 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Formation of hybrid poly(styrene-co-maleic anhydride)–silica microcapsules
Judith
van Wijk
a,
Nedine
van Deventer
b,
Elrika
Harmzen
b,
Jan
Meuldijk
a and
Bert
Klumperman
*b
aEindhoven University of Technology, Department of Chemical Engineering and Chemistry, P. O. Box 513, 5600 MB Eindhoven, The Netherlands
bStellenbosch University, Dept Chemistry and Polymer Science, Private Bag X1, Matieland 7602, South Africa. E-mail: bklump@sun.ac.za
First published on 15th May 2014
Abstract
In this contribution we report the synthesis of hybrid poly(styrene-co-maleic anhydride)–SiO2 microcapsules by cross-linking of the stabilizing particles of an inverse Pickering emulsion droplet at the interface. This was achieved by the ring-opening aminolysis reaction of the maleic anhydride residue of poly(St-co-MAh) with amine-functionalized silica particles that stabilized the Pickering emulsion. The cross-linking reaction is clearly shown by labeling the polymer with a green dye and the silica particles with a red dye, followed by confocal fluorescence microscopy analysis. Because poly(St-co-MAh) is a versatile polymer that can react with different other polymers, this opens the possibility of producing microcapsules with versatile properties. Encapsulation of delicate matter, e.g., live cells or enzymes, might be suitable as a result of the straightforward synthesis method.
Introduction
In view of different controlled release applications for microcapsules, the synthesis of microcapsules has gained increased attention over the past decade, e.g., in the food industry or in the drug industry.1–4 In the process of microencapsulation, typically a solid shell is formed around a micrometer-sized droplet. For controlled release applications, the shell material should be semi-permeable and should possess a certain strength and toughness, so that it does not easily break. This strength is among others necessary upon exposure to external forces, e.g., during re-dispersion of the capsules in a different medium.Pickering emulsion droplets are suitable templates for microencapsulation because of their extreme stability against coalescence.5,6 A Pickering emulsion is an emulsion solely stabilized by solid particles. The cross-linking of inorganic particles at the interface of a Pickering emulsion droplet proved to be a powerful method to produce microcapsules.7–10 In that situation, the interstitial space among the stabilizing particles of a Pickering emulsion droplet can be filled with a polymer. In order to synthesize microcapsules by templating Pickering emulsion droplets, a polymer is necessary that can interconnect the particles at the interface, also referred to as cross-linking of the microparticles at the interface. A very useful feature of the particles that stabilize the Pickering emulsion is that they should be easy to chemically modify. This will make it straightforward to tune the hydrophobicity and to make the particles suitable for stabilization of the emulsion.11–13 In addition, the same feature will allow the introduction of reactive groups for the cross-linking (interconnecting) reaction. Alternatively, the cross-linking polymer may also be able to react with a different polymer that can add responsive properties, and so the capsules are responsively permeable.
Poly(styrene-co-maleic anhydride) is a versatile polymer that can react with different other polymers, which results in tunable properties of the polymer material formed.14–16 Silica particles are known to be suitable for Pickering stabilization.11,17,18 This is largely caused by their tunable hydrophobicity upon modification.19,20 Modification of silica particles is possible because of the large number of reactive silanol groups at the surface. These silanol groups are also suitable for the introduction of reactive groups for the cross-linking reaction on the particle surface.
In this contribution, we report the facile synthesis of hybrid poly(St-co-MAh)–SiO2 microcapsules by cross-linking of the stabilizing particles of an inverse Pickering emulsion droplet, see Fig. 1. This was achieved by the ring-opening aminolysis reaction of the maleic anhydride residue of poly(St-co-MAh) with amine-functionalized stabilizing silica particles (Fig. 2 and 3), which produces an amide and a carboxylate group. The location of the components is clearly shown by labeling the polymer with a green dye and the silica particles with a red dye, followed by confocal fluorescence microscopy analysis. The cross-linking reaction is shown via a redispersion experiment, in aqueous media, followed by observation via light microscopy analysis. After redispersion in water, the capsules remain intact. If cross-linking of the particles that initially stabilized the interface had not happened, redispersion would result in a dispersion of silica microparticles and microcapsules would not be observed.
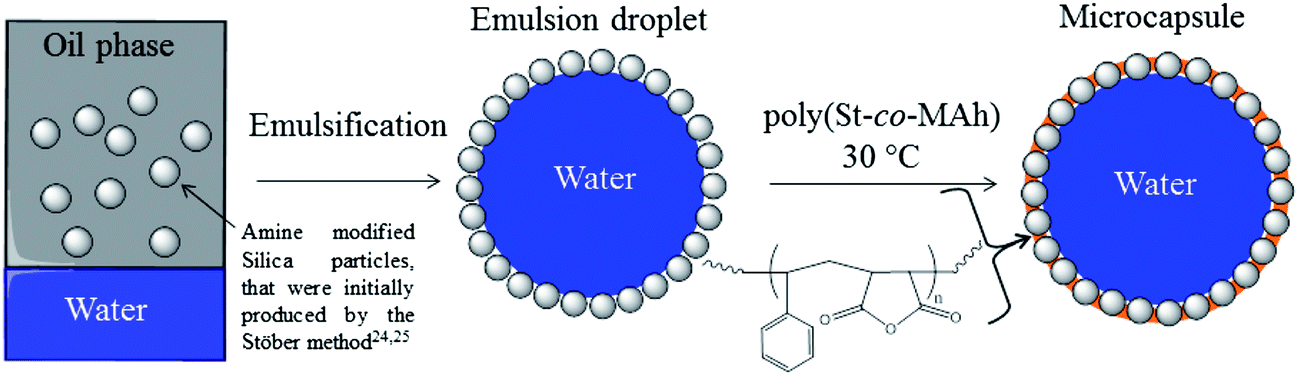 | ||
| Fig. 1 Overview of the synthesis route to produce hybrid poly(St-co-MAnh) SiO2 microcapsules. | ||
 | ||
| Fig. 2 Silica microparticles produced by the Stöber mechanism, followed by a seeded polymerization technique.25,26 | ||
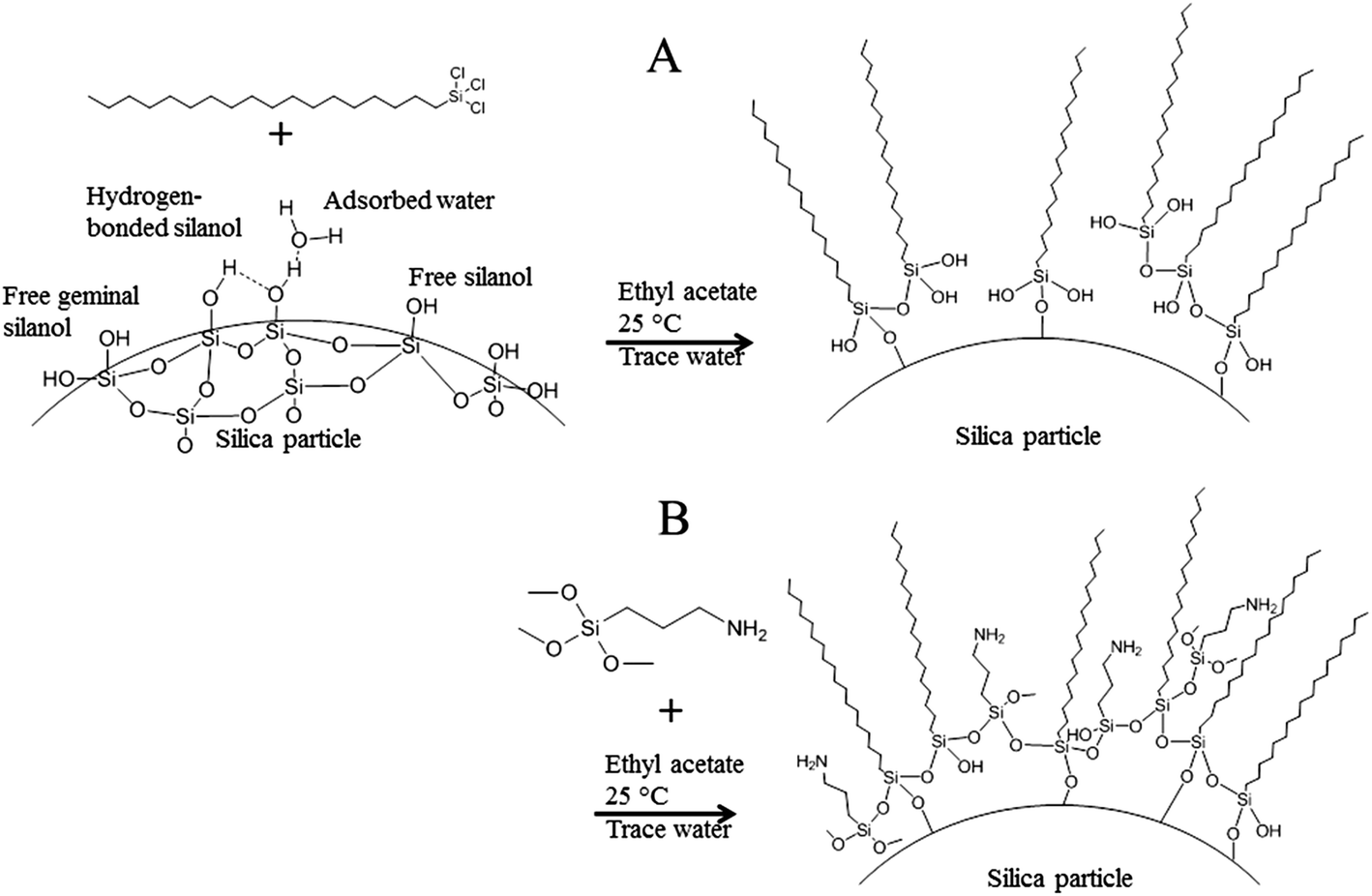 | ||
| Fig. 3 Modification steps of the silica microparticle surface. The reaction of octadecyltrichlorosilane (OTC) with the reactive silanol groups on the silica surface, with adsorbed water and with each other (A) to make the particles more hydrophobic. The reaction of residual hydroxy groups on the hydrophobized silica microparticles with aminopropyl trimethoxysilane (APTMS) (B). | ||
