 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light-triggered CO release from nanoporous non-wovens†
Carmen
Bohlender
a,
Steve
Gläser
a,
Moritz
Klein
c,
Jürgen
Weisser
b,
Susanne
Thein
b,
Ute
Neugebauer
cf,
Jürgen
Popp
cef,
Ralf
Wyrwa
b and
Alexander
Schiller
*ade
aFriedrich Schiller University Jena, Institute for Inorganic and Analytical Chemistry (IAAC), Humboldtstr. 8, 07743 Jena, Germany. E-mail: alexander.schiller@uni-jena.de; Fax: +49 3641 948 102; Tel: +49 3641 948 113
bINNOVENT e.V., Biomaterials Department, Pruessingstr. 27 B, 07745 Jena, Germany
cLeibniz Institute of Photonic Technology, Albert-Einstein-Straße 9, 07745 Jena, Germany
dFriedrich Schiller University Jena, Jena Center for Soft Matter (JCSM), Humboldtstr. 10, 07743 Jena, Germany
eInstitute of Physical Chemistry and Abbe Center of Photonics, Friedrich Schiller University Jena, Max Wien Platz 1, 07743 Jena, Germany
fCenter for Sepsis Control and Care, Jena University Hospital, Erlanger Allee 101, 07747 Jena, Germany
First published on 11th February 2014
Abstract
The water insoluble and photoactive CO releasing molecule dimanganese decacarbonyl (CORM-1) has been non-covalently embedded into poly(L-lactide-co-D/L-lactide) fibers via electrospinning to enable bioavailability and water accessibility of CORM-1. SEM images of the resulting hybrid non-wovens reveal a nanoporous fiber morphology. Slight CO release from the CORM-1 in the electrospinning process induces nanoporosity. IR spectra show the same set of carbonyl bands for the CORM-1 precursor and the non-woven. When the material was exposed to light (365–480 nm), CO release from the incorporated CORM-1 was measured via heterogeneous myoglobin assay, a portable CO electrode and an IR gas cuvette. The CO release rate was wavelength dependent. Irradiation at 365 nm resulted in four times faster release than at 480 nm. 3.4 μmol of CO per mg non-woven can be generated. Mouse fibroblast 3T3 cells were used to show that the hybrid material is non-toxic in the darkness and strongly photocytotoxic when light is applied.
A Introduction
Carbon monoxide (CO), a byproduct of heme catabolism via heme oxygenases (HO), has been recognized as an important gasotransmitter in mammals akin to nitric oxide (NO) and hydrogen sulfide (H2S) and plays versatile roles in tissue protection via anti-inflammatory, antiproliferative, and antiapoptotic effects.1–4 CO relaxes smooth muscles at low concentrations and is involved in wound healing and cardiovascular protection.5,6 Furthermore, HO-deficient mice show an increased lethality during polymicrobial sepsis,7 suggesting that the clearance of heme and/or the production of CO is beneficial. With regard to the second point CO attracts particular attention as a potential therapeutic agent. CO inhalation appears to protect vital organs, including the brain, heart, lung, and liver, during ischemia/hypoxia and organ transplantation.8–11 However, its practical clinical use is currently hampered by the methods to deliver CO precisely and safely to target locations.4CO releasing molecules (CORMs) as carrier systems that release CO only after directed triggering appear as a convenient alternative to the application of pure CO gas.1 CORMs can be generated from main or transition metal carbonyl complexes as well as from organic molecules. Recent developments have been summarized in excellent reviews.4,12–18 The manifold impact of CO on regulatory processes facilitates broad possibilities for medicinal CORM application.1 For example, [Ru(CO)3Cl(glycinate)] (CORM-3) has been successfully tested in models of vascular dysfunction, ischemic injury and inflammation.19 Poole et al. demonstrated that CORM-3 can enter cells, transfer CO intracellularly and inhibit bacterial growth by complex interactions with the respiratory chain in Pseudomonas aeruginosa and Escherichia coli.20 Importantly, it has been recently shown that CORM-3 derived CO and not post-release metal fragments (inactive CORM, iCORM) caused the observed effects on respiration. Administration of CORM-2 (tricarbonyldichloro-ruthenium(II) dimer) in rat aortic smooth muscle cells showed contrasting context-dependent effects on the vessel tone demonstrating the complex actions of CO during muscle relaxation.21 CORM-2, just as CORM-3, has versatile effects on pathogenic bacteria, such as E. coli and Staphylococcus aureus, resulting in rapid cell death. This effect on bacterial cell viability is lost in the presence of a CO scavenger, revealing that released CO and not metal degradation products are responsible for the antimicrobial action.22 Dimanganese decacarbonyl (CORM-1) can play an important role in vascular control as shown with isolated rat hearts.5 It has long been recognized that dissociative CO loss and the Mn–Mn bond break are primary photoprocesses of Mn2(CO)10.23,24 Light stimulated CORM-1 (just as gaseous CO) is able to activate calcium-dependent potassium channels in smooth muscle cells.21,25,26 Further, CORM-1 derived CO showed a positive effect on renal circulation in rats and anti-inflammatory effects were demonstrated in a mesenteric microcirculation model.19
