 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioinspired silica as drug delivery systems and their biocompatibility†
Christopher R.
Steven‡
ab,
Grahame A.
Busby‡
ac,
Craig
Mather
a,
Balal
Tariq
ac,
Maria Lucia
Briuglia
d,
Dimitrios A.
Lamprou
d,
Andrew J.
Urquhart§
*d,
M. Helen
Grant
*c and
Siddharth V.
Patwardhan
*a
aChemical and Process Engineering, University of Strathclyde, 75 Montrose Street, Glasgow, G1 1XJ, UK. E-mail: Siddharth.Patwardhan@strath.ac.uk
bPure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK
cDepartment of Biomedical Engineering, University of Strathclyde, 106 Rottenrow, Glasgow, G4 0NW, UK. E-mail: m.h.grant@strath.ac.uk
dStrathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, 161 Cathedral Street, Glasgow, G4 0RE, UK
First published on 27th June 2014
Abstract
Silica nanoparticles have been shown to have great potential as drug delivery systems (DDS), however, their fabrication often involves harsh chemicals and energy intensive laborious methods. This work details the employment of a bioinspired “green” method for the controlled synthesis of silica, use of the products to entrap and release drug molecules and their cytotoxicity in order to develop novel DDS. Bioinspired silica synthesis occurs at pH 7, room temperature and in less than 5 minutes, resulting in a rapid, cheaper and greener route. Drugs were loaded into silica during the silica formation, thus allowing a one step and one pot method for simultaneous silica synthesis and drug loading. We established that the drug release profile can be modulated by synthetic parameters, which can allow design of tailored DDS. A systematic investigation using a two level factorial design was adopted in order to identify the key synthetic parameters and quantify their effects on silica formation, drug loading and drug release. The observation that these new DDS are considerably less cytotoxic than their current counterparts, and exhibit additional benefits such as green synthesis and ease of functionalization, strengthens the argument for their future use in DDS and other biomedical applications.
1. Introduction
The nanotechnology market is expanding rapidly,1 however, understanding of the toxicity of nanostructured materials has started to emerge only recently due to their biomedical use such as in drug delivery systems (DDS). Nanomaterial based biomedical devices, such as DDS, are of growing importance in effective clinical treatment2 and cover a wide range of chemistries.3 Despite the extensive efforts on nanomaterial based DDS over decades, a relatively small number have been approved and are under the clinical trials stage for clinical use.4 Silica based nanomaterials as DDS have attracted considerable attention due to their ability to control drug loading, porosity and surface chemistry (through functionalisation).5 Silica has been approved by the US Food and Drug Administration as “Generally Recognised As Safe” and by the EU for its use in cosmetics and food additives.4,6 Furthermore, syntheses of a range of distinct types of silica are well-established.4,6–8 Finally, silica has excellent biodegradation properties9 and dissolved silica has been suggested as beneficial to bones.10Most of the studies use amorphous xerogels, fumed silica nanoparticles or mesoporous silica such as MCM-41 or SBA-15. In particular mesoporous silica nanoparticles (MSN) have a relatively large pore volume, a unique mesoporous structure and provide a high surface area for drug molecule adsorption, as a result they have been of wide interest in DDS.4,11 For example, MSN loaded with the anticancer drug doxorubicin (DOX) was administered to mice and caused a significant reduction in mean tumour weight, beyond the effect observed with free DOX treatment.12,13 However, since the drug was loaded on silica by physisorption post-synthesis, it limits any control of sustained release parameters. Alternatively, post-synthesis functionalization can be used to modulate the drug release but this severely has impact on other aspects such as the pore volume of the silica used14 and increased complexity in the synthesis of such DDS. Typical strategies to offer controlled release, and, in some cases, stimuli responsive release, involve capping of MSN pores. Song et al. used mesoporous silica (SBA-15) which can be functionalized by amine surface groups prior to loading of bovine serum albumin (BSA) into the silica followed by encapsulation in polyacrylic acid.15 Another study used MCM-41 to load vancomycin and adenosine triphosphate (ATP) and capped the pores with toxic cadmium sulphide (CdS) to trap drugs thus avoiding unwanted drug release.16 Although the strategies discussed above can produce effective DDS, they typically involve two to four steps in the fabrication of DDS: silica synthesis, surface functionalisation of silica, drug loading and capping of the pores. Recently, a one-step synthesis of silica gels for in situ drug encapsulation was investigated where propanolol and persantin were dissolved with the silica precursor and the fabrication required 24 hours and acidic pH (0.5–5.5).17
The current use of silica in this way has some limitations, however. Silica, in particular, MSN tend to be synthesised under harsh conditions (elevated temperature, high pressure, strongly alkaline or acidic solutions), with multistep preparations taking several days. Furthermore, they require either a high temperature (∼600 °C) calcination step or template extraction using concentrated acid prior to loading a drug molecule resulting in additional cost, time and complexity in fabricating DDS.18 With limited flexibility, the synthesis of the mesoporous silica remains difficult to manipulate, and control over the drug release rate is difficult to achieve. The release of any hydrophilic drug appears very rapid but controlling this release seems more of an issue. Burst release from the silica will confer no advantage over free drug. For example, one study showed that the control of the pore size to slow drug release19 also reduced the drug loading by leaving only a tortuous path into the matrix for the drug molecules.20 Clearly, the fabrication of silica DDS is an area warranting further investigation where milder and simpler routes can be established for DDS preparation, while also offering control over physical properties of DDS which in turn controls the drug loading and release. Furthermore, confusion regarding their safe use is widespread, especially because the interactions at the nano-bio interface are unknown.21 For example, MSN have been shown to be toxic, especially above 25 μg ml−1 concentrations for submicron sized particles.4,22 Although the risk of significant reduction in cell viability in the presence of MSN can be minimised by functionalisation, this is known to lead to complex cell responses.23 It is therefore of utmost importance to carefully and thoroughly investigate the biocompatibility and any toxic effects of nanomaterials that are intended for use in biomedical applications. Taking all these factors into consideration, the potential negative impact can outweigh the benefits MSN can offer.
In recent years, there has been significant growth in the research on biological and biologically inspired silica formation.24–27 For example, researchers have probed mechanisms of biosilica formation and it has become apparent that certain biomolecules (typically rich in amines) play a crucial role in biosilica deposition, where the chemistry and architecture of these biomolecules are important. Learning from nature, in vitro experiments on silica formation have developed bioinspired green routes to nanostructured and porous silica by utilising a wide variety of “additives” (analogues of biomolecules).24,28 Green nanosilica (GN) synthesis is far safer, routinely performed on bench-top at room temperature in water and takes about 5 minutes. A large number of reports have recently discussed the advantages of employing this bioinspired route in the synthesis of silica (extensively reviewed in ref. 24). Furthermore, control over properties of GN, including particle size and porosity, is possible.29 This control in green synthesis can be readily achieved by the ‘correct’ choice of an additive and synthesis conditions. It is clear that the utilisation of this approach for the fabrication of silica nanoparticles as DDS has the potential to offer a one step, “green” synthesis in contrast to the time, energy and material intensive methods which are detailed above for traditional materials.24,28–30
The bioinspired silica synthesis has been employed to support or encapsulate quantum dots,31 catalysts,32 enzymes33–35 and fluorescent molecules,36,37 however, there appears to be only one literature report on drug delivery.38 This report immobilised a dual-function protein which was able to both precipitate silica and act as an anti-cancer agent. The release of protein from the silica was thought to be achieved by the hydrolysis of the silica giving a sustained release. The one step fabrication relied upon the dual functionality of the protein used, however, this approach may not be applicable to a broad set of drug molecules and thus further investigations are warranted.
In this work, we aim to further explore the possibility of using GN for DDS by systematically and extensively focusing on the synthesis of the DDS with a view to understand their fabrication, drug loading and drug release. Since the method presented here achieves drug loading while silica formation occurs, controlling silica chemistry in the presence of the drug is of great importance in order to keep both the drug and silica minimally affected, while simultaneously allowing the drug to be released when desired. In this report we aim to (1) establish the use of GN as DDS and understand the drug release mechanisms using a model drug-like compound (Sections 3.1 and 3.2); (2) use design of experiments to identify key synthetic conditions and quantify their effects on DDS fabrication and drug release (Section 3.3); and (3) apply GN DDS for loading and releasing several therapeutically useful drugs (Section 3.4). We note that the toxicity of bioinspired silica has not been previously reported despite their proven potential in drug delivery, biocatalysis, carbon capture and environmental decontamination.39–42 Therefore, we also aim to (4) investigate cytotoxicity of bioinspired silica (Section 3.5). Drug delivery and biocompatibility in biological systems form part of future investigations.
2. Experimental
2.1 Chemical reagents
The following chemical reagents were used and are listed along with their indicated purity, where available, and supplier. Sodium metasilicate (Na2SiO3·5H2O, ≥95%, Sigma Aldrich), tetraethylorthosilicate (TEOS, Sigma-Aldrich), 3-aminopropyl-trimethoxysilane (APTMOS, Sigma-Aldrich), 2-(4-chlorosulphonylphenyl)ethyltrimethoxysilane (CSTMOS, Fluorochem), cetyltrimethylammonium bromide (CTAB, Sigma-Aldrich) calcein (Sigma Aldrich), Nile red (technical grade, Sigma Aldrich), diethylenetetramine DETA (99%, Sigma Aldrich), triethylenepentamine TEPA (technical grade, Sigma Aldrich), pentaethylenehexamine PEHA (technical grade, Sigma Aldrich), polyallylamine hydrochloride PAH (15 kDa, ≥95%, Sigma Aldrich), 1 M HCl (0.950–1.050 M, Sigma Aldrich), and phosphate buffered saline (PBS) tablets (137 mM NaCl, 2.7 mM KCl and 10 mM phosphate buffer solution, pH 7.4 at 25 °C, Sigma Aldrich). Pindolol, atenolol and ibuprofen were purchased from Sigma Aldrich. Pindolol is a nonselective beta blocker with partial beta-adrenergic receptor agonist activity, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 1.75, pKa = 9.25, λ = 210 nm, water solubility = 7.8 mg ml−1. Atenolol is a selective beta1 receptor antagonist, which is used for the treatment in cardiovascular disease, logP = 0.16, pKa = 9.6, λ = 230 nm, water solubility = 13.5 mg ml−1. Ibuprofen (sodium salt) is an NSAID (non-steroidal anti-inflammatory drug) that is commonly used for the relief of symptoms of arthritis, fever, primary dysmenorrhea, and as an analgesic, logP = 3.9, pKa = 4.9, λ = 214 nm, water solubility = 100 mg ml−1. The chemical structures of the additives used in silica formation and the drug/drug-like molecules are shown in Scheme 1.
P = 1.75, pKa = 9.25, λ = 210 nm, water solubility = 7.8 mg ml−1. Atenolol is a selective beta1 receptor antagonist, which is used for the treatment in cardiovascular disease, logP = 0.16, pKa = 9.6, λ = 230 nm, water solubility = 13.5 mg ml−1. Ibuprofen (sodium salt) is an NSAID (non-steroidal anti-inflammatory drug) that is commonly used for the relief of symptoms of arthritis, fever, primary dysmenorrhea, and as an analgesic, logP = 3.9, pKa = 4.9, λ = 214 nm, water solubility = 100 mg ml−1. The chemical structures of the additives used in silica formation and the drug/drug-like molecules are shown in Scheme 1.