The main advantages of the synthesis procedure described above, e.g. Pickering emulsion polymerization, to produce microcapsules21 is firstly that the core is not contaminated in the process of capsule formation and secondly that the polymer is pre-synthesized before addition, which allows independent control of the polymer properties. Because the synthesis method is straightforward without severe condition changes in temperature or chemicals inside the microdroplets and microcapsules, this opens the possibility to encapsulate delicate materials. In addition, all Pickering emulsions were produced manually to avoid severe conditions, in this case due to high shear emulsification. Besides that, there is an option to use versatile polymers to cross-link the particles together at the oil–water interface, which opens the possibility to produce microcapsules that are responsive to external stimuli, which is a relatively new concept within the state of the art of microencapsulation.7,8
Results and discussion
Fig. 1 schematically shows the approach for the formation of poly(St-co-MAh)–SiO2 microcapsules. Initially, an inverse Pickering emulsion is produced, which is a water-in-oil emulsion that is stabilized by solid particles.5 The continuous phase of the inverse Pickering emulsion is chosen to be ethyl acetate and the stabilizing particles are modified silica particles. A strong indicator towards the stability of a Pickering emulsion is the three-phase contact angle that the particles have with the oil–water interface.22–24 The closer this contact angle is to ninety degrees, the higher the energy of detachment of the particles from the interface, which results in an emulsion with high stability.22 More than half of the volume of a particle is in the oil phase when its contact angle at the water–oil interface is larger than 90°. In that case, predominantly for geometric reasons, an inverse emulsion (w/o) is the most stable. The particles are then more hydrophobic in nature and from the literature it is known that an inverse Pickering emulsion is the most stable when the stabilizing particles have a three-phase contact angle with the interface between 94° and 110°.22In this work, silica particles that stabilize the inverse Pickering emulsion had to be modified for two reasons. Firstly, silica particles are very hydrophilic and as a consequence, their three phase contact angle is smaller than 10°.20 In the present study, the particles were hydrophobized to make them suitable for the stabilization of an inverse Pickering emulsion. Secondly, the silica particles were modified to provide them with primary amine containing groups for the reaction with MAh residues of poly(St-co-MAh).
The basic concept is that the poly(St-co-MAh) before ring-opening has a more hydrophobic nature and will therefore be oil-soluble. After ring-opening, the generated acid groups make the polymer much more hydrophilic. However, since ring-opening takes place during the reaction that links the polymer to the primary particles, the polymer does not have the opportunity to migrate into the aqueous phase. Reaction of the polymer with the primary amine functional groups on the SiO2 particles results in microcapsules with hydrophilic properties, which makes the capsules easy to re-disperse in water or other hydrophilic fluids at a later stage. In practice, first, water is added to a dispersion of hydrophobized and amine-functionalized silica microparticles in ethyl acetate. After emulsification, a stable inverse Pickering emulsion is produced. To the Pickering emulsion, poly(St-co-MAh) is added, which reacts with the amine functional groups on the particle surfaces and this process links the particles together, see Fig. 1.
The silica particles were synthesized using the well-known Stöber mechanism to produce monodisperse seed silica particles, followed by a seeded polymerization technique.25,26 A seeded technique was used since it was proven to be difficult to produce monodisperse silica particles in the micron-size range via a one-step Stöber process.25 The silica particles were then dried, re-dispersed in ethyl acetate and modified by two different modification agents. After the modification steps, the particles were re-dispersed twice in ethyl acetate to get rid of residual modification agents. Fig. 2 shows typical SEM images of the produced monodisperse silica particles. A narrow particle size distribution is a prerequisite, since the surface of the silica particles is the basis for calculation of the amount of modification agents in the formulation. The diameter of the silica particles is also necessary to calculate the recipe of the inverse Pickering emulsion.
To tune the hydrophobicity of the silica microparticles, octadecyltrichlorosilane (OTC) was used as the first modification agent, see Fig. 3A. OTC is known to be a suitable modifier.19,20 To determine the surface concentration of OTC that results in the most stable Pickering emulsion, the silica particles were reacted with different concentrations of OTC. Subsequently, the silica particles with varying hydrophobicities were used to produce an inverse Pickering emulsion. The particle surface concentration of OTC that resulted in the most stable inverse Pickering emulsions was determined to be 18 μmol m−2, Fig. 4. For this system, phase separation did not take place within days after emulsification. The average concentration of reactive silanol groups on the surface of silica particles that are produced by the Stöber method is 8 μmol m−2, Fig. 3A.27,28 This means that the introduction of OTC on the surface of the particles proceeds via polymerization of the trihydroxy silicate moiety. A hydroxy group is produced by the reaction of a chloride moiety of OTC with water.29 There is plenty of water present in the particle dispersion mixture, since the silica particles were not dried under vacuum or/and high temperature and water physically adsorbs onto amorphous silica, Fig. 3A.30 Besides that, the ethyl acetate that was used was not dried before use. These two drying steps are unnecessary, since the particles will subsequently be used in a water–oil emulsion. In addition, the three-phase contact angle of the particles could be tuned satisfactorily although the reaction was not performed under dry conditions. As a result of the reaction of OTC, not only with the particle surface, but also with water and with each other, a monolayer of OTC will not be formed on the surface.31,32 Instead, an OTC-based network structure will be produced and sufficient hydroxy groups will be left for the next modification step, Fig. 3.