However, the water insoluble CORM-1 is much less used in medical experiments compared to its soluble analogues. Previous experiments always required the use of DMSO as a (co)solvent. On the other hand, CORM-1 is a highly loaded CO storage molecule (10 mole CO per mole CORM). The desired water accessibility of CORM-1 for medical applications can be achieved by a pharmaceutical formulation: CORM-1 is incorporated into a biocompatible support matrix (Fig. 1a). Subsequently, the resulting hybrid material can be attached to the desired environment/tissue.27–29 This novel concept reveals also a possible solution to the metal fragment problem (Fig. 1a): CORM degradation products of water soluble CORMs (or even the CORMs themselves) can cause dramatic side effects within cells.1,21,30 Instead, the leftover metal fragments would be kept trapped within the scaffold after release of the biologically active drug.4,16,27,31 Other groups used immobilization strategies of CORMs to address specific biological targets, novel release mechanisms, and stability issues in physiological medium.32–38 To our knowledge, only three examples have been published where Mn2(CO)10 has been introduced into a cyclodextrin, cellulose or polymer matrix, but never with the intention to use the CORM-1 derived CO.39–41 However, fibrous non-wovens with NO donors have already been obtained via electrospinning.27,42,43
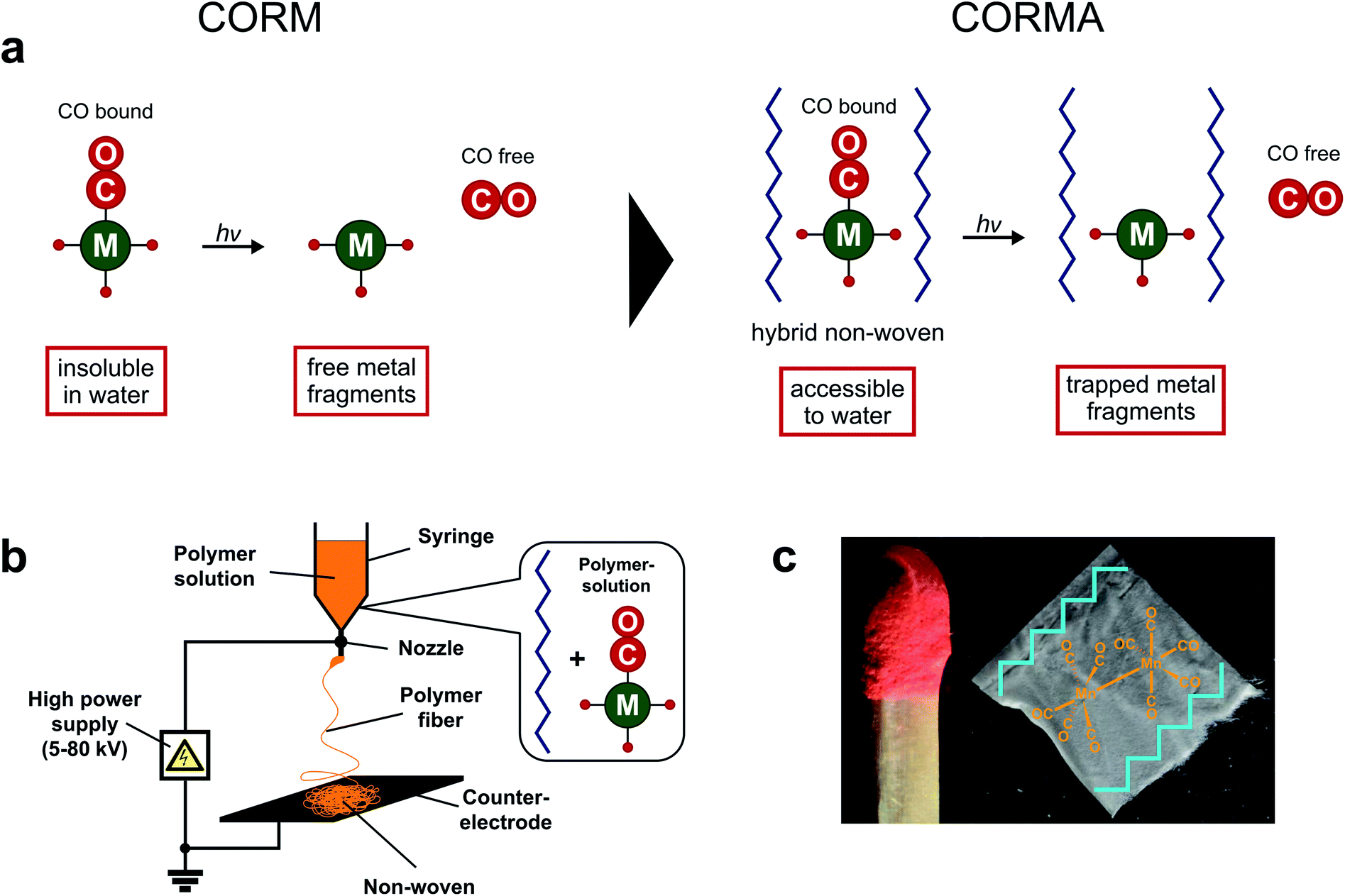 | ||
| Fig. 1 (a) Concept of CORM incorporation into a polymeric non-woven (resulting in a hybrid CORMA) to achieve water compatibility and retain the metal fragment (inactive CORM, iCORM) from the biological medium; (b) electrospinning of a dissolved mixture of polymer and CORM to produce the hybrid non-woven; (c) a sample of electrospun hybrid material CORMA-1–PLA1 (m ≈ 0.6 mg). | ||
Electrospun materials are highly desirable, since the hybrid matrices are rapidly obtained from the support material and the embedded substance without covalent attachment strategies. The electrospinning technique allows the formation of materials with complex properties and morphologies (e.g. high surface area) that match specific requirements (e.g. efficient gas exchange).44,45
In terms of nanofiber morphology, porous instead of smooth fibers are advantageous for a variety of medical or industrial applications, such as tissue engineering, drug delivery, catalysis or filtration.44,46 Matrix porosity and the resulting high specific surface area allow the tuning of drug release profiles from such materials. In the field of sensor development a highly porous structure is required to ensure high sensitivity and fast sensor response. Electrospun carbon nanofibers have proved their increased sensor ability and sensitivity towards NO and CO gases due to their porous structure.47
In the present work CORM-1 was introduced into a polymeric support material to render the compound accessible to water and biological environment (Fig. 1). With this novel procedure not only a drug container was created, but also the advantageous CO release properties of CORM-1 were used to generate nanoporous polymeric materials.31 This dual use of CORM-1 provides a new way for the production of porous electrospun fibers44 and their application as CO releasing materials (CORMAs).4,16
B Results and discussion
Synthesis and characterization of CORMA-1–PLA
Water insoluble CORM-1 has been non-covalently incorporated into a nanofibrous poly(L-lactide-co-D/L-lactide) (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, PLA) non-woven via the electrospinning technique (Fig. 1b).44,45,48 The polymer PLA was chosen due to its known cytocompatibility as well as for its ability to form stable nanofibers under electrospinning conditions.27–29,49 For the preparation of the hybrid material PLA was dissolved in chloroform at room temperature to obtain a 3 wt% polymer solution. Afterwards, 1, 10 or 20 wt% of CORM-1 (based on PLA) was added and stirred under exclusion of light for 20 min yielding yellow homogeneous solutions. These mixtures have been electrospun at daylight. We finally obtained the loaded fleece materials CORMA-1–PLA1, CORMA-1–PLA10, and CORMA-1–PLA20. These hybrid polymers were of yellowish color caused by the embedded CORM-1 (Fig. 1c). The color intensity was increasing from 1 wt% to 20 wt% loading. Attempts to prepare films from casting solutions of PLA, PMMA and PS with CORM-1 as control samples failed due to phase separation, crystallization and finally decomposition of Mn2(CO)10 during film formation. This emphasizes even more that electrospinning is an ideal process for preparing CORM-containing hybrid materials.