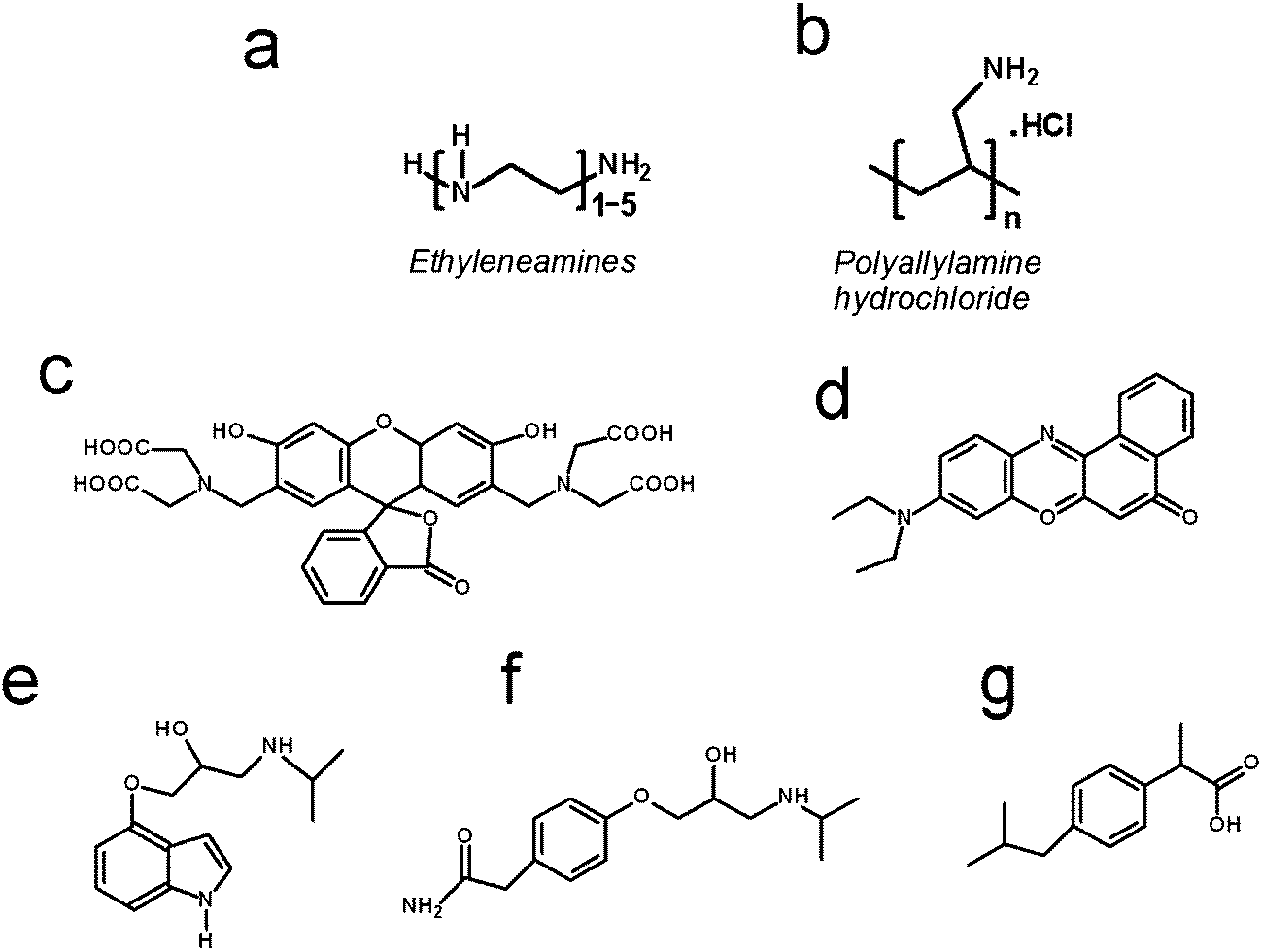 | ||
| Scheme 1 Chemical structures of the additives used in silica formation (a and b). For ethyleneamines, n = 2 is DETA, n = 4 is TEPA and n = 5 is PEHA. Chemical structures of (c) calcein, (d) nile red, (e) pindolol, (f) atenolol and (g) ibuprofen. | ||
2.2 Silica synthesis
A co-condensation reaction was used in the synthesis of basic aminopropyl or sulfonic acid functionalised MSN producing APMSN or SAMSN respectively. TEOS was used as a silica precursor. Silane coupling agents CSTMOS and APTMOS were used to produce SAMSN and APMSN respectively. The surfactant used was CTAB. The first stage of this method is to heat 480 ml of DI water to 80 °C followed by the addition of CTAB and 2 M NaOH (7 ml of water, 14 mmol of NaOH), and the mixture was stirred constantly at 80 °C. After 30 minutes, 10 ml TEOS (51.4 mmol) was added to the solution followed by immediate addition of either 1 mmol of CSTMOS (0.237 ml) for SAMSN synthesis, or 1 mmol of APTMOS (0.18 ml) for APMSN synthesis. The solution remained at 80 °C and was vigorously stirred for 2 hours. To separate the synthesised material, the reaction mixture was poured into the filter funnel whilst still hot and was washed with copious amounts of methanol. The solid obtained on the filter paper was dried in a vacuum oven for approximately 8 hours at 80 °C.
After vacuum drying, the surfactant CTAB was required to be removed by methanol extraction. To pre-heated 100 ml methanol (60 °C) 1 g of solid MSN was added with vigorous stirring. 1 ml concentrated hydrochloric acid was then added and the solution was vigorously stirred at 60 °C for 6 hours. The solid with the surfactant removed was obtained through hot filtration and again was washed with a copious amount of methanol. The solid collected was then vacuum dried for approximately 8 hours at 90 °C. The complete removal of CTAB was confirmed by FTIR analysis.
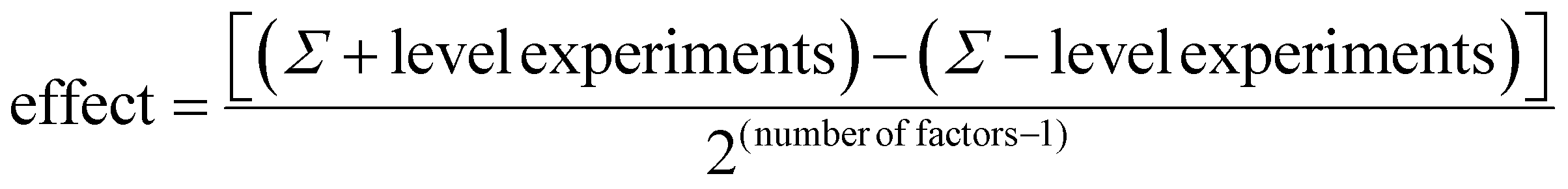 | (1) |
It was intended to use a two tailed Student's t-test to test the hypothesis that both populations of the plus and minus levels had the same mean. Unfortunately due to the splitting of the population at each level by the other factors in the experiments, a large variance was obtained causing the t-test to predict that effects were less significant than they were in practice. Thus, in its place we used the result from eqn (1) along with a 95% confidence interval (CI) to estimate whether the factor was significant. A factor which caused an effect greater than either CI was likely to have significant influence on the process. In contrast, one which gave a result less than the CI was not likely to have significant influence, with any observed change due to natural variation in one, or both, factors.
2.3 Materials characterisation
Dried samples were mounted on SEM sample holders with double sided sticky carbon tape. Upon gold coating by sputtering, samples were analysed on a HITACHI SU-6600 Field Emission Scanning Electron Microscope (FE-SEM) at 10 kV. The measurement of the ζ-potential of silica particles was performed using a Zetasizer Nano ZS from Malvern Instruments using a disposable capillary cell. Typically, 1 ml aliquots of each sample were injected into the capillary cell and 5–10 measurements per sample were performed at 20 °C. Prior to analysis, silica particles were suspended in water by sonication.Thermo-gravimetric analysis (TGA) was used in order to determine the organic content of the samples. Samples were subjected to temperatures from 25–900 °C under a nitrogen atmosphere with the temperature increasing at a rate of 10 °C min−1. Porosity measurements were performed by nitrogen adsorption using a Micromeritics ASAP 2420 porosimeter on samples degassed for 6 h at 200 °C. The data were processed using the BET model to yield surface areas while the BJH model was adopted for obtaining pore sizes and pore volumes. Infrared spectra were obtained in the 4000–400 cm−1 region with a resolution of 4 cm−1, by accumulating 32 scans using an attenuated total reflectance (diamond with ZnSe lens reflection ATR plate) Fourier transform infrared (ABB, MB3000 FTIR) spectrometer. The presence of ordered pores was analysed by X-ray powder diffraction using a PANalytical X'Pert Powder diffractometer in the theta–theta geometry. These data were collected under ambient conditions using Cu Kα radiation over the range 1° ≤ 2θ ≤ 100° using a single step size of ∼0.0170 2θ.
The surface functionalisation of MSN samples was also studied using titrations. 0.5 g of APMSN or SAMSN were suspended in 10 ml of deionised water. To this solution 20 ml of 0.1 M of HCl or 0.1 M NaOH were added in 1 ml aliquots with the pH of the solution continuously recorded. Titration using commercially available precipitated silica was used as a control. Prior to their use, this sample underwent an identical methanol extraction procedure. The treated control sample was then titrated as above.
2.4 Cytotoxicity studies
:15 and 1:20. 3T3 mouse fibroblasts were cultured in DMEM (catalogue number 12-604F, Lonza) also supplemented with 10% (v/v) foetal bovine serum, 50 units per ml penicillin, 50 μg ml−1 streptomycin and 1% (v/v) non-essential amino acids. The 3T3 cells were maintained as monolayers in 25 or 75 cm2 flasks in an incubator (37 °C and 5% CO2 in air) and were routinely passaged every 3–4 days using a split ratio of between 1:10 and 1:15.
Immediately prior to their use in assays the prepared nanoparticles were suspended in the appropriate growth medium by sonication to ensure their colloidal distribution. The cells in the 96-well plates were then exposed to the nanoparticles in the appropriate growth medium solutions and incubated at 37 °C and 5% CO2 for 24 or 48 h. At each culture end-point the medium was collected and immediately frozen at −80 °C for later analysis (ELISA). MTT or NR assays were then carried out on the cells.
:1:49. Once the medium had been removed from the 96-well plates 100 μl NR solution was added to each well. The plates were then incubated for a further 3 hours at 37 °C. NR solution was then removed and the cells were washed once with PBS (200 μl per well). At this stage 100 μl destain was added to each well and the plate shaken until a homogenous colour was achieved in all wells. Again the absorbance was read at 540 nm. Results were compared with ANOVA followed by Dunnett's test.
Acridine orange (AO) and propidium iodide (PI) were prepared at 20 μg ml−1 and 100 μg ml−1 respectively in PBS pH 7.4. They were then mixed together to form a 1:1 AO + PI solution. The medium was first removed from the Petri dishes and the samples washed three times with PBS pH 7.4. 1 ml of the AO + PI solution was then added to samples and they were incubated in the dark for 1 min at room temperature. Samples were given a further three washes with PBS before viewing in PBS with a ×20 (0.5 NA) water lens using a Zeiss AxioImager Z1 wide field fluorescence microscope and the digital imaging system. Fluorescence was excited using a mercury lamp and emission was recorded using a fluorescein isothiocyanate (FITC)/rhodamine filter block (485/515–530 nm; 546/580–563 nm).
