 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBiocompatible, hyperbranched nanocarriers for the transport and release of copper ions†
S.
Nowag
a,
C.
Frangville
b,
G.
Multhaup
c,
J.-D.
Marty
b,
C.
Mingotaud
b and
R.
Haag
*a
aInstitut für Chemie und Biochemie, Organische Chemie, Freie Universität Berlin, Takustr. 3, 14195 Berlin, Germany. E-mail: haag@chemie.fu-berlin.de
bLaboratoire des Interactions Moléculaires et Réactivités Chimiques et Photochimiques, Université de Toulouse, CNRS UMR 5623, 31062 Toulouse Cedex 09, France
cDepartment of Pharmacology and Therapeutics, McGill University, 3655 Promenade Sir-William-Osler, Montréal, QC H3G 1Y6, Canada
First published on 16th April 2014
Abstract
Core–shell and core–multishell nanocarriers were designed to transport copper ions into cells. Herein, we present their synthesis and physicochemical characterization and demonstrate the high influence of their architectures on the loading and release of copper. Their low toxicity may open a new way to balance the Cu-homeostasis in neurodegenerative diseases.
Copper (Cu) is an important biological cofactor for a number of metalloenzymes and plays a key role in many neurodegenerative diseases,1e.g., Alzheimer's disease (AD) and Parkinson's disease. Particularly in the case of AD, disturbed homeostasis of Cu leads to Cu deficiency in the central nervous system (CNS). Exogenous supply of Cu to the brain or CNS is one of the many therapeutic approaches for addressing such deficiency symptoms.2 For such a purpose, copper supply needs to bypass the blood–brain barrier (BBB), which remains an unsolved problem. Additionally, hyperperfusion in the AD brain leads to decreased pH and inflammatory processes in the CNS.3 All the previous information helps in the design of smart nanocarriers capable of performing Cu pH-triggered delivery across the BBB (see Scheme 1). Hyperbranched polymers are promising representatives of size controllable nanoscaffolds for transport and drug delivery.4–7 In previous studies, hyperbranched poly(ethyleneimine) (PEI) has been used to develop Cu-encapsulating nanocarriers.8 However, the poor biocompatibility due to the presence of a large number of amine groups within PEI limits the applications of such systems.
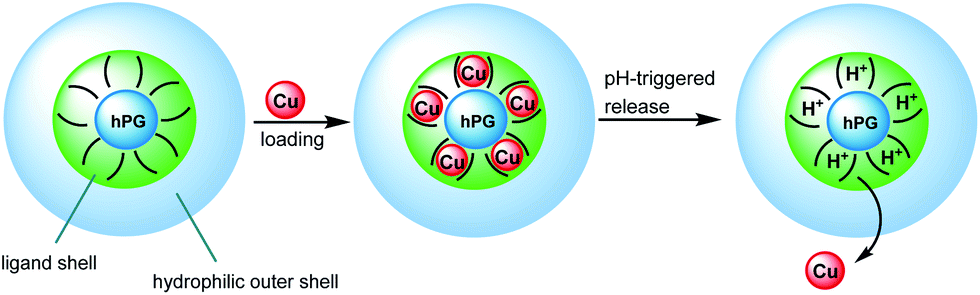 | ||
| Scheme 1 Encapsulation and release of Cu ions in nanocarrier systems. | ||
To enhance biocompatibility, new core–multishell nanocarriers with lower toxicity are required, e.g., functionalized, hyperbranched polyglycerol (hPG). As a macromolecule, hPG possesses a highly branched, globular dendritic structure of a stable polyether scaffold and can be synthesized in a one-step process.9 It exhibits similar properties to the well-used and FDA-approved poly(ethylene glycol) (PEG), as it is non-toxic, biocompatible, and not recognized by the immune system.10 Furthermore, modification of hPG is facile due to its large number of peripheral hydroxyl groups. These functionalized hPG can therefore be tailored to required properties such as size, charge, or molecular weight, respectively.5,11–15 By the attachment of shells, specialized architectures can be designed for a wide variety of applications.16–20
