 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A general and versatile procedure for coating of hydrophobic nanocrystals with a thin silica layer enabling facile biofunctionalization and dye incorporation†
J.
Cichos
and
M.
Karbowiak
*
University of Wrocław, Faculty of Chemistry, ul. F. Joliot-Curie 14, 50-383 Wroclaw, Poland. E-mail: miroslaw.karbowiak@chem.uni.wroc.pl
First published on 14th November 2013
Abstract
Here we report a method for coating of hydrophobic nanocrystals (NCs) with a thin silica layer. By using this approach 11 nm NaGdF4 NCs were coated with a mesoporous silica shell thinner than 20 nm resulting in highly monodisperse core–shell nanocomposite particles smaller than 50 nm. The synthetic protocol used here is based on the two-phase system that ensures high repeatability of the nanoparticle morphology and can be conveniently adapted for large-scale production. The use of an organic solvent as a diluent for tetraethoxysilane allows us to control not only the thickness, but also the porosity of the uniform silica shell. This procedure can be applied for coating NCs of various sizes and can be generalized to encapsulate other nanocrystals, e.g. semi-conductor quantum dots, into silica spheres. Furthermore, it allows facile surface modification with –NH2 groups that can be then used to conjugate biomolecules to the nanoparticle surface, as well as straightforward incorporation of various organic molecules (e.g. dyes or drugs) into the silica shell during its growth. To demonstrate potential applications up-converting NaGd0.80Yb0.18Er0.02F4 NCs were used as cores and zinc phthalocyanine was incorporated into the silica shell as a photosensitizer. Under irradiation with a 980 nm laser diode efficient generation of singlet oxygen was observed indicating that such nanocomposite particles have the capability to be used for photodynamic therapy.
1. Introduction
Biocompatible, monodisperse, up-converting nanoparticles (UCNPs) doped with rare-earth ions are attracting appreciable attention due to their unique optical properties and possibility of use in many technological fields.1 The rare earth doped hexagonal-phase sodium lanthanide (Ln) fluorides (β-NaLnF4) are one of the most efficient up-converting phosphors to date.2 The most successful and in consequence the most widely used method of synthesis of NaLnF4 nanocrystals (NCs) is based on the thermal decomposition of trifluoroacetate precursors in the presence of a capping ligand (e.g. oleic acid) in a high boiling point solvent (e.g. 1-octadecene).3 The NCs obtained in this way are crystalline, have a narrow size distribution in the range of nanometers, but are not dispersible in water. The solubility is a crucial issue for biological applications, therefore, in recent years considerable efforts have been undertaken to render the NaLnF4 nanocrystals dispersible in water.To render NaYF4 NCs hydrophilic a number of compounds were tested as bio-compatible ligands for surface modification, e.g. PEG phosphate,4 mercaptopropionic acid,5,6 diphosphonic acid,7 phospholipids8 as well as polymer molecules such as poly(acrylic acid),9 poly(allylamine),10 polyethylenimine,11 polyvinylpyrrolidone12 and amorphous silica.12–16 The latter appears as the most widely accepted approach. Silica coating provides shielding of NCs from the water environment, thereby preventing emission quenching. At the same time silica facilitates introduction of reactive groups onto the nanoparticle (NP) surface for further bio-functionalization using well-established protocols. The most popular reagents used for surface modification are (3-aminopropyl)triethoxysilane (APTES) and (3-mercaptopropyl)trimethoxysilane (MPTES) which introduce NH2 and SH groups, respectively, enabling the covalent binding of biological molecules to the surface of NPs.
The two most common methods in the literature for coating of nanofluoride NCs are analogous to those widely used for preparation of monodisperse colloidal silica particles, i.e. the Stöber approach and the microemulsion based method. The first group of methods originates from the procedure described in 1968 by Stöber17 including its subsequent various modifications. There are numerous examples of using this method for silica coating of various NCs, for example Fe3O4,18 NaYF4:Ln3+,13,15,19 or LnF3.20 The main limitation of the Stöber method is that it can be employed for NCs forming colloidal solutions in water–alcohol media, whereas the vast majority of synthetic methods yields NCs with a hydrophobic surface which enables them to be suspended in chloroform or toluene, but not in water or ethanol. Moreover, the Stöber method, which was originally devised for preparation of colloidal silica nanoparticles, is very efficient in the synthesis of monodisperse silica spheres with diameters larger than 200–250 nm. Even though it is possible to obtain much smaller particles (<30 nm), their polydispersity typically exceeds 20%.21 Consequently, the silica shells obtained on NCs by Stöber related methods are relatively thick and achievement of thin uniform layers with controllable thickness is still quite challenging.
An alternative route for synthesis of small silica nanoparticles (<50 nm) with better polydispersity is the microemulsion approach.22 Microemulsion based methods have been employed for fabrication of silica shells in various NCs (quantum dots,23 noble metals,24,25 FexOy,26 NaYF4 nanofluorides,12,14,27 and many others) yielding, in general, superior results compared to the Stöber method. However, the distinct disadvantage of microemulsion methods is the use of large amounts of surfactants, which require extensive washing, as well as problems with obtainment and sustainment during the synthesis of uniform micelles.21,28 Moreover, these methods allow obtaining silica layer with thickness that is rather discrete than variable in a continuous range.29 The efficacy and repeatability of the microemulsion method is strongly conditioned by controlling many strictly defined parameters of the process, such as the temperature and concentration of water, catalysts, SiO2 precursors, coated nanoparticles as well as surfactants and co-surfactants. Deviation from optimal conditions makes the control of silica shell thickness significantly more difficult and results, in a most fortunate case, in agglomeration and/or embodying of many cores into one silica shell.30 Another important limitation results from the fact that microemulsion methods cannot be conveniently adapted for large-scale production.
For some applications, such as photodynamic therapy (PDT), it is important to be able to obtain the mesoporous structure of the silica layer and to control the porosity degree. Most of the methods hitherto reported in the literature require additional steps in the synthesis procedure to achieve the desired porosity.31,32 Moreover, photosensitization of NPs usually entails a separate procedure of chemical attachment of photosensitizing molecules to the surface of modified (e.g. using (3-aminopropyl)triethoxysilane NH2(CH2)3Si(OC2H5)3) silica. The simple impregnation of NPs in the solution of photosensitizer usually leads to agglomeration and leakage of the dye outside NPs during PDT treatment.
The demonstrated disadvantages of Stöber and microemulsion methods indicate that coating of hydrophobic NaLnF4 NCs with a thin and uniform silica layer with controllable degree of porosity still remains quite challenging. Despite recent advances the morphology and monodispersity of silica coated NCs synthesized using existing protocols are still far from ideal. The important breakthrough in synthesis of monodisperse silica particles with sizes below 50 nm was made by Yokoi et al.33,34 who carried out hydrolysis and polycondensation of TEOS in the two-phase system in the presence of an amino acid (lysine or arginine) as a base catalyst. Under such conditions, silicate species are slowly delivered from an organic to an aqueous phase, whereas buffering properties of the amino acid keep the pH value constant (pH = 9–10) during the synthesis. Modification of the method of Yokoi et al. enabled Hartlen et al.21 to obtain silica particles with the controllable size ranging from 15 to 200 nm. We used these approaches which proved to be the most efficient in the synthesis of silica spheres as a starting point for exploiting the synthetic route for coating of nanocrystals with silica shells.