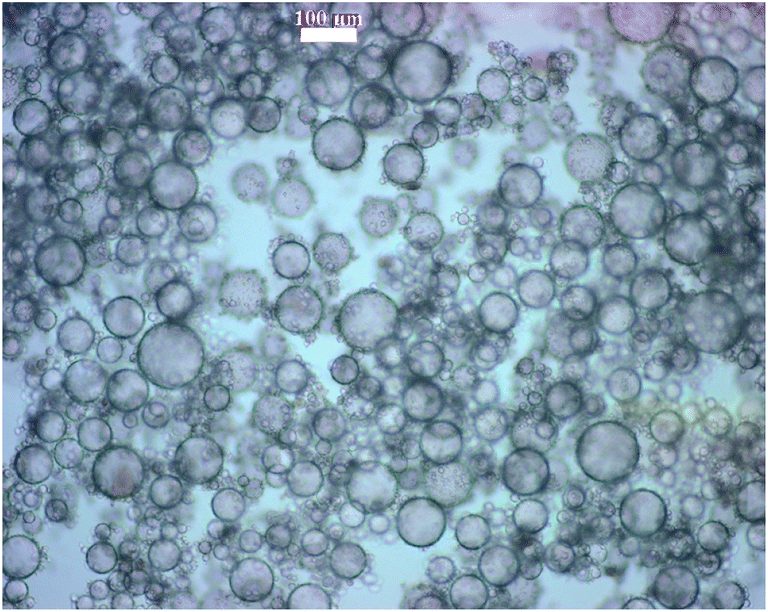 | ||
| Fig. 4 Light microscopy image of an inverse Pickering emulsion. The Pickering emulsion was produced by the addition of 3 mL distilled water to 0.35 g silica particles (dp = 740 nm) dispersed in 40 mL ethyl acetate. The particles were functionalized with 18 μmol m−2 OTC and 90 μmol m−2 APTMS. | ||
To give the particles reactive functional groups for the cross-linking with poly(St-co-MAh), aminopropyl trimethoxysilane (APTMS) was added. An excess of APTMS was used (90 μmol m−2) to ensure that sufficient amine groups were present at the surface of the silica microparticles for the cross-linking reaction, Fig. 3B. After the second modification step, the particles were still suitable for inverse Pickering stabilization. After redispersion of the modified silica particles in ethyl acetate, to get rid of the unreacted agents, water was added and upon emulsification of this mixture, the Pickering emulsion was produced, Fig. 4.
The different concentrations in the inverse Pickering emulsion were calculated according to eqn (1), in which the droplet diameter is always set at 50 μm, unless indicated otherwise.33
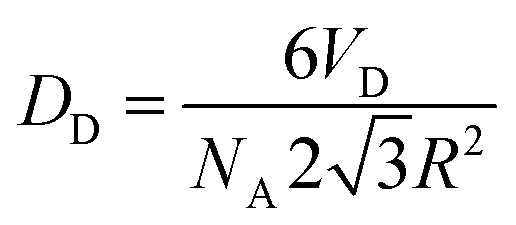 | (1) |
In eqn (1), NA is the total number of particles attached to the interface of the total amount of droplets and when the particles are hexagonally close-packed. R is the radius of the modified silica particles, the area that a particle occupies is 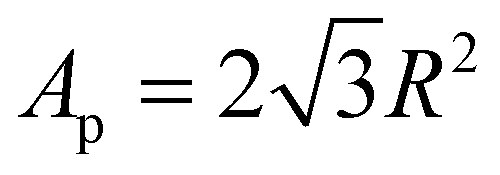 . VD is the total volume of the dispersed phase and D is the diameter of the emulsion droplets. After the addition of water, emulsification was achieved by manually shaking the mixture for 15 to 30 seconds.
. VD is the total volume of the dispersed phase and D is the diameter of the emulsion droplets. After the addition of water, emulsification was achieved by manually shaking the mixture for 15 to 30 seconds.
Since the Pickering emulsions were produced manually, exact control of the droplet size and the droplet size dispersity proved to be difficult, compared to emulsification using high shear, see Fig. 4.33 It takes a certain force to position a particle at the interface and a certain time to position them all at the interface. Manual emulsification was selected to avoid severe conditions, since high shear is not desirable in the case of encapsulation of delicate materials, e.g., live bacteria.
After a stable inverse Pickering emulsion was produced, it was charged into a three-neck round bottom flask with overhead stirring. To the stable inverse Pickering emulsion, a certain amount of a poly(St-co-MAh) solution in ethyl acetate was added and cross-linking took place within several minutes.34 This could be observed, since some flocculation of the microcapsules in the continuous oil phase occurred. The hydrophobized silica particles that initially stabilize the inverse Pickering emulsion are now covered by the polymer that has a more hydrophilic character, which is deemed responsible for the flocculation. In order for the cross-linking reaction to be successful, the concentration of poly(St-co-MAh) had to be optimized. If an insufficient amount of polymer is added, not all silica microparticles at the interface of the emulsion droplets will be linked together. However, if an excess of polymer is used, every amine group will just react with one polymer chain, which would also lead to unsuccessful cross-linking of the particles, Fig. 5B. The alternating poly(St-co-MAh) that was used had a number average molecular weight (Mn) of 48.4 × 103 g mol−1, the average number of MAh residues in one chain therefore is 239 (MSTY = 104 and MMAh = 98 g mol−1). To every inverse Pickering emulsion, 0.14 μmol poly(St-co-MAh) chains per m2 of particle interface was added. An amount of 90 μmol aminopropyl trimethoxysilane per m2 of particle interface was used to provide the silica microparticles with reactive groups. Consequently, it can be assumed that one polymer chain will react with more than one amine group, and bridge between particles, which results in cross-linking of the particles, Fig. 5A.