3. Results and discussion
3.1 GN as a drug delivery system
In order to engineer GN for drug delivery applications, it is important to understand the effects of DDS fabrication parameters on drug loading and release. Initially, calcein was chosen as a model hydrophilic drug molecule. The bioinspired silica synthesis is known to be facilitated by ‘additives’24 and we selected diethylenetriamine (DETA), tetraethylenepentamine (TEPA), pentaethylenehexamine (PEHA) and poly(allylamine hydrochloride) (PAH); these additives had been shown to catalyse the formation of silica from a solution of silicates, while offering variation in the properties of silica produced.24,28Fig. 1a inset shows images of GN prepared with or without the calcein and the presence of drug in the orange silica is clear when compared to white silica powder that is typically produced without any drug. Fig. 1a shows percentage weight of drug in drug–silica samples (i.e. calcein content) and the fraction of initially added drug which loaded (i.e. loading efficiency). It can be seen that both drug content and drug loading efficiency is heavily influenced by the presence of additives, although the additive with the smallest molecular weight (DETA) showed the least influence. Additionally, a clear correlation between the number of nitrogen atoms per additive molecule and drug content was not observed. The highest drug content (30 wt%) was obtained for samples prepared in the presence of TEPA while the highest loading efficiency of ∼40% was recorded for TEPA and PAH mediated silica. These loading figures are significantly higher than those reported in the literature for unfunctionalised or functionalised mesoporous silicas (typically 2–5 and ∼20 wt% respectively).15,16 This result, which is not unexpected, highlights one of the advantages of using GN for drug delivery. The reason behind such high drug loading is that the drug is present during GN formation and can easily occupy pores. This also enables slow release by modulation of drug release rates using GN synthetic chemistry, as detailed in Sections 3.2 and 3.3.
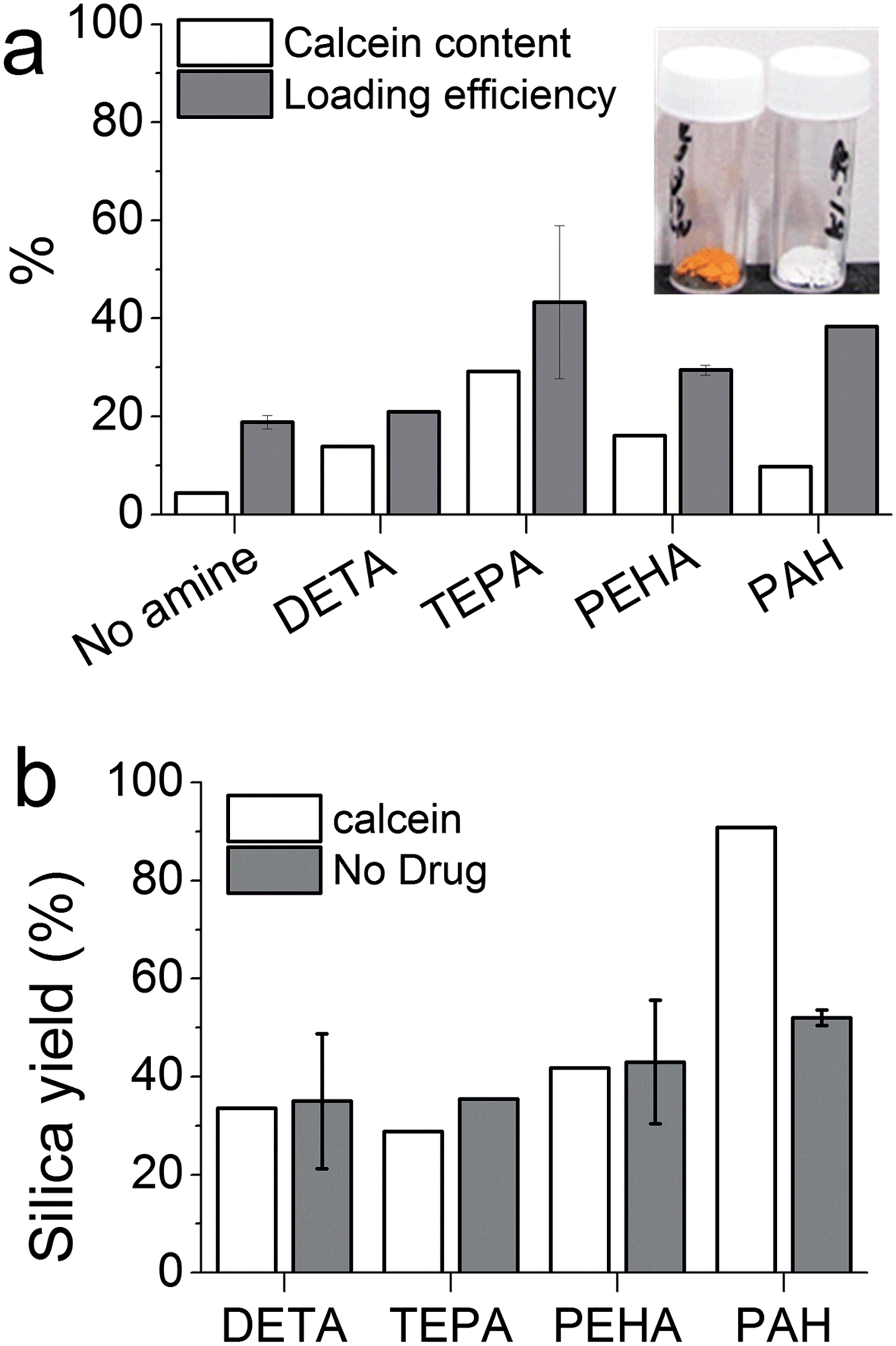 | ||
| Fig. 1 (a) Loading and loading efficiency of drug for each bioinspired additive (see text for details), inset shows images of final products with (left) and without calcein. (b) The effect of the presence of drug molecules on silica yield. | ||
It is interesting to note that both the drug content and loading efficiency for samples prepared with PEHA were lower than those prepared with TEPA. It has been reported that PEHA catalyses silica formation about 3 times faster than TEPA, while the silica prepared in the presence of PEHA is essentially non-porous compared with TEPA–silica which is porous.29,30 It then appears that PEHA does not allow the drug molecules to enter silica as much as TEPA due to rapid silica condensation, while the rate of silica formation with TEPA appears to be optimum for maximising drug loading.
In order to examine if the presence of drug affected silica precipitation, the amounts of silica precipitated in the presence and absence of the drug were measured by weighing the final product and subtracting the amount of drug loaded. For small additives (DETA, TEPA and PEHA), the presence of the drug did not affect the silica yield significantly (compare white and grey bars for each additive in Fig. 1b). Similarly, the silica yield of the control sample (prepared without any additive, essentially a gel collected after 7 days) was not influenced by the presence of the drug (data not shown). However, when a polymeric additive (PAH) was used, a significant increase in silica yield was observed in the presence of the drug. This is probably due to a co-operative effect from the interactions of negatively charged calcein and positively charged PAH molecules under synthetic conditions (pH 7). Such interactions have been previously reported in the one-step fabrication of the drug in silica gels.17 Although a similar effect may be expected with small additives, the higher charge density in polymeric amines (PAH in this case) is known to exhibit stronger interactions with oppositely charged biomolecules, micelles, colloidal particles, etc.47–50
The release of calcein (Fig. 2) shows that the particles prepared with longer polyamines gave more sustained release. It is evident that the chemistry of the synthesis of GN, the additives used in particular, was able to modulate the drug release profiles all the way from instantaneous release to sustained release over weeks. The control sample prepared with no additive exhibited a release profile reminiscent of the dissolution profile of free calcein, which means that the control sample did not offer any regulation over release. Furthermore, the total amount of drug released had a strong correlation with the number of nitrogen atoms present in each additive (Fig. 3a). It is important to note here that the final concentration of nitrogen was kept constant for each additive used. This means that the number of additive molecules decreased as the additive size increased, while the amine/charge density increased significantly with the size of the additive molecule. For example, at the same nitrogen concentration, there were twice as many DETA molecules (three amines) than PEHA (six amines). The effects of change in amine size on silica formation have been extensively studied and reported in the literature,29,30 while the effects on the amine–drug interactions are discussed in subsequent sections. An important point to note here from the drug release results is that the chemistry of the additives can be used to control the release profile by regulating the structure of GN produced and/or its interactions with the drug.
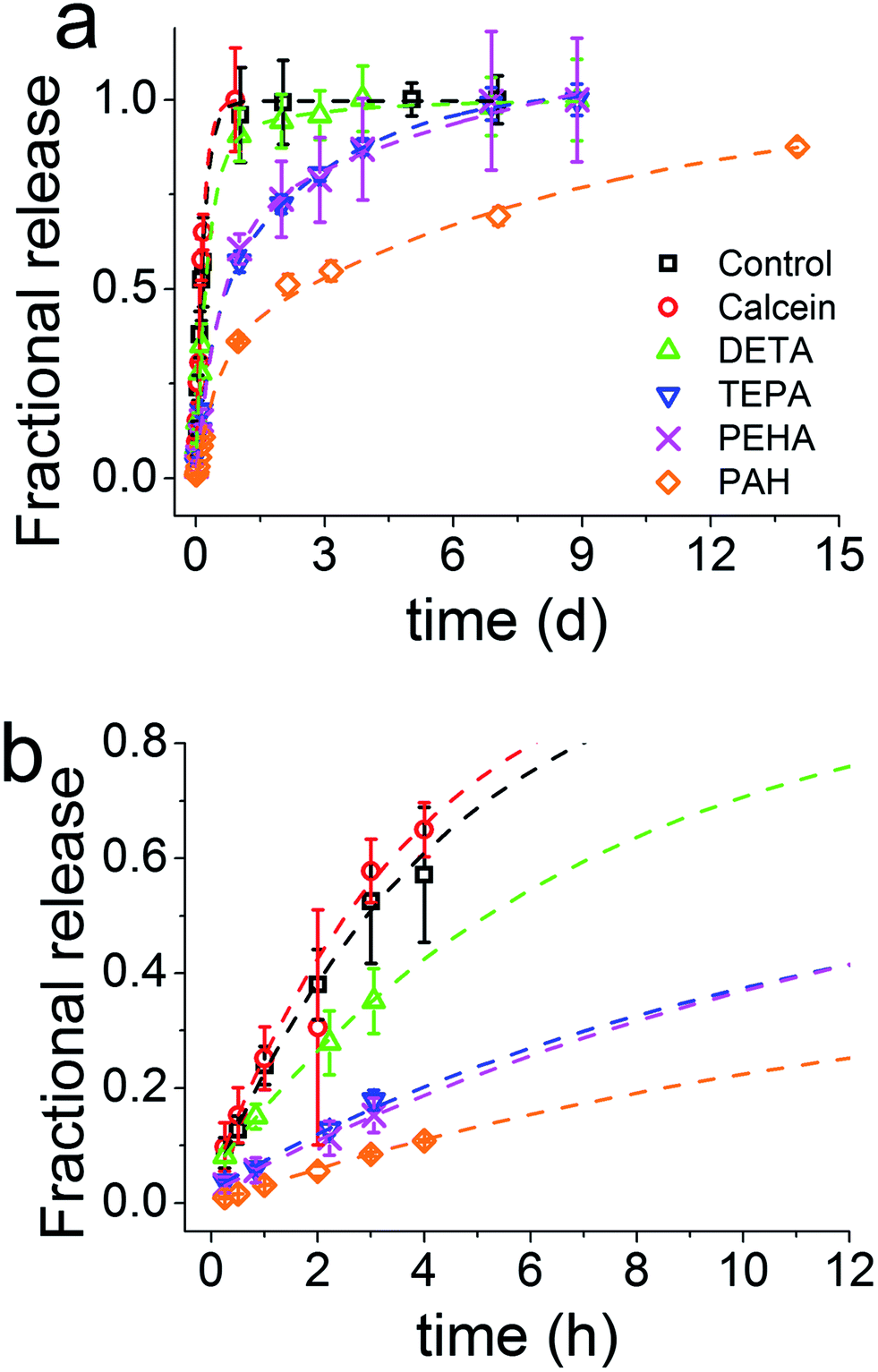 | ||
| Fig. 2 Overall (a) and initial (b) release profiles of calcein from GN synthesised using several additives. Note the difference in X-axis units between (a) and (b). The fitted lines are not showing the Korsmeyer–Peppas model, see Section 3.2 for details. | ||
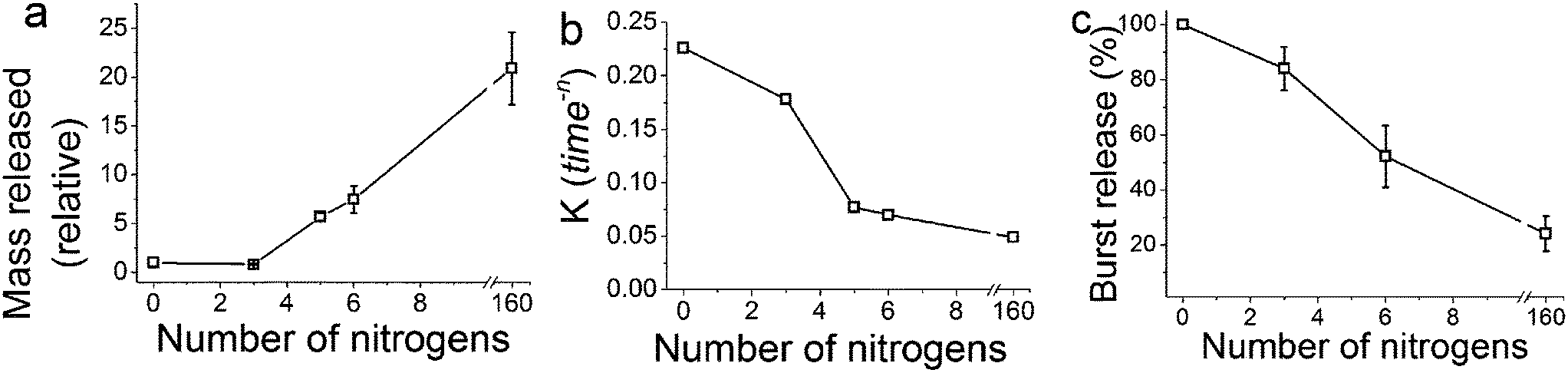 | ||
| Fig. 3 (a) Mass of drug released, normalised to the control sample (n = 0) as a function of nitrogen atoms per additive molecule. (b) The release rate constant (K) obtained from fitting the release data to the Korsmeyer–Peppas Equation (eqn (2)). (c) The amount of burst release as a function of nitrogen atoms per additive molecule obtained by fitting the release data to a double exponential eqn (3). Note that the final concentration of nitrogen was kept constant for each additive used. | ||
3.2 Drug release mechanisms
Using the Korsmeyer–Peppas model (a single exponential), we can fit the above release data and obtain parameters which correspond to physical characteristics of the system and enable quantitative comparison between samples.51,52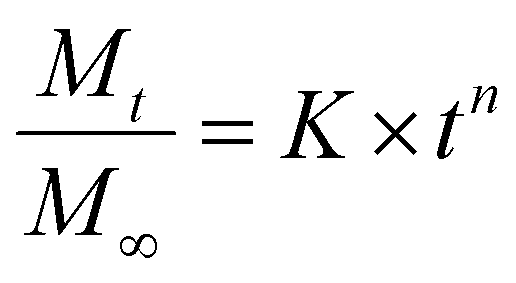 | (2) |
The values of the rate constant K and exponent n are presented in Table S1.† From Fig. 3b once again we see that the drug release (release rate constant K) was modulated by the chemistry of the additive used in silica synthesis. Values of n correspond to the release mechanism as per Table S2† reproduced from Ritger and Peppas.51 In all cases n was between 0.5 and 1, which means that the drug release followed non-Fickian diffusion. These values are typical of those given in the literature.53 This release pattern expected to be Fickian diffusion only applies where the flux is limited by the diffusion through the medium and is less likely to be applicable in our case where the loaded drug must first diffuse from the internal pores of the silica matrix followed by its diffusion from the external surface into the bulk liquid, consistent with the literature.18 DDS prepared in the presence of TEPA, PEHA and PAH exhibited similar exponents, suggesting that the release mechanisms were identical. For TEPA, PEHA and PAH the release is much slower with much higher exponents (n) of release. It is therefore possible that another step in the release process is limiting for these samples.