Herein, we report a novel, versatile and universal procedure for direct coating of hydrophobic nanocrystals with a uniform mesoporous silica layer resulting in monodisperse NC@mSiO2 core–shell nanocomposite particles smaller than 60 nm. The coating is carried out in a biphasic system in which TEOS is delivered heterogeneously from a top organic layer to the bottom aqueous layer containing CTAB stabilized NCs and L-arginine as a catalyst, which ensures high repeatability of NP morphology. The method can be used for coating of various kinds of NCs, such as nanofluorides (e.g. NaGdF4) or quantum dots (e.g. PbS). Coating of hydrophobic NCs with a layer of silica makes them dispersible in water and other polar solvents. Furthermore, the proposed procedure allows facile surface modification with various functional groups (e.g., amine, thiol, carboxyl) which can be then used to conjugate biomolecules to the NP surface using some well established protocols. The synthetic parameters allowing control of the porosity and thickness of the silica layer are also indicated and discussed. The method is particularly suitable for straightforward incorporation of dyes (e.g. photosensitizers) or other organic molecules into the silica layer during its growth. If photon up-converting NCs, e.g. NaLnF4:Er3+,Yb3+, are used as cores, the incorporated photosensitizer can be excited with radiation in the NIR region.
2. Experimental section
2.1. Chemicals
Gd2O3, Er2O3 and Yb2O3 (all 99.99%) were purchased from Stanford Materials, and trifluoroacetic acid (TFA, peptide grade) was from Iris Biotech. Oleic acid (OA, 90% technical grade), octadecene (ODE, 90% technical grade) and all other chemicals (analytical grade) were from Sigma Aldrich and were used without further purification.2.2. Synthesis of NGF@OA cores
NaGd0.80Yb0.18Er0.02F4 nanocrystals (NGF@OA) were synthesized through thermal decomposition of trifluoroacetate precursors using a slightly modified protocol described previously.35 Ln2O3 (Ln = Gd, Er or Yb) was placed in a beaker, moistened with water and some concentrated TFA was added. The beaker was heated under cover until a transparent solution was obtained; afterwards the excess of water and TFA was evaporated nearly to dryness and the crystals of trifluoroacetate were placed in a vacuum desiccator over P2O5 for 3 days. According to a reported protocol35 the appropriate amounts of the TFA salts were placed in a three neck round bottom flask and a mixture of OA and ODE (30 ml, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) was added. The suspension was heated to 110 °C and degassed using a vacuum pump at 110–130 °C for 60 min with periodic Ar refills. Afterwards the flask was placed in a salt bath composed of NaNO3 and KNO3 (1:1 wt) preheated to 355 °C, kept in it for 30 min and subsequently cooled down by addition of 15 ml of ODE. Nanocrystals were precipitated with ethanol and centrifuged. The product was washed by dissolution in toluene and precipitation with ethanol 5 times and then dried in a N2 stream and stored in a tightly closed vial. The nanocrystals were redispersed in chloroform before use.
:1 v/v) was added. The suspension was heated to 110 °C and degassed using a vacuum pump at 110–130 °C for 60 min with periodic Ar refills. Afterwards the flask was placed in a salt bath composed of NaNO3 and KNO3 (1:1 wt) preheated to 355 °C, kept in it for 30 min and subsequently cooled down by addition of 15 ml of ODE. Nanocrystals were precipitated with ethanol and centrifuged. The product was washed by dissolution in toluene and precipitation with ethanol 5 times and then dried in a N2 stream and stored in a tightly closed vial. The nanocrystals were redispersed in chloroform before use.
2.3. Coating procedure yielding NGF@mSiO2 NPs
In a standard procedure 0.65 cm3 of solution of oleic acid stabilized monodispersed NGF@OA nanoparticles in chloroform (10 mg cm−3) was added to 5 cm3 of 0.05 M CTAB solution in water and mixed vigorously for about 1 h to obtain a homogeneous oil-in-water microemulsion. Afterwards the vial was placed under N2 and the temperature was raised to about 70–80 °C to evaporate CHCl3, which yielded aqueous-phase dispersed nanoparticles. The as prepared transparent solution was transferred into a round bottom flask followed by addition of 25 cm3 of DI water. The solution of 40 mg of L-arginine in 1 cm3 of DI water was added into the flask and the solution was warmed up to 70 °C and stirred gently with a magnetic stirrer (about 400–500 rpm). At this temperature the solution of 0.25 cm3 of tetraethoxysilane in 2 cm3 of ethyl acetate was added slowly to avoid mixing of the phases. Afterwards the flask was closed tightly with a glass plug sealed with Teflon tape (after 5 min the pressure was equalized via unplugging the flask for a second). During coating the bottom aqueous phase should be mixed, but care should be taken to leave the top organic phase nearly undisturbed. It is easily manageable by using a magnetic stirrer and adjusting appropriately the stirring rate of the standard cylinder-shaped magnetic stirring rod. Under laboratory conditions the synthesis can be easily scaled up to the total volume of about 500 cm3. In this case the appropriate mixing can be simply realized by the use of dumbbell or ellipsoidal magnetic stirring rods of appropriate size. After 24 h the product (NGF@SiO2) was collected by centrifugation and washed with water and ethanol (5 times each). Nanoparticles were then redispersed in ethanol–water or kept dry for other applications.2.4. Surface modification with –NH2 groups yielding NGF@mSiO2–NH2 NPs
Nanoparticles functionalized with –NH2 groups were obtained by addition of 50 μl of APTES ((3-aminopropyl)triethoxysilane) in 2 ml of cyclohexane or ethyl acetate to the top organic layer after 6–24 h of reaction time. All other synthesis steps were the same as those described for NGF@SiO2.2.5. Photosensitizer incorporation yielding NGF@mSiO2(Pc) or NGF@mSiO2(Pc)–NH2 NPs
To incorporate zinc phthalocyanine (ZnPc) the coating method was modified by using a saturated solution of ZnPc in ethyl acetate instead of a pure solvent. All other treatments, including the washing protocol of the product, remained the same as in the coating procedure yielding NGF@mSiO2 NPs. Addition of APTES (50 μl in 2 ml of ethyl acetate) after 6–24 h of reaction time to the top layer yields NPs with ZnPc incorporated into the silica shell and –NH2 groups present on the surface (NGF@mSiO2(Pc)–NH2).2.6. FITC labeling
Conjugation of a fluorescent dye to the amine-modified surface of silica-coated nanoparticles (NGF@mSiO2–NH2) was carried out as follows: 100 μl of freshly prepared fluorescein isothiocyanate FITC solution (1 mg cm−3) in carbonate buffer (pH = 9) was added to the Eppendorf vials containing a colloidal solution of 5 mg silica coated nanoparticles in 1 cm3 of carbonate buffer (pH = 9). The vial was then protected from light and mixed gently for 2 hours at room temperature using rotary shaker (R = 10 cm, full rotation every 5 sec). Afterwards to remove the non-conjugated fluorescein FITC-modified nanoparticles were centrifuged and washed 6 times with carbonate buffer until no fluorescence could be detected in the supernatant under a UV-lamp, and then once with DI water. Nanoparticles were dispersed in 2 cm3 PBS (pH = 7.4) for emission measurements.2.7. Synthesis of PbS@mSiO2 NPs
PbS quantum dots were synthesized using modified reported methods.36,37 Lead(II) oleate stock solution (0.2 M) was prepared by heating the PbO (10 mmol, 2.2320 g) in a mixture of oleic acid (80 mmol, 28.16 cm3) and octadec-1-ene (21 cm3) at 130 °C in an argon atmosphere, and a sulfur stock solution (0.2 M) was prepared by dissolving sulfur (10 mmol, 320 mg) in 50 cm3 of ODE in an argon atmosphere. The stock solutions were stored under Ar in septum closed vials. 1 cm3 of lead(II) oleate and 5 cm3 of ODE were placed in a three-neck flask purged with argon and heated to 130 °C. In the temperature range between about 100 °C and 130 °C the mixture was degassed using a vacuum pump and periodically purged with argon. After about 30 min the flask (filled with Ar) was transferred to a salt bath consisting of the NaNO3–KNO3 mixture (1:1 wt) preheated to 310 °C. After the temperature of the reaction mixture reached 310 °C 1 cm3 of sulfur solution was injected. After 5 min of reaction time the black solution was cooled down by placing the flask in cold water and QDs were precipitated with ethanol and washed by dispersing in chloroform and precipitation with ethanol, repeatedly (5 cycles). The as prepared PbS was dispersed in CHCl3 for further application. The procedure used for silica coating of the obtained PbS@OA was in general the same as that for the NaGdF4 nanocrystals, except that the amount of PbS@OA NCs was 3 mg.