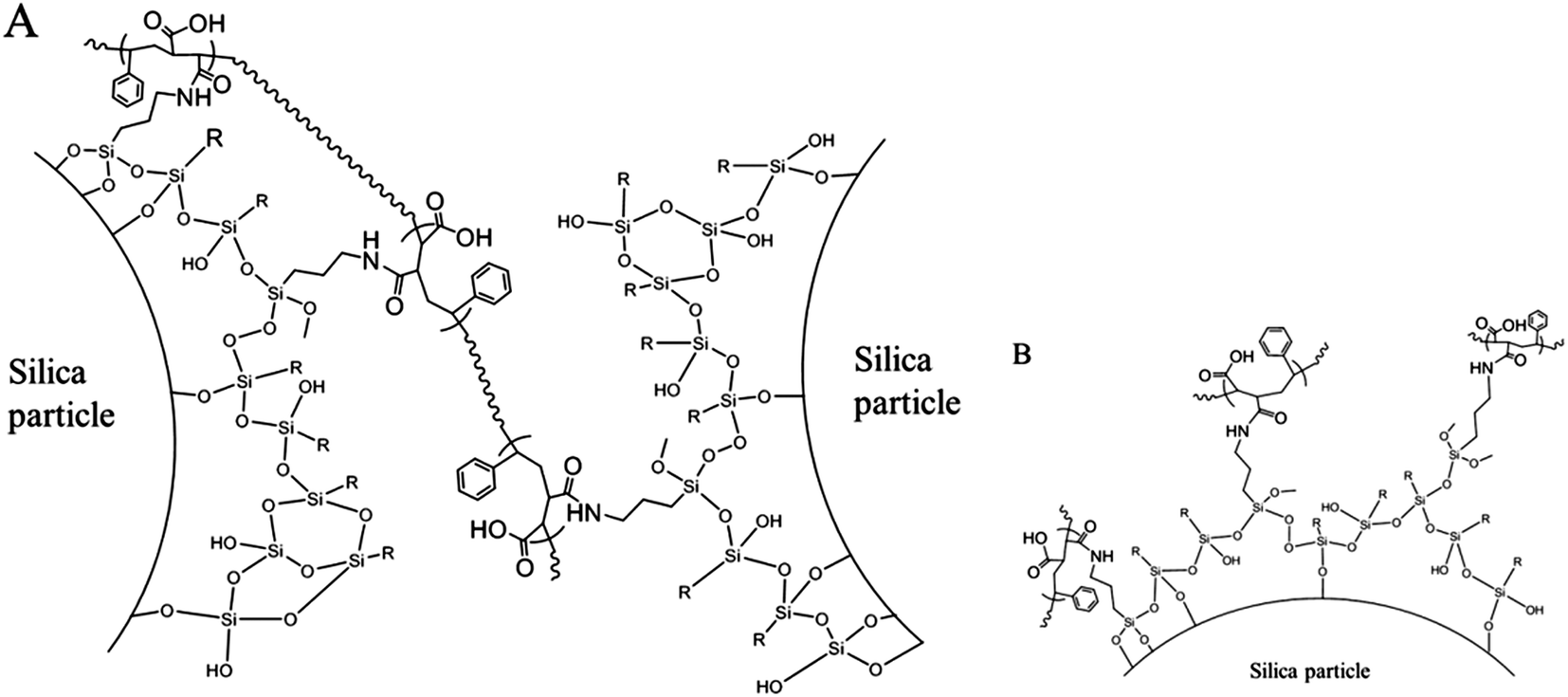 | ||
| Fig. 5 Schematic illustration of the cross-linking of amine and octadecyl (–R) functionalized silica microparticles with poly(St-co-MAh). Successful cross-linking will be the result if one polymer chain reacts with more than one amine group on different particles (A), otherwise cross-linking will not take place (B). | ||
After addition of poly(St-co-MAh) to an inverse Pickering emulsion stabilized by OTC and APTMS-modified silica microparticles, hybrid poly(St-co-MAh)–SiO2 microcapsules were produced. When the stabilizing silica particles were not amine-functionalized, the capsule forming reaction did not take place. This is evidenced by an additional experiment in which non-amine functionalized microparticles were used to stabilize the Pickering emulsion and all other conditions were kept the same. In the case of amine-functionalized particles, after the aminolysis reaction of poly(St-co-MAh), the produced carboxylic acid residues provide the polymer with more hydrophilic properties. These carboxylic acid groups consequently result in the aggregation of the produced hybrid microcapsules. Aggregation can be visually observed by a clean transparent continuous phase. When the primary stabilizing particles were not amine-functionalized, this was not observed. Also, when light microscopy was used as an analysis method, no microcapsules were detected when using SiO2 particles that are not amine-functionalized.
Fig. 6A shows scanning electron microscopy (SEM) images of the produced microcapsules. The SEM images are unable to reveal the presence of poly(St-co-MAh) on the surface of the microcapsules, apart from a few exceptions. In contrast to the formation of a robust shell with a thickness of (at least) a few nanometers,35 in this case only a new polymer is covering the silica microparticles and bonding them together, which is virtually impossible to detect with SEM. The most important observation from the SEM images is that the microcapsules remain intact, even though they usually collapse under high vacuum conditions. In the absence of cross-linking, the microcapsules do not form and the SEM images only show individual SiO2 particles, or clusters, but no microcapsules. In order to collect additional evidence for the successful cross-linking of the particles, light microscopy (LM) analysis was used. Before LM analysis, the microcapsules were superficially dried and redispersed in water with a trace of acetone (1 wt%). Isolation of the capsules was relatively easy, since they settled readily due to the gravity (water droplets of 50 μm in diameter, dispersed in a lower density organic solvent), after which the continuous phase was decanted. Redispersion of the capsules in water was favored by their hydrophilic properties. Although the microcapsules can be re-dispersed in pure water, the process can be (and was in the current study) further enhanced by the addition of some acetone to increase the solubility of the residual ethyl acetate. Due to the permeability of the capsules, the fluid is the same inside and outside the capsules, after redispersion. As a consequence, they settle much slower after redispersion, namely hours relative to seconds. However, the most important observation from this redispersion experiment is that the capsules remain intact. If cross-linking of the particles that initially stabilized the interface had not happened, redispersion in aqueous media would result in a dispersion of microparticles, and microcapsules would not be observed with LM. Fig. 6B shows LM images of poly(St-co-MAh)–SiO2 microcapsules that were redispersed in water with a trace of acetone. Evidently, the cross-linking reaction took place as intact microcapsules were observed, Fig. 6B. The debris observed in Fig. 6B is most probably due to particle aggregates. This can be caused by, e.g., insufficient re-dispersion of the microparticles in ethyl acetate before modification. Furthermore, since emulsification took place manually, the size distribution in the primary Pickering emulsion was broad in comparison to the scenario where high shear would have been used. Therefore, residual microparticles were present in the continuous phase and aggregated with the microparticles at the interface of the microdroplets.
 | ||
| Fig. 6 Scanning electron microscopy (A) and light microscopy (B) images of hybrid poly(St-co-MAh)–SiO2 microcapsules, produced by poly(St-co-MAh)-induced cross-linking of the stabilizing silica microparticles of an inverse Pickering emulsion at the interface. | ||
Although it can now be concluded that poly(St-co-MAh)–SiO2 microcapsules have been produced (Fig. 6), it is not exactly clear which fraction of the polymer reacted with the amine-functionalized particles. It is possible that only part of the polymer reacted, which would lead to a residual polymer in the oil phase, or as a result of ring-opening, the polymer may have diffused into the dispersed water phase. In an additional experiment, silica microparticles were labeled with a red fluorescent dye and were cross-linked with poly(St-co-MAh) that was labeled with a green fluorescent dye in order to synthesize microcapsules. Confocal Fluorescence Microscopy (CFM) allowed the determination of the individual locations of the microparticles and the polymer, see Fig. 8. The red fluorescently labeled silica microparticles were synthesized by the addition of a very small amount of labeled silica precursor during the synthesis procedure. The labeled silica precursor in turn was synthesized by the addition reaction of rhodamine B isothiocyanate with aminopropyl trimethoxysilane, see Fig. 7.36 The green fluorescent poly(St-co-MAh) was produced by the partial aminolysis of MAh with 5-aminofluorescein, see Fig. 7. An amount of 1 mol% of 5-aminofluorescein was used relative to the MAh groups in the polymer. Hence, sufficient MAh groups were left for the cross-linking reaction.
