N. P. Reddy and P. K. Sehgal, J. Biomed. Mater. Res., Part B, 2010, 92, 5–12 CrossRef PubMed.
- S. Balaji, R. Kumar, R. Sripriya, U. Rao, A. Mandal, P. Kakkar, P. N. Reddy and P. K. Sehgal, Polym. Adv. Technol., 2012, 23, 500–507 CrossRef CAS.
- P. Hill, H. Brantley and M. Van Dyke, Biomaterials, 2010, 31, 585–593 CrossRef CAS PubMed.
- J. A. Rippon, The structure of wool in Wool dyeing, Society of Dyers and Colourists, 1992, pp. 1–52 Search PubMed.
- W. Astbury and H. Woods, Philos. Trans. R. Soc. London, 1934, 232, 333–394 CrossRef.
- L. Pauling and R. B. Corey, Proc. Natl. Acad. Sci. U. S. A., 1951, 37, 729–740 CrossRef CAS.
- L. Pauling and R. B. Corey, Nature, 1953, 171, 59–61 CrossRef CAS.
- A. Tachibana, Y. Furuta, H. Takeshima, T. Tanabe and K. Yamauchi, J. Biotechnol., 2002, 93, 165–170 CrossRef CAS.
- V. Verma, P. Verma, P. Ray and A. R. Ray, Biomed. Mater., 2008, 3, 025007 CrossRef PubMed.
- M. E. Furth, A. Atala and M. E. Van Dyke, Biomaterials, 2007, 28, 5068–5073 CrossRef CAS PubMed.
- M. J. Humphries, A. Komoriya, S. K. Akiyama, K. Olden and K. M. Yamada, J. Biol. Chem., 1987, 262, 6886–6892 CAS.
- S. Hamasaki, A. Tachibana, D. Tada, K. Yamauchi and T. Tanabe, Mater. Sci. Eng., C, 2008, 28, 1250–1254 CrossRef CAS PubMed.
- L. A. Michaelis, J. Biol. Chem., 1934, 106, 605–614 Search PubMed.
- J. Maclaren, Aust. J. Chem., 1962, 824–831 CrossRef CAS.
- I. O'Donnell and E. Thompson, Aust. J. Biol. Sci., 1969, 22, 471–488 CrossRef.
- C. Earland and C. Knight, Biochim. Biophys. Acta, 1956, 9, 405–411 CrossRef.
- J. H. Buchanan, Biochem. J., 1977, 167, 489–491 CAS.
- G. L. Ellman, Archives of Biochemistry and Biophysics, 1959, 82, 70–77 CrossRef CAS.
- R. Silva, H. Ferreira, C. Little and A. Cavaco-Paulo, Ultrason. Sonochem., 2010, 17, 628–632 CrossRef CAS PubMed.
- O. C. Lowry and N. Rosebrough, J. Biol. Chem., 1951, 193, 265–275 CAS.
- K. Katoh, M. Shibayama, T. Tanabe and K. Yamauchi, Biomaterials, 2004, 25, 2265–2272 CrossRef CAS PubMed.
- M. Feughelman, Keratin in Encyclopedia of Polymer Science and Engineering, Wiley, New York, 1985, pp. 566–600 Search PubMed.
- K. Katoh, T. Tanabe and K. Yamauchi, Biomaterials, 2004, 25, 4255–4262 CrossRef CAS PubMed.
- J. Ming and B. Zuo, Polym. Eng. Sci., 2013, 1–8 Search PubMed.
- P. I. Haris and F. Severcan, J. Mol. Catal. B: Enzym., 1999, 7, 207–221 CrossRef CAS.
- K. K. Chittur, Biomaterials, 1998, 19, 357–369 CrossRef CAS.
- A. Dong, P. Huang and W. S. Caughey, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS.
- D. M. Byler and H. Susi, Biopolymers, 1986, 25, 469–487 CrossRef CAS PubMed.
- T. J. Mason, Ultrason. Sonochem., 2007, 14, 476–483 CrossRef CAS PubMed.
- J. O. S. M. J. Paulusse and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5445–5453 CrossRef CAS.
- J. Rae, M. Ashokkumar, O. Eulaerts, C. Von Sonntag, J. Reisse and F. Grieser, Ultrason. Sonochem., 2005, 12, 325–329 CrossRef CAS PubMed.
- J. H. Bang and K. S. Suslick, Adv. Mater., 2010, 22, 1039–1059 CrossRef CAS PubMed.
- K. S. Suslick and M. W. Grinstaff, Protein microencapsulation of nonaqueous liquids, J. Am. Chem. Soc., 1990, 112, 7807–7809 CrossRef CAS.
- S. Suslick and M. Fang, Acoustic cavitation and its chemical consequences, 1999, pp. 335–353 Search PubMed.
- M. Boguń and A. Łącz, J. Therm. Anal. Calorim., 2011, 106, 953–963 CrossRef PubMed.
- W. L. Tham, W. S. Chow and Z. A. Mohad Ishak, eXPRESS Polym. Lett., 2010, 4, 517–528 CrossRef CAS.
- C. Zhou and H. Ma, J. Agric. Food Chem., 2006, 54, 2223–2228 CrossRef CAS PubMed.
- S. Caridade, E. G. Merino, N. M. Alves, V. de Zea Bermudez, A. R. Boccaccini and J. F. Mano, J. Mech. Behav. Biomed. Mater., 2012, 20, 173–183 CrossRef PubMed.
- C. Joshi, B. Karumuri, J. J. Newman and M. A. Decoster, Curr. Microsc. Contrib. to Adv. Sci. Technol., ed. A. Méndez-Vilas, 2012, pp. 756–762 Search PubMed.
- Y. A. Ioannou and F. W. Chen, Nucleic Acids Res., 1996, 24, 992–993 CrossRef CAS PubMed.
- S. M. Short, G. A. Talbott, R. L. Juliano, N. Carolina, C. Hill and N. Carolina, Mol. Bio. Cell., 1998, 9, 1969–1980 CAS.
- E. Chavakis and S. Dimmeler, J. Am. Heart Assoc., 2002, 22, 887–893 CAS.
- T. Korff and H. G. Augustin, J. Cell Biol., 1998, 143, 1341–1352 CrossRef CAS.
- J. E. Leadsham, V. N. Kotiadis, D. J. Tarrant and C. W. Gourlay, Cell Death Differ., 2009, 17, 754–762 CrossRef PubMed.
- C. W. Gourlay and R. K. Ayscough, Nat. Rev. Mol. Cell Biol., 2005, 6, 583–589 CrossRef CAS PubMed.
- S. Kidoaki and H. Sakashita, PLoS One, 2013, 8, 1–10 Search PubMed.
- Y. Wen, H. Wang, S.-h. Kho, S. Rinkiko, X. Sheng, H.-m. Shen and Y.-z. Zhu, PLoS One, 2013, 8, 1–10 Search PubMed.
| This journal is © The Royal Society of Chemistry 2014 |