Herein, we present the synthesis and properties of different modified hPG to develop biocompatible Cu-nanocarriers as potential candidates in the therapy of Cu deficiency disorders. An hPG with a molecular weight of 10 kDa was selected for the core in order to obtain nanocarriers of a few tens of nanometers. Cu-binding ligands to be attached on hPG cores were then reviewed to fit in our specifications. An example of Cu-binding ligands are triazacyclononanes, which are known to form very strong complexes with Cu that are irreversible under physiological conditions.21 Clearly, copper-release into CNS acidic conditions requires a weaker ligand for a pH-triggered release of Cu. Tri- or dimethylethylene diamine moieties (TMEDA or DMEDA, respectively) have been chosen as suitable candidates.22 They form square planar bidentate Cu-complexes with moderate interactions, which enable pH-dependent Cu release. Furthermore, in order to increase the biocompatibility, to enhance the retention time within blood circulation, or to hide Cu from the environment, attachment of poly(ethylene glycol) (PEG) chains in the outer sphere is required (see Scheme 1). Two kinds of attachments were chosen to investigate the influence of structural design of nanocarriers on Cu-loading and release, as well as their biocompatibility. Thus, hydrophilic PEG chains were bound to amine building blocks leading to a core–multishell structure (CMS) or directly to the hPG core for a core–random shell architecture (CRS) (see Scheme 2). Finally, hPG fully modified by TMEDA (CS structure in Scheme 2) was used as a reference system.23
 | ||
| Scheme 2 Structure and synthesis scheme of CS, CMS and CRS. | ||
For the CMS synthesis, boc-protected DMEDA was first reacted with hPG to cover 70% of the hydroxyl groups of hPG's surface. After deprotection, PEG1000-OM chains were attached to the amino ligands. For the CRS, reaction of hPG with TMEDA was controlled to yield a degree of functionalization on hPG's surface of 70% (similar to that of CMS). The remaining mesylates were then substituted by azides, which could be used in a click reaction to attach propargylated PEG5000 (see characterization of CMS and CRS in the ESI†).
UV/vis spectroscopic experiments were then carried out to determine the maximum Cu-loading capacity of the nanocarrier in water. A stock solution of the polymer was mixed with an increasing amount of Cu(II) to achieve different Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :polymer molar ratios. Regardless of the polymer structure, an increase of the absorbance at 580 nm was observed and should be related to the d–d transition of copper ions complexed by amine functions (see Fig. S1 in the ESI†). This demonstrated the complexation of Cu inside the nanocarriers. Above a certain concentration of copper, the absorbance at 580 nm became constant and a new band around 800 nm related to aqueous copper ions grew in size. This behavior is evidence for the saturation of nanocarriers and that the later added copper ions remained free in solution. The plot of the absorbance at 580 nm for the increasing Cu:polymer ratio reveals that the CMS structure can load more copper ions than the CRS polymer (see Fig. 1a). The close fit of these curves (see ESI†) leads to a more quantitative evaluation of this maximum loading (see Table 1).
:polymer molar ratios. Regardless of the polymer structure, an increase of the absorbance at 580 nm was observed and should be related to the d–d transition of copper ions complexed by amine functions (see Fig. S1 in the ESI†). This demonstrated the complexation of Cu inside the nanocarriers. Above a certain concentration of copper, the absorbance at 580 nm became constant and a new band around 800 nm related to aqueous copper ions grew in size. This behavior is evidence for the saturation of nanocarriers and that the later added copper ions remained free in solution. The plot of the absorbance at 580 nm for the increasing Cu:polymer ratio reveals that the CMS structure can load more copper ions than the CRS polymer (see Fig. 1a). The close fit of these curves (see ESI†) leads to a more quantitative evaluation of this maximum loading (see Table 1).