The work of Peppas et al. was originally composed for soft polymers which are close to their glass transition temperatures and any non-Fickian behaviour was attributed to polymer swelling and relaxation in the solution.54 This will not apply to the structure of the silica formed in this work, given that at 37 °C silica remains a hard, covalently bound, network. Another commonly used model for describing drug release is the Higuchi equation.55 This model is essentially a special case of the Peppas model where n = 0.5. However, from Table S1† it can be seen that for GN DDS, n > 0.5 and therefore the Higuchi model is unlikely to fit the release data from Fig. 2. Although, this model describes diffusion of the drug from smooth surfaces and typically only fits the first 15% of the data, it has been widely applied to drug release from mesoporous silicas, which are far from smooth surfaces.19,20,56
It is clear that neither the Higuchi nor Peppas models can adequately describe the entire profile for drug release (100% of the release data), with PAH being especially poorly modelled (Fig. S1†). In addition, these models are insufficient to fully describe non-ideal porous materials such as silica. Therefore an empirical double exponential function was employed of the form:
| Mt = M0 + M1(1 − e−x/t1) + M2(1 − e−x/t2) | (3) |
Fitted curves are shown as dashed lines in Fig. 2 and a number of advantages were apparent with this model. The R2 values were always greater than 0.99 and the equation modelled the entire release profile, unlike only parts modelled by the Korsmeyer–Peppas model (modelled 60% of the data54) or the Higuchi model (modelled 15% of data55). Furthermore, it affords the quantification of distinct release mechanisms that are typically observed with non-ideal materials arising from internal porosities.57 This double exponential suggests that calcein release is a two stage process for GN, which can afford a clear distinction between burst and sustained release. We attribute the first, faster release stage to the drug that is weakly physisorbed on the external surface of GN. The slower and sustained release of the drug in the second stage could be attributed to drug–additive interactions, internal porosity of GN or both. As a result, drug release will be expected to be a two stage process, which can be accurately modelled with a double exponential equation. Although perhaps new to the drug delivery field, such models are commonly applied in many other areas of research such as reaction kinetics and dissolution processes.58
The release of the drug from within the network of pores, which is a common feature of GN,41 would present an additional barrier to mass transfer for the release of drug molecules when compared to the drug adsorbed simply on external surfaces of silica particles. This is particularly important to note since the drug is loaded as silica forms and therefore it is highly likely that the drug molecules are located in the internal voids between primary particles. Interactions between the model drug and the additive are highly likely, as reported previously for a silica gel DDS,17 and were also evident from the results discussed in Section 3.1. A stronger interaction would explain a slower second exponential stage of release. This also explains why the control sample, where no amine groups were present for interactions, was well described by a single exponential function (i.e. Korsmeyer–Peppas model), which represents drug loading via physisorption on the surface of the control sample. Applying eqn (3) to decouple burst and sustained release, we observed that GN allows control over the amount of drug released in a sustained fashion (Fig. 3c). The fraction of the drug that was weakly physisorbed on silica (burst release) decreased with increasing amine chain length. This observation further strengthens the tunability of GN for drug delivery and also highlights the importance of the interaction of the drug with amines occluded in silica, which were further investigated and the results are presented in Section 3.3 below.
3.3 The role of chemistry in controlling drug delivery
A closer inspection of the drug loading and release data indicates that entrapping the drug with PAH showed a promising loading capacity as well as a release profile in terms of sustained release when compared to other additives studied herein. Therefore PAH was selected for further systematic investigations in order to study the effect of synthesis parameters on drug loading and release. A two level full factorial experiment was set up using eqn (1) as described in Section 2.2.4 (ref. 46) with the three key parameters (reaction time, [Si] :[N] molar ratio r and silica precursor concentration [Si]) at “high” (+) and “low” (−) levels as shown in Table 1. The effects of these parameters were quantified by measuring silica yield, drug loading and drug release. Such factorial design of experiments and analysis of results allow the identification of single, or a group of, parameters that significantly affect the outcomes. Fig. S2† shows the effect of these factors on the silica yield for each experiment. Assessing the graph first we can see that in all experiments silica yields were similar (70–80%) with the exception of experiment D6 where the yield never exceeded 60%. No grouping of the bars in Fig. S2† is obvious, suggesting that no large changes in silica yield are afforded by changing the parameters under study. To account for any possible skewing of the data towards any single parameter, we can discount one factor at a time and establish a series of second level two factor tests. This analysis suggests that there were no differences observed beyond the 95% CI (Fig. S3†). It is therefore likely that the silica yield is uninfluenced by the factors under study.
| Experiment | t | [Si]b | r | t ↔ [Si] interaction | t ↔ r interaction | [Si] ↔ r interaction | t ↔ [Si] ↔ r interaction |
|---|---|---|---|---|---|---|---|
|
a + level = 5 minutes, − level = 1 minute.
b + level = 50 mM, − level = 30 mM.
c + level = 2:1, − level = 1:1.
|
|||||||
| D1 | − | − | − | + | + | + | − |
| D2 | − | − | + | + | − | − | + |
| D3 | − | + | − | − | + | − | + |
| D4 | − | + | + | − | − | + | − |
| D5 | + | − | − | − | − | + | + |
| D6 | + | − | + | − | + | − | − |
| D7 | + | + | − | + | − | − | − |
| D8 | + | + | + | + | + | + | + |
Fig. 4a shows the effect of experimental parameters on the entrapment of calcein within the optimization experiments. A clear periodic trend was observed with higher drug loading associated with experiments with higher [Si] :[N] ratios (r). This is confirmed by further analysis which showed a large magnitude of the effect of changing the ratio r, far in excess of the 95% CI (Fig. 4b). In this case the molar ratio r had a significant effect with a ratio of 2 giving a much higher entrapment than 1:1 ratio. None of the other synthetic parameters appeared to significantly affect drug loading. In order to investigate whether the absolute nitrogen concentration had any effect (which could be convoluting the apparent effect of r), four nitrogen concentrations used were explored and their effects on drug entrapment were measured (Fig. 4c). The effect of nitrogen concentration was clear: below a critical concentration of 25 mM, enhanced drug loading was observed. The changes caused by r or nitrogen concentration are likely to be due to the well-known effect of amines on the physical properties of GN such as porosity30 and also perhaps drug–additive interactions. While identifying conditions for achieving high drug loading efficiencies without affecting the yield of silica formation, these results suggest that both the [Si] :[N] ratio and the absolute nitrogen concentration are important.