2.8. Characterization
TEM images were recorded on the FEI Tecnai G2 20 X-TWIN transmission electron microscope at an acceleration voltage of 200 kV. Colloidal solutions of nanoparticles with a concentration of about 5 mg ml−1 in chloroform (before coating) or in ethanol (after coating) were drop-cast on a 3 mm TEM grid and dried under an IR lamp for 15 min.The emission and excitation spectra were recorded on the Edinburgh Instruments FLSP920 spectrometer equipped with a Xe900 xenon lamp (450 W).
Zeta potential measurements were performed with a Malvern Zetasizer Nano ZS90 instrument.
2.9. Measurement of singlet oxygen generation
Singlet oxygen generation was determined quantitatively using ABDA (9,10-anthracenediylbis(methylene)dimalonic acid) as a probe. A quartz cuvette (1 cm × 1 cm) containing nanoparticle water dispersion and ABDA was placed in a spectrophotometer holder equipped with a magnetic stirrer. During all measurements the ABDA concentration was set to 20 μM and the amount of nanoparticles was variable. The decrease in ABDA emission intensity (excited at 382 nm) was monitored after 0, 5, 10, 40 and 70 min of irradiation with a 980 nm laser diode (400 mW). Alternatively, the kinetic measurement mode of the spectrophotometer was used and emission spectra of ABDA were collected every minute over an hour under continuous irradiation of the sample with a 980 nm laser diode.3. Results and discussion
3.1. Synthesis and characterization of NGF@mSiO2 nanoparticles
The core up-converting β-NaGd0.80Yb0.18Er0.02F4 NCs (NGF@OA) were synthesized by thermal decomposition of sodium and lanthanide trifluoroacetates dissolved in a mixture of oleic acid and 1-octadecene following the procedure described by Murray et al.35 TEM images show that this one-pot method enables synthesis of NGF@OA NCs which are spherical in shape with a uniform size of 12 nm (Fig. S1 in the ESI†). The obtained oleic acid stabilized NCs form transparent colloidal solutions when dispersed in toluene, chloroform or other nonpolar organic solvents. By using the same method but extending the reaction time larger nanocrystals in the form of hexagonal prisms were obtained and also used in this study.The difficulty in obtaining an aqueous solution of hydrophobic core NCs was an important problem which had to be tackled. Fortunately, it has been previously demonstrated that hydrophobic NCs can be successfully transferred to an aqueous solution using an appropriate surfactant, e.g. CTAB.38,39 Following this approach Kim et al.40,41 transferred hydrophobic Fe3O4@OA NCs to an aqueous phase and coated them with a mesoporous silica (mSiO2) shell, using TEOS as a silica precursor and NaOH as a base catalyst. Using this approach in the next step of our procedure a microemulsion of NGF@OA NCs in water was prepared. To transfer hydrophobic NCs from a nonpolar solvent (chloroform) to an aqueous phase CTAB, Tween 80, SDS (sodium dodecyl sulfate), Triton X-100 and Igepal-CO 520 surfactants were tested. Among them only CTAB and SDS allowed us to obtain stable O/W microemulsions with good repeatability. Evaporation of chloroform from O/W microemulsion droplets yields a transparent colloidal solution of NGF@OA/CTAB (NGF@OA/SDS) NCs in water.
NGF@OA/CTAB NCs were coated with a mesoporous silica layer using the biphasic system – the bottom water phase and the top organic phase. The latter was composed of ethyl acetate or cyclohexane in which TEOS was dissolved as the silica precursor. The water phase contained dispersed NGF@OA/CTAB (NGF@OA/SDS) NCs and L-arginine (L-Arg) as a base catalyst. Such a biphasic system slows down the TEOS hydrolysis, since it is delivered with restricted rate from the organic phase to the aqueous phase where it hydrolyzes.21 In several trials it was found that the coating process proceeded much better for the CTAB stabilized microemulsion. In the case of using SDS as the surfactant only highly agglomerated, multi-core and shapeless agglutinations were observed after coating. Besides stabilizing hydrophobic NGF@OA NCs in the aqueous phase, the CTAB surfactant serves also as a template enabling formation of a mesoporous silica layer.40,41
Washing with water and ethanol (5 times each) is sufficient for removal of all unreacted species and CTAB. In FTIR spectra (Fig. S2†) only very weak bands attributed to CH vibrations can be detected at about 2900 cm−1. Additional washing with HCl solution (pH = 1–2) or heating at 130 °C in air for 12 h decreases only slightly and to the same extent the intensity of CH bands, due to removal of residual ethoxy groups (resulting from partially hydrolyzed TEOS) and/or solvent molecules from pores. Then, the residual CH bands observed in the FTIR spectrum should be attributed to OA which was not completely replaced by hydrolyzed TEOS on the NGF@OA NCs' surface. The band of N(CH3)R+ expected at 2800–3200 cm−1 is not observed in FTIR spectra. Moreover, also in the EDX spectrum (Fig. S3†) no signal from Br was detected, which indicates that CTAB is absent in the obtained material. Therefore, in our synthetic protocol no additional steps, such as treatment with HCl solution41 or calcination at 500 °C,31 are required for removal of the surfactant.
Fig. 1A and B show TEM images of the silica coated NGF@mSiO2 NPs obtained using ethyl acetate or cyclohexane, respectively, as the top organic layer. The resulting core–shell NGF@mSiO2 NPs are highly monodisperse, with empty or multicore particles being practically absent. No agglomeration is observed and the porous structure of the silica shell is clearly noticeable. The average size of particles obtained from ethyl acetate and cyclohexane is 72 and 61, whereas the shell thickness is 30 and 25 nm, respectively. NPs shown in Fig. 1 were synthesized using the procedure described in the Experimental section, which hereafter will be referred to as the standard procedure.
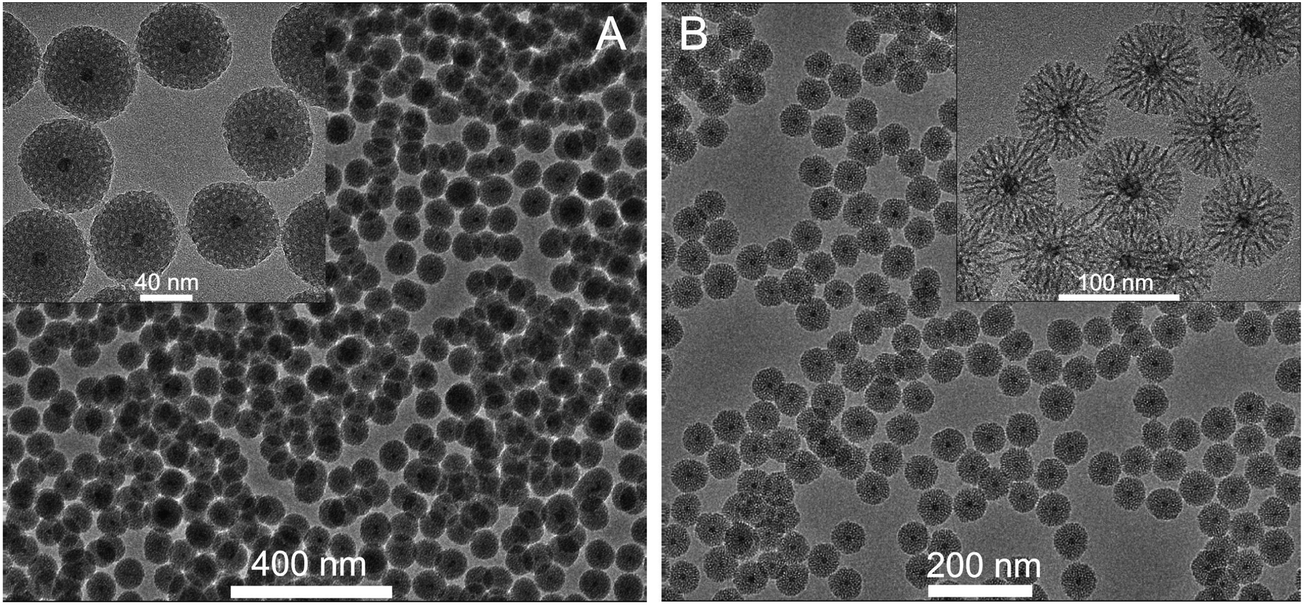 | ||
| Fig. 1 Silica coated nanoparticles prepared with the use of ethyl acetate (A) or cyclohexane (B) as the diluent for TEOS. | ||
EDX maps confirm the core–shell structure of NGF@mSiO2 NPs. Fig. 2 proves that Gd and Na are only present in the core of NPs, whereas the shell is composed of Si and O. The HR-TEM image and SAED pattern indicate that the monocrystallinity of the core is retained after silica coating.