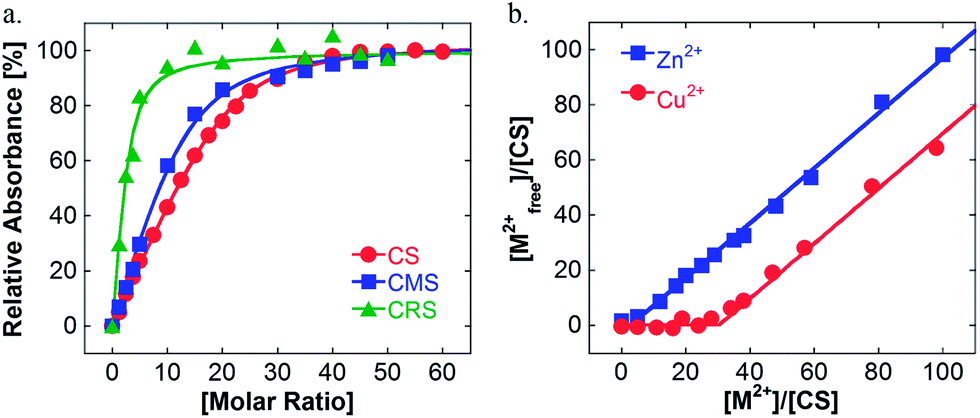 | ||
| Fig. 1 Determination of the maximum loading of polymers by UV-visible measurements. (a) Influence of the Cu:nanocarrier molar ratio on the relative absorbance at 580 nm at room temperature with a constant polymer concentration (80 μM). (b) Free metal ion concentration depending on metal:CS molar ratios at room temperature with CS concentration (6.2 μM). | ||
| CS | CMS | CRS | |
|---|---|---|---|
| a Determined by UV-visible spectroscopy (see text). b Ratio between the experimental loading capacity and the maximum theoretical number of sites per polymer estimated as half the number of ligand ethylene diamine per polymer. c Determined by UV-visible spectroscopy (see ESI). d Hydrodynamic radius (nanocarriers/Cu-loaded nanocarriers) determined by DLS with an analysis in number from 80 μM nanocarrier solutions. e Aggregation. f Determined on human neuroblastoma cell line SH-SY5Y. | |||
| Loading capacitya | 22 ± 4 | 14 ± 3 | 5 ± 2 |
| Percentage of active sitesb | 46 ± 9% | 42 ± 9% | 15 ± 6% |
| Relative intake kineticsc | 1 | 11 | 36 |
| Sized (nm) | 12/8 | 8/5 | 360e/6 |
| pH for 10% of copper release | 4 | 4 | 7 |
| Relative pH-release kineticsc | 1 | 2 | 3 |
| Cytotoxicity of the polymer loaded with copper (IC50)f | 38.1 mg Cu per L | 4.8 mg Cu per L | 4.1 mg Cu per L |
| 220 mg CS per L | 415 mg CMS per L | 2.9 g CRS per L | |
To check the loading capacity, we developed a second method based on centrifugal filtration of the metal ion/polymer solution. Titration of the metal ion was performed on the filtrate using a well known spectroscopic method based on the Zincon indicator (see Fig. S2 in the ESI†).24 As seen in Fig. 1b, the CS system retains copper ions until a maximum loading of approx. 30. This is quite comparable with the direct evaluation by UV/vis spectroscopy. Assuming a binding site of two ligands per copper, one can estimate the maximum theoretical number of sites per polymer and therefore the percentage of active sites. As seen in Table 1, similar values were found for CS and CMS structures. In contrast, the architecture of the CRS polymer is less favorable for complexing Cu metal ions, which may be due to the higher steric hindrance from the PEG-chains attached between TMEDA-ligand units as well as from the aggregation of CRS. The binding kinetics were fast and equilibrium was reached only after a few minutes (see Fig. S1 in the ESI†). All these results indicated a behavior highly related to the architecture of the nanocarrier.
After addition of copper ions into a polymer solution, reaching the equilibrium was a slower process for CRS than CMS or CS (see Table 1). This may be partially due to the PEG layer (larger in CRS than in CMS), which should have decreased the accessibility of ions toward the ligand shell.
The ability for such structures to interact with other ions may be providential for the delivery of copper in a controlled manner. To evaluate such a possibility, a large excess of different ions (Ca, Mg, Mn, Co, Zn, Ni) was added to Cu-loaded nanocarrier solutions (see Fig. S6 in the ESI†). None of the metal ions were able to displace copper ions from the hyperbranched structures.
Moreover, no direct complexation could be evidenced for zinc ions, another ion of interest in the case of AD,25 as shown in Fig. 1b. Thus, the obtained nanocarriers were of special interest for the specific complexation and delivery of copper.
Blood clearance for molecules below 60 kDa and 6 nm is fast, while bigger particles remain longer in the blood.26,27 However, particles bigger than 200 nm are likely renally excreted.28 Since size also has a big impact on the permeability of the BBB of substances,29–31 the size of the empty and Cu-loaded nanocarriers was investigated by dynamic light scattering (see Fig. S4 in the ESI†). Empty nanocarriers in solution as a unimer were found to have a hydrodynamic diameter close to 10 nm (see Table 1). When CMS was only in the form of unimer, CS and CRS nanocarriers were more or less present as polydisperse aggregates in solutions (Rh above 100 nm). When Cu was stabilized inside nanocarriers, a significant decrease of the aggregation and a slight change in the size of the unimer were observed. Thus, the nanocarriers presented here having molecular weights above 80 kDa and diameters d ≥ 8 nm are in the right range for application as a delivery system. However, permeation through the BBB might be critical, since it has been postulated that most molecules bigger than 400 Da are not able to pass the BBB.29 On the other hand, it was observed that the ability to cross the BBB is much higher for NPs with cationic character.30–32 Due to this, zeta potentials of empty and Cu-loaded nanocarriers were studied at pH values ranging from 7 to 3 in buffer solution. At pH 7, the zeta potential of empty CS was found to be 30 ± 9 mV. As expected, grafting of PEG chains induced a significant decrease of zeta potential that remained slightly positive for both CRS (+13.9 ± 0.8 mV) and CMS (+9.2 ± 1.0 mV) (see Fig. S5 in the ESI†). Partial protonation of amine groups may have been responsible for this observation. Therefore, the highest zeta potentials were measured at pH 3 due to the increase of the protonation level at lower pH. Interestingly, the surface charge at all the pH values did not significantly evolve when Cu ions were stabilized inside the nanocarriers. Therefore, these positively charged nanocarriers have high potential to cross the BBB.
To prove whether nanocarriers were able to release copper ions at low pH, UV/vis spectroscopy experiments were performed. Cu-loaded nanocarriers were transferred in Britton Robinson universal buffer aliquots with pH values from 8 to 2 and the absorbance was monitored (see Fig. S3 in the ESI†). Fig. 2 shows the percentage of Cu-release as a function of pH. As a result, Fig. 2 shows that CRS nanocarriers continuously released Cu with decreasing pH. This profile of release highlights the weakness of these nanocarriers due to the random architecture as discussed above and might be too prompt, since the release already started at pH 7. For application in biological systems a controlled release is therefore not possible presumably due to Cu leakage before the required target was reached. On the other hand, Cu-complexes' integrity was retained in CS and CMS structures until lower pH values. This indicates a stronger Cu binding in this architecture which makes it a good candidate for carrying its entire load to the target, after which a sharp and fast Cu release could occur according to Fig. 2 and kinetics data (see Fig. S3 in the ESI†).