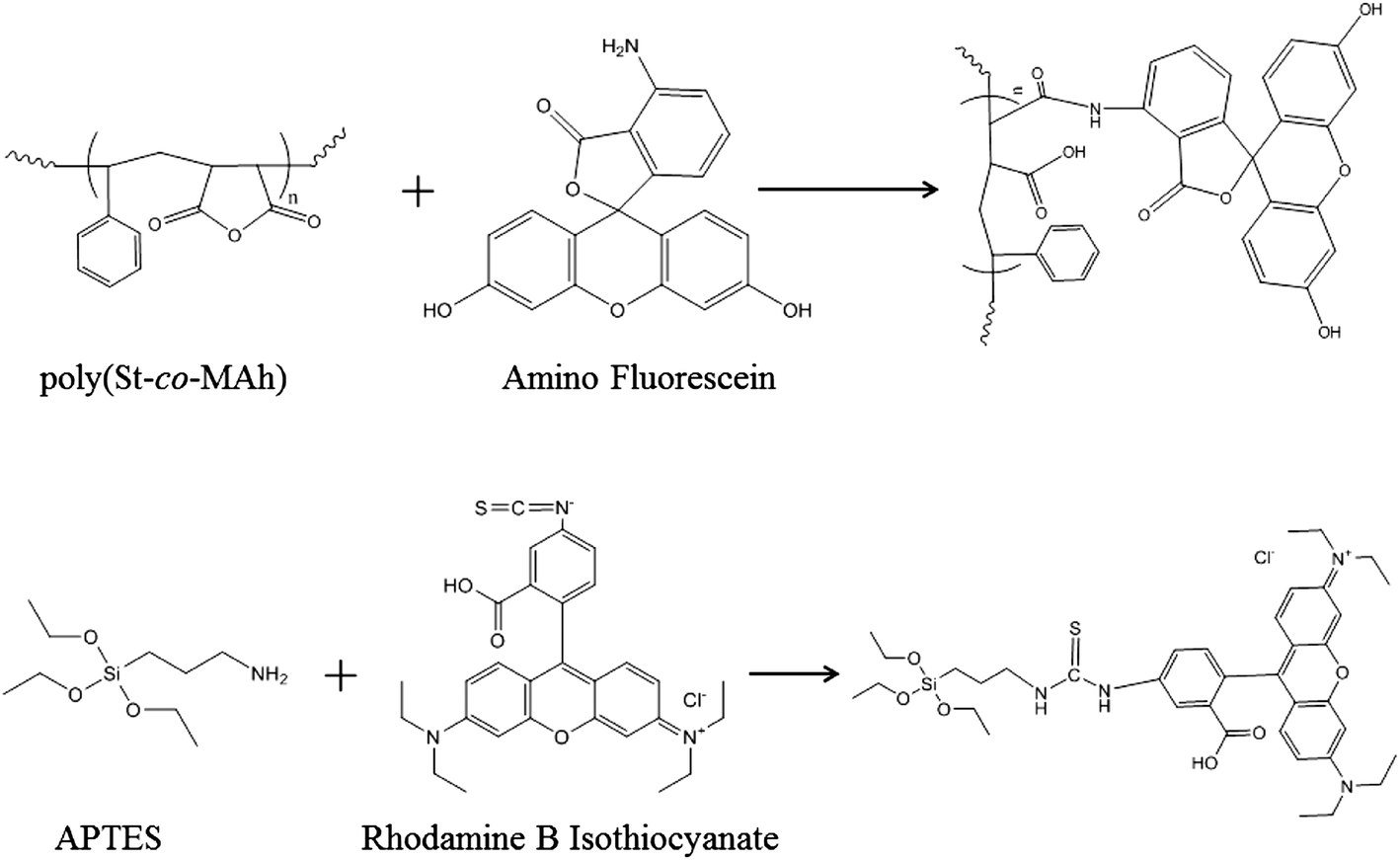 | ||
| Fig. 7 Synthetic scheme for the labeling of poly(St-co-MAh) with 5-aminofluorescein and of APTMS with rhodamine B isothiocyanate. | ||
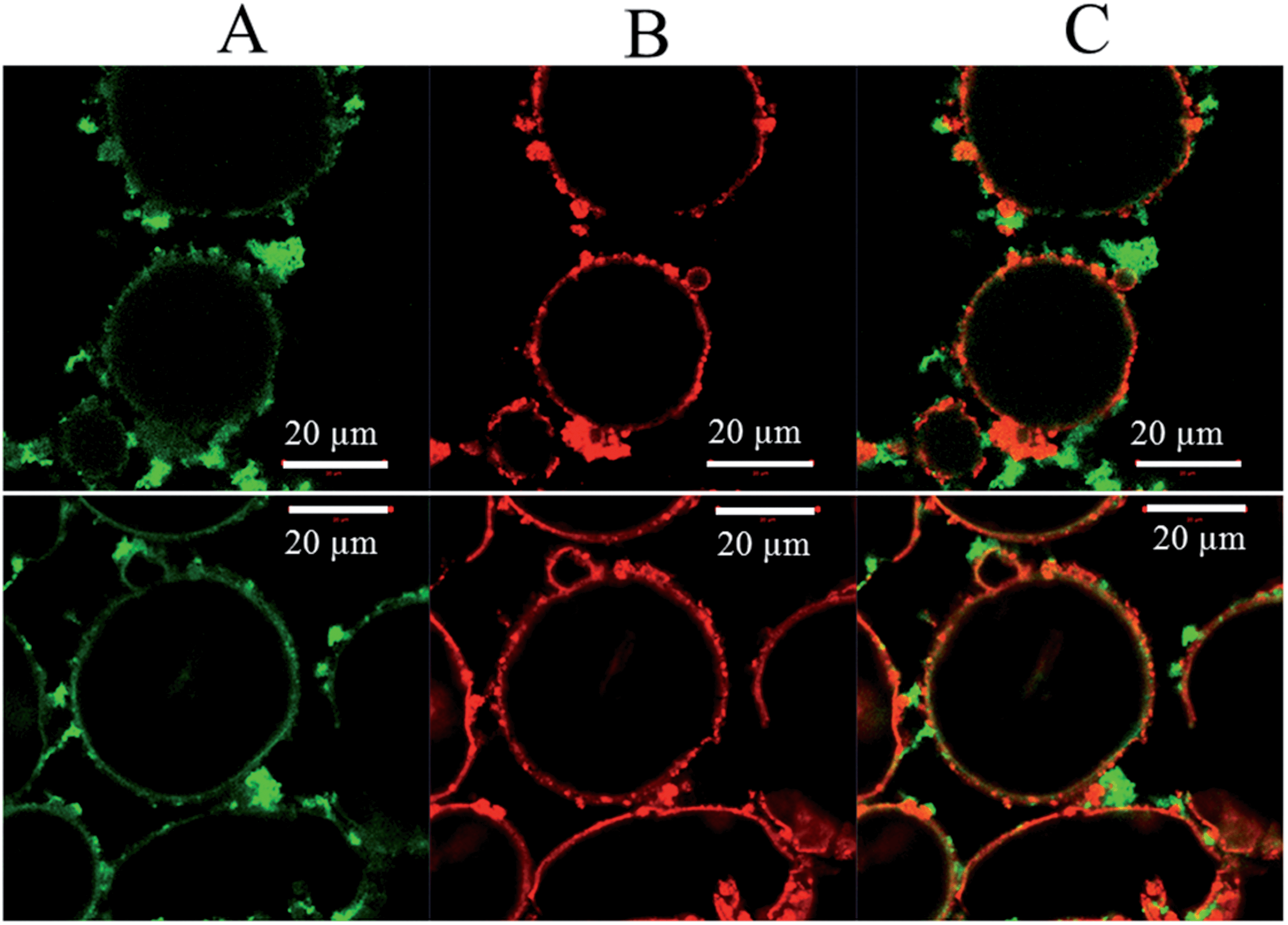 | ||
| Fig. 8 Confocal fluorescence microscopy images of hybrid poly(St-co-MAh)–SiO2 microcapsules (C). Produced with green fluorescein-labeled poly(St-co-MAh) (A) and red rhodamine B-labeled silica microparticles (B). | ||
As a result, after the microcapsule synthesis with the labeled precursors, the silica particles that initially stabilized the emulsion could be observed, see Fig. 8B. Also, the labeled polymer could be observed, Fig. 8A. When the two individual images are overlaid, it can be concluded that the majority of the polymer is in contact with the amine-functionalized particles, see Fig. 8C. In addition, hardly any green poly(St-co-MAh) was observed in the oil or water phase. This means that almost all the polymer is in close proximity of the silica shell around the water droplets.
When excess polymer was added, this could be observed by the green dye in the continuous ethyl acetate phase, Fig. 9. In that case, most probably all the amine groups on the modified primary stabilizing silica particles had reacted. It is noticeable that in this specific experiment, the poly(St-co-MAh) does not seem to react with water. Such a hydrolysis reaction would increase the hydrophilicity of the polymer, which would then be expected to migrate into the water phase. Instead it resides in the continuous oil phase, Fig. 9A and C.