 | ||
| Fig. 2 EDX maps measured for NGF@mSiO2 NPs (synthesized using ethyl acetate) shown in the TEM image presented in the upper inset. The bottom inset presents the HR-TEM image and SAED pattern of silica coated nanoparticles. | ||
The synthesis of uniform NaYF4:Tm3+/Yb3+/Gd3+@mSiO2 nanocomposites with the shell thickness between 15 and 45 nm was reported by Liu et al.14 The authors used CTAB to transfer hydrophobic cores into an aqueous phase and then TEOS was hydrolyzed using NaOH as the catalyst in the mixture of water and ethyl acetate. Although the quality of NPs reported in ref. 14 and here seems to be comparable, our method presents significant advantages. First, the synthesis presented here is far more straightforward. In ref. 14 removal of an excess amount of CTAB surfactant by cooling the solution overnight in an ice bath at 0 °C was required. Moreover, to avoid aggregation the coating process had to be carried out under continuous ultrasonication (600 W, 53 kHz) at a temperature maintained precisely at 25 °C. In addition, to obtain a uniform silica layer TEOS was added at especially low rate of one drop every ten minutes using microsyringes. Here, we use an organic phase for controllable delivery of TEOS at a reduced rate, which is favorable in regard of process reproducibility. Second, our process allows us to control not only the thickness, but also the porosity of the shell. Third, our method, as we shall present below, can be easily adopted for coating of other NCs, for example PbS QDs, which would hydrolyze in aqueous solution under prolonged ultrasonic treatment. Fourth, our process is easier for scaling up to large scale production.
Since in the biphasic system used in the present study the process of TEOS hydrolysis and condensation runs at significantly slower rate as compared to the Stöber or microemulsion method, the synthesis is very controllable and repeatable. High repeatability has been confirmed by over 60 independent trials that yielded coated NPs with very similar monodispersity as well as porosity and thickness of the silica layer. Moreover, nanocrystals with different sizes and shapes can be effectively coated. For example, Fig. 3D presents 12 nm cores coated with a silica shell of about 32 nm, whereas Fig. 3E and F show 50 nm cores coated with silica shells of 12 and 35 nm, respectively. The process is not much sensitive to small changes in reaction conditions, but at the same time there are a number of parameters, as is discussed in more detail below, which can by altered to control and optimize the properties of obtained shells. Moreover, the relatively long reaction time enables straightforward and controllable dosage of some other species in the course of silica growth, which can be incorporated into the shell or used to modify its surface for further biofunctionalization.
 | ||
| Fig. 3 TEM images of NGF@mSiO2 NPs. Top panel – obtained with the use of ethyl acetate in the process carried out for 24 h at 70 °C: (A) using 300 μl of TEOS (standard procedure); (B) using 250 μl of TEOS; (C) using 300 μl of TEOS and two times the number of core NCs used for the particles shown in (A). Bottom panel – obtained with the use of 300 μl of TEOS in cyclohexane in the process carried out at RT: (D) for 72 h using 12 nm cores; (E) for 24 using 12 nm cores; (F) for 72 h using 50 nm cores. The thickness of the silica shell is (in nm): (A) 31.1 ± 1.4; (B) 21.8 ± 1.2; (C) 11.9 ± 3.9; (D) 32.3 ± 2.0; (E) 12.0 ± 2.0 and (F) 35.2 ± 2.3. | ||
Hence, the developed method provides the complete procedure for synthesis and coating of hydrophobic NCs with a mesoporous silica shell, biofunctionalization of the surface and/or incorporation of dye molecules, which can be completed under mild conditions in simple steps represented in Scheme 1.
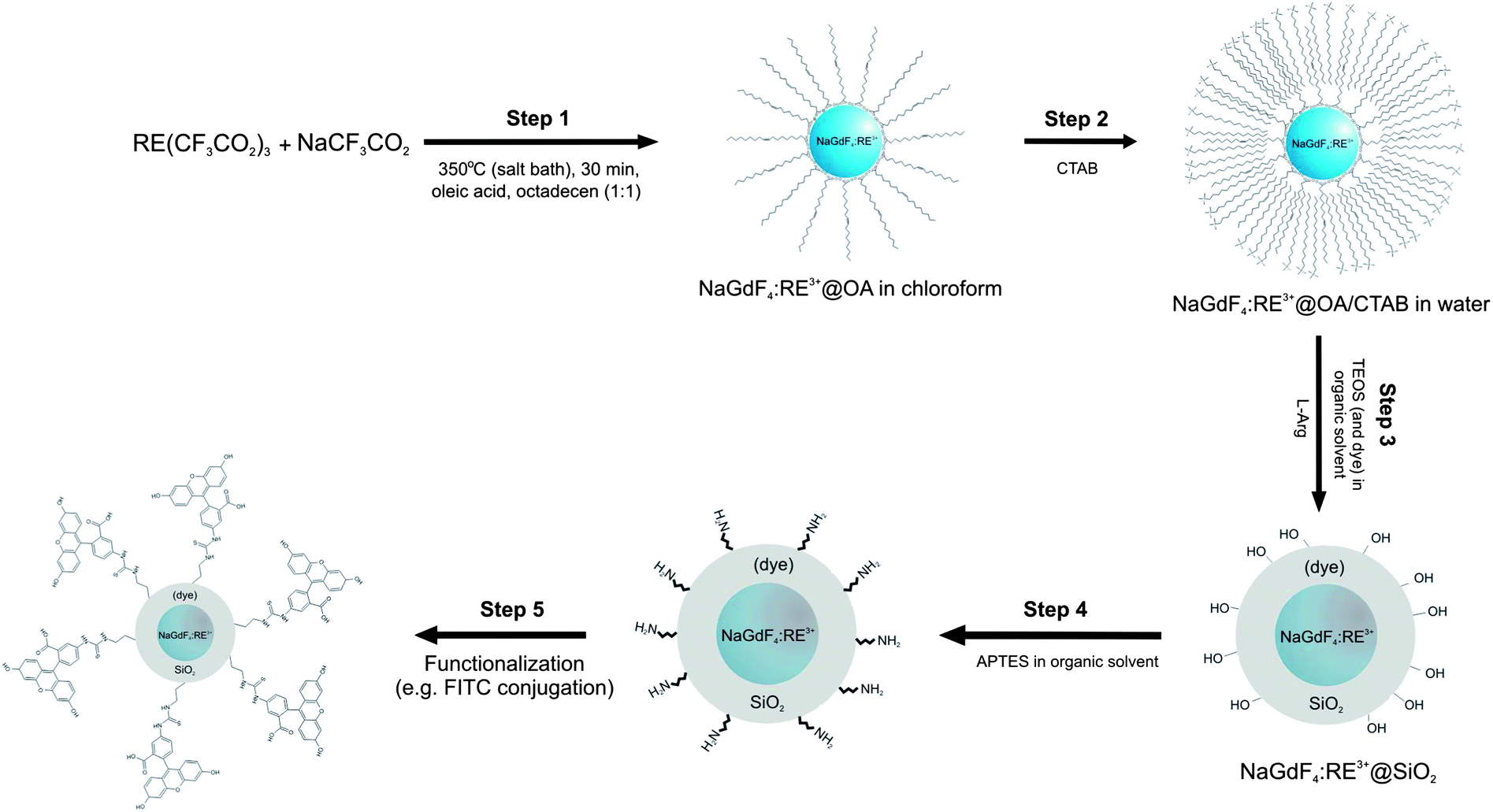 | ||
| Scheme 1 Schematic representation of synthetic steps for NGF@mSiO2, NGF@mSiO2(Pc), NGF@mSiO2–NH2, NGF@mSiO2(Pc)–NH2, NGF@mSiO2–FITC or NGF@mSiO2(Pc)–FITC nanoparticles. | ||
3.2. Influence of coating parameters
Ethyl acetate, cyclohexane and amyl acetate were tested as suitable solvents for TEOS. However, only for the first two solvents formation of silica shells was observed. The solvent forming the top organic layer does not participate directly in hydrolysis and condensation of TEOS, which takes place in the water phase or at the W–O interface. Therefore, it is somewhat surprising that the composition of the organic layer has a pronounced effect on the porosity of the silica shell (Fig. 1). If ethyl acetate is used the surface of nanoparticles becomes golf ball like (inset in Fig. 1A). The wormhole-like mesopores of the silica shell are smaller, around 2–4 nm in diameter, and more spherical than in the case of cyclohexane. The usage of cyclohexane yields spiked-nanoparticles, with a more dendrimeric structure of the layer. The pores are very big, with diameters of around 6–10 nm, and extend from the core to the surface forming irregular channels (inset in Fig. 1B).