 | ||
| Fig. 2 %Cu-release based on the relative absorbance at 580 nm of the CMS or CRS polymer (80 μM) in Britton Robinson Buffers, 0.1 M NaCl at 37 °C. | ||
The toxicity of Cu-loaded and empty nanocarriers in human neuroblastoma (SH-SY5Y) cells was then investigated by MTT assay (see experimental procedures in the ESI†). The IC50 value of CRS was found to be 22.4 g L−1, which was much higher and therefore less toxic than CMS (IC50ca. 415 mg L−1). It is known that long PEG-chains lead to higher biocompatibility.33 Hence, we assume that CRS with longer mPEG-chains shows less toxicity than CMS, i.e., shielding of the amino ligands by mPEG-chains determines the toxicity of the cargo in living cells. Both nanocarriers in the present study were less toxic than the previously described PEI-based CMS8 which supported our earlier assumption that the hPG-based nanocarrier has a higher compatibility than nanocarriers with a PEI core. Investigation on the toxicity of Cu-loaded nanocarriers showed the same result for CMS as for empty particles regarding the nanocarrier concentration (IC50 [Cu:CMS = 15:1] = 415 mg L−1). In contrast, the cytotoxicity of Cu-loaded CRS increased to IC50 [Cu:CRS = 5:1] = 2.9 g L−1 in comparison to unloaded CRS. Regarding the Cu concentration, however, Cu-loaded CMS was in the same range as Cu:CRS in terms of cytotoxicity (see Table 1). Both nanocarriers could reduce the toxicity of Cu by a factor of 7.8 The compatibility at high loading is especially of importance for in vivo applications when a high local concentration of Cu is required. According to these results, it seems that CMS is a better candidate for the treatment of Cu deficiency disorders.
Conclusions
By designing new core–shell polymeric structures, we were able to obtain nanocarriers that had a high binding affinity for copper ions and were able to release these ions at low pH. We demonstrate that the exact architecture of the core–shell system is a paramount parameter to control the maximum loading, the strength of complexation, and the release profile of copper into the solution. The CMS structure proved to be the most promising structure for Cu complexation and release. The CMS structure will be studied to estimate its transport through the BBB and tested in vivo for the release of copper.Acknowledgements
This work was financially supported by the ANR-DFG program (ANR-11-INTB-1004, “Dendrion-Bio”). The authors would like to thank the SFB 1112 and the Helmholtz portfolio project “Technologie und Medizin” for financial support and Christina Kühl for technical assistance.Notes and references
- M. A. Telpoukhovskaia and C. Orvig, Chem. Soc. Rev., 2013, 42, 1836–1846 RSC.
- D. J. Waggoner, T. B. Bartnikas and J. D. Gitlin, Neurobiol. Dis., 1999, 6, 221–230 CrossRef CAS PubMed.
- G. J. Broussard, J. Mytar, R. C. Li and G. J. Klapstein, Inflammopharmacology, 2012, 20, 109–126 CrossRef CAS PubMed.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- M. A. Quadir and R. Haag, J. Controlled Release, 2012, 161, 484–495 CrossRef CAS PubMed.
- F. Aulenta, W. Hayes and S. Rannard, Eur. Polym. J., 2003, 39, 1741–1771 CrossRef CAS.
- M. Krämer, J. F. Stumbe, H. Turk, S. Krause, A. Komp, L. Delineau, S. Prokhorova, H. Kautz and R. Haag, Angew. Chem., Int. Ed., 2002, 41, 4252–4256 CrossRef.
- C. Treiber, M. A. Quadir, P. Voigt, M. Radowski, S. J. Xu, L. M. Munter, T. A. Bayer, M. Schaefer, R. Haag and G. Multhaup, Biochemistry, 2009, 48, 4273–4284 CrossRef CAS PubMed.
- D. Wilms, S.-E. Stiriba and H. Frey, Acc. Chem. Res., 2009, 43, 129–141 CrossRef PubMed.
- R. K. Kainthan, J. Janzen, E. Levin, D. V. Devine and D. E. Brooks, Biomacromolecules, 2006, 7, 703–709 CrossRef CAS PubMed.
- D. Gröger, F. Paulus, K. Licha, P. Welker, M. Weinhart, C. Holzhausen, L. Mundhenk, A. D. Gruber, U. Abram and R. Haag, Bioconjugate Chem., 2013, 24, 1507–1514 CrossRef PubMed.
- S. Reichert, P. Welker, M. Calderon, J. Khandare, D. Mangoldt, K. Licha, R. K. Kainthan, D. E. Brooks and R. Haag, Small, 2011, 7, 820–829 CrossRef CAS PubMed.
- D. Steinhilber, S. Seiffert, J. A. Heyman, F. Paulus, D. A. Weitz and R. Haag, Biomaterials, 2011, 32, 1311–1316 CrossRef CAS PubMed.
- Z. Chen, L. Zhou, F. Zhang, C. Yu and Z. Wei, Appl. Surf. Sci., 2012, 258, 5291–5298 CrossRef CAS PubMed.
- J. Khandare, A. Mohr, M. Calderón, P. Welker, K. Licha and R. Haag, Biomaterials, 2010, 31, 4268–4277 CrossRef CAS PubMed.
- M. Seiler, Fluid Phase Equilib., 2006, 241, 155–174 CrossRef CAS PubMed.
- Y. Xu, C. Gao, H. Kong, D. Yan, P. Luo, W. Li and Y. Mai, Macromolecules, 2004, 37, 6264–6267 CrossRef CAS.
- C. R. Yates and W. Hayes, Eur. Polym. J., 2004, 40, 1257–1281 CrossRef CAS PubMed.
- D. Appelhans, H. Komber, M. A. Quadir, S. Richter, S. Schwarz, J. van der Vlist, A. Aigner, M. Muller, K. Loos, J. Seidel, K. F. Arndt, R. Haag and B. Voit, Biomacromolecules, 2009, 10, 1114–1124 CrossRef CAS PubMed.
- D. Steinhilber, T. Rossow, S. Wedepohl, F. Paulus, S. Seiffert and R. Haag, Angew. Chem., Int. Ed., 2013, 52, 13538–13543 CrossRef CAS PubMed.
- G. Gasser, L. Tjioe, B. Graham, M. J. Belousoff, S. Juran, M. Walther, J.-U. Kunstler, R. Bergmann, H. Stephan and L. Spiccia, Bioconjugate Chem., 2008, 19, 719–730 CrossRef CAS PubMed.
- X. Wang and D.-S. Yang, J. Phys. Chem. A, 2006, 110, 7568–7576 CrossRef CAS PubMed.
- M. A. Quadir, Dendritic Polymers for Nanoscale Delivery of Metal Ion and Drugs (Doctoral Dissertation), Freie Universität, Berlin, 2010 Search PubMed.
- R. M. Rush and J. H. Yoe, Anal. Chem., 1954, 26, 1345–1347 CrossRef CAS.
- H. Kozlowski, M. Luczkowski, M. Remelli and D. Valensin, Coord. Chem. Rev., 2012, 256, 2129–2141 CrossRef CAS PubMed.
- H. Soo Choi, W. Liu, P. Misra, E. Tanaka, J. P. Zimmer, B. Itty Ipe, M. G. Bawendi and J. V. Frangioni, Nat. Biotechnol., 2007, 25, 1165–1170 CrossRef PubMed.
- M. J. Knauf, D. P. Bell, P. Hirtzer, Z. P. Luo, J. D. Young and N. V. Katre, J. Biol. Chem., 1988, 263, 15064–15070 CAS.
- P. J. Crouch, L. W. Hung, P. A. Adlard, M. Cortes, V. Lal, G. Filiz, K. A. Perez, M. Nurjono, A. Caragounis, T. Du, K. Laughton, I. Volitakis, A. I. Bush, Q. X. Li, C. L. Masters, R. Cappai, R. A. Cherny, P. S. Donnelly, A. R. White and K. J. Barnham, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 381–386 CrossRef CAS PubMed.
- W. M. Pardridge, Drug Discovery Today, 2007, 12, 54–61 CrossRef CAS PubMed.
- W. Lu, J. Wan, Z. She and X. Jiang, J. Controlled Release, 2007, 118, 38–53 CrossRef CAS PubMed.
- L. H. Reddy, R. K. Sharma, K. Chuttani, A. K. Mishra and R. R. Murthy, AAPS J., 2004, 6, e23 Search PubMed.
- L. Kozlovskaya and D. Stepensky, J. Controlled Release, 2013, 171, 17–23 CrossRef CAS PubMed.
- H. Petersen, P. M. Fechner, D. Fischer and T. Kissel, Macromolecules, 2002, 35, 6867–6874 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section, Cu and Zn loading capacity, pH-triggered Cu release, colloidal behavior in water, and metal ion competition. See DOI: 10.1039/c4tb00454j |
| This journal is © The Royal Society of Chemistry 2014 |