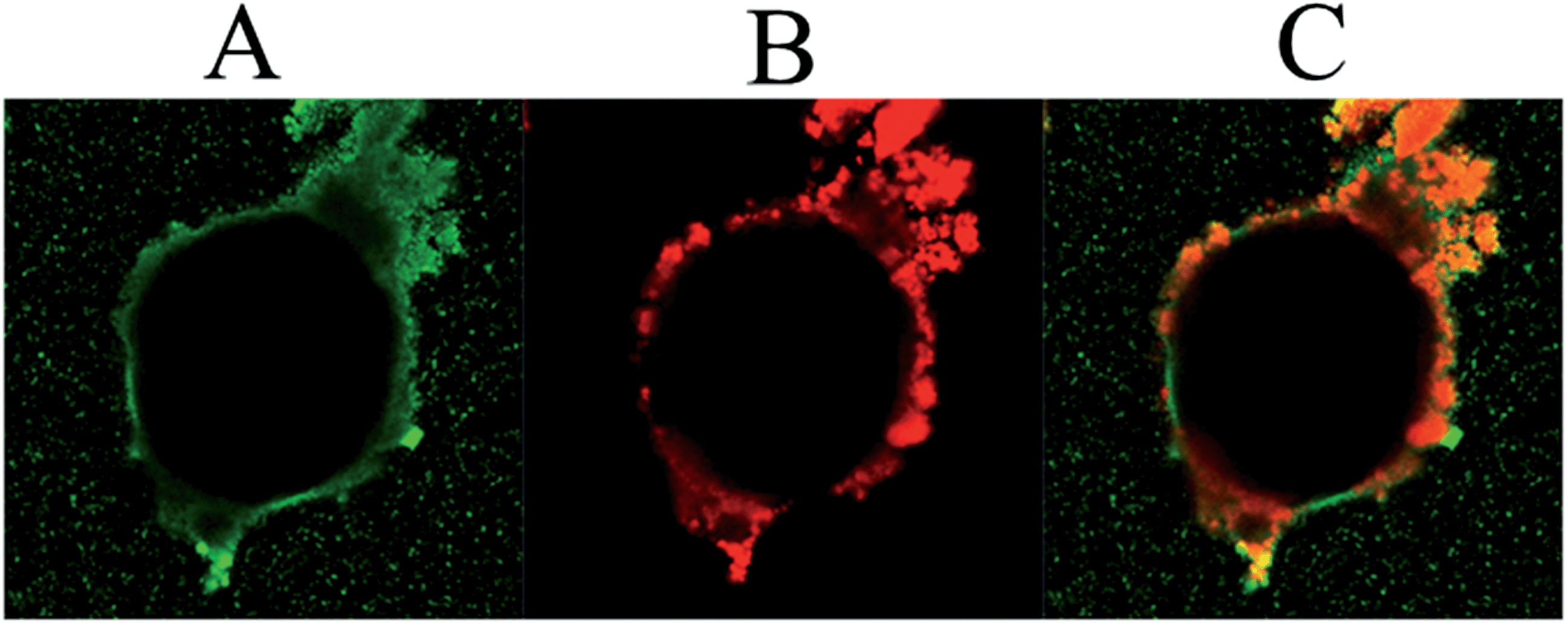 | ||
| Fig. 9 Confocal fluorescence microscopy images of hybrid poly(St-co-MAh)–SiO2 microcapsules (C). Produced with excess green fluorescein-labeled poly(St-co-MAh) (A) and red rhodamine B-labeled silica microparticles (B). | ||
Conclusion
Hybrid poly(styrene-co-maleic anhydride)–silica microcapsules were successfully synthesized by templating inverse Pickering emulsion droplets. The inverse Pickering emulsion droplets were stabilized by surface-modified silica microparticles. The surface modification was carried out in a two-step process. Initially, the surface was hydrophobized using octadecyl trichlorosilane. Subsequently, the remaining hydroxy groups were reacted with aminopropyl trimethoxysilane, which resulted in reactive amine groups on the silica surface. After addition of poly(styrene-co-maleic anhydride) to an inverse Pickering emulsion stabilized with amine-functionalized silica microparticles, cross-linking of the microparticles at the interface took place and microcapsules were produced.Since poly(styrene-co-maleic anhydride) is a versatile polymer that can be reacted with different other polymers, this opens the possibility of producing microcapsules with versatile properties. Furthermore, because of the straightforward synthesis method the procedure might be applicable to the encapsulation of delicate materials, for example live cells or enzymes. Future work will also combine the present work with the use of microfluidic devices to allow the formation of uniform capsule sizes under low-shear conditions.37,38
Materials
All chemicals were used as received, unless indicated otherwise. Tetraethyl orthosilicate (TEOS), (3-aminopropyl)trimethoxysilane (99%) (APTMS), octadecyl trichlorosilane (>90%), 6-aminofluorescein (C20H13NO5, λex 590 nm, λem 520 nm in 0.1 Tris pH 9) and rhodamine B isothiocyanate (C29H30ClN3O3S, λex 543 nm, λem 580 nm in methanol) were purchased from Sigma-Aldrich. Ethanol anhydrous (99.8%) and ammonia 32% were purchased from Merck-Chemicals. Ethanol absolute (dehydrated AR) was purchased from Biosolve. The water used was double de-ionized water from an Elix Millipore purification system. Styrene monomer was purchased from Fluka Chemika (99.5%). Maleic anhydride (99%), methyl ethyl ketone (≥99.7%), tetrahydrofuran (CHROMASOLV® Plus, for HPLC, ≥99.9%) and deuterated acetone (99.9 atom%, acetone-d6) were purchased from Sigma-Aldrich. 2,2′-Azo-bis(isobutyronitrile) (AIBN) was purchased from Riedel de Haen, recrystallized twice using methanol and dried under vacuum before use.Methods
The silica microparticles were produced by an initial synthesis of monodisperse seed silica microparticles (±500 nm) using the well-known Stöber technique.26 A three-neck round bottom flask was charged with ethanol (100 g), TEOS (7 g) and water (5 mL). The mixture was left to stir for 10 minutes with a magnetic stirrer at 30 °C. Subsequently, an ammonia solution (15 mL, 25%) was added and the reaction was conducted for 6 hours. The particles were grown through a seeded polymerization technique to the required diameter, while retaining a narrow size distribution.25 The seeded polymerization entailed the addition of five aliquots of TEOS (2 g), which were added at 6 hour intervals.The precursor for the dye-labeled silica particles was synthesized by the addition reaction of rhodamine B isothiocyanate with aminopropyl trimethoxysilane, according to a procedure described by van Blaaderen et al.36,39 Briefly, in 5 mL anhydrous ethanol, aminopropyl trimethoxysilane (0.2 mmol, 44 mg) was reacted with rhodamine B isothiocyanate (0.1 mmol, 53 mg). The reaction was allowed to proceed for 17 hours in a nitrogen atmosphere.
The labeled silica microparticles were synthesized by the addition of 0.05 mL of the above-described mixture (0.1 mmol, 97 mg, labeled silica precursor in 5 mL EtOH) during the synthesis of the seed silica microparticles.36,39
Modification of the surface of the microparticles proceeded in a three-neck round bottom flask under continuous magnetic stirring at 30 °C. Before modification, centrifugation was used to separate the microparticles from the ethanol solution, after which the ethanol solution was decanted and the particles were air-dried. In a typical modification procedure, silica microparticles (0.5 g) with an average diameter of 1 μm were dispersed in ethyl acetate (10 g). If a density of 2.15 [g cm−3] is used, the total surface area of the particles in the reaction mixture is 2.79 m2.40 To the microparticles, OTC (18 μmol m−2, i.e. 50 μmol, 19 mg) was added. The reaction was allowed to proceed for 24 hours. Subsequently, APTMS (90 μmol m−2, i.e. 251 μmol, 55 mg) was added to the mixture, and the reaction was continued for 24 hours. After modification, centrifugation was again used to isolate the particles and the ethyl acetate phase was decanted. The particles were air-dried, after which they were redispersed in ethyl acetate, to get rid of the unreacted modification agents.