:30, PLA) non-woven via the electrospinning technique (Fig. 1b).44,45,48 The polymer PLA was chosen due to its known cytocompatibility as well as for its ability to form stable nanofibers under electrospinning conditions.27–29,49 For the preparation of the hybrid material PLA was dissolved in chloroform at room temperature to obtain a 3 wt% polymer solution. Afterwards, 1, 10 or 20 wt% of CORM-1 (based on PLA) was added and stirred under exclusion of light for 20 min yielding yellow homogeneous solutions. These mixtures have been electrospun at daylight. We finally obtained the loaded fleece materials CORMA-1–PLA1, CORMA-1–PLA10, and CORMA-1–PLA20. These hybrid polymers were of yellowish color caused by the embedded CORM-1 (Fig. 1c). The color intensity was increasing from 1 wt% to 20 wt% loading. Attempts to prepare films from casting solutions of PLA, PMMA and PS with CORM-1 as control samples failed due to phase separation, crystallization and finally decomposition of Mn2(CO)10 during film formation. This emphasizes even more that electrospinning is an ideal process for preparing CORM-containing hybrid materials.
SEM images of electrospun non-wovens were recorded to determine the influence of CORM-1 on the fiber structure of PLA during incorporation (Fig. 2a–d). All non-wovens displayed roughly the same mean fiber diameter of about 1 μm. The content of CORM-1 did not significantly influence the fiber diameters. Interestingly, the images show porous nanofibers (Fig. 2a–c and S1†). In contrast, a control sample of CORMA-1–PLA20 (electrospun under exclusion of daylight) exhibited no comparable porosity (Fig. 2d). PLA fibers without CORM-1 displayed a smooth morphology (Fig. S2c†).29 The number of uniformly distributed pores increases from CORMA-1–PLA1 to CORMA-1–PLA20. It is important to note that already 1 wt% of Mn2(CO)10 was enough to produce a porous morphology in PLA fibers (Fig. 2a). In addition, we performed Kr-BET and Hg porosimetry measurements on the CORMA-1–PLA non-wovens. Surface areas of 1–3 m2 g−1 from a control PLA non-woven, CORMA-1–PLA10, and CORMA-1–PLA20 have been determined (Table S1†). These values can be expected from typical fiber diameters of around 1 μm in electrospun non-wovens.45 Unfortunately, we were not able to corroborate the BET measurements of CORMA-1–PLA10 and CORMA-1–PLA20 with SEM pictures (see Table S1†). However, light-induced CO release from Mn2(CO)10 in the electrospinning process is necessary to form these porous hybrid materials (for reproduced batches and their SEM images, see Fig. S2a and b†). Until now, porous fibers within non-wovens have been produced by phase-separation of polymer blends and solvent mixtures in the electrospinning process.45 By using a photoCORM,15 we found a novel method to generate reliable nanoporous fibers by triggering it with visible light.
 | ||
| Fig. 2 SEM images of electrospun PLA nanofibrous non-wovens ((a) CORMA-1–PLA1, (b) CORMA-1–PLA10, (c) CORMA-1–PLA20 and (d) CORMA-1–PLA20 electrospun in the dark); (e) EDX-SEM maps of CORMA-1–PLA20 after electrospinning: the “structure” in these pictures do not show the fiber pores but is formed by the different layers of fibers; (f) Infra red difference spectra (CORMA-1–PLAx minus pure PLA) exhibit 3 CO vibration bands (downwards) next to PLA vibration bands (upwards) for CORMA-1–PLA20 and CORMA-1–PLA10. | ||
SEM-EDX elemental mapping was conducted on a CORMA-1–PLA20 non-woven to determine the distribution of CORM-1 throughout the fibers. Fig. 2e shows cross-sectional SEM-EDX elemental maps of Si (from underground Si wafer), C, O and Mn (see also Fig. S3†). The pictures reveal a homogeneous distribution of manganese, and therefore CORM-1, within the polymeric fibers. In addition, DSC measurements of CORMA-1–PLA have been performed. The thermogram displayed the expected glass transition temperature Tg of PLA at 59 °C and two exothermic conversions of Mn2(CO)10 at 137 and 174 °C (Fig. S7†). The content of the carbonyl complex before thermal degradation and fiber porosity did not have any significant influence on the Tg of the carrier polymer.29
UV-Vis and IR analyses were used to examine the identity of the incorporated manganese complex. UV-Vis spectra from the CORM-1 itself and the dissolved hybrid non-woven CORMA-1–PLA20 showed a comparable absorption band at 343 nm and a shoulder at 395 nm for both samples (Fig. S4†). Thus, CORM-1 was substantially retained during high-voltage electrospinning. The actual amount of incorporated manganese carbonyl was also estimated. Samples of CORMA-1–PLA20 were dissolved in deaerated CHCl3 and UV-Vis spectra were immediately recorded. The obtained absorbance was compared to the absorbance of a solution of pure CORM-1, its concentration was representing the theoretical amount of 20 wt% within the matrix sample. We found that the absorption of the material samples corresponded to 56–59% of the initial CORM-1 concentration within the polymer (Fig. S4†). For CORMA-1–PLA10 we obtained similar values. These results were combined with another UV-Vis experiment, where CORM-1 was left standing in a non-deaerated CHCl3 solution (analogue to the solution applied for electrospinning) in the dark. A decrease of the CORM-1 absorption band at 343 nm was monitored within 2 hours (Fig. S5†). Therefore, we assume that the porous fiber morphology originates from decomposition (and concomitant CO-gas generation) of CORM-1 in the chloroform solution during electrospinning by the influence of daylight and air.
To determine the amount of incorporated Mn2(CO)10, we dissolved batches of CORMA-1–PLA10 and CORMA-1–PLA20 in aqueous nitric acid and measured the manganese content by ICP-MS analysis. For samples where 10 wt% of CORM-1 was originally embedded into PLA, a Mn amount was found that represents 7.9 wt% (σ = 0.3 wt%) of containing CORM-1. For CORMA-1–PLA20 the measured Mn content was 14.8 wt% (σ = 0.3 wt%) CORM-1 (see Table S2†). Comparing UV-Vis and ICP-MS, we measured incorporation efficiencies from 56% to 79% of CORM-1 in PLA nanofibers via the electrospinning process.
The structure of the incorporated CORM-1 was examined by attenuated total reflection infrared spectroscopy (ATR-IR). We measured a spectrum of pure PLA fibers (Fig. S6†) and then subtracted it from spectra of the CORMA-1–PLA materials. The resulting difference ATR-IR spectra showed the appearance of three ν(C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O) vibration bands at 2046, 2031 and 2004 cm−1, whereas ν(Mn–CO) vibrations were found at 645 and 465 cm−1 next to the PLA bands after incorporation of CORM-1 (Fig. 2f). Those bands were comparable to pure CORM-1 (Fig. S6†). However, in CORMA-1–PLA1 the content of Mn2(CO)10 was too small to be detected by ATR-IR and the porous morphology of CORMA-1–PLA1 displayed also the CO loss from the Mn carbonyl (Fig. 2a). The IR results from CORMA-1–PLA10 and CORMA-1–PLA20 indicated that the major manganese carbonyl species retained is CORM-1.
O) vibration bands at 2046, 2031 and 2004 cm−1, whereas ν(Mn–CO) vibrations were found at 645 and 465 cm−1 next to the PLA bands after incorporation of CORM-1 (Fig. 2f). Those bands were comparable to pure CORM-1 (Fig. S6†). However, in CORMA-1–PLA1 the content of Mn2(CO)10 was too small to be detected by ATR-IR and the porous morphology of CORMA-1–PLA1 displayed also the CO loss from the Mn carbonyl (Fig. 2a). The IR results from CORMA-1–PLA10 and CORMA-1–PLA20 indicated that the major manganese carbonyl species retained is CORM-1.
CO release from CORMA-1–PLA
Light induced release of CO bubbles from CORMA-1–PLA20 with its high surface area (see Fig. 2c) was directly observed with a laser scanning microscope (LSM). The non-woven was covered with a cover lid to retain the CO gas. The formation of gas bubbles was observed within 25 seconds during illumination with the internal LSM lamp (405 nm). Fig. 3d shows the sample before illumination and after 50 seconds of light exposure (for a video of the CO release see ESI†). After exhaustive illumination of the CORMA-1–PLA samples they turned dark yellow to brown. Manganese(IV) oxide (MnO2) was qualitatively identified from an acidic CORMA solution with hydrochloric acid and iodine water. | ||
| Fig. 3 (a) CO release profile of CORMA-1–PLA20 under exposure to 440–480 nm light (dentist lamp). Irradiation was accomplished in a sealed beaker (Fig. S8a†) in 10 second intervals (CO release) followed by an equilibration time of 6 minutes; (b) irradiation of CORMA-1–PLA20 at 365 nm (blue) and 480 nm (red) at a similar intensity of 10 mW cm−2 shows the wavelength dependency of the CO release rate; (c) total amount of CO released into the gas phase during irradiation at 365 nm determined via gas phase IR. Data points are means with a 95% confidence interval (inset: the rotationally resolved CO vibration band of measured IR spectra at different time intervals); (d) LSM pictures of light-triggered CO release from CORMA-1–PLA20: the sample before (left) and after illumination (right) at 405 nm in the microscope. The formed CO bubbles are shown in black patches; (e) difference spectra (the CORMA-1–PLA20 spectrum subtracted from the pure PLA spectrum) indicate the loss of CO vibration bands after irradiation at 365 nm. The PLA stays intact during UV-illumination. | ||
The photo-induced loss of CO as the origin of the formed bubbles was confirmed by IR spectroscopy. A sample of CORMA-1–PLA20 was irradiated at 365 nm for 60 minutes and an ATR-IR spectrum was subsequently taken and depicted as the difference spectrum (Fig. 3e). The comparison between the irradiated and the non-irradiated sample of CORM-1–PLA20 showed a loss of the CO vibration bands at 2046, 2031, 2004, 645 and 465 cm−1 after illumination (Fig. 3e, bands downwards). In contrast, no change in the PLA vibration bands could be observed (Fig. 3e, bands upwards).
CO detection under solvent free conditions was performed using a portable CO detector (Draeger Pac7000).32 CO release from CORMA-1–PLA20 samples was achieved by irradiation at three different wavelengths. Irradiation with a dental LED lamp at 440–480 nm (Translux lamp) allowed rapid application for CO release (Fig. S8b†). The CO release was tested in a closed vessel with fixed duration of illumination (10 seconds ON) and 6 minutes OFF to reach equilibrium (see Fig. S8a† for the experimental setup). Fig. 3a shows that CO release was exclusively achieved through illumination, while no further CO was formed when the light source was shut off. Illumination at 365 nm and 480 nm was used to demonstrate CO release ability at UV-A and at wavelengths of visible light (see Fig. S9† for the experimental setup).23,24,50 Comparison of these two wavelengths showed the energy dependency of the CO release mechanism. Both wavelengths were fixed at the same light intensity (10 mW cm−2), but the CO release occurred more rapidly at 365 nm compared to 480 nm. This is shown by the calculated t1/2 values of 309 ± 51 and 1289 ± 16 seconds (Fig. 3b).50 These data impressively revealed how our CORMAs can be tuned in terms of their CO release kinetics. Simply varying the wavelength of irradiation allowed a difference of the CO release rate by a factor of four.
The quantification of the formed CO amount was accomplished via the gas IR technique. An IR gas cuvette was equipped with the non-woven sample (CORMA-1–PLA-20) and irradiated from the outside through a quartz window (Fig. S12†). Repeated measurements resulted in an average value of 3.4 ± 0.3 μmol CO per mg sample (Fig. 3c).
CORMA-1–PLA10 was applied to a heterogeneous myoglobin assay in aqueous solution. When attached to a paperclip and introduced in a sealable fluorescence cuvette, the non-woven could be irradiated from one direction while absorbance measurements could be applied in the orthogonal direction (Fig. S10†). CO was released during irradiation at 365 nm (and 480 nm) and reacted with reduced horse heart myoglobin (Mb) under oxygen free conditions to form carboxy-myoglobin (Mb-CO, Fig. 4a). We quantified the rate of CO release by a half-life value.51 It has recently been shown by Poole et al. that water-soluble CORMs exhibited variable CO release rates dependent on the amount of sodium dithionite (reducing agent for Mb) in the myoglobin assay.52 It was suggested that sodium dithionite can directly react with a CORM and enhance the CO release rate. Therefore, we checked the effect of three different amounts of the reducing agent compared to the initial Mb in the heterogeneous assay (Fig. S11†). Using 2, 20 and 200 equivalents of sodium dithionite we found release half-lives of 965 ± 63, 1305 ± 290, and 1180 ± 154 seconds, respectively. These data show that the given Mb-CO concentrations were formed in comparable time intervals. Thus, the amount of sodium dithionite did not significantly influence the rate of Mb conversion (Fig. S11†). An additional myoglobin experiment was performed during 480 nm irradiation. The results in Fig. 4b show that Mb conversions occurred much slower at 480 nm compared to 365 nm illumination, which confirms the wavelength dependency of the CO release.23,24,50
 | ||
| Fig. 4 CORMA-1–PLA10 in a heterogeneous myoglobin assay; (a) CO is released during irradiation and reacts with reduced horse heart myoglobin (Mb) under oxygen free conditions to form carboxy-myoglobin (Mb-CO); (b) irradiation wavelengths (365 nm in blue or 480 nm in red) influence the rate of CO release and myoglobin conversion. | ||
Leaching of CORM-1 from CORMA-1
ICP-MS was used to determine the loss of CORM-1 from CORMA-1–PLA10 and CORMA-1–PLA20 in an aqueous environment. In a typical experiment samples were soaked in water and agitated for 3 hours or 3 days. The amount of manganese in the aqueous supernatant was measured by ICP-MS. The amount of leached CORM-1 was recalculated from these data (see Table S2 in the ESI†). We found that around 0.5% of CORM-1 was lost from CORMA-1–PLA10, while CORMA-1–PLA20 only leaches approximately 0.1% after 3 hours of soaking. After three days the amounts of loss were less than 0.8% for CORMA-1–PLA10 and less than 0.3% for CORMA-1–PLA20. We also checked whether the extent of leaching was influenced during and after irradiation with light. The samples were irradiated at 365 nm during agitation for one hour. Then the samples were removed and soaked in a fresh aqueous solution to determine leaching of the degradation products. It was found that during irradiation up to 2.4% of CORM-1 was lost. However, agitation after irradiation showed again a lower leaching of the photolytic products of 0.8% maximum. These results showed that water insoluble CORM-1 and its metal fragment photoproduct(s) were very efficiently retained by the nanoporous PLA fibers.Cell experiments and (photo)cytoxicity
To ensure that CORMA-1–PLA materials do not cause toxic effects in a biological environment before CO is released, the cytoxicity on 3T3 mouse fibroblast cells was determined in the dark. Criteria of evidence for cytotoxicity will be given: due to the 3-dimensional fiber frameworks loss of detached dead cells could occur. Therefore, cytoxicity was not determined by quantities of dead cells. Instead, we analyzed changes in cell morphology and rough quantities that reveal non-toxic (below ∼5% of dead cells) and toxic effects (above 5%). The incorporation of different amounts of CORM-1 to the PLA did not significantly influence the cell morphology compared to a control sample (see Fig. S13†). After one and four days of cell incubation the cells were still adherent and only a small amount of cells appeared more spherical than the control cells. Less than 5% of dead cells were found. Therefore, the samples were categorized as non-toxic. The cell number clearly increased and a dense cell population was found after the incubation intervals (not shown) so that the proliferation of the cells was nearly not affected (Fig. S13†).In contrast, phototoxicity was observed during illumination of the samples at 365 nm (surveyed after 0, 15, and 60 minutes of irradiation; Fig. 5a). Samples of CORMA-1–PLA10 and CORMA-1–PLA20 were used to test the cell response during exposure to high concentrations of CO. A direct correlation between ongoing irradiation and toxic effects on cells was found for CORMA-1–PLA10 (Fig. 5b) and CORMA-1–PLA20 (Fig. S14†). For both samples the amount of dead cells increased during illumination; the increase was more pronounced for CORMA-1–PLA20 (Fig. S14†). Both experiments led to a change in cell morphology towards spherically shaped cells. These results indicate a strong phototoxic effect of the incorporated CORM-1 on 3T3 mouse fibroblasts compared to a pure PLA control sample. The contribution of UV-A light itself to the phototoxicity was negligible (see PLA control in Fig. 5b).
 | ||
| Fig. 5 Phototoxicity experiments with 3T3 mouse fibroblast cells; (a) experimental setup where cells and control (left part) or sample (right part) have been illuminated within the same well; (b) microscopic images of PLA control (60 min at 365 nm) and CORMA1–PLA-10 after 0, 15 and 60 minutes of exposure to UV-A light; (c) experimental setup with separated cells and samples. The released CO gas from the sample can diffuse to uncovered cells (right part), while control cells are protected from CO (left part); (d) microscopic images of a protected control and an unprotected cell sample after exposure of CORMA1–PLA-20 to UV-A light for 60 minutes. | ||
To evince that truly CO is responsible for the observed cell death, the phototoxicity experiments at 365 nm were repeated with a different setup. Instead of keeping cells and samples in the same well the non-woven sample was stored in one well and the cells were cultivated in the neighboring one, while a third control well was protected from CO intrusion (Fig. 5c). A cover lid over the whole well-plate avoided the loss of released CO into the atmosphere and allowed the diffusion of CO from the sample to the cells. Images in Fig. 5d show that more spherical and dead cells were found in the cell samples subjected to CO compared to the blank sample demonstrating the toxic effect of the pure CO gas on 3T3 mouse fibroblasts. It has to be noted here that the toxic effect of CO after 60 minutes of illumination was lower when the material and cells were separated instead of being stored together during irradiation. The reason for this difference can be found in the CO diffusion between two wells.
C Conclusion
Embedding the photosensitive dimanganese decacarbonyl (CORM-1) into nanoporous fibrous non-wovens of polylactic acid has been achieved in this work. The manganese carbonyl is of dual advantage. In the electrospinning process at ambient light slight decomposition of the complex via the loss of CO induces a nanoporous structure in the fibers. The resulting high surface area facilitates the interaction with light to generate an efficient triggerable CO delivery platform. In addition to the CO electrode and IR cuvette assay, a heterogeneous myoglobin assay was developed to characterize the light-induced CO release. Cytotoxicity of the biocompatible CORMAs on mouse fibroblast cells was very low in the dark. Exposure of the hybrid polymers to UV-A and visible light resulted in a rapid release of CO bubbles while the photoproducts were retained in the polymer matrix. Leaching of CORM-1 from the polymer matrix was negligible under physiological conditions due to the very low solubility of CORM-1 in water. The CO delivery platform has been tested by light-induced eradication of grown mouse fibroblast cells on the non-wovens. The efficacy was dependent on the content of dimanganese decacarbonyl. The versatility of the presented CORMAs in future applications is outstanding. The material can be wrapped around biological targets (skin, etc.) or attached to optical fibers. Currently we are investigating the non-wovens as a triggerable antibacterial platform53 and as a possible CO storage system for onboard calibration of atmospheric CO sensors.D Experimental
Materials
Chemicals and solvents were obtained from Fluka and VWR and used as received. Crystalline Mn2(CO)10 (CORM-1) was bought from ABCR GmbH & Co. KG (Germany). Horse heart myoglobin was obtained from Sigma-Aldrich. Poly(L-lactide-co-D/L-lactide) 70/30 (PLA, Resomer LR 708, Boehringer Ingelheim Pharma GmbH & Co. KG, Germany) was used for electrospinning. A weight-average molar mass of 1.3 × 106 g mol−1 was determined for the polymer by size exclusion chromatography using CHCl3 as the solvent and polystyrene as the external standard.Generation of CORMA-1–PLA non-wovens
The electrospinning process was carried out using the electrospinning apparatus E-Spintronic (Erich Huber GmbH, Germany). A stainless-steel straight-end hollow needle (0.4 mm) was used as the nozzle. A glass mirror of 3 mm thickness (30 × 30 cm) was used as a collector plate for collecting the electrospun non-woven fibers. The distance between the needle tip and the mirror was maintained at 20 cm. The voltage was adjusted to 25 kV. A 3 wt% solution of the PLA in CHCl3 containing 1, 10, and 20 wt% of CORM-1 related to the applied polymer was employed (CORMA-1–PLA1, CORMA-1–PLA10, and CORMA-1–PLA20). The polymer solution was fed at a constant rate of 1.5 ml h−1 through the syringe to the needle tip in the dark, resulting in the formation of fibers with diameters of about 500 nm to 3 μm. The dimension of the obtained electrospun non-wovens was approximately 30 cm2 with white (CORMA-1–PLA1), slightly yellow (CORMA-1–PLA10), and yellow (CORMA-1–PLA20) color, depending on the content of Mn2(CO)10.31Analysis of the manganese content of CORMA-1–PLA fibers
The overall manganese content within CORMA-1–PLA samples was determined after sample destruction in 65% ultrapure nitric acid and subsequent dilution with MilliQ water. The quantity of manganese in the aqueous solutions was determined by inductively coupled plasma-mass spectrometry (ICP-MS) on an XSeriesII from Thermo Fisher Scientific, Germany (isotope 55Mn). Calibration was performed with a multielement solution containing manganese ICP standard from Merck KGaA, Germany (CertiPUR). Measurements were performed against 101Ru as the internal standard (c = 20 μg l−1 in all samples).Leaching of CORM-1 from CORMA-1–PLA fibers
To determine the loss of CORM-1 from CORMA-1–PLA within an aqueous environment the non-woven samples were agitated in MilliQ water for three hours or three days (with or without previous 365 nm illumination) and the solution was subsequently mixed with nitric acid. The quantity of manganese in the aqueous solutions was determined by ICP-MS.Spectroscopic characterization of CORM-1 and CORMA
Absorption spectra of the materials were recorded on an Analytik Jena Specord S 600 UV-Vis spectrometer. IR spectra were recorded in a range of 400–4000 cm−1 on an Equinox 55 FTIR/FTNIR Spectrometer from Bruker Optik GmbH, Germany.Other measurements
Scanning electron microscopy (SEM) was performed with a field-emission scanning electron microscope Supra 55VP (Carl Zeiss AG, Germany). A Si wafer was used as the substrate material for the fibers and Au was sputtered on the specimens to ensure sufficient electric conductivity for the analysis. The images were taken with an InLens-Detector using 5 keV excitation energy.Energy dispersive X-ray spectroscopy (EDX) was investigated using the SEM equipped with an EDX-system (Quantax with Si(Li)-detector, Bruker Nano GmbH, Germany). The non-woven mats were coated with evaporated carbon. For the measurements, an excitation energy of 2 keV was used. Spectra were recorded from crossed fiber regions of the non-woven mat to get a larger excitation volume for EDX.
For optical investigation of CO release the laser scanning microscope LSM 700 (Carl Zeiss Microscopy GmbH, Germany) was applied. The non-woven mat (CORMA-1–PLA-20) was placed on an object slide, moistened with PBS buffer solution (pH = 7.4) and covered with a cover slip in the dark. After adjustment the sample was irradiated with light of 405 nm of the integrated light emitting diode. Every 2 s an image was collected for over 50 s.
CO release profile of CORMAs
For heterogeneous myoglobin assays a given amount of CORMA-1–PLA10 was accurately weighed and fixed on a paper clip. The clip and a small stirring bar were introduced into a fluorescence cuvette that was sealed with a rubber septum. Horse heart myoglobin was dissolved in 2.5 ml phosphate buffer (pH 7.4), filtered and added to the cuvette. Nitrogen gas was bubbled gently through the solution for 5 minutes. Sodium dithionite was weighed into a Schlenk flask, dissolved in degassed phosphate buffer and 2, 20 or 200 equivalents compared to met-myoglobin were added to the cuvette. The non-woven was irradiated at 365 nm (or 480 nm) in a fixed distance of 10 cm (1.4 mW cm−2) and UV-Vis absorption spectra were recorded in 5 minute intervals. The absorptions were recalculated to the corresponding Mb-CO concentrations and release half-life values were determined for quantification that is defined by the time taken for a solution of CORM (c = x μM) to produce a solution of carboxy-myoglobin with c = x/2 μM.51 Accordingly, the t1/2 release value was used, where CORM-1 (resembling 20 μM if it was dissolved) produces 10 μM of Mb-CO. LED excitation was performed with a HD-LED module consisting of 5 vertically aligned LED lamps (INNOTAS Elektronik GmbH, Germany) in a sealable illumination box. The intensity of the lamps was determined with the Photodiode Power Sensor S120VC equipped with the USB Power and Energy Meter Interface PM100USB from Thorlabs GmbH, Germany.CO release from CORMA in air was monitored with a portable CO detector from Draeger Safety Austria GmbH, Austria (Draeger Pac7000). CO release at 440–480 nm was achieved with a dental LED polymerization lamp (Translux Power Blue, Heraeus Holding GmbH, Germany). Further experiments at 365 and 480 nm were performed using the HD-LED modules applied in the myoglobin assay. The lamp intensities were adjusted to 10 mW cm−2.
Quantification of CO release in air was achieved via gas IR spectroscopy within a self-made IR gas cuvette. A Varian 670-IR FTIR spectrometer (Agilent Technologies Deutschland GmbH, Germany) was used to detect CO. Four UV LEDs (365 nm; Nitride Semiconductors Co., Ltd., Japan) were used to irradiate the non-woven from outside the cuvette through the glass window during the IR measurement. The applied light power (1 mW cm−2) was determined with a FieldMaxII laser power meter (Coherent Inc., Germany).
Live/dead assay
Mouse fibroblast 3T3 cells were seeded onto the non-woven mats fixed in cell crowns (Sigma-Aldrich, Germany) in daylight to a density of about 25000 cells cm−2 (50000 cells cm−2 in (photo)cytotoxicity experiments) using DMEM culture medium (Biochrom-Seromed KG, Germany) supplemented by 10% fetal bovine serum and Pen/Strep. For different experiments on cytocompatibility and on (photo)cytotoxicity (see below) cells were stained on viability with 15 μg ml−1 fluorescein diacetate and 1:10000 diluted GelRed® stock solution (VWR International, Germany). An Axiovert 25 microscope (Carl Zeiss Jena GmbH, Germany) with Zeiss filter sets 44 (excitation: BP 475/40, beamsplitter: FT 500, emission: BP 530/50) and 14 (excitation: BP 510-560, beamsplitter: FT 580, emission: LP 590) and a halogen lamp were used to monitor green and red fluorescence. Photomicrographs were recorded immediately after staining using a CCD fluor microscope imager MP 5000 (Intas Science Imaging Instruments GmbH, Germany). Imaging was supported by Image-Pro Plus software (Media Cybernetics Inc., USA). The total number of cells and the percentage of living cells were calculated after counting red-fluorescent nuclei of dead cells and green-fluorescent living cells.
Cytocompatibility of CORMAs
To assess cytocompatibility under prevention of CO release, the non-woven mats CORMA-1–PLA1, CORMA-1–PLA10, CORMA-1–PLA20 and a PLA blank were incubated with the 3T3 fibroblast cell line in the dark for one and four days.(Photo)cytotoxicity of CORMAs
To investigate the behavior of cells under CO release from the fibers, the non-woven mats CORMA-1–PLA10, CORMA-1–PLA20 and a PLA control (blank sample spun from an acetone solution) were incubated with the 3T3 fibroblast cell line in the dark for one day. For induction of CO release materials were irradiated for 15 min as well as 1 h with UV light using an UV transilluminator (365 nm, 4 mW cm−2) within a dark box (Gel Imager, Intas Science Imaging Instruments GmbH, Germany). Afterwards the cells were cultivated for further 60 min and then stained with fluorescein diacetate and GelRed®. In a control experiment seeded cells and the test material were stored in different adjacent wells and covered under one lid to examine the influence of CO gassing from the sample wells into the cell wells. Blank cells were seeded in another well row and covered with foil to avoid CO gas influences on the cells. The well plate was then incubated at 37 °C for one day and the non-wovens were subsequently irradiated at 365 nm.Acknowledgements
We thank the Deutsche Forschungsgemeinschaft (DFG) for supporting FOR 1738. A.S. is grateful to the Carl Zeiss foundation for a Junior Professor fellowship and the EC for financial support through the FP7 project ‘Novosides’ (grant agreement no. KBBE-4-265854). The German Federal Ministry of Economics and Technology (BMWi) is gratefully acknowledged for financial support (grant no. MF110109). We also acknowledge the support of the Center of Medical Optics and Photonics (CEMOP), the Abbe Center of Photonics (ACP) and the Jena Center of Soft Matter (JCSM) at the Friedrich Schiller University Jena. We thank Dr. M. Schweder (INNOVENT e.V.) for SEM investigation.References
- R. Motterlini and L. E. Otterbein, Nat. Rev. Drug Discovery, 2010, 9, 728–743 CrossRef CAS PubMed.
- B. E. Mann and R. Motterlini, Chem. Commun., 2007, 4197–4208 RSC.
- S. W. Ryter and L. E. Otterbein, BioEssays, 2004, 26, 270–280 CrossRef CAS PubMed.
- S. H. Heinemann, T. Hoshi, M. Westerhausen and A. Schiller, Chem. Commun., 2014 10.1039/C3CC49196J.
- R. Motterlini, J. E. Clark, R. Foresti, P. Sarathchandra, B. E. Mann and C. J. Green, Circ. Res., 2002, 90, e17–24 CrossRef CAS.
- L. E. Otterbein, B. S. Zuckerbraun, M. Haga, F. Liu, R. Song, A. Usheva, C. Stachulak, N. Bodyak, R. N. Smith, E. Csizmadia, S. Tyagi, Y. Akamatsu, R. J. Flavell, T. R. Billiar, E. Tzeng, F. H. Bach, A. M. K. Choi and M. P. Soares, Nat. Med., 2003, 9, 183–190 CrossRef CAS PubMed.
- S. W. Chung, X. Liu, A. A. Macias, R. M. Baron and M. A. Perrella, J. Clin. Invest., 2008, 118, 239–247 CAS.
- J. Kohmoto, A. Nakao, T. Kaizu, A. Tsung, A. Ikeda, K. Tomiyama, T. R. Billiar, A. M. K. Choi, N. Murase and K. R. McCurry, Surgery, 2006, 140, 179–185 CrossRef PubMed.
- G. Li Volti, L. F. Rodella, C. Di Giacomo, R. Rezzani, R. Bianchi, E. Borsani, D. Gazzolo and R. Motterlini, Nephron Exp. Nephrol., 2006, 104, e135–e139 CrossRef CAS PubMed.
- R. Motterlini, A. Gonzales, R. Foresti, J. E. Clark, C. J. Green and R. M. Winslow, Circ. Res., 1998, 83, 568–577 CrossRef CAS.
- A. Nakao, D. J. Kaczorowski, Y. Wang, J. S. Cardinal, B. M. Buchholz, R. Sugimoto, K. Tobita, S. Lee, Y. Toyoda, T. R. Billiar and K. R. McCurry, J. Heart Lung Transplant., 2010, 29, 544–553 CrossRef PubMed.
- F. Zobi, Future Med. Chem., 2013, 5, 175–188 CrossRef CAS PubMed.
- R. Alberto and R. Motterlini, Dalton Trans., 2007, 1651–1660 RSC.
- C. C. Romao, W. A. Blattler, J. D. Seixas and G. J. L. Bernardes, Chem. Soc. Rev., 2012, 41, 3571–3583 RSC.
- U. Schatzschneider, Inorg. Chim. Acta, 2011, 374, 19–23 CrossRef CAS PubMed.
- D. Crespy, K. Landfester, U. S. Schubert and A. Schiller, Chem. Commun., 2010, 46, 6651–6662 RSC.
- B. E. Mann, Organometallics, 2012, 31, 5728–5735 CrossRef CAS.
- R. D. Rimmer, A. E. Pierri and P. C. Ford, Coord. Chem. Rev., 2012, 256, 1509–1519 CrossRef CAS PubMed.
- R. Motterlini, Biochem. Soc. Trans., 2007, 35, 1142–1146 CrossRef CAS PubMed.
- J. L. Wilson, H. E. Jesse, B. Hughes, V. Lund, K. Naylor, K. S. Davidge, G. M. Cook, B. E. Mann and R. K. Poole, Antioxid. Redox Signaling, 2013, 19, 497–509 CrossRef CAS PubMed.
- A. Marazioti, M. Bucci, C. Coletta, V. Vellecco, P. Baskaran, C. Szabó, G. Cirino, A. R. Marques, B. Guerreiro, A. M. L. Gonçalves, J. D. Seixas, A. Beuve, C. C. Romão and A. Papapetropoulos, Arterioscler. Thromb. Vasc. Biol., 2011, 31, 2570–2576 CrossRef CAS PubMed.
- L. S. Nobre, J. D. Seixas, C. C. Romão and L. M. Saraiva, Antimicrob. Agents Chemother., 2007, 51, 4303–4307 CrossRef CAS PubMed.
- R. S. Herrick and T. L. Brown, Inorg. Chem., 1984, 23, 4550–4553 CrossRef CAS.
- A. Rosa, G. Ricciardi, E. J. Baerends and D. J. Stufkens, Inorg. Chem., 1996, 35, 2886–2897 CrossRef CAS.
- S. Hou, R. Xu, S. H. Heinemann and T. Hoshi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 4039–4043 CrossRef CAS PubMed.
- W. J. Wilkinson and P. J. Kemp, J. Physiol., 2011, 589, 3055–3062 CrossRef CAS PubMed.
- C. Bohlender, M. Wolfram, H. Goerls, W. Imhof, R. Menzel, A. Baumgaertel, U. S. Schubert, U. Mueller, M. Frigge, M. Schnabelrauch, R. Wyrwa and A. Schiller, J. Mater. Chem., 2012, 22, 8785–8792 RSC.
- P. J. Kluger, R. Wyrwa, J. Weisser, J. Maierle, M. Votteler, C. Rode, M. Schnabelrauch, H. Walles and K. Schenke-Layland, J. Mater. Sci.: Mater. Med., 2010, 21, 2665–2671 CrossRef CAS PubMed.
- M. Reise, R. Wyrwa, U. Müller, M. Zylinski, A. Völpel, M. Schnabelrauch, A. Berg, K. D. Jandt, D. C. Watts and B. W. Sigusch, Dent. Mater., 2012, 28, 179–188 CrossRef CAS PubMed.
- J. D. Seixas, A. Mukhopadhyay, T. Santos-Silva, L. E. Otterbein, D. J. Gallo, S. S. Rodrigues, B. H. Guerreiro, A. M. L. Goncalves, N. Penacho, A. R. Marques, A. C. Coelho, P. M. Reis, M. J. Romao and C. C. Romao, Dalton Trans., 2013, 42, 5985–5998 RSC.
- R. Wyrwa, M. Schnabelrauch, C. Altmann and A. Schiller, Kohlenstoffmonoxid freisetzende Materialien und deren Verwendung, DE 10 2012 004 132 A1, 2012.
- U. Hasegawa, A. J. van der Vlies, E. Simeoni, C. Wandrey and J. A. Hubbell, J. Am. Chem. Soc., 2010, 132, 18273–18280 CrossRef CAS PubMed.
- B. J. Heilman, J. St. John, S. R. J. Oliver and P. K. Mascharak, J. Am. Chem. Soc., 2012, 134, 11573–11582 CrossRef CAS PubMed.
- J. B. Matson, M. J. Webber, V. K. Tamboli, B. Weber and S. I. Stupp, Soft Matter, 2012, 8, 6689–6692 RSC.
- M. Ma, H. Noei, B. Mienert, J. Niesel, E. Bill, M. Muhler, R. A. Fischer, Y. Wang, U. Schatzschneider and N. Metzler-Nolte, Chem. – Eur. J., 2013, 19, 6785–6790 CrossRef CAS PubMed.
- G. Dördelmann, T. Meinhardt, T. Sowik, A. Krueger and U. Schatzschneider, Chem. Commun., 2012, 48, 11528–11530 RSC.
- G. Dördelmann, H. Pfeiffer, A. Birkner and U. Schatzschneider, Inorg. Chem., 2011, 50, 4362–4367 CrossRef PubMed.
- P. C. Kunz, H. Meyer, J. Barthel, S. Sollazzo, A. M. Schmidt and C. Janiak, Chem. Commun., 2013, 49, 4896–4898 RSC.
- M. Shimada, Y. Morimoto and S. Takahashi, J. Organomet. Chem., 1993, 443, C8–C10 CrossRef CAS.
- M. Cheiky and W. Hago, Method for manufacture of films containing insoluble solids embedded in cellulose-based films, US Pat., US 6929884 B2, 2005.
- A. Shaver, J. P. Gao and I. S. Butler, Appl. Organomet. Chem., 1988, 2, 9–15 CrossRef CAS.
- C. Bohlender, K. Landfester, D. Crespy and A. Schiller, Part. Part. Syst. Charact., 2013, 30, 138–142 CrossRef CAS.
- K. A. Wold, V. B. Damodaran, L. A. Suazo, R. A. Bowen and M. M. Reynolds, ACS Appl. Mater. Interfaces, 2012, 4, 3022–3030 CAS.
- A. Greiner and J. Wendorff, Angew. Chem., Int. Ed., 2007, 46, 5670–5703 CrossRef CAS PubMed.
- J. H. Wendorff, S. Agarwal and A. Greiner, Electrospinning: Materials, Processing, and Applications, Wiley-VCH, Weinheim, 2012 Search PubMed.
- J. Fang, X. Wang and T. Lin, Functional Applications of Electrospun Nanofibers, Nanofibers - Production, Properties and Functional Applications, InTech, Shanghai, 2011 Search PubMed.
- J. S. Im, S. C. Kang, S.-H. Lee and Y.-S. Lee, Carbon, 2010, 48, 2573–2581 CrossRef CAS PubMed.
- C. J. Luo, S. D. Stoyanov, E. Stride, E. Pelan and M. Edirisinghe, Chem. Soc. Rev., 2012, 41, 4708–4735 RSC.
- R. Wyrwa, B. Finke, H. Rebl, N. Mischner, M. Quaas, J. Schaefer, C. Bergemann, J. B. Nebe, K. Schroeder, K.-D. Weltmann and M. Schnabelrauch, Adv. Eng. Mater., 2011, 13, B165–B171 CrossRef.
- T. Kobayashi, H. Ohtani, H. Noda, S. Teratani, H. Yamazaki and K. Yasufuku, Organometallics, 1986, 5, 110–113 CrossRef CAS.
- A. J. Atkin, J. M. Lynam, B. E. Moulton, P. Sawle, R. Motterlini, N. M. Boyle, M. T. Pryce and I. J. S. Fairlamb, Dalton Trans., 2011, 40, 5755–5761 RSC.
- S. McLean, B. E. Mann and R. K. Poole, Anal. Biochem., 2012, 427, 36–40 CrossRef CAS PubMed.
- A. Melaiye, Z. Sun, K. Hindi, A. Milsted, D. Ely, D. H. Reneker, C. A. Tessier and W. J. Youngs, J. Am. Chem. Soc., 2005, 127, 2285–2291 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: SEM, EDX, UV-Vis, IR, DSC, BET, Hg porosity measurements, detection of CO release, myoglobin assay, and ICP-MS. See DOI: 10.1039/c3tb21649g |
| This journal is © The Royal Society of Chemistry 2014 |