Besides larger porosity, also the growth rate of the silica layer is significantly faster if cyclohexane instead of ethyl acetate is used as a diluent for TEOS. As we shall discuss below, this effect may be attributed to a smaller decrease of pH in the course of synthesis, which drops from the initial pH = 10.5 to pH = 5.0 when using ethyl acetate, while remaining almost unchanged (pH ∼ 9) when using cyclohexane. Consequently, the coating process, which is typically carried out at 70 °C in the presence of ethyl acetate, proceeds at a sufficient rate even at room temperature (RT) with the use of cyclohexane. This makes the process of silica layer growth more controllable. If the coating process is carried out for 24 h at 70 °C, the cores are incorporated into homogeneous silica spheres of about 50–60 nm in diameter, regardless of whether ethyl acetate or cyclohexane is used. After 24 h of reaction with the use of ethyl acetate as the top layer (the standard procedure), the 12 nm cores are coated with a silica shell of 29 nm, resulting in NPs with the total diameter of 70 nm (Fig. 3A). The shell thickness can be further reduced by decreasing the amount of TEOS (Fig. 3B) and/or increasing the amount of NCs (Fig. 3C). In the case when cyclohexane is used as the diluent for TEOS the reaction time appears to be another parameter suitable for controlling the layer thickness. NPs with the thickness of silica shell and the total size practically the same as those shown in Fig. 3A (ethyl acetate, 24 h, 70 °C) can be produced using cyclohexane and carrying out the process at RT, provided that it is prolonged to 72 h (Fig. 3D). Shortening of the reaction time from 72 h to 24 h allows reduction of the thickness of the layer below 15 nm (Fig. 3E). The larger cores (50 nm) were used in this experiment; nevertheless, a comparison of NPs shown in Fig. 3D and F demonstrates that the core size exerts only minor influence on the layer thickness. Under the same conditions 50 nm and 12 nm cores were coated with the silica layers of about 32 and 35 nm, respectively. The presented results indicate that using cyclohexane instead of ethyl acetate as the top layer enables easier regulation of the shell thickness.
CTAB stabilizes NGF@OA NCs in the aqueous phase, and also acts as a template for the nanometer-sized pore formation in the silica structure. Hence, the pronounced effect of the solvent on the shell porosity may result from different pH values at which the process occurs depending on whether cyclohexane or ethyl acetate is used as the top layer. In the latter case the more acidic conditions, owing to hydrolysis of ethyl acetate during the process, may decrease the amount of CTAB present on the surface of NGF@OA/CTAB NCs resulting in formation of a less porous structure.
Since the influence of other synthetic parameters is similar regardless of whether cyclohexane or ethyl acetate constitutes the top layer, only NPs obtained with the use of the latter are discussed below.
 | ||
| Fig. 4 TEM images of NGF@mSiO2 nanoparticles obtained using the standard procedure (the same as that used for NPs shown in Fig. 1A) except that: (A) 1.95 cm3 of chloroform solution of NGF@OA was used (three times the number of NCs used for the particles shown in Fig. 1A and 3A); (B) L-Arg concentration was 0.17 mg cm−3; (C) 50 μl of TEOS was used; (D) 100 μl of TEOS was used; (E) CTAB concentration was 0.03 M; (F) CTAB concentration was 0.04 M. | ||
3.3. Surface modification
The essential advantage of our method is the possibility of very straightforward modification of NPs' surface with –NH2 groups during silica growth, which does not require any additional steps. A simple addition of APTES solution in cyclohexane or ethyl acetate to the top organic layer during the course of reaction results in a –NH2 functionalized surface. The particle zeta potential is expected to change after functionalization. Zeta potential measurements show that unmodified NPs dispersed in water are positively charged under acidic, neutral and slightly basic solutions, whereas they are negatively charged at higher pH. For example at pH = 5 and pH = 10 the zeta potentials are +37.9 mV and −17.5 mV, respectively. The isoelectric point is at pH = 8.9. For –NH2 modified NPs the isoelectric point shifts, as expected, towards lower pH values, to pH = 5.0, whereas at pH = 10 the zeta potential is −26.3 mV. This indicates that the surface is modified with –NH2 groups. The DLS determined hydrodynamic radius of unmodified NPs is 95 nm, and after modification it increases to 170 nm (from TEM the mean sizes of NPs used in this experiment are 84 and 90 nm, respectively).The presence of –NH2 groups was further confirmed by labeling NPs with FITC. This step illustrates also the possibility of biofunctionalization of NH2-modified NPs. FITC reacts with the primary amino groups on the surface of NPs to form stable conjugates through thiourea linkage. After washing out of the non-conjugated fluorescein the FITC-labeled NPs can be detected by fluorescence spectroscopy. PL and PLE spectra recorded for FITC-modified nanoparticles dispersed in PBS buffer (pH = 7.4) confirm that NPs were successfully modified with FITC (Fig. S5, ESI†). Under the UV lamp the strong green fluorescence of FITC-modified NPs is easily visible even with the naked eye (inset in Fig. S5, ESI†).
Favorably, a slow rate of the coating process enables controllable introduction of APTES at an appropriate stage of reaction. Addition of APTES in a mixture with TEOS at the beginning of the reaction leads to formation of voids around the cores. In some NPs these empty spaces extend from the core to the surface yielding cracked spheres (Fig. 5A). A considerable number of empty spheres are also formed. A similar TEM image is observed for NGF@mSiO2–NH2 NPs obtained with APTES added after 6 h of the reaction time, although the number of cracked or void containing NPs is smaller (Fig. 5B). The observed effect of APTES is analogous to that leading to obtainment of silica nanoparticles with a rough surface44 and results from the presence of aminopropyl groups which do not participate in polycondensation. Consequently, the defective sites are formed yielding an unperfected –O–Si–O– network, which can collapse and sink causing that NPs may locally shrink when C2H5OH and H2O molecules are removed due to drying. Interestingly, addition of APTES at the initial reaction stage exerts a pronounced effect also on the porosity of formed silica shells. The inset in Fig. 5B shows that the porosity of shells obtained with APTES added after 6 h of the reaction time is much smaller compared to those obtained with APTES added after 24 h (Fig. 5C). In the latter case the porosity is practically the same as that of NPs with the unmodified surface (synthesized in the absence of APTES, Fig. 1A). Hence, we can conclude that most suitably APTES should be added after 24 h of the reaction time that results in mesoporous NPs with the surface functionalized with NH2 groups, but at the same time without any noticeable influence of APTES on the NP morphology.
 | ||
| Fig. 5 TEM images of modified NGF@mSiO2–NH2 NPs obtained with APTES added (A) in a mixture with TEOS at the beginning of the reaction (270 μl of TEOS and 30 μl of APTES); (B) after 6 h of the reaction time (50 μl of APTES in 1 cm3 of ethyl acetate); (C) after 24 h of the reaction time (50 μl of APTES in 2 cm3 of ethyl acetate). In the case of A and B the reaction lasted for 24 h, and in C it was prolonged for an additional 6 h. | ||
3.4. Photosensitization
If a solution of zinc phthalocyanine (ZnPc) is used instead of a pure organic solvent as the top organic layer, ZnPc species are found to be incorporated into the silica shell (NGF@mSiO2(Pc)). Addition of APTES after 24 h of reaction time yields NH2-modified NGF@mSiO2(Pc)–NH2 hybrid NPs (Scheme 1). To the best of our knowledge, such a method to obtain a photosensitized, up-converting and biocompatible core–shell material in one-pot synthesis, without additional procedures required for dye incorporation or surface functionalization, has not been reported until now.The method developed here presents great advantages in comparison with the soaking method.31 Using the soaking procedure it was observed that ZnPc could be released out of the silica layer in ethanol.31 In order to prove that in our case no release of ZnPc occurs, the ZnPc incorporated NPs were dispersed in ethanol under continuous stirring for 1 h. Then, after centrifugation of NPs the UV/vis absorption spectra of the supernatants were measured showing no absorption bands of ZnPc expected in the 590–680 nm range. Importantly, NPs preserve their favorable properties, such as monodispersity, regular shape and porosity, during the photosensitization procedure proposed here (Fig. 6A).
 | ||
| Fig. 6 TEM images of (A) NGF@mSiO2(Pc) nanoparticles with ZnPc incorporated during growth of the silica layer; (B) NGF@mSiO2 nanoparticles soaked in solution of ZnPc in pyridine. | ||
3.5. Potential applications
Up-converting NPs with ZnPc incorporated into the silica layer can be considered as photosensitizers for photodynamic therapy (PDT),31 excitable by radiation in the infrared region, where biological tissue penetration is optimal (the so-called therapeutic window) and the risk of photo-damage is reduced.45,46 For NaGd0.80Yb0.18Er0.02F4 core NCs used in this study, one of the most efficient photon up-conversion processes has been reported.47 The infrared radiation is absorbed by Yb3+ ions and then the excitation energy is transferred efficiently to Er3+ ions through the multiphonon process (Fig. S6A, ESI†). Excited Er3+ ions emit visible radiation in green (about 547 nm) and red (about 660 nm) spectral regions (Fig. S6B, ESI†). Owing to the good overlap of the red emission of Er3+ with the ZnPc absorption Q-band (Fig. S6B, ESI†) the photosensitizing molecules may get efficiently excited and interact with molecular oxygen, generating 1O2 (Fig. S6C, ESI†).48The efficiency of singlet oxygen generation was evaluated by using a photochemical method based on oxidation of 9,10-anthracenediylbis(methylene)dimalonic acid (ABDA)49 (Fig. S6, ESI†). In the presence of 1O2 the ABDA fluorescence is quenched, because it is oxidized to its nonfluorescent endoperoxide form (Fig. S6C and D†).
In a typical experiment 10 mg of NGF@mSiO2(Pc) nanoparticles was suspended in 1 ml of 20 μM ABDA solution in PBS or DI water and placed in a quartz cuvette. After irradiation with a 980 nm laser diode for a specific period of time, the fluorescence of ABDA was measured under excitation at 382 nm. Fig. 7 shows the changes in the ABDA fluorescence intensity (λex = 382 nm) as a function of exposure time of the sample to NIR laser radiation. Analogous dispersion of NGF@mSiO2 NPs (without a photosensitizer) was also employed as the control in the experiment. A continuous decrease of ABDA fluorescence intensity is observed in NGF@mSiO2(Pc) colloidal solution during the exposure time, which is indicative of singlet oxygen generation. The same results were obtained regardless of whether DI water or PBS solution was used as a dispersion medium. After 70 minutes of irradiation the ABDA fluorescence intensity drops to 60% of its initial value, whereas a decrease by less than 10% is observed in the control experiment.
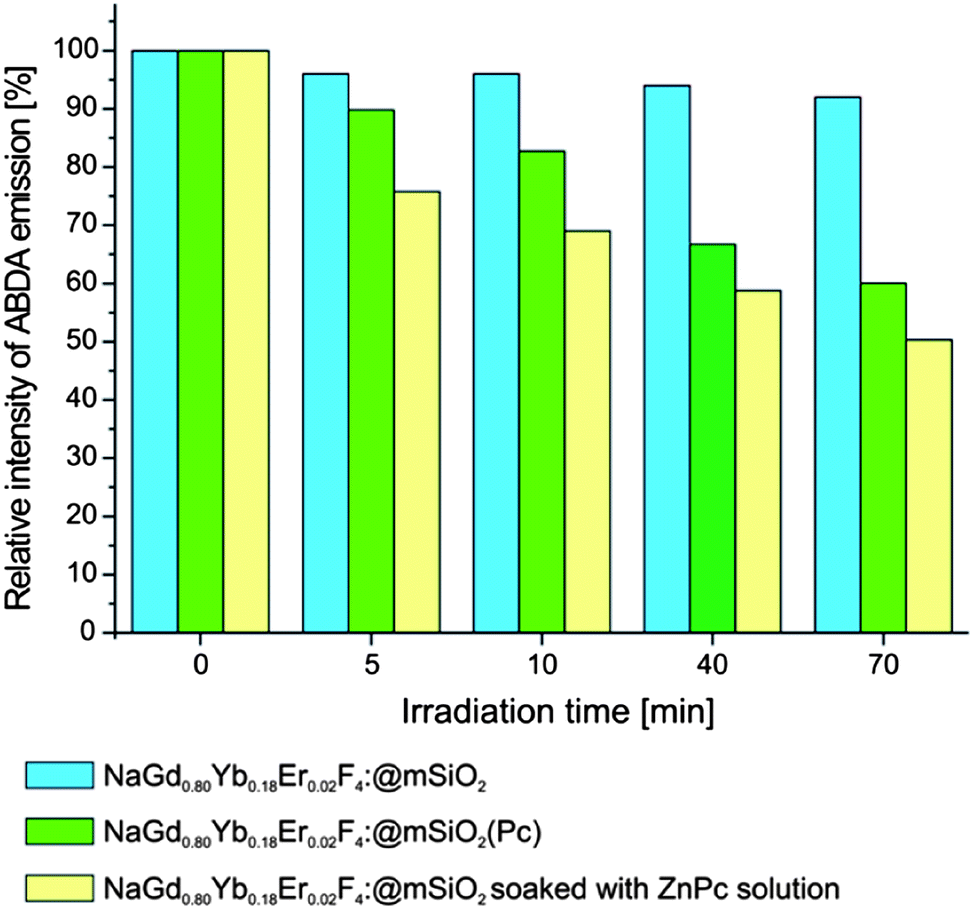 | ||
| Fig. 7 ABDA fluorescence intensity (λex = 382 nm) as a function of exposure time of the sample to NIR laser irradiation (980 nm); all samples were prepared by dispersing 10 mg of appropriate nanoparticles in 1 ml of 20 μM ABDA solution in DI. | ||
Qian et al.31 obtained NaGdF4:Yb3+,Er3+ up-converting NPs coated with porous silica using the microemulsion method and then incorporated ZnPc into the silica layer by soaking. The decrease of ABDA fluorescence intensity after exposure to NIR laser radiation was comparable to that observed in our case (for details see the ESI†). The major important difference is, however, that in the procedure reported here the photosensitizer is incorporated into the silica layer during its growth. This prevents its leakage even if NPs are dispersed in a solvent (e.g., ethanol) in which the photosensitizer dissolves. Qian et al.31 observed that when ZnPc is loaded by soaking nanoparticles in a solution of the photosensitizer in pyridine, ZnPc could be released out from silica in ethanol within 1 hour. Obviously, the photosensitizer was not released in DI water or PBS buffer, since ZnPc does not dissolve in these solvents.
For comparison of two methods of photosensitizer incorporation, we prepared NPs loaded with ZnPc, employing the same protocol as in ref. 31 (however, using 10 mg instead of 100 mg of NGF@mSiO2 to preserve comparability with our results obtained for NGF@mSiO2(Pc)), which includes soaking of NPs in a solution of ZnPc in pyridine (1 ml, 0.5 mg ml−1) for 24 h at room temperature, centrifuging and washing with PBS solution. Apparently, a larger load of ZnPc into nanoparticles can be obtained in this way. Fig. 7 shows that such soaked NPs generate 1O2 more efficiently than those with ZnPc incorporated during silica growth.
However, even simple visual observation of the stability of NP colloidal suspension demonstrates obviously that soaking in ZnPc solution followed by several washings with PBS leads to a considerable agglomeration of NPs. After centrifugation a thin layer of pyridine solution of ZnPc stays on the surface of NPs causing their aggregation in PBS buffer. This is owing to the insolubility of ZnPc in PBS since a similar effect was observed if some diluted pyridine solution of ZnPc was added to the PBS buffer – just a gentle centrifugation (2000 rpm, 318g) was sufficient to separate the ZnPc from the solution. TEM images prove these visual observations. Incorporation of ZnPc during growth of the silica layer yields monodisperse NPs (Fig. 6A), forming stable colloidal solutions. In contrast, strongly agglomerated NPs, with ZnPc present between grains, are observed if a soaking protocol was employed (Fig. 6B). Then, in spite of the higher efficiency of 1O2 generation, formation of NP aggregates excludes application of soaked NPs in PDT.
The preliminary results show that even a larger amount of 1O2 is produced by NGF@mSiO2(Pc) NPs obtained with the use of cyclohexane, which can be attributed to their higher porosity. Quantitative results and more detailed studies of the kinetics of 1O2 generation (Fig. S7†) as a function of NP porosity will be presented in a forthcoming communication.
3.6. Coating of hydrophobic quantum dots
Coating of hydrophobic quantum dots (QDs) with a silica layer makes them dispersible in aqueous solution, prevents agglomeration, reduces cytotoxicity and extends photostability. The protocols based on the modified Stöber process and W/O reverse microemulsion systems have been used as two main methods for incorporation of QDs into silica spheres, both suffering from the same disadvantages as indicated above while discussing their application for coating of nanofluorides. The synthetic protocol proposed here, based on using the biphasic system for silica coating of hydrophobic nanofluorides, can be also successfully employed for encapsulation of other nanocrystals, for example QDs, with mesoporous silica. For instance, following the same coating procedure as for nanofluorides, the OA stabilized PbS QDs (5 nm) were incorporated into silica spheres. TEM images (Fig. 8) show that this resulted in monodisperse 65 nm silica spheres, each incorporating a single QD. Only about 2% of the NPs are empty and less than 5% contain more than one core. The coating is homogeneous with a high degree of porosity. Moreover, the –NH2 groups can be easily attached to the surface of QD@mSiO2, using an analogous procedure as described for NGF@mSiO2, without any degradation of the morphology of resulting nanoparticles. | ||
| Fig. 8 TEM images of (A) silica coated PbS@mSiO2, and (B) modified PbS@mSiO2–NH2 nanoparticles. | ||
Further optimization of the procedure should enable obtainment of thinner shells, with a reduced number of empty or multicore NPs. The influence of various synthetic parameters on the morphology and optical properties of silica coated QDs will be discussed in more detail in one of our forthcoming papers. Here we can conclude that the presented results demonstrate the large versatility of our procedure. It is likely that it can be applied for incorporation of other hydrophobic nanoparticles, such as magnetic oxides (Fe2O3) or metal nanocrystals (Au), into mesoporous silica spheres.
4. Conclusion
In conclusion, we have developed a facile one-pot method for coating of hydrophobic NaLnF4 nanocrystals with a thin layer of mesoporous silica. Importantly, the synthetic protocol used here, based on the biphasic system, ensures high repeatability of the NP morphology. This procedure can be applied for coating NCs of various sizes and the thickness of the silica coating layer can be varied by changing synthetic parameters, such as the amount of NCs and TEOS or reaction temperature and time. The usage of different solvents as diluents for TEOS allows control of the shell porosity. The silica shell can be easily modified with –NH2 groups enabling further biofunctionalization of silica coated NPs. The procedure ensures also a facile incorporation of organic molecules (dyes, drugs, etc.) into a silica shell, eliminating important drawbacks such as dye (drug) leakage or the necessity of a multistep protocol. The method can be generalized for silica coating of other OA-stabilized NCs, as is demonstrated by successful encapsulation of PbS@OA QDs with a mesoporous silica layer. The core–shell NPs composed of a luminescent (up-converting) core and a mesoporous silica layer present their potential use as fluorescence imaging agents or drug delivery vehicles and are likely to find many biomedical applications in diagnosis and therapy. Here, we demonstrated that nanocomposite particles composed of up-converting cores and silica shell incorporating ZnPc have potential to be used in photodynamic therapy.References
- M. Haase and H. Schäfer, Angew. Chem., Int. Ed., 2011, 50, 5808–5829 CrossRef CAS PubMed.
- A. Aebischer, S. Heer, D. Biner, K. Krämer, M. Haase and H. U. Güdel, Chem. Phys. Lett., 2005, 407, 124–128 CrossRef CAS.
- J. C. Boyer, F. Vetrone, L. A. Cuccia and J. Capobianco, J. Am. Chem. Soc., 2006, 128, 7444–7445 CrossRef CAS PubMed.
- J. C. Boyer, M. P. Manseau, J. I. Murray and F. C. J. M. van Veggel, Langmuir, 2010, 26, 1157–1164 CrossRef CAS PubMed.
- R. Kumar, M. Nyk, T. Y. Ohulchanskyy, C. A. Flask and P. N. Prasad, Adv. Funct. Mater., 2009, 19, 853–859 CrossRef CAS.
- D. Wawrzynczyk, A. Bednarkiewicz, M. Nyk, J. Cichos, M. Karbowiak, D. Hreniak, W. Strek and M. Samoc, J. Nanosci. Nanotechnol., 2012, 3, 1886–1891 CrossRef.
- H. Schafer, P. Ptacek, K. Kompe and M. Haase, Chem. Mater., 2007, 19, 1396–1400 CrossRef.
- L.-L. Li, R. Zhang, L. Yin, K. Zheng, W. Qin, P. R. Selvin and Y. Lu, Angew. Chem., Int. Ed., 2012, 51, 6121–6125 CrossRef CAS PubMed.
- W. Lin, K. Fritz, G. Guerin, G. R. Bardajee, S. Hinds, V. Sukhovatkin, E. H. Sargent, G. D. Scholes and M. A. Winnik, Langmuir, 2008, 24, 8215–8219 CrossRef CAS PubMed.
- X. Liu, X. Kong, Y. Zhang, L. Tu, Y. Wang, Q. Zeng, C. Li, Z. Shi and H. Zhang, Chem. Commun., 2011, 47, 11957–11959 RSC.
- F. Wang, D. K. Chatterjee, Z. Q. Li, Y. Zhang, X. P. Fan and M. Q. Wang, Nanotechnology, 2006, 17, 5786–5791 CrossRef CAS.
- N. J. J. Johnson, N. M. Sangeetha, J. C. Boyer and F. C. J. M. van Veggel, Nanoscale, 2010, 2, 771–777 RSC.
- X. Kang, Z. Cheng, C. Li, D. Yang, M. Shang, P. Ma, G. Li, N. Liu and J. Lin, J. Phys. Chem. C, 2011, 115, 15801–15811 CAS.
- J. Liu, W. Bu, S. Zhanh, F. Chen, H. Xing, L. Pan, L. Zhou, W. Peng and J. Shi, Chem.–Eur. J., 2012, 18, 2335–2341 CrossRef CAS PubMed.
- J. Yang, Y. Deng, Q. Wu, J. Zhou, H. Bao, Q. Li, F. Zhang, F. Li, B. Tu and D. Zhao, Langmuir, 2010, 26, 8850–8856 CrossRef CAS PubMed.
- Z. Li, Y. Zhang and S. Jiang, Adv. Mater., 2008, 20, 4765–4769 CrossRef CAS.
- W. Stöber, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- P. Yang, Z. Quan, Z. Hou, C. Li, X. Kang, Z. Cheng and J. Lin, Biomaterials, 2009, 30, 4786–4795 CrossRef CAS PubMed.
- G. Yi, H. Lu, S. Zhao, Y. Ge, W. Yang, D. Chen and L. H. Guo, Nano Lett., 2004, 4, 2191–2196 CrossRef CAS.
- S. Sivakumar, P. R. Diamente and F. C. J. M. van Veggel, Chem.–Eur. J., 2006, 12, 5878–5884 CrossRef CAS PubMed.
- K. D. Hartlen, A. P. T. Athanasopoulos and V. Kitaev, Langmuir, 2008, 24, 1714–1720 CrossRef CAS PubMed.
- K. Osseo-Asare and F. Arriagada, Colloids Surf., 1990, 50, 321–339 CrossRef CAS.
- J. Pan, D. Wanand and J. Gong, Chem. Commun., 2011, 47, 3442–3444 RSC.
- T. Li, J. Moon, A. A. Morrone, J. J. Mecholsky, D. R. Talham and J. H. Adair, Langmuir, 1999, 15, 4328–4334 CrossRef CAS.
- M. Alejandro-Arellano, T. Ung, Á. Blanco, P. Mulvaney and L. M. Liz-Marzán, Pure Appl. Chem., 2000, 72, 257–267 CrossRef CAS.
- Y. H. Lien and T. M. Wu, J. Colloid Interface Sci., 2008, 326, 517–521 CrossRef CAS PubMed.
- R. A. Jail and Y. Zhang, Biomaterials, 2008, 29, 4122–4128 CrossRef PubMed.
- J. Wang, A. Sugawara-Narutaki, M. Fukao, T. Yokoi, A. Shimojima and T. Okubo, ACS Appl. Mater. Interfaces, 2011, 3, 1538–1544 CAS.
- Y. Jin, S. Lohstreter, D. T. Pierce, J. Parisien, M. Wu, C. Hall and J. X. Zhao, Chem. Mater., 2008, 20, 4411–4419 CrossRef CAS.
- M. Stjerndahl, M. Andersson, H. E. Hall, D. M. Pajerowski, M. W. Meisel and R. S. Duran, Langmuir, 2008, 24, 3532–3536 CrossRef CAS PubMed.
- H. S. Qian, H. C. Guo, P. C. L. Ho, R. Mahendran and Y. Zhang, Small, 2009, 5, 2285–2290 CrossRef CAS PubMed.
- D. K. Yi, S. S. Lee, G. C. Papaefthymiou and J. Y. Ying, Chem. Mater., 2006, 18, 614–619 CrossRef CAS.
- T. Yokoi, Y. Sakamoto, O. Terasaki, Y. Kubota, T. Okubo and T. Tatsumi, J. Am. Chem. Soc., 2006, 128, 13664–13665 CrossRef CAS PubMed.
- T. Yokoi, J. Wakabayashi, Y. Otsuko, W. Fan, M. Iwama, R. Watanabe, K. Aramaki, A. Shimojima, T. Tatsumi and T. Okubo, Chem. Mater., 2009, 21, 3719–3729 CrossRef CAS.
- X. Ye, J. E. Collins, Y. Kang, J. Chen, D. T. N. Chen, A. G. Yodh and C. B. Murray, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22430–22435 CrossRef CAS PubMed.
- H. Shen, H. Wang, X. Li, J. Z. Niu, H. Wang, X. Chen and L. S. Li, Dalton Trans., 2009, 10534–10540 RSC.
- J. Liu, H. Yu, Z. Wu, W. Wang, J. Peng and Y. Cao, Nanotechnology, 2008, 19, 345602–345611 CrossRef PubMed.
- H. Fan, E. W. Leve, C. Scullin, J. Gabaldon, D. Tallant, S. Bunge, T. Boyle, M. C. Wilson and C. J. Brinker, Nano Lett., 2005, 5, 645–648 CrossRef CAS PubMed.
- F. Bai, D. Wang, Z. Huo, W. Chen, L. Liu, X. Liang, C. Chen, X. Wang, Q. Peng and Y. Li, Angew. Chem., Int. Ed., 2007, 46, 6650–6653 CrossRef CAS PubMed.
- T. Kim, E. Momin, J. Choi, K. Yuan, H. Zaidi, J. Kim, M. Park, N. Lee, M. T. McMahon, A. Quinones-Hinojosa, J. W. M. Bulte, T. Hyeon and A. A. Gilad, J. Am. Chem. Soc., 2011, 133, 2955–2961 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438–8441 CrossRef CAS PubMed.
- C. J. Brinker and G. W. Scherer, Sol-Gel Science, Academic Press, New York, 1990 Search PubMed.
- K. S. Birdi, Handbook of Surface and Colloid Chemistry, CRC Press, Boca Raton, FL, 1997 Search PubMed.
- S. Xu, S. Hartvickson and J. X. Zhao, ACS Appl. Mater. Interfaces, 2011, 3, 1865–1872 CAS.
- H. Sertchook and D. Avnir, Chem. Mater., 2003, 15, 1690–1694 CrossRef CAS.
- J. F. Suyver, A. Aebischer, D. Biner, P. Gerner, J. Grimm, S. Heer, K. W. Kramer, C. Reinhard and H. U. Gudel, Opt. Mater., 2005, 27, 1111–1130 CrossRef CAS.
- S. Heer, K. Kompe, H. U. Gudel and M. Haase, Adv. Mater., 2004, 16, 2102–2105 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233, 351–371 CrossRef.
- B. A. Lindig, M. A. J. Rodgers and A. P. Schaap, J. Am. Chem. Soc., 1980, 102, 5590–5593 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Figures presenting additional TEM images, FTIR, EDX, PL and PLE spectra, schematic representation of singlet-oxygen generation under irradiation of NPs with a NIR laser diode, quenching of the ABDA fluorescence in the presence of 1O2 and kinetics of singlet oxygen generation. See DOI: 10.1039/c3tb21442g |
| This journal is © The Royal Society of Chemistry 2014 |