A typical Pickering emulsion was produced by the addition of water (1 mL) to modified silica microparticles (8 × 10−2 g) dispersed in ethyl acetate (15 g). The droplets were calculated to have a diameter of 50 μm, see eqn (1). The Pickering emulsion was produced manually by vigorous shaking of the mixture for about 30 s.
The poly(styrene-co-maleic anhydride)–silica microcapsules were synthesized by the addition of poly(St-co-MAh) (0.14 mol per m2 of silica particles) to a stable Pickering emulsion. So, if the above described Pickering emulsion was used, the total surface area of the modified silica microparticles (8 × 10−2 g), with a diameter of 1 μm, is equal to 0.45 m2. Therefore, poly(St-co-MAh) (6.3 × 10−2 μmol, 3 mg) was added to the emulsion. The reaction was continued for 1 hour under continuous overhead stirring at 30 °C. The poly(St-co-MAh) was solubilized in ethyl acetate (10 mL) before addition.
Alternating poly(styrene-co-maleic anhydride) was synthesized by conventional radical copolymerization of styrene and maleic anhydride monomers in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of styrene:maleic anhydride.41
:1 molar ratio of styrene:maleic anhydride.41
In methyl ethyl ketone (250 mL) were dissolved maleic anhydride (192 mmol, 18.82 g), styrene (192 mmol, 19.99 g) and 2,2′-azobis(isobutyronitrile) (3.96 mmol, 0.65 g). The reaction mixture was purged with argon (45 min) at room temperature and placed in a preheated oil bath (60 °C). Subsequently, the argon needle was taken out of the solution, but kept in the round bottom flask for another 50 min. The reaction was conducted for an additional 15 hours. The polymer solution was allowed to cool to room temperature, after which the polymer was precipitated in diethyl ether. The polymer was dried under vacuum at 80 °C for 1.5 hours and left under vacuum for 16 hours to remove any unreacted monomer and residual solvent. Analyses were done using SEC, which resulted in Mn = 48.4 × 103 [g mol−1], Đ = 4.09 and a MAh content of 50 mol%. 1H-NMR analysis was used to calculate the respective maleic anhydride contents, this was done by integrating the ratio of the aromatic protons of the styrene residue to the methine protons of the maleic anhydride residue. It was determined that the poly(St-co-MAnh) was alternating, i.e., a 1:1 ratio of maleic anhydride to styrene (50%) was found.
Analysis
Scanning Electron Microscopy (SEM) was used for imaging of the microparticles and microcapsules. SEM was performed using a Zeiss Evo MA15VP scanning electron microscope. Samples were prepared by placing a droplet of the sample on a sample holder, which was covered by double sided carbon tape. After the sample had dried, it was coated with a thin layer of gold. The gold was used to ensure good conductivity during the measurements.Light microscopy (LM) was used for the imaging of the inverse Pickering emulsion and the microcapsules. An Olympus CX31 Light Microscope was used to perform light microscopy. The samples were prepared by placing a droplet of sample on a glass slide after which it was placed on the specimen stage.
The Confocal Fluorescence Microscopy (CFM) images of the labelled microcapsules were visualized with a Carl Zeiss LSM 780 with an Elyra S.1 superresolution platform. The images were taken using a 561 nm (100 mW) laser (red) and a 488 nm (100 mW) laser (green). Sample preparation was done by placing a droplet of sample in a glass microscope dish, for the sample to be measured in solution.
Size Exclusion Chromatography (SEC) was used to determine the molar mass and dispersity of poly(styrene-co-maleic anhydride). Two PLgel (Polymer Laboratories) 5 μm Mixed-C (300 × 7.5 mm) columns and a pre-column (PLgel 5 μm Guard, 50 × 7.5 mm) were used. The SEC instrument consists of a Waters 1515 isocratic HPLC pump, a Waters 717plus auto-sampler, Waters 600E system controller (run by Breeze Version 3.30 SPA) and a Waters in-line Degasser AF. A Waters 2414 differential refractometer was used at 30 °C in series with a Waters 2487 dual wavelength absorbance UV/vis detector operating at variable wavelengths. Tetrahydrofuran (THF, HPLC grade, stabilized with 0.125% BHT) was used as the eluent at a flow rate of 1 mL min−1. The column oven was kept at 30 °C and the injection volume was 100 μL. Calibration was done using narrow polystyrene standards ranging from 580 to 2 × 106 g mol−1. All molecular weights were reported as polystyrene equivalents. Sample preparation was done by dissolving the sample in BHT stabilized THF (2 mg mL−1). Sample solutions were filtered via a syringe through 0.45 μm nylon filters before being subjected to analysis.
Nuclear magnetic resonance spectroscopy (1H-NMR) was used to verify whether the synthesis of poly(styrene-co-maleic anhydride) resulted in an alternating copolymer. Therefore, the spectra were obtained on a Varian Unity INOVA 400 MHz spectrometer with a pulse width of 3 μs (45°) and a 2 second acquisition time. The samples were prepared using deuterated acetone.
References
- V. Nedovic, A. Kalusevic, V. Manojlovic, S. Levic and B. Bugarski, An Overview of Encapsulation Technologies for Food Applications, Procedia Food Sci., 2011, 1, 1806–1815 CrossRef CAS PubMed.
- J.-X. Wang, Z.-H. Wang, J.-F. Chen and J. Yun, Direct Encapsulation of Water-Soluble Drug into Silica Microcapsules for Sustained Release Applications, Mater. Res. Bull., 2008, 43, 3374–3381 CrossRef CAS PubMed.
- B. Mishra, B. B. Patel and S. Tiwari, Colloidal Nanocarriers: A Review on Formulation Technology, Types and Applications toward Targeted Drug Delivery, Nanomedicine, 2010, 6, 9–24 CrossRef CAS PubMed.
- G. Orive, E. Santos, J. L. Pedraz and R. M. Hernández, Application of Cell Encapsulation for Controlled Delivery of Biological Therapeutics, Adv. Drug Delivery Rev., 2013, 65, 0–12 Search PubMed.
- S. U. Pickering, Pickering: Emulsions, J. Chem. Soc., 1907, 91, 2001–2021 RSC.
- W. Ramsden, Of Solids in the Surface-Layers of Solutions and Suspensions (Observations on Surface-Membranes, Bubbles, Emulsions, and Mechanical Coagulation), Proc. R. Soc. London, Ser. A, 1903, 72, 156–164 CrossRef CAS.
- M. Li, R. L. Harbron, J. V. M. Weaver, B. P. Binks and S. Mann, Electrostatically Gated Membrane Permeability in Inorganic Protocells, Nat. Chem., 2013, 5, 529–536 CrossRef CAS PubMed.
- M. Li; R. L. Harbron; J. V. M. Weaver; B. P. Binks; S. Mann, in Inorganic Protocells, 2013, vol. 5, pp. 529–536 Search PubMed.
- M. F. Hsu, M. G. Nikolaides, A. D. Dinsmore, A. R. Bausch, V. D. Gordon, X. Chen, J. W. Hutchinson, D. a Weitz and M. Marquez, Self-Assembled Shells Composed of Colloidal Particles: Fabrication and Characterization, Langmuir, 2005, 21, 2963–2970 CrossRef CAS PubMed.
- K. L. Thompson and S. P. Armes, From Well-Defined Macromonomers to Sterically-Stabilised Latexes to Covalently Cross-Linkable Colloidosomes: Exerting Control over Multiple Length Scales, Chem. Commun., 2010, 46, 5274–5276 RSC.