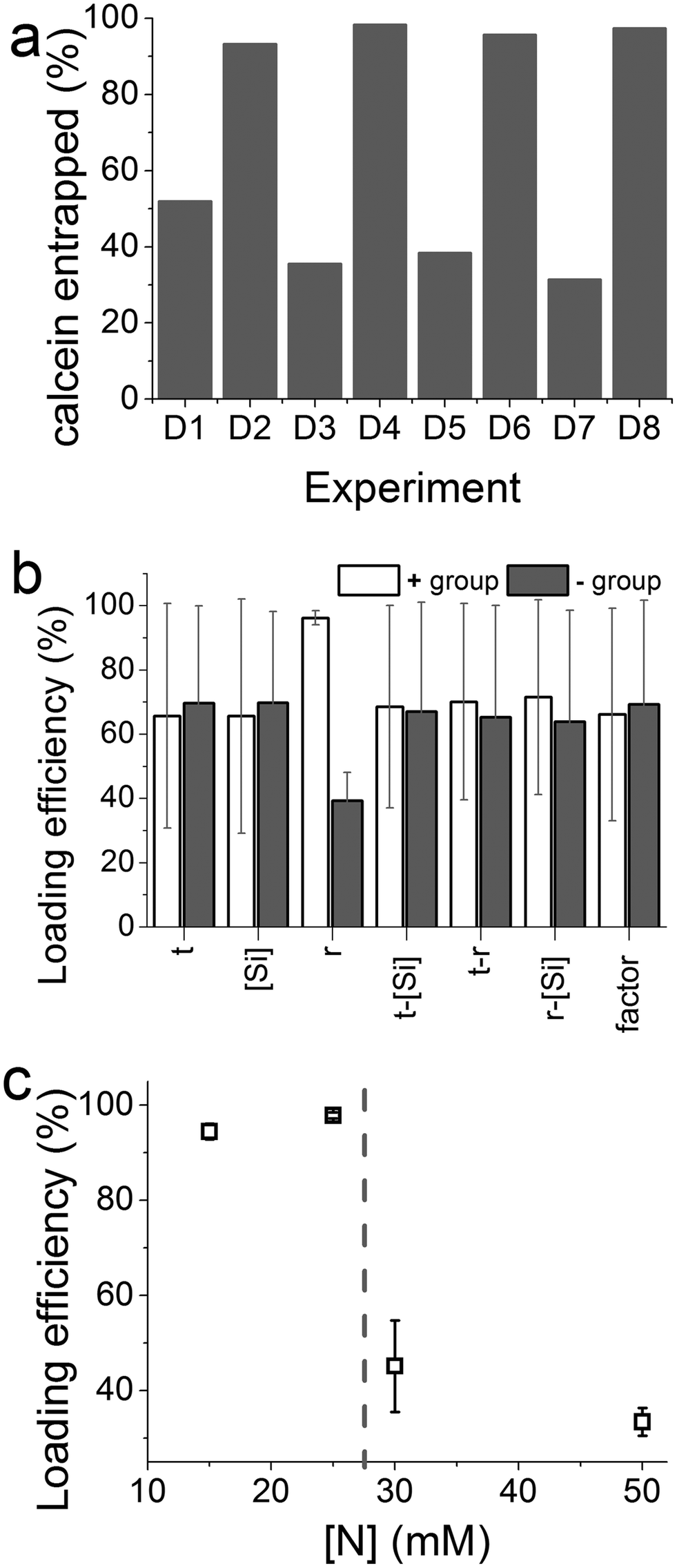 | ||
| Fig. 4 (a) Drug loading achieved in the factorial experiments. (b) Results from factorial experiments designed to establish key parameter(s) that have a significant effect on drug loading. (c) Further exploration of the effect of amine concentration on drug loading. | ||
The release of calcein from these optimization experiments was also recorded (Fig. 5) where two groupings of release profiles were obvious. The four curves above the dashed line, where the higher amounts of the drug were released, correspond to higher concentration of the silica precursor (and in turn higher r). Although the exact mechanism of the dependence of drug release on silica precursor concentration is not clear at this point of time, we speculate the following from the information available in the literature. At lower [Si] the formation of silica is slow, which allows sufficient opportunity for the drug to be deeply entrapped and “sealed” inside silica. On the other hand, at high concentrations of the silica precursor, silica formation is very rapid where the drug is only loosely bound and hence available for release. Nonetheless, it is clear that higher r is beneficial for both drug loading efficiency and drug release. This again clearly supports the notion that the additive has a pronounced effect on both silica formation and drug loading/release.
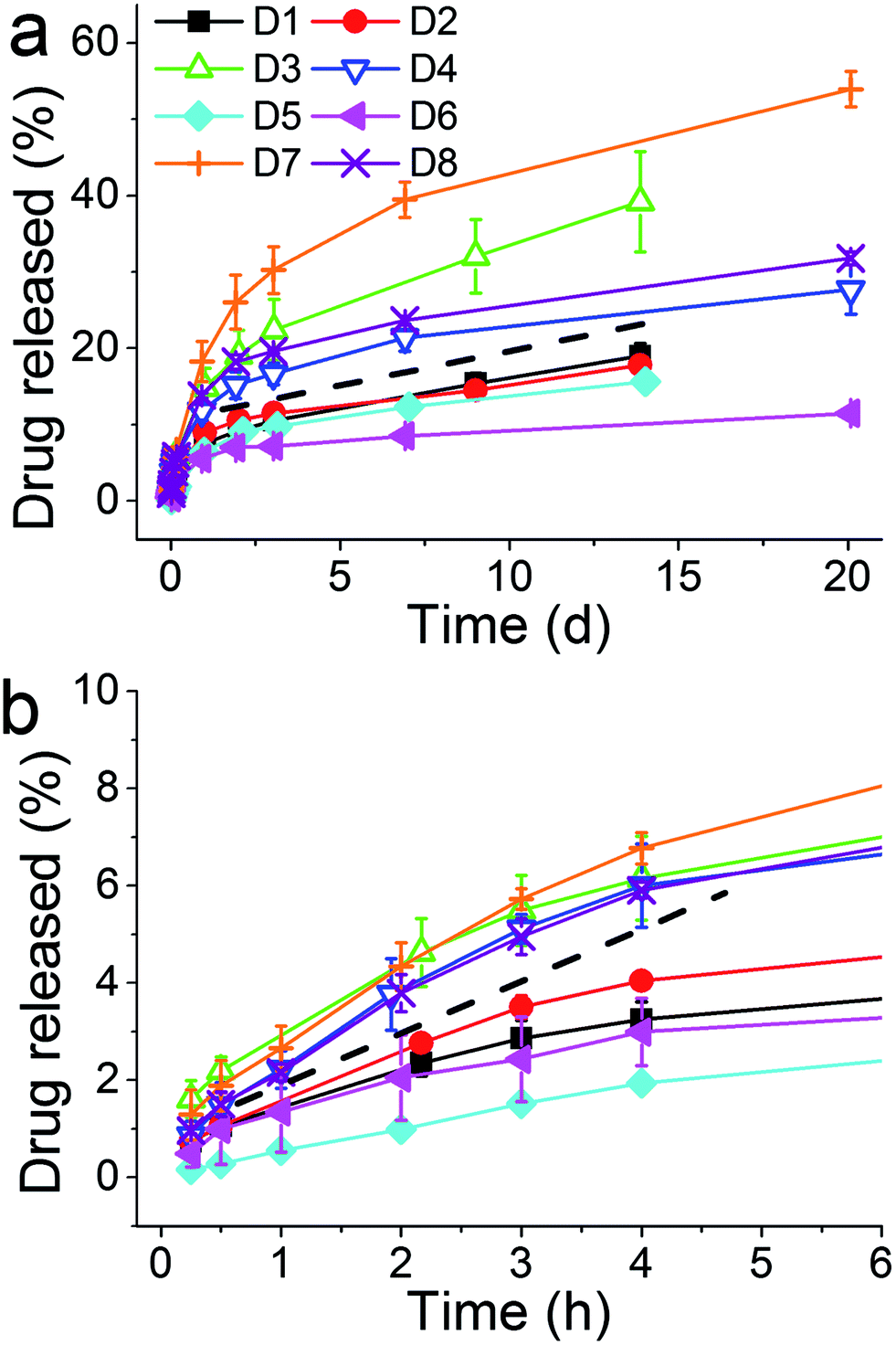 | ||
| Fig. 5 Overall (a) and initial (b) release of the drug from the samples prepared in the factorial experiments. Note the different X-axis units in (a) and (b). The dashed line separates two sets of results, see the text. | ||
Having identified the key parameters controlling the fabrication of the delivery system, we sought to investigate the effects of the synthesis procedures on the fabrication as a potential factor which could affect the product. It is clear that in GN synthesis, the interactions between the amine and silicates are important, while the results above indicate that there is also a possibility of the occurrence of amine–calcein interactions. In particular, the order of addition of precursor, additive and drug was investigated systematically and the experimental design is detailed in Table 2. The silica yields and drug loading efficiencies for these experiments are shown in Fig. 6.
| Step 1 | Step 2 | Step 3 | Step 4 | |
|---|---|---|---|---|
| O1 | Drug + silicate solution | Dissolved PAH added to solution from step 1 | pH adjusted to 7 | N/A |
| O2 | Drug + PAH solution | Dissolved silicate added to solution from step 1 | pH adjusted to 7 | N/A |
| O3 | PAH + silicate solution | pH of solution from step 1 adjusted to 7 | Dissolved drug added immediately to solution from step 2 | N/A |
| O4 | Drug + silicate solution | pH of solution from step 1 adjusted to 7 | pH of PAH solution adjusted to pH 7 | Solutions from steps 2 and 3 mixed |
| O5 | Drug + PAH solution | pH of solution from step 1 adjusted to 7 | Silicate solution acidified to pH 7 | Solutions from steps 2 and 3 mixed |
| O6 | pH of PAH solution adjusted to pH 7 | Silicate solution acidified to pH 7 | Solutions from step 1 and 2 mixed | N/A |
| O7 | Identical to O5 but PAH replaced with PEHA. | |||
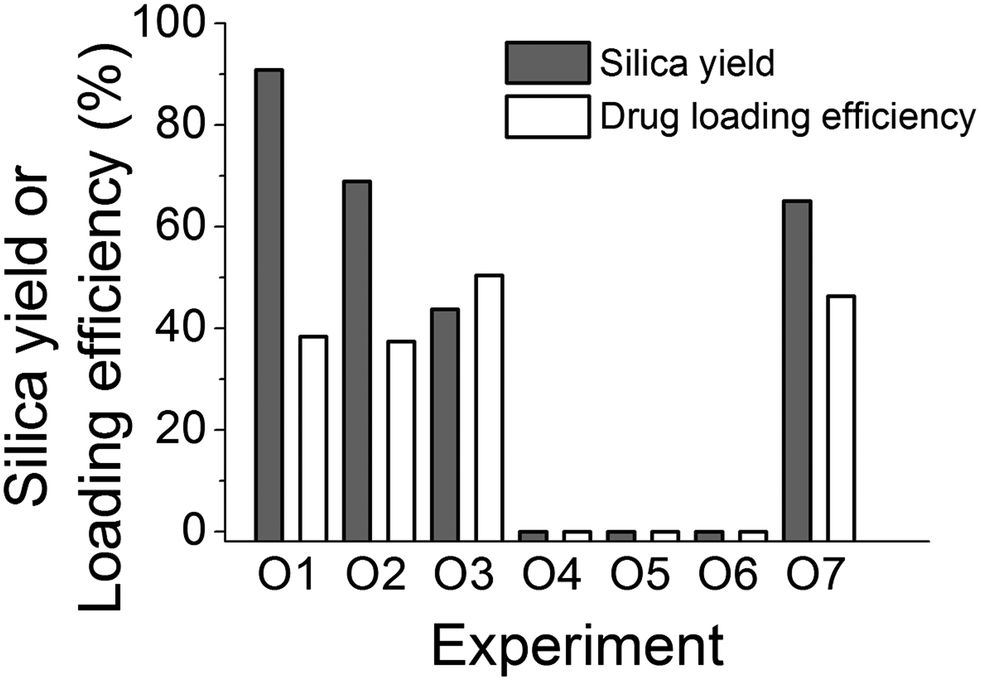 | ||
| Fig. 6 The effect of the order of adding reactants on the silica yield and drug loading efficiency. | ||
It is evident that the order of adding each component to the reaction system clearly has a profound effect on both silica yield (modulated from 0 to 90%) and drug loading. Surprisingly, neutralizing additive and silicate solutions before mixing the reactants did not produce any silica recoverable by centrifugation (experiments O4–O6). If we consider experiments O1, O2 and O3, the mixing of PAH and silicate occurs in a basic solution at around pH 12. The conjugate acid–base equilibrium for PAH, calcein and orthosilicic acid can be written as below along with their reported pKa values:
| [CH2CH(CH2NH+3)]n ⇌ [CH2CH(CH2NH2)]n + nH+ pKa: 9.5 (ref. 59) | (4) |
| C30H26N2O13 ⇌ [C30H25N2O13]− + H+ pKa: 6.67 (ref. 60) | (5) |
| SiO4H4 ⇌ SiO4H3− + H+ pKa: 9.8 (ref. 61 and 62) | (6) |
Applying the Henderson–Hasselbalch equation at pH 12, it appears that PAH is not protonated (>99% as –NH2), the majority of calcein is deprotonated (>99% as –COO−) and silicic acid is deprotonated (>99% as ![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO−). Therefore, there will not be any electrostatic interactions between the amine and calcein, while it is known that at pH 12, the formation of the silica particles will not occur.62 As the pH is adjusted to 7, PAH will become extensively protonated (>99% as –NH3+), and the protonated amines will be available to interact with silica and will not be associated or “blocked” by calcein prior to silica formation. Strong electrostatic interactions between PAH and silicates occur, leading to rapid silica precipitation. In experiments O4, O5 and O6 the protonation states of amine, calcein and silicic acid are significantly different before the solutions are mixed. Each solution is first neutralised to pH 7 and then mixed, which can cause significant association between protonated amines (>99% protonation) and deprotonated calcein (68% as –COO−). This can result in no silica precipitation by the restriction of amine–silicate interactions before the silica formation can start. Interestingly, under identical conditions, when PAH is replaced by PEHA (experiment O7), a significant amount of silica formation was observed (Fig. 6), which can be explained by the differences in the protonation between PAH and PEHA. It has been reported that at pH 7, PEHA is only 50% protonated29,30 in contrast to >99% protonated PAH. This leaves PEHA–calcein interactions weak and allows PEHA–silicate interactions to occur, resulting in silica precipitation. These results have helped optimise the order of mixing of reagents in fabricating DDS.
SiO−). Therefore, there will not be any electrostatic interactions between the amine and calcein, while it is known that at pH 12, the formation of the silica particles will not occur.62 As the pH is adjusted to 7, PAH will become extensively protonated (>99% as –NH3+), and the protonated amines will be available to interact with silica and will not be associated or “blocked” by calcein prior to silica formation. Strong electrostatic interactions between PAH and silicates occur, leading to rapid silica precipitation. In experiments O4, O5 and O6 the protonation states of amine, calcein and silicic acid are significantly different before the solutions are mixed. Each solution is first neutralised to pH 7 and then mixed, which can cause significant association between protonated amines (>99% protonation) and deprotonated calcein (68% as –COO−). This can result in no silica precipitation by the restriction of amine–silicate interactions before the silica formation can start. Interestingly, under identical conditions, when PAH is replaced by PEHA (experiment O7), a significant amount of silica formation was observed (Fig. 6), which can be explained by the differences in the protonation between PAH and PEHA. It has been reported that at pH 7, PEHA is only 50% protonated29,30 in contrast to >99% protonated PAH. This leaves PEHA–calcein interactions weak and allows PEHA–silicate interactions to occur, resulting in silica precipitation. These results have helped optimise the order of mixing of reagents in fabricating DDS.
3.4 GN performance in delivering therapeutically useful drugs
The mechanisms underpinning the drug release were investigated and we presented how fine tuning the chemistry of synthesis of GN–DDS can allow us to achieve desired designs for drug delivery systems. In this section, we sought to utilise these results and apply them for developing DDS for a number of therapeutically useful drugs while also exploring any potential limitations to the GN-based DDS. A series of pharmaceutically active small molecules were chosen to reflect a range of hydrophobicities: atenolol (logP = 0.16), pindolol (logP = 1.75), ibuprofen (logP = 3.97) and nile red (logP = 5.0). Ibuprofen is a non-steroidal anti-inflammatory agent, atenolol and pindolol are betaadrenoceptor antagonists (beta blockers) and nile red is a dye which was used to emulate a very hydrophobic molecule. The loading for each of these molecules was very high (between 87 and 100%), while there were differences in both the release profiles and amounts released were observed (Fig. 7a).
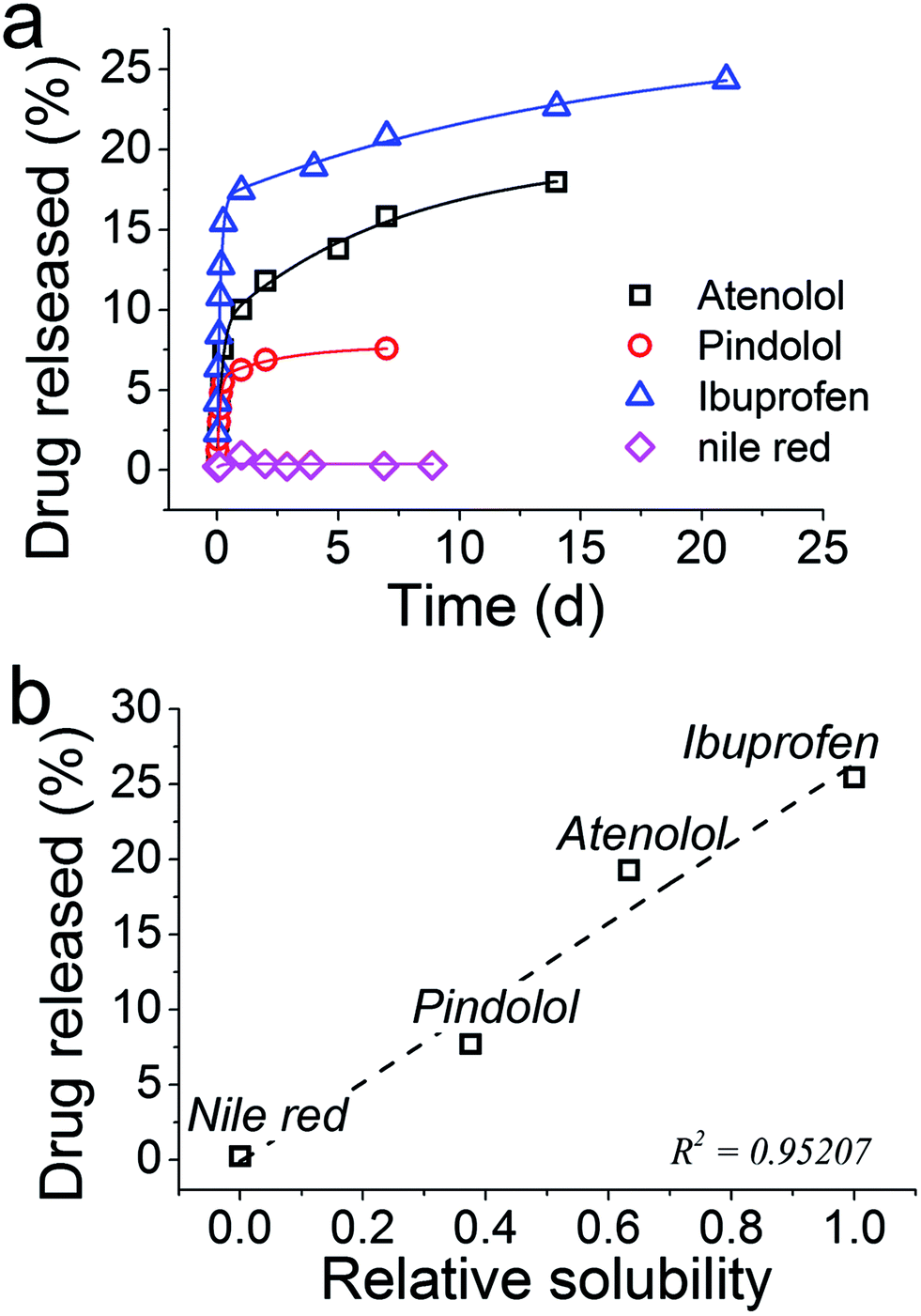 | ||
| Fig. 7 (a) Release profiles for several drugs from GN. (b) The total amount of drug released from GN as a function of the solubility of each drug molecule, relative to Nile red. | ||
With an exception of Nile red, the drug release was sustained over several days. Although the amount of drug released could be improved, differences in the release profiles were observed between the drugs under consideration. Since the synthesis of GN–drug systems occurred entirely in aqueous solutions, the hydrophobicity or water solubility of each drug may be regulating their release. In order to test this hypothesis, solubility data for each compound were obtained from the literature,63 and plotted with the per cent drug released (Fig. 7b). From these data, it is evident that the water solubility of the drugs strongly influences the amount of drug released, with the least soluble drug, Nile red, preferring to stay inside silica with no release occurring. These results will be of immense importance in designing GN as DDS in future, while they also highlight a limitation of the GN systems such that the chemistry of GN will require further consideration in future work in order to efficiently load and release hydrophobic drugs – an important class of drugs (e.g. anticancer drugs).
3.5 Toxicity results
In order to test the cytotoxicity of GN, PAH–GN and PEHA–GN were selected due to their encouraging release profiles shown in Fig. 2. The results were compared with a colloidal Stöber silica sample, and two MSN: APMSN and SAMSN (amino- and sulfonic acid-functionalised MCM-41 respectively, see Experimental section). Stöber silicas form excellent comparative standards. The functionalised MCM-41 have been preferred DDS over unfunctionalised silicas due to their reported enhanced drug loading capacity from strong interactions with drug molecules, improved biocompatibility and potential in targeted delivery.4,6The toxicity of nanomaterials can result in a number of effects including damage to membranes, changes in protein folding, DNA mutation, blood abnormalities and oxidative stress injuries.21,64 Although extensive analysis is necessary to fully understand the toxic effects, measurements of cell viability and proliferation can provide an indication of the safety of nanomaterials. Hence cell viability assays were employed in this study using both murine 3T3 fibroblast cells, a standard cell line widely used for in vitro cytotoxicity evaluation as recommended by ISO 10993, and U937 human monocyte-like cells in both rested and activated states. U937 cells were activated to phagocytotic macrophage characteristics, and comparison of toxic responses in resting and activated cells allows the contribution of nanoparticle internalisation by phagocytosis to be determined.
In the case of fibroblasts, Stöber particles reduced cell viability to ∼80% of the control (cell lines without silica particles) even at concentrations as low as 5 μg ml−1 (Fig. 8a). The observed mild toxicity of Stöber silica at low concentrations is consistent with the literature.65,66 Upon increasing the silica dose, the cell response was unchanged and hence appeared to be independent of Stöber silica concentration. On the other hand, Stöber particles were not found to reduce cell viability of the human monocytes (Fig. 8b and c). Such cell-line dependent responses have been well-documented. For example, Yu et al. reported that a range of silica nanoparticles did not have any toxic effects on tumour cells while a dramatic reduction in cell viability was recorded for macrophages, probably reflecting intracellular uptake of particles by phagocytosis in the latter cells.67 We observed that the Stöber silica concentration to reduce cell viability to 50% (IC50) was >500 μg ml−1 (Table 3).
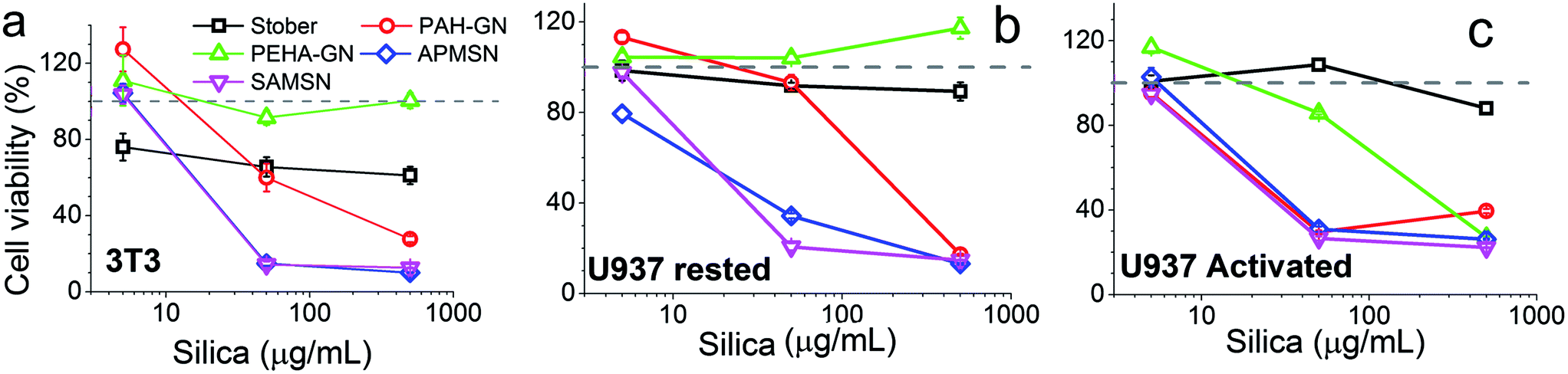 | ||
| Fig. 8 Cell viability upon incubation of silica particles with (a) 3T3, (b) U937 in the resting state and (c) U937 in the activated state. | ||
| Cell lines | |||
|---|---|---|---|
| Samples | 3T3 | U937 rested | U937 activated |
| Stöber | >500 | ≫500 | ≫500 |
| AP–MSN | 20 | 20 | 25 |
| SA–MSN | 20 | 20 | 25 |
| PAH–GN | 100 | 200 | 25 |
| PEHA–GN | ≫500 | ≫500 | 200 |
Surprisingly, in the current study, PEHA–GN did not show any toxic effects, to either fibroblasts or human monocytes in the resting state, even when silica concentrations as high as 500 μg ml−1 were used (Fig. 8a and b). This observation is particularly interesting since PEHA (i.e. without silica) was toxic to the same cell lines (Fig. S4†), thus suggesting that when trapped in silica, PEHA becomes less toxic. In contrast, PAH–GN, and both MSN samples, reduced cell viability significantly in both cell lines. These differences in toxicity are illustrated in the micrographs presented, where the cell number is markedly depleted in samples treated with both MSN particles (Fig. 9a), and the fluorescent staining shows an increased number of dead cells, their nuclei stained red with propidium iodide (Fig. 9b and c). The IC50 values listed in Table 3 imply that MSN were clearly more toxic than Stöber silicas or either of the GN.
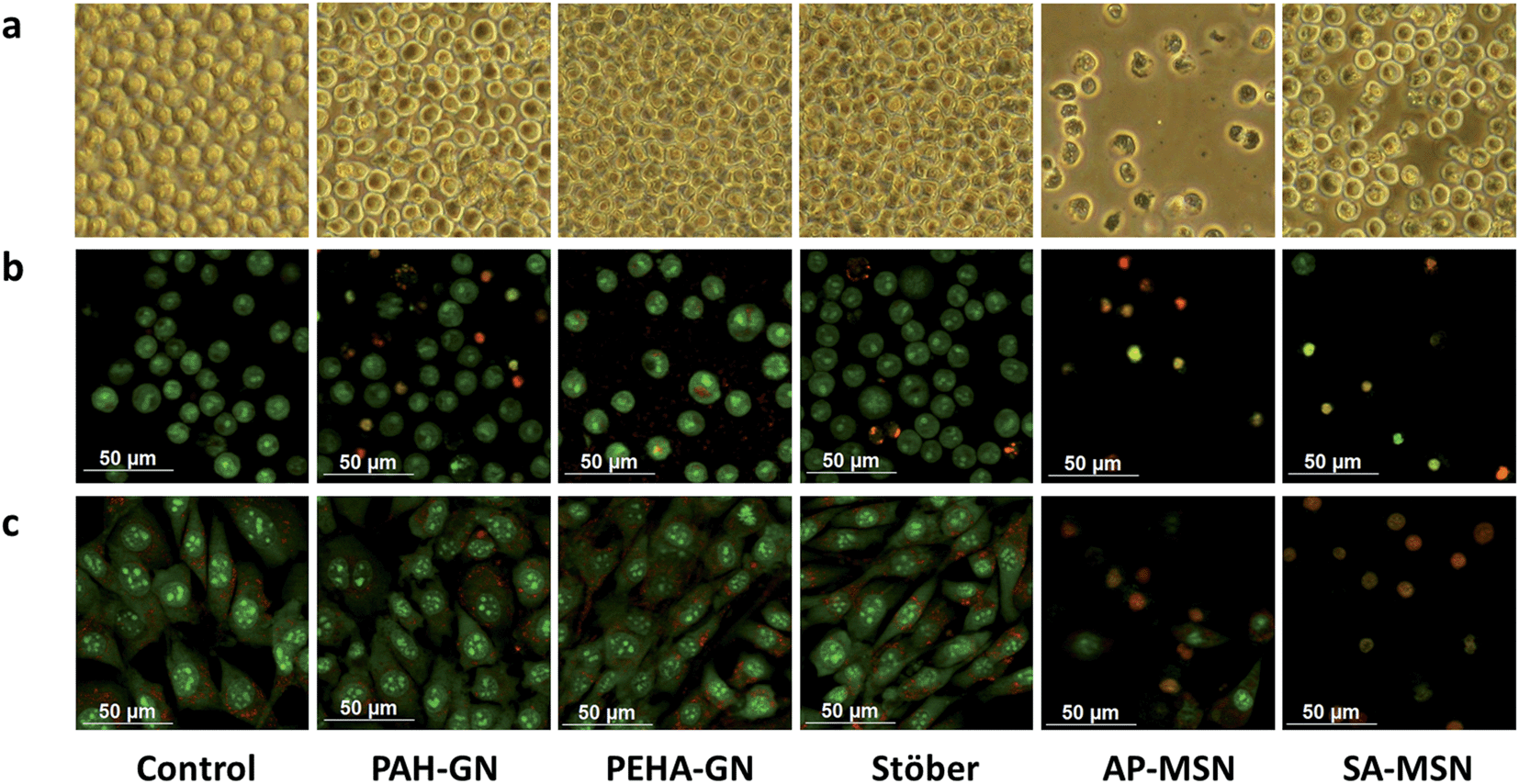 | ||
| Fig. 9 Optical (a) and fluorescent (b and c) microscopy images of cells with (50 μg ml−1) and without silica particles for U937 cells in the resting state after 5 days (a) or 1 day (b) in culture, and 3T3 cells (c) after 1 day in culture. Images in (a) are presented at 300× magnifications. | ||
No significant differences in cell viability for Stöber, MSN and PAH–GN samples were observed when the human monocytes were activated, even at the highest silica concentrations investigated. However, reduced cell viability was observed for activated monocytes in the presence of PEHA–GN, which had previously shown no toxicity to resting monocytes (Fig. 8c). Although the IC50 of 200 μg ml−1 for PEHA–GN indicates that toxicity is not a realistic safety issue for the exposures encountered in most biomedical uses, it is comparable to values reported for most silica particles.65,67 The increased sensitivity of activated monocytes to PEHA–GN probably occurs due to intracellular access of particles following phagocytosis, leading to some specific toxic response inside the cells.64,68
Inflammatory responses to nanoparticles are known to cause considerable adverse effects in vivo, and in order to predict the likelihood of these the release of inflammatory cytokines, interleukins, tumour necrosis factor-α (TNF-α) and interferon-γ, in response to nanoparticle exposure are commonly measured by specific ELISA.21 TNF-α secretion has been reported to increase by 20-fold in the presence of silica particles.69 However, in this study, only at high levels of exposure ≥500 μg ml−1 detectable cytokine release was measured at either 24 or 48 h treatment, although transient trace secretion of all four mediators was induced by PAH–GN at 24 h, but not at 48 h (data not shown). It is evident that none of the samples used induced secretion of inflammatory cytokines at the concentrations proposed for use in DDS.
Physicochemical properties of nanomaterials such as chemical composition, particle size, porosity and surface functionalisation, are important when considering cell responses.23 “Silica” represents a vast number of materials with distinct structures and properties, including crystalline (e.g. quartz), amorphous, porous, colloidal, gels, etc.7,8 They differ from one another in the sizes of particles and aggregates, porosities, the level of hydration, and the surface chemistry. These seemingly subtle differences which mainly arise from the processing conditions, can manifest themselves in significant ways such as drastic differences in the biodegradation9 and biological response.70,71 For Stöber silicas, there was no evidence of the presence of organics from Infrared spectroscopy (FTIR), while small amounts of organics were detected in all other samples, as expected (also verified from thermal analysis, Fig. S5†). A combination of surface sensitive ATR-FTIR, zeta potential measurements and titrations revealed that Stöber silica possessed only silanol (Si–OH) and siloxide (Si–O−) groups on the surface with a zeta potential of −48 mV (Fig. S5 and Table S3†). SAMSN possessed acidic surfaces and negative zeta potential, which was also confirmed by titration, while APMSN had a basic surface with positive zeta potential, as expected (Fig. S6 and Table S3†). Interestingly, PEHA–GN exhibited a slightly positive zeta potential (ζ = +10 mV), perhaps from surface-bound PEHA, while PAH–GN had a slightly negative zeta potential (ζ = −18 mV), indicating that the majority of PAH remains inside the sample. It has been reported that positively charged silica particles are relatively less toxic,67,72 which explains the differences in toxicity between PEHA–GN and PAH–GN. However, AP–MSN particles, which were also positively charged were found to be highly toxic (IC50 ≈ 20 μg ml−1) compared with the positively charged PEHA–GN (IC50 ≫ 500 μg ml−1 for both 3T3 and resting U937 cell lines).
The only differences between both positively charged silica samples (PEHA–GN and APMSN) were their porosities and particle sizes. It was found that MSN samples exhibited large surface areas (≈500–700 m2 g−1) and pore volumes with narrow and ordered mesopores of ∼2.6 nm (Fig. S7–9 and Table S3†). In contrast, GN appeared to have low surface areas (<55 m2 g−1), broad pore size distribution and disordered pores, which is consistent with the literature reports.41 Despite low surface areas, the drug loading in GN was more than twofold higher compared to that reported in the literature (see Section 3.1). Although low surface area may affect drug release, it has been reported that surface area of GN can be readily controlled.30 Furthermore, there were variations in the particle sizes between samples, which ranged from 130 nm (PAH–GN) to 720 nm (SAMSN) (Fig. 10). SEM revealed that the particles were present as aggregates, which was also confirmed by DLS analysis (data not shown) with a broad distribution of particle size, suggesting aggregated systems (with an exception of Stöber particles). It is therefore clear that in this study, the cells were not interacting with individual nanoparticles but rather with aggregates of nanoparticles which were several hundreds of nanometers or even micrometers in size. This observation is not unexpected since nanoparticles are unlikely to avoid aggregation in complex biological media containing a wide variety of ions and biomolecules11,73 and hence our systems are thought to closely represent realistic conditions. There was no apparent trend between particle size and their toxicity, which is contrary to most literature reports.65 However, the particles reported in the literature are much smaller with negligible aggregation compared to those reported here.
 | ||
| Fig. 10 Scanning electron micrographs of silica particles used: (a) Stöber silica, (b) PEHA–GN, (c) PAH–GN, (d) APMSN and (e) SAMSN. Scale bars = 500 nm except 1 μm for (b). | ||
4. Conclusions
The main conclusions from this work are threefold, as listed below.(1) We have shown that for the model hydrophilic drug molecule calcein, additive chemistry and synthesis/loading method can be used to tailor the release profile, particularly in the burst time frame (first few hours). This shows that GN DDS have the potential to be tailorable in release profile terms without the need for extensive synthetic routes/functionalisations.
(2) We have shown that GN DDS have the potential to perform sustained release with drug molecules which cover a range of logP values and this, coupled with point 1, and the ease of synthesis, makes GN a viable alternative to MSN systems which have traditionally dominated the DDS field with regard to silica.
(3) GN were least toxic silica particles in most experiments, while, both types of MSN particles showed substantially reduced cell viability, even at low concentrations. Furthermore, GN exhibit additional benefits such as green synthesis and ease of functionalization. This strengthens the argument for future use of GN in DDS and other biomedical applications.
Further studies using a wider expanse of cell lines, organ baths and animal models are required to fully understand the GN and optimise administrative routes of GN DDS but current in vitro cell data appear to be highly promising.
Acknowledgements
The authors thank the Department of Chemical and Process Engineering, Strathclyde Institute of Pharmacy and Biological Science, The Bridging the Gap award and the EPSRC Doctoral Training Centre in Medical Devices for funding. We also thank the Advanced Materials Research Laboratory for the SEM and TGA analyses and the Department of Pure and Applied Chemistry for access to XRD facility. Dr Thomas Yip and Colin McKinstry are thanked for their help with XRD and nitrogen adsorption.References
- A. Nel, T. Xia, L. Madler and N. Li, Science, 2006, 311, 622 CrossRef CAS PubMed.
- C. A. Schutz, L. Juillerat-Jeanneret, H. Mueller, I. Lynch and M. Riediker, Nanomedicine, 2013, 8, 449 CrossRef PubMed.
- T. Doane and C. Burda, Adv. Drug Delivery Rev., 2013, 65, 607 CrossRef CAS PubMed.
- F. Q. Tang, L. L. Li and D. Chen, Adv. Mater., 2012, 24, 1504–1534 CrossRef CAS PubMed.
- S. Simovic, N. Ghouchi-Eskandar, A. M. Sinn, D. Losic and C. A. Prestidge, Curr. Drug Discovery Technol., 2011, 8, 250–268 CrossRef CAS.
- A. E. Garcia-Bennett, Nanomedicine, 2011, 6, 867 CrossRef CAS PubMed.
- C. J. Brinker and G. W. Scherer, Sol-Gel Science, Academic Press, Boston, 1990 Search PubMed.
- R. K. Iler, The Chemistry of Silica, John Wiley & Sons, New York, 1979 Search PubMed.
- Q. J. He, J. L. Shi, M. Zhu, Y. Chen and F. Chen, Microporous Mesoporous Mater., 2010, 131, 314 CrossRef CAS PubMed.
- E. M. Carlisle, Science, 1970, 167, 279 CAS.
- F. Sharif, F. Porta, A. H. Meijer, A. Kros and M. K. Richardson, Int. J. Nanomed., 2012, 7, 1875 CrossRef CAS PubMed.
- S. Tan, Q. X. Wu, J. Wang, Y. L. Wang, X. L. Liu, K. K. Sui, X. Y. Deng, H. F. Wang and M. H. Wu, Microporous Mesoporous Mater., 2011, 142, 601–608 CrossRef CAS PubMed.
- T. T. Wang, F. Chai, Q. Fu, L. Y. Zhang, H. Y. Liu, L. Li, Y. Liao, Z. M. Su, C. A. Wang, B. Y. Duan and D. X. Ren, J. Mater. Chem., 2011, 21, 5299–5306 RSC.
- P. Horcajada, A. Rámila, G. Férey and M. Vallet-Regí, Solid State Sci., 2006, 8, 1243–1249 CrossRef CAS PubMed.
- S. W. Song, K. Hidajat and S. Kawi, Chem. Commun., 2007, 4396–4398 RSC.
- C. Y. Lai, B. G. Trewyn, D. M. Jeftinija, K. Jeftinija, S. Xu, S. Jeftinija and V. S. Y. Lin, J. Am. Chem. Soc., 2003, 125, 4451–4459 CrossRef CAS PubMed.
- G. Paul, J. Heimink and H. Koller, Chem. Mater., 2008, 20, 5083–5089 CrossRef CAS.
- W. Shaobin, Microporous Mesoporous Mater., 2009, 117, 1–9 CrossRef PubMed.
- I. Izquierdo-Barba, Á. Martinez, A. L. Doadrio, J. Pérez-Pariente and M. Vallet-Regí, Eur. J. Pharm. Sci., 2005, 26, 365–373 CrossRef CAS PubMed.
- P. Horcajada, A. Rámila, J. Pérez-Pariente and R. Vallet, Microporous Mesoporous Mater., 2004, 68, 105–109 CrossRef CAS PubMed.
- H. Meng, T. Xia, S. George and A. E. Nel, ACS Nano, 2009, 3, 1620 CrossRef CAS PubMed.
- S. P. Hudson, R. F. Padera, R. Langer and D. S. Kohane, Biomaterials, 2008, 29, 4045 CrossRef CAS PubMed.
- P. Roach, T. Parker, N. Gadegaard and M. R. Alexander, Surf. Sci. Rep., 2010, 65, 145 CrossRef CAS PubMed.
- S. V. Patwardhan, Chem. Commun., 2011, 47, 7567–7582 RSC.
- D. E. Morse, Trends Biotechnol., 1999, 17, 230–232 CrossRef CAS.
- P. J. Lopez, C. Gautier, J. Livage and T. Coradin, Curr. Nanosci., 2005, 1, 73–83 CrossRef CAS.
- M. Hildebrand, Chem. Rev., 2008, 108, 4855–4874 CrossRef CAS PubMed.
- S. V. Patwardhan, S. J. Clarson and C. C. Perry, Chem. Commun., 2005, 1113–1121 RSC.
- D. J. Belton, S. V. Patwardhan, V. V. Annenkov, E. N. Danilovtseva and C. C. Perry, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5963–5968 CrossRef CAS PubMed.
- D. J. Belton, S. V. Patwardhan and C. C. Perry, J. Mater. Chem., 2005, 15, 4629–4638 RSC.
- S. V. Patwardhan and C. C. Perry, Silicon, 2010, 2, 33–39 CrossRef CAS PubMed.
- M. R. Knecht and D. W. Wright, Chem. Mater., 2004, 16, 4890–4895 CrossRef CAS.
- L. Betancor and H. R. Luckarift, Trends Biotechnol., 2008, 26, 566–572 CrossRef CAS PubMed.
- C. Forsyth and S. V. Patwardhan, J. Mater. Chem. B, 2013, 1, 1164–1174 RSC.
- C. Forsyth, T. W. S. Yip and S. V. Patwardhan, Chem. Commun., 2013, 49, 3191–3193 RSC.
- R. H. Jin and J. J. Yuan, Chem. Mater., 2006, 18, 3390–3396 CrossRef CAS.
- R.-H. Jin and J.-J. Yuan, in Advances in Biomimetics, ed. M. Cavrak, 2011, p. 159 Search PubMed.
- K. I. Sano, T. Minamisawa and K. Shiba, Langmuir, 2010, 26, 2231–2234 CrossRef CAS.
- K.-I. Sano, T. Minamisawa and K. Shiba, Langmuir, 2010, 26, 2231 CrossRef CAS PubMed.
- C. Forsyth and S. V. Patwardhan, J. Mater. Chem. B, 2013, 1, 1164 RSC.
- A. M. Ewlad-Ahmed, M. A. Morris, S. V. Patwardhan and L. T. Gibson, Environ. Sci. Technol., 2012, 46, 13354–13360 CrossRef CAS.
- C. Forsyth, T. W. S. Yip and S. V. Patwardhan, Chem. Commun., 2013, 49, 3191 RSC.
- A. Vinu, K. Z. Hossain and K. Ariga, J. Nanosci. Nanotechnol., 2005, 5, 347 CrossRef CAS.
- Y. L. Huang, B. G. Trewyn, H. T. Chen and V. S. Y. Lin, New J. Chem., 2008, 32, 1311 RSC.
- S. Shylesh, A. Wagner, A. Seifert, S. Ernst and W. R. Thiel, Chem.–Eur. J., 2009, 15, 7052 CrossRef CAS.
- R. Mee, A Comprehensive Guide to Factorial Two-Level Experimentation, Springer, New York, 2009 Search PubMed.
- G. Decher, Science, 1997, 277, 1232–1237 CrossRef CAS.
- S. Stoll and P. Chodanowski, Macromolecules, 2002, 35, 9556–9562 CrossRef CAS.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- R. de Vries and M. C. Stuart, Curr. Opin. Colloid Interface Sci., 2006, 11, 295–301 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 23–36 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25–35 CrossRef CAS.
- Y.-f. Zhu, J.-l. Shi, Y.-s. Li, H.-r. Chen, W.-h. Shen and X.-p. Dong, Microporous Mesoporous Mater., 2005, 85, 75–81 CrossRef CAS PubMed.
- P. L. Ritger and N. A. Peppas, J. Controlled Release, 1987, 5, 37–42 CrossRef CAS.
- T. Higuchi, J. Pharm. Sci., 1963, 52, 1145–1149 CrossRef CAS.
- J. C. Doadrio, E. M. B. Sousa, I. Izquierdo-Barba, A. L. Doadrio, J. Perez-Pariente and M. Vallet-Regi, J. Mater. Chem., 2006, 16, 462–466 RSC.
- A. L. Doadrio, E. M. B. Sousa, J. C. Doadrio, J. Pérez Pariente, I. Izquierdo-Barba and M. Vallet-Regí, J. Controlled Release, 2004, 97, 125–132 CrossRef CAS PubMed.
- S. V. Patwardhan, G. E. Tilburey and C. C. Perry, Langmuir, 2011, 27, 15135–15145 CrossRef CAS PubMed.
- S. R. Lewis, S. Datta, M. Gui, E. L. Coker, F. E. Huggins, S. Daunert, L. Bachas and D. Bhattacharyya, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 8577–8582 CrossRef PubMed.
- R. W. Sabnis, Handbook of Acid-Base Indicators, CRC Press, Florida, 2008 Search PubMed.
- C. Lad, N. H. Williams and R. Wolfenden, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5607–5610 CrossRef CAS PubMed.
- R. K. Iler, The chemistry of silica: Solubility, polymerization, colloid and surface properties and biochemistry, Wiley, New York, 1979 Search PubMed.
- C. Knox, V. Law, T. Jewison, P. Liu, S. Ly, A. Frolkis, A. Pon, K. Banco, C. Mak, V. Neveu, Y. Djoumbou, R. Eisner, A. C. Guo and D. S. Wishart, Nucleic Acids Res., 2011, 39, D1035–D1041 CrossRef CAS PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133 CrossRef CAS.
- D. Napierska, L. C. J. Thomassen, V. Rabolli, D. Lison, L. Gonzalez, M. Kirsch-Volders, J. A. Martens and P. H. Hoet, Small, 2009, 5, 846 CrossRef CAS PubMed.
- J.-S. Chang, K. L. B. Chang, D.-F. Hwang and Z.-L. Kong, Environ. Sci. Technol., 2007, 41, 2064 CrossRef CAS.
- T. Yu, A. Malugin and H. Ghandehari, ACS Nano, 2011, 5, 5717 CrossRef CAS.
- N. J. Hallab and J. J. Jacobs, Bull. NYU Hosp. Jt. Dis., 2009, 67, 182 Search PubMed.
- M. Lucarelli, A. M. Gatti, G. Savarino, P. Quattroni, L. Martinelli, E. Monari and D. Boraschi, Eur. Cytokine Network, 2004, 15, 339 CAS.
- S. V. Patwardhan, F. S. Emami, R. J. Berry, S. E. Jones, R. R. Naik, O. Deschaume, H. Heinz and C. C. Perry, J. Am. Chem. Soc., 2012, 134, 6244 CrossRef CAS PubMed.
- G. J. Hickman, A. Rai, D. J. Boocock, R. C. Rees and C. C. Perry, J. Mater. Chem., 2012, 22, 12141 RSC.
- M. N. V. Ravi Kumar, M. Sameti, S. S. Mohapatra, X. Kong, R. F. Lockey, U. Bakowsky, G. Lindenblatt, H. Schmidt and C. M. Lehr, J. Nanosci. Nanotechnol., 2004, 4, 876 CrossRef CAS PubMed.
- M. Akbar, A. R. Fraser, G. J. Graham, J. M. Brewer and M. H. Grant, J. R. Soc., Interface, 2012, 9, 2109 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4tb00510d |
| ‡ Shared first authorship. |
| § Present address: Technical University of Denmark, Department of Micro- and Nanotechnology, Ørsteds Plads, Building 345Ø, 2800 Kgs. Lyngby, Denmark. Email: anur@nanotech.dtu.dk |
| This journal is © The Royal Society of Chemistry 2014 |