- A. K. F. Dyab, Destabilisation of Pickering Emulsions Using pH, Colloid. Surface. Physicochem. Eng. Aspect., 2012, 402, 2–12 CrossRef CAS PubMed.
- J. W. O. Salari, F. a M. Leermakers and B. Klumperman, Pickering Emulsions: Wetting and Colloidal Stability of Hairy Particles a Self Consistent Field Theory, Langmuir, 2011, 27, 6574–6583 CrossRef CAS PubMed.
- B. P. Binks and S. O. Lumsdon, Influence of Particle Wettability on the Type and Stability of Surfactant-Free Emulsions, Langmuir, 2000, 16, 8622–8631 CrossRef CAS.
- J.-J. Lin and Y.-C. Hsu, Temperature and pH-Responsive Properties of Poly(Styrene-co-Maleic Anhydride)-Grafting Poly(oxypropylene)-Amines, J. Colloid Interface Sci., 2009, 336, 82–89 CrossRef CAS PubMed.
- S. M. Henry, M. E. H. El-Sayed, C. M. Pirie, A. S. Hoffman and P. S. Stayton, pH-Responsive Poly(Styrene-alt-Maleic Anhydride) Alkylamide Copolymers for Intracellular Drug Delivery, Biomacromolecules, 2006, 7, 2407–2414 CrossRef CAS PubMed.
- B. Klumperman, Mechanistic Considerations on Styrene–maleic Anhydride Copolymerization Reactions, Polym. Chem., 2010, 1, 558 RSC.
- B. P. Binks and S. O. Lumsdon, Catastrophic Phase Inversion of Water-in-Oil Emulsions Stabilized by Hydrophobic Silica, Langmuir, 2000, 16, 2539–2547 CrossRef CAS.
- B. P. Binks and C. P. Whitby, Silica Particle-Stabilized Emulsions of Silicone Oil and Water: Aspects of Emulsification, Langmuir, 2004, 20, 1130–1137 CrossRef CAS.
- S. Brandriss and S. Margel, Synthesis and Characterization of Self-Assembled Hydrophobic Monolayer Coatings on Silica Colloids, Langmuir, 1993, 100, 1232–1240 CrossRef.
- S. a Kulkarni, S. B. Ogale and K. P. Vijayamohanan, Tuning the Hydrophobic Properties of Silica Particles by Surface Silanization Using Mixed Self-Assembled Monolayers, J. Colloid Interface Sci., 2008, 318, 372–379 CrossRef CAS PubMed.
- K. Zhang, W. Wu, H. Meng, K. Guo and J.-F. Chen, Pickering Emulsion Polymerization: Preparation of Polystyrene/Nano-SiO2 Composite Microspheres with Core–Shell Structure, Powder Technol., 2009, 190, 393–400 CrossRef CAS PubMed.
- G. Kaptay, On the Equation of the Maximum Capillary Pressure Induced by Solid Particles to Stabilize Emulsions and Foams and on the Emulsion Stability Diagrams, Colloid. Surface. Physicochem. Eng. Aspect., 2006, 282–283, 387–401 CrossRef PubMed.
- N. Yan, M. R. Gray and J. H. Masliyah, On Water-in-Oil Emulsions Stabilized by Fine Solids, Colloid. Surface. Physicochem. Eng. Aspect., 2001, 193, 97–107 CrossRef CAS.
- J. Schulman and J. Leja, Control of Contact Angles at the Oil–Water–Solid Interfaces. Emulsions Stabilized by Solid Particles (BaSO4), Trans. Faraday Soc., 1954, 598–605 RSC.
- G. Bogush, M. Tracy and C. Z. Iv, Preparation of Monodisperse Silica Particles: Control of Size and Mass Fraction, J. Non-Cryst. Solids, 1988, 104, 95–106 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, Controlled Growth of Monodisperse Silica Spheres in the Micron Size Range, J. Colloid Interface Sci., 1968, 69, 62–69 CrossRef.
- L. T. Zhuravlev, The Surface Chemistry of Amorphous Silica. Zhuravlev Model, Colloid. Surface. Physicochem. Eng. Aspect., 2000, 173, 1–38 CrossRef CAS.
- L. T. Zhuravlev, Concentration of Hydroxyl Groups on the Surface of Amorphous Silicas, Langmuir, 1987, 3, 316–318 CrossRef CAS.
- A. N. Parikh, M. A. Schivley, E. Koo, K. Seshadri, D. Aurentz, K. Mueller, D. L. Allara, V. Pennsyl, S. Uni and U. V. Park, N-Alkylsiloxanes: From Single Monolayers to Layered Crystals. The Formation of Crystalline Polymers from the Hydrolysis of N-Octadecyltrichlorosilane, J. Am. Chem. Soc., 1997, 7863, 3135–3143 CrossRef.
- L. T. Zhuravlev, Surface Characterization of Amorphous Silica—a Review of Work from the Former USSR, Colloid. Surface. Physicochem. Eng. Aspect., 1993, 74, 71–90 CrossRef CAS.
- M. Jin, S. Li, J. Wang, M. Liao and Y. Zhao, Controllable Fabrication of Organosilane Nano-Architectured Surfaces with Tunable Wettability, Appl. Surf. Sci., 2012, 258, 7552–7555 CrossRef CAS PubMed.
- A. Y. Fadeev and T. J. McCarthy, Self-Assembly Is Not the Only Reaction Possible between Alkyltrichlorosilanes and Surfaces: Monomolecular and Oligomeric Covalently Attached Layers of Dichloro- and Trichloroalkylsilanes on Silicon, Langmuir, 2000, 16, 7268–7274 CrossRef CAS.
- J. W. O. Salari, G. T. Jemwa, H. M. Wyss and B. Klumperman, Reconstruction of the 3D Structure of Colloidosomes from a Single SEM Image, Soft Matter, 2011, 7, 2033–2041 RSC.
- G. H. Hu and J. T. Lindt, Amidification of Poly(Styrene-co-Maleic Anhydride) with Amines in Tetrahydrofuran Solution: A Kinetic Study, Polym. Bull., 1992, 363, 357–363 CrossRef.
- J. Van Wijk, J. W. O. Salari, N. Zaquen, J. Meuldijk and B. Klumperman, Poly(Methyl Methacrylate)–Silica Microcapsules Synthesized by Templating Pickering Emulsion Droplets, J. Mater. Chem. B, 2013, 1, 2394–2406 RSC.
- A. Blaaderen van and A. Vrij, Synthesis and Characterization of Colloidal Dispersions of Fluorescent, Monodisperse Silica Spheres, Langmuir, 1992, 81, 2921–2931 CrossRef.
- Z. Nie, J. I. Park, W. Li, S. a F. Bon and E. Kumacheva, An “Inside-Out” Microfluidic Approach to Monodisperse Emulsions Stabilized by Solid Particles, J. Am. Chem. Soc., 2008, 130, 16508–16509 CrossRef CAS PubMed.
- G. M. Whitesides, The Origins and the Future of Microfluidics, Nature, 2006, 442, 368–373 CrossRef CAS PubMed.
- N. Verhaegh and A. Blaaderen, Dispersions of Rhodamine-Labeled Silica Spheres: Synthesis, Characterization, and Fluorescence Confocal Scanning Laser Microscopy, Langmuir, 1994, 96, 1427–1438 CrossRef.
- E. J. A. Pope and J. D. Mackenzie, Sol–Gel Processing of Silica, J. Non-Cryst. Solids, 1986, 87, 185–198 CrossRef CAS.
- H. De Brouwer, M. a. J. Schellekens, B. Klumperman, M. J. Monteiro and A. L. German, Controlled Radical Copolymerization of Styrene and Maleic Anhydride and the Synthesis of Novel Polyolefin-Based Block Copolymers by Reversible Addition–Fragmentation Chain-Transfer (RAFT) Polymerization, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 3596–3603 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |