 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePolyglycerol-based amphiphilic dendrons as potential siRNA carriers for in vivo applications†
Ariane
Tschiche‡
a,
Anna M.
Staedtler‡
b,
Shashwat
Malhotra‡
a,
Hannah
Bauer
c,
Christoph
Böttcher
d,
Soroush
Sharbati
c,
Marcelo
Calderón
a,
Markus
Koch
b,
Thomas M.
Zollner
b,
Anna
Barnard
e,
David K.
Smith
e,
Ralf
Einspanier
c,
Nicole
Schmidt
*b and
Rainer
Haag
*a
aInstitute of Chemistry and Biochemistry, Freie Universität Berlin, Takustrasse 3, Berlin 14195, Germany. E-mail: haag@chemie.fu-berlin.de; Web: http://www.polytree.de Fax: +49-30-838-53357; Tel: +49-30-838-52633
bBayer Pharma AG, Global Drug Discovery, Therapeutic Research Group Oncology/Gynecological Therapy, Muellerstrasse 178, Berlin 13353, Germany. E-mail: nicole.schmidt1@bayer.com; Tel: +49-30-468-194360
cInstitute of Veterinary Biochemistry, Freie Universität Berlin, Oertzenweg 19b, Berlin 14163, Germany
dResearch Center of Electron Microscopy, Institute of Chemistry and Biochemistry, Freie Universität Berlin, Fabeckstrasse 36a, Berlin 14195, Germany
eDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK
First published on 7th March 2014
Abstract
The development of nonviral synthetic vectors for clinical application of gene therapy using siRNA transfection technology is of particular importance for treatment of human diseases, which is yet an unsolved challenge. By employing a rational design approach, we have synthesized a set of well-defined, low-molecular-weight dendritic polyglycerol-based amphiphiles, which are decorated peripherally with the DAPMA (N,N-di-(3-aminopropyl)-N-(methyl)amine) moiety. The main differences that were introduced in the structural motif relate to dendron generation and the type of linker between the hydrophilic and hydrophobic segment. The synthesized amphiphiles were then characterized for their aggregation behaviour and further evaluated with respect to their siRNA transfection potential by comparing their physico-chemical and biological features. Our findings demonstrated that all four synthesized amphiphiles yielded high gene binding affinities. Furthermore, the ester-linked compounds (G1-Ester-DAPMA, G2-Ester-DAPMA) revealed noticeable gene silencing in vitro without affecting the cell viability in the tumor cell line 786-O. Remarkably, neither G1-Ester-DAPMA nor G2-Ester-DAPMA induced inflammatory side effects after systemic administration in vivo, which is noteworthy because such highly positively charged compounds are typically associated with toxicity concerns which in turn supports their prospective application for in vivo purposes. Therefore, we believe that these structures may serve as new promising alternatives for nonviral siRNA delivery systems and have great potential for further synthetic modifications.
Introduction
Gene therapy holds substantial promise for the treatment of inherited and acquired diseases such as cystic fibrosis, HIV, arthritis, cancer, and other medical conditions.1–3 It is based on correcting the origin of diseases by delivery and subsequent expression of exogenous genetic materials, either RNA or DNA, to replace defective genes, substitute missing ones, silence unwanted gene expression, or introduce new cellular functions.4,5 In recent years, the antisense approach has been regarded as particularly promising, which involves the intracellular delivery of antisense constructs that usually lead to a reduction of target activity, termed gene silencing.6 Nonviral gene carriers including the main classes of cationic polymers and lipids have been extensively studied for two decades now as alternatives to viral vectors due to their improved safety, greater synthetic tunability and versatility, larger capacity for therapeutic genes, and more facile manufacturing.7–15 However, low transfection efficiencies and toxicity issues still remain major barriers for clinical application of nonviral gene therapy.16–18 Among the nonviral nanocarriers, dendrimers have been utilized and examined in biomedical fields for drug and gene delivery systems because of their defined molecular structures, versatility with regard to molecular size and surface chemistry, intrinsic multivalent features, and high cargo payload at nanosized dimensions.19–24 Developing dendrimers as nanovectors to deliver RNA therapeutics, especially synthetic small interfering RNAs (siRNAs), and to employ them for the specific inhibition and knockdown of disease genes by RNA interference (RNAi) is one of the most promising pathways within the field of gene therapy.25–30 The main challenge lies in the safe and efficient delivery of siRNA. An optimal gene vector has to fulfill specific requirements in order to successfully transfect cells both in vitro and in vivo. Foremost, the transporter needs to complex and condense the genetic material, protect it from nucleolytic degradation, enable cellular uptake, and finally release it from the endosomal pathway into the cytosol, where the RNAi machinery is located.31Although high molecular weight dendrimers like PAMAM (poly(amidoamine)),7,32,33 PPI (poly(propylene imine)),34–36 and PEI (poly(ethylenimine))37–39 can lead to high transfection efficiencies, they also exhibit problematic toxicity profiles,40 which can be partially attributed to their cellular accumulation after gene delivery has taken place.41 An alternative approach, mainly established and advanced by Florence,42 Safinya,43 Diederich,44 and Smith,45 was to modify low-molecular-weight, amine-functionalized dendritic arrays with hydrophobic portions to promote their self-assembly into supramolecular dendrimers (“pseudodendrimers”). In this way, positively charged large multivalent arrays are generated that are suitable for gene delivery applications (Fig. 1).
 | ||
| Fig. 1 Concept of self-assembling amphiphiles used for gene delivery applications. | ||
Our research group has previously proven that such a strategy can enhance gene delivery and lead to synergistic effects if aspects of both classes of nonviral gene delivery, i.e., polymers and lipids, are combined,27 which also has been demonstrated with related architectures.17,45–48 In fact, along with a recent report by Peng and co-workers,28 this was one of the first successful examples of in vitro siRNA transfection using dendritic amphiphiles.27 More specifically, we developed glycine-functionalized amphiphiles based on polyglycerol dendrons that had successfully delivered luciferase and GAPDH siRNA into HeLa cells.27 Interestingly, one candidate (G2-octaamine) with eight amine groups on its surface and a hydrophobic C-18 alkyl chain at the core acted as an efficient siRNA vector and still remained non-toxic, even at high N/P ratios (e.g., N/P of 100).
In an endeavor to create a gene vector which is efficient and safe for both in vitro and in vivo applications, we have now adopted the same structural motif but incorporated distinctive structural variations with respect to the employed amine moiety as well as the type of linker between the hydrophilic and hydrophobic domains (Fig. 2).
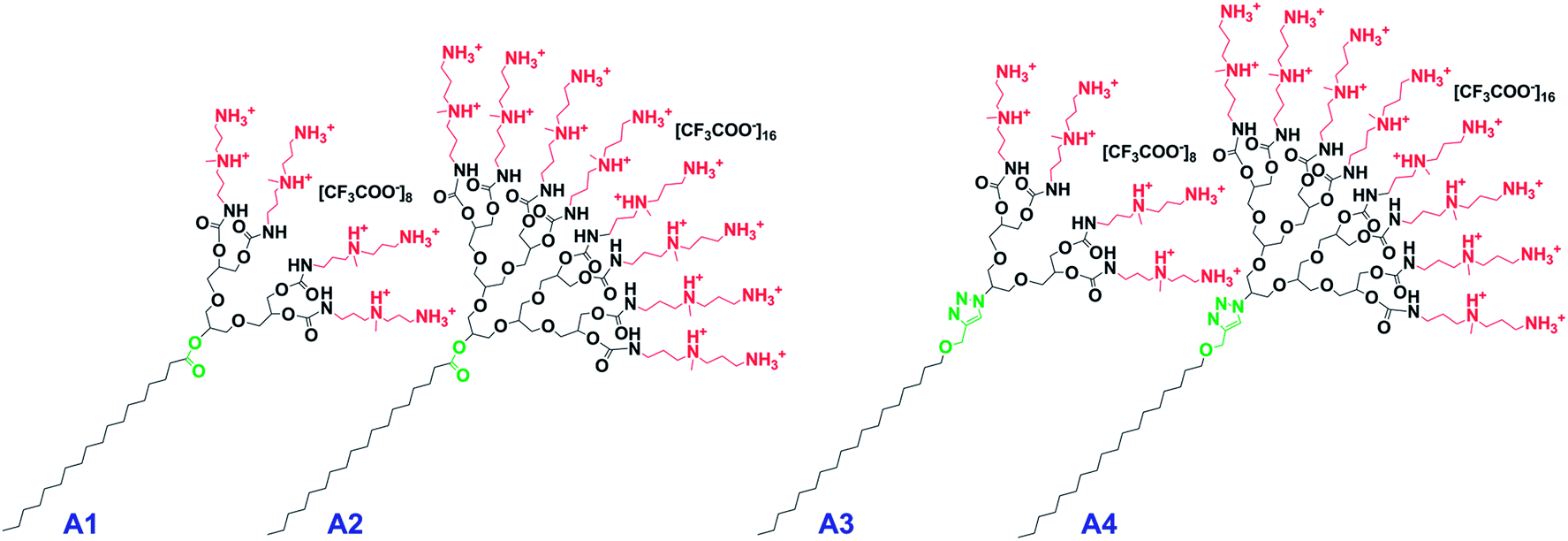 | ||
| Fig. 2 Dendritic amphiphiles investigated in this study (A1 = G1-Ester-DAPMA, A2 = G2-Ester-DAPMA, A3 = G1-Trz-DAPMA, A4 = G2-Trz-DAPMA). | ||
On the surface of the dendritic head group of the amphiphiles, we attached DAPMA, an amine functionality that has already proven to be an effective entity for gene delivery applications.41,49 With reference to the linkage, we decided to integrate flexible, biodegradable ester functionalities (A1, A2) in contrast to rigid, non-degradable triazole groups (A3, A4), to assist the effective release of the genetic cargo. It is known that micelles with biodegradable features can enhance polyplex dissociation and thus improve transfection efficiency.50,51 The integration of rigid elements has also been shown to have a beneficial effect with respect to biocompatibility and transfection properties.44,52
Systematically, we investigated these amphiphilic dendrons for their aggregation behaviour and further evaluated their siRNA transfection potential by employing different kinds of physico-chemical and biological techniques. The obtained results demonstrate that all constructs lead to a comparably strong gene binding. However, only the ester-bridged amphiphiles (A1, A2) revealed no cytotoxic effects and caused reasonable gene silencing in 786-O tumor cells in vitro. Notably, systemic administration of A1 and A2 in mice demonstrated that these constructs do not induce inflammatory side effects, which renders these novel architectures highly interesting for siRNA delivery in future applications of therapeutic RNAs.
Experimental section
Materials
Air and moisture sensitive reactions were carried out in dried glassware under an argon atmosphere. Anhydrous solvents were either commercially purchased from Acros Organics™ in septum-sealed bottles or chemically dried using a MBRAUN SPS 800 solvent purification system. Compounds 2, 3, and 10 were synthesized according to reported procedures.49,53 All other chemicals were of reagent grade quality and used without further purification from the suppliers Acros Organics™, Fluka®, Sigma-Aldrich®, Roth®, Invitrogen™, and Merck™.Measurements
NMR spectra were recorded on a Bruker™ ECX 400 (1H: 400 MHz, 13C: 100.5 MHz), a Jeol™ Eclipse (1H: 500 MHz, 13C: 125.8 MHz), or on a Bruker™ Biospin (1H: 700 MHz, 13C: 176.1 MHz) spectrometer at 25 °C in deuterated chloroform (CDCl3), methanol (CD3OD), or water (D2O). Chemical shifts (δ) are given in parts per million (ppm) according to calibration to the corresponding solvents CDCl3 (1H: 7.26 ppm, 13C: 77.00 ppm), CD3OD (1H: 4.87 ppm, 13C: 49.05 ppm), and D2O (1H: 4.79 ppm). 13C NMR spectra were recorded with 1H broadband decoupling. Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), and m (multiplet). Coupling constants (J) are given in Hz.
Electrospray-ionization time-of-flight mass spectrometry (ESI-TOF-MS) experiments were carried out using an Agilent 6210 ESI-TOF, Agilent Technologies™. The calculated masses refer to the respective isotopes with the highest intensity. The measured masses typically represent the main MS peak, while the found isotope pattern intensities matched those calculated.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and an accelerating voltage of 160 kV (FEG-illumination). Images were recorded using an EAGLE 4k-CCD camera (FEI Company, Oregon, USA) operated with binning factor 2 (2048 by 2048 pixel). The defocus was chosen in all cases to be 2 μ. It is important to note that although the determined diameter measurements were prone to error due to the very small size of the assemblies, more reliable diameter values could be derived from sample areas where micelles were densely packed. Fourier transforms of corresponding images revealed a diffraction pattern which indicates repetitive distances which can be said to correlate with the diameter of the micelles (cf.Fig. 3B).
000 and an accelerating voltage of 160 kV (FEG-illumination). Images were recorded using an EAGLE 4k-CCD camera (FEI Company, Oregon, USA) operated with binning factor 2 (2048 by 2048 pixel). The defocus was chosen in all cases to be 2 μ. It is important to note that although the determined diameter measurements were prone to error due to the very small size of the assemblies, more reliable diameter values could be derived from sample areas where micelles were densely packed. Fourier transforms of corresponding images revealed a diffraction pattern which indicates repetitive distances which can be said to correlate with the diameter of the micelles (cf.Fig. 3B).
 | ||
| Fig. 3 Representative cryo-TEM images of G1-Ester-DAPMA (A1). (A) Small spherical particles with diameters of 3–5 nm coexist with (B) extended two-dimensional arrays possessing a hexagonal ultrastructure. Fourier transform of these arrays reveals a repetitive distance of 4.44 nm indicating a highly ordered packing of A1. | ||
The cell viability was additionally investigated in 786-O-Luc tumor cells following amphiphile treatment. Cells were cultured in 96-well plates at a density of 1 × 104 cells per well. After 24 hours the growth medium was discarded and replaced by 100 μL Opti-MEM (Gibco). The amphiphiles G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2) were dissolved in water and complexed with luciferase specific (GCAAGAUCGCCGUGUAAUAUU, Dharmacon) or non-targeting siRNA (ON-TARGETplus Non-targeting siRNA, Dharmacon) by incubating 5 pmol siRNA with corresponding amounts of amphiphiles depending on N/P ratios 10, 20, and 30. Lipofectamine RNAiMAX transfection reagent (Invitrogen) was used as the positive control following the manufacturer's protocol. Untreated cells were used as the negative control. In a total volume of 50 μL the polyplexes were added to the cells and incubated for 24 h at 37 °C, which was then replaced by RPMI growth medium and incubated for another 24 h. To determine the number of viable cells within this study, the CellTiter-Glo Luminescent Cell Viability Assay (Promega) was used according to the manufacturer's protocol.
Synthesis
:3) to remove the acetal groups. The reaction mixture was then evaporated under reduced pressure and purified via column chromatography (eluent CHCl3–MeOH, 90:20) to afford the desired amphiphiles 6 and 7 as viscous oils in 75% and 65% yields, respectively.
:2, 64 mL min−1). Due to the relative instability and since thorough purification was not required at this stage, the intermediates 8 and 9 were used in their crude form for further synthesis.
:9:1) and size exclusion chromatography (SEC) using a Sephadex™ LH-20 (CHCl3–MeOH 1:1). Drying under high vacuum yielded the products 11 and 12 as yellowish oils.
Results and discussion
Synthesis
Four distinct low-molecular-weight amphiphilic constructs (cf.Fig. 2) have been synthesized differing in linker chemistry and dendron generation. Thus, specific properties with reference to biodegradability, size, and the number of multivalent interactions were introduced in order to create an efficient gene carrier with a particular attributed structural motif. The potential gene vectors were designed to utilize hydrophilic polyglycerol dendrons of two different generations [G1, G2] combined with a simple C-18 alkyl chain representing the hydrophobic domain, in order to unite the advantageous features of both lipid and dendritic gene delivery vehicles. As mentioned above, it is necessary to modify the dendritic polyglycerol scaffold with a hydrophobic unit to allow the formation of multivalent supramolecular architectures and thus facilitate efficient complexation and condensation of the genetic material, which is an important prerequisite for transfection ability.27Due to their water solubility and physiological safety, which is based on a very biocompatible glycerol building block,59 polyglycerol dendrons were selected to serve as the hydrophilic part within the amphiphilic structure. As a dendritic scaffold, they provide multiple hydroxyl groups which can be synthetically modified, thus offering the opportunity for multivalent interactions with biological substrates,60 resulting in considerably stronger binding affinities compared to monovalent interactions.61 In addition, the degree of functionality and thus the size can be varied by choosing a certain dendron generation.
Here, the amphiphiles were functionalized at their peripheral positions with a particular triamine: N,N-di-(3-aminopropyl)-N-(methyl)amine (DAPMA) that already proved to be an effective entity for gene delivery purposes.49 It is known for a few decades now that in the presence of multivalent cations, such as the naturally occurring oligoamines spermine and spermidine, DNA undergoes a dramatic condensation to a compact, usually highly ordered structure,62 which is essential for the gene delivery process, since as one of the first steps the genetic material needs to be transported efficiently through the cellular membrane. In the specific case of DAPMA, it has been suggested that the sterically hindered tertiary amine deforms DNA in order to achieve the interactions, resulting in strong local binding and thus in efficient DNA compaction.49 Moreover, this specific triamine ligand has been shown to lower the toxicity when compared with diamine- and tetraamine modified dendrons.49 In addition to that, it is believed that primary amine groups generally participate in nucleic acid binding, condense it into nanoscale particles and promote its cellular uptake, while tertiary amines may act as “proton sponge” entities in endosomes and enhance the release of the genetic material into the cytoplasm.63 Therefore, we reason that this specific amine unit (DAPMA) is an ideal candidate for gene delivery applications.
Besides the dissimilarity in the size of the hydrophilic head group by using different generations of polyglycerol dendrons (G1, G2), variations were also introduced regarding their degradation profile that may assist the efficient release of the nucleic acids from the polyplex. Therefore, two amphiphiles were equipped with a biodegradable ester bond at the focal point [G1-Ester-DAPMA (A1), G2-Ester-DAPMA (A2)] in contrast to a non-degradable rigid triazole linker within the other two scaffolds [G1-Trz-DAPMA (A3), G2-Trz-DAPMA (A4)] (cf.Fig. 2).
For the synthesis of G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2) (Scheme 1), we first reacted each of the acetal group protected polyglycerol dendrons [G1.0]-OH (2) and [G2.0]-OH (3) with commercially available stearoyl chloride (1), thereby forming an ester bond at the focal point which represents a biodegradable connection between both domains of the amphiphile. After removing the 1,2-diol protecting groups under acidic conditions, the peripheral hydroxyl entities were converted into activated carbonate functionalities by using p-nitrophenyl chloroformate. In the next step, mono-Boc-protected DAPMA units (10) were attached via nucleophilic substitution to the dendritic backbone of compounds 8 and 9 in 50% and 40% yield over two steps, respectively, involving the liberation of p-nitrophenol. The Boc-protected species 11 and 12 were then treated overnight with TFA to afford G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2) in quantitative yields, as their trifluoroacetate salts (Scheme 1).
 | ||
| Scheme 1 Synthesis of G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2). | ||
Regarding the synthesis of the amphiphiles G1-Trz-DAPMA (A3) and G2-Trz-DAPMA (A4), the hydroxylated, non-degradable precursors (13, 14; cf. ESI, Scheme S1†) were initially generated according to our previously published procedure.27 The DAPMA functionalization of the dendron's deprotected surface occurred in a similar fashion to those for the compounds 6 and 7. Correspondingly, the free hydroxyl groups of the triazole-bridged amphiphiles (13, 14; cf. ESI, Scheme S1†) were converted into the respective carbonate species (15, 16; cf. ESI, Scheme S1†), followed by the addition of mono-Boc-protected DAPMA (10) in the presence of DMAP and DIPEA, to give the aminated triazole amphiphiles (17, 18; cf. ESI, Scheme S1†) in 39% and 27% yields over two steps, respectively. After the final treatment with trifluoroacetic acid, the two triazole-based target molecules G1-Trz-DAPMA (A3) and G2-Trz-DAPMA (A4) were obtained in quantitative yields (cf. ESI, Scheme S1†).
Physico-chemical characterization of amphiphiles A1–A4
Amphiphiles generally tend to aggregate towards micellar assemblies in aqueous media due to the hydrophobic effect.64 By suitable synthetic modification of the head group and balancing the hydrophobic tail, even structurally persistent supramolecular architectures can be generated,65,66 which stand in contrast to conventional micelles that form loose, short-lived dynamic aggregates. This particular behavior can, for instance, be exploited for transporting therapeutic material like genes, particularly if the head group exposes a positive net charge that enables electrostatic interactions with negatively charged nucleic acids.To examine the physico-chemical characteristics of the synthesized dendritic amphiphiles A1–A4 and to evaluate their applicability as potential gene delivery systems, the aggregation behavior as well as size and surface charge were studied using different physical characterization methods such as critical micelle concentration (CMC) determination, cryogenic transmission electron microscopy (cryo-TEM), dynamic light scattering (DLS), and zeta potential measurements (Table 1).
| Sample | CMCa,b [M] | d H (DLS)c [nm] | d (TEM)a,d [nm] | Zeta potentiala [mV] |
|---|---|---|---|---|
| a HEPES saline buffer (pH 7.4, 9.4 mM NaCl). T = 25 °C. b Pyrene concentration: 0.5 μM. c Phosphate buffer (pH 7.4, 10 mM); c = 4.5 mM. T = 25 °C. Size distribution by volume, PDI = 0.2–0.4. d c = 4.5 mM. TEM diameter values of micelles are estimated from individual micelles in the case of G2 derivatives; in the case of the G1 species, areas of two-dimensionally ordered spherical micelles were analyzed in reciprocal space (Fourier analysis), where diffraction rings indicate the mean micelle diameter. e G1 derivatives turned out to assemble towards extended two-dimensionally ordered structures besides spherical micelles. Because of this structural bimodality DLS measurements are not applicable.66 | ||||
| A1 | 2.9 × 10−3 | n.a.e | 4.0 (± 0.2) | 51.7 (± 5.2) |
| A2 | 1.6 × 10−3 | 3.4 ± 0.2 | 3.0 (± 1.0) | 41.5 (± 4.2) |
| A3 | 2.7 × 10−3 | n.a.e | 5.0 (± 0.2) | 52.9 (± 5.3) |
| A4 | 1.3 × 10−3 | 3.8 ± 0.5 | 3.0 (± 1.0) | 42.7 (± 4.3) |
Initially, the aggregation of the amphiphiles was studied via determination of the CMC values, since self-assembly of amphiphilic entities and thus formation of thermodynamically stable micellar aggregates only occurs, when the concentration of the amphiphile exceeds a critical concentration, known as the CMC. In fact, the CMCs were identified by means of fluorescence spectroscopy using pyrene as a fluorescence probe in aqueous solution. This hydrophobic dye is a preferred fluorescent probe due to its strong fluorescence in nonpolar domains and its weak radiation in polar media. Upon micellization, pyrene partitions preferentially into the hydrophobic microdomain of the micelles which is accompanied by a sharp increase in fluorescence.
The determined CMC values given in Table 1 show that all four amphiphiles (A1–A4) self-assemble at a millimolar level ranging from 1.3–2.9 mM. These rather high CMC values can be ascribed to the high cationic charge density, which is the result of the conjugated oligoamine surface ligand DAPMA. This is especially true, when comparing the CMCs of G1-Trz-DAPMA (A3) and G2-Trz-DAPMA (A4) to similar constructs27 that only differ in the attached amine unit and exhibit CMC values in the micromolar range (10–60 μM). Interestingly, the G2 amphiphiles (A2, A4) possess slightly lower CMC values than their G1 counterparts (A1, A3). A similar observation has been made by Trappmann et al., who showed that in their case a larger degree of hydrophilicity can lead to lower CMC values.66 As expected, the different spacer units only had a small effect on the CMC, since G1-Trz-DAPMA (A3) and G2-Trz-DAPMA (A4), which comprise a triazole ether linkage, exhibited slightly lower CMC values than the ester-bridged, less hydrophobic analogs, G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2).
The aggregation behavior was further studied by DLS, which allows determination of hydrodynamic diameters (dH). As shown in Table 1, the G2 amphiphiles A2 and A4 formed very small micelles with diameters between 3 and 4 nm. Surprisingly, these values are in the same range as the theoretical size of the amphiphilic monomer units G2-Ester-DAPMA (A2) and G2-Trz-DAPMA (A4) (∼3.3–3.6 nm), which indicates that the formed spherical particles are only composed of a small number of self-assembled amphiphiles, presumably dimers. In the case of generation one amphiphiles G1-Ester-DAPMA (A1) and G1-Trz-DAPMA (A3), the obtained DLS values could not be used because cryo-TEM measurements signified a bimodal aggregation behavior (see below).
In order to supplement the described aggregation phenomena in the case of A2 and A4 by a direct structural analysis and to elucidate the self-assembly behavior of G1-Ester-DAPMA (A1) and G1-Trz-DAPMA (A3), cryo-TEM measurements were performed on aqueous sample solutions at a general concentration of 4.5 mM, which is well above the amphiphiles' corresponding CMC values. All amphiphiles (A1–A4) showed a tendency to form small spherical particles, though the respective G1 derivatives A1 and A3 revealed a second supramolecular species by showing a more complex two-dimensionally ordered hexagonal aggregation pattern (cf.Fig. 3). This finding renders DLS measurements non-applicable in the particular case of A1 and A3. The relevance of the second assembly phenomenon is not yet fully understood and remains to be clarified in future investigations.
The spherical particles formed by all four amphiphiles (A1–A4) showed diameters in the range of 3–5 nm and did not denote internal structural differentiations. Interestingly, the diameters are far below the values obtained from the dendritic non-ionic analogs,66 where dimensions and density profiles indicate the formation of molecular bilayer structures. Due to the presence of the strongly hydrated amine sphere and hence the corresponding large head group volume, we suppose that small assemblies consisting of only very few molecules are obtained. For geometrical reasons this might even lead to the formation of dimeric assemblies in the case of the G2 derivatives A2 and A4. It is further assumed that in such a dimeric assembly the diameter of the head groups (2–4 nm) determines the whole size of the assemblies, whereas hydrophobic alkyl chains can be easily accommodated within the core volume without a significant impact on the overall assembly size.
In accordance with the large number of peripheral amine groups, the conducted zeta measurements (Table 1) showed that all four compounds displayed high positive surface charges between 41 and 53 mV, which supports the applicability of these systems as prospective gene carriers. Interestingly, the G1 amphiphiles A1 and A3 exhibited slightly higher surface charges, which presumably relates to the different aggregation patterns observed with these compounds. Moreover, this can be explained by less repulsion of the positive charges within the hydrophilic head groups of the generation one amphiphiles, as already observed for related structures.27,47
Oligonucleotide binding and condensation
The process of efficient binding and condensation of oligonucleotides represents an elemental step within the gene delivery route, since it enables the cellular internalization of the genetic material. Due to the fact that cell membranes are typically negatively charged owing to their content of glycoproteins, proteoglycans, and glycerolphosphates,67,68 the complex between the gene carrier and the nucleic acid, termed polyplex, should preferentially exhibit a positive net charge for effectively penetrating the cell membrane. Moreover, the induced complexation of the genetic material causes its collapse and condensation due to the decrease of the repulsive forces between the anionic charges. As a result, much smaller oligonucleotide particles are formed which can be endocytosed by the cell.Notably, increasing the net positive surface charge of polyplexes will promote the uptake but also raise cytotoxicity due to membrane destabilization. Accordingly, the complexes have to strike a balance between cellular uptake and cytotoxicity to achieve optimal delivery efficiencies. Hence, only the actually required amount of nitrogen atoms or rather cationic charges should be employed. This can be realized by finding the appropriate N/P ratio which describes the molar ratio of amine (N) to phosphate (P) groups. Relevant N/P ratios have to be determined individually since they are dependent on a variety of factors such as size, shape, charge, and ligand modification of the used gene carrier.
In order to examine the ability of the synthesized compounds A1–A4 to efficiently complex and condense the genetic material, the binding affinity to DNA was studied first by using the ethidium bromide (EthBr) displacement assay (cf. ESI, S11–S13 for additional information†). Although the resulting data rather reflect a competition assay than an absolute binding strength, it is a powerful comparative method, because it gives valuable information about the gene carriers' general tendency to bind oligonucleotides.55 According to the obtained, very low CE50 values (∼0.3) (cf. ESI, Table S1†) – that generally reflect the relative ability to bind anionic DNA per cationic charge – all scaffolds (A1–A4) proved to be overall strong DNA binders. This is supported by CE50 values of dendritic amphiphiles that were decorated with primary amine groups.27 There, the DNA binding was significantly weaker as CE50 values of 0.8–1.3 were obtained. In contrast to the currently employed triamine unit (DAPMA), a simple glycine unit (monoamine) was connected to each dendron branching in that case, thereby indicating the beneficial effect of using an inherently multivalent amine group, such as DAPMA.
The binding capacity of all four amphiphiles (A1–A4) was further investigated using the agarose gel electrophoresis retardation assay that directly measures interactions between oligonucleotides and potential gene carriers. As a sign of successful DNA or RNA neutralization and condensation, the constructs should either reduce or completely retard the electrophoretic mobility of the genetic material. The obtained results (Fig. 4) show that all four amphiphiles (A1–A4) are able to efficiently complex and condense siRNA at N/P ratios between 10 and 20. Complex-free siRNA and a complex of siRNA and Lipofectamine 2000 were loaded as controls. In accordance with the results obtained by the EthBr assay, the G2 derivatives (A2, A4) are slightly more efficient than their lower generation counterparts (A1 and A3), i.e., amphiphile G2-Ester-DAPMA (A2) reduces the siRNA mobility at N/P 10 to a more pronounced extent than G1-Ester-DAPMA (A1).
 | ||
| Fig. 4 Binding capacity and migration of amphiphile–siRNA complexes using agarose gel electrophoresis. Complexes of FAM-labeled siRNA and the amphiphiles G1-Ester-DAPMA (A1), G2-Ester-DAPMA (A2), G1-Trz-DAPMA (A3), and G2-Trz-DAPMA (A4) with increasing N/P ratios (lane 1–7: 5, 10, 20, 30, 50, 70, and 100, each) are shown. Complex-free FAM-siRNA (siRNA) and Lipofectamine 2000 complexed FAM-siRNA (Lipo) are displayed in the last two slots on the right. Images were acquired by detecting the green fluorescence signal. | ||
As mentioned above, in order to facilitate efficient gene expression, on the one hand, slightly positively charged polyplexes are believed to promote interactions with predominantly negatively charged cell membranes.67,68 On the other hand, the process of DNA/siRNA condensation plays a vital role in the transfection pathway. Therefore, the hydrodynamic diameters plus surface charges of the amphiphile–DNA polyplexes were then determined by DLS and zeta potential measurements.
The obtained data (Fig. 5) indicate that all four compounds (A1–A4) which already exhibited high DNA binding affinities, as revealed by the EthBr assay as well as by gel electrophoresis studies, in turn condense DNA effectively into polyplexes of ∼200 nm in diameter, at N/P ratios 10–20. Predictably, on raising the N/P ratios, the complexed oligonucleotides are condensed to a more pronounced extent which leads to smaller polyplexes. However, relatively large polyplex particles have been observed with lower N/P ratios (N/P 2–5), which underlines the necessity of applying a minimum N/P ratio of 10 for all four amphiphiles (A1–A4) to yield sufficiently condensed polyplexes that are able to be internalized by cells (ca. 200 nm).
 | ||
| Fig. 5 Hydrodynamic diameter (size) and zeta potential of DNA polyplexes obtained with amphiphiles A1–A4 using varying N/P ratios (2, 5, 10, and 20). Amphiphile–DNA polyplexes were measured in HEPES saline buffer (pH 7.4, 9.4 mM NaCl). DLS size distribution by volume, PDI = ∼0.1–0.4. Zeta potential of DNA = −29.5 ± 3.2 mV (size not determinable, due to high polydispersity). | ||
The conducted zeta potential measurements give overall positive net charges ranging from 12–28 mV. Interestingly, even at the lowest N/P ratio of 2, positive net charges can still be detected with every amphiphilic transporter (A1–A4) which reflects the strong interaction and as a result leads to a reasonable neutralization of the DNA. In accordance with the DLS results, the identified zeta potentials of the polyplexes illustrate the general tendency that the zeta potential rises with increasing N/P ratios, which is in agreement with theoretical expectations.27 Although the concentrations of the sample solutions were convenient for zeta potential and DLS measurements, they turned out to be too low for cryo-TEM investigations, so that direct structural data of polyplexes could not be obtained under the experimental conditions used.
In vitro cytotoxicity
Cytotoxicity is an important aspect to be considered when evaluating nanocarriers for their specific transfection potency. Therefore, the cytotoxic potential of all four amphiphiles (A1–A4) was determined by the WST-1 assay as well as by the xCELLigence system (Roche) using non-targeting (nT) siRNA and HeLa cells. As mentioned earlier, the most appropriate N/P ratio has to be a compromise between sufficient complexation and release of the siRNA together with low cytotoxicity and high transfection efficiency. In order to cover a broad range of possible N/P ratios and in accordance with the findings extracted from the gel retardation assay, amphiphile–siRNA polyplexes with N/P 10, 30, and 50 were evaluated.The results of the WST-1 assay (Fig. 6A–D) relative to nT siRNA treated cells (control cells which were treated with nT siRNA but without transfection reagent; henceforth referred to as “nT siRNA cells”) illustrate that all examined amphiphiles (A1–A4) showed no toxicity while using an N/P ratio of 10. Furthermore, siRNA polyplexes at N/P 30 formulated with G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2) did not show considerable toxic effects. However, G2-Trz-DAPMA (A4) induced a decrease of cell viability at N/P 30 (61% reduction) as well as to some extent G1-Trz-DAPMA (A3) (31% reduction), indicating a higher cytotoxic potential for the triazole derivatives when compared to the ester-linked analogs (A1: 7% and A2: 28% reduction). At N/P 50 all amphiphiles led to a reduction of cell viability by at least 54% (i.e., A1) (A2: 70%), which was again found to be more pronounced in the case of the triazole-bridged amphiphiles (A3: 79%, A4: 99%). Therefore, an N/P ratio well below 50 was used for subsequent transfection studies with all four nanotransporters (A1–A4).
 | ||
| Fig. 6 Cytotoxicity determined by the two different methods: WST-1 assay (left) and xCELLigence (right). HeLa cells were transfected with non-targeting (nT) siRNA. The results of the WST-1 assay (A–D) and of the xCELLigence system (E–H) are shown relative to nT siRNA control cells that are treated with nT siRNA, but without transfection reagent (WST-1 assay = 1 (dashed line); xCELLigence = 0 (baseline)). The control group “Control” (integrated within the xCELLigence graphics) characterizes untreated cells. N/P ratios 10, 30, and 50 were analyzed for all four amphiphiles. G1-Ester-DAPMA (A1) is shown in panels A and E, G2-Ester-DAPMA (A2) in B and F, G1-Trz-DAPMA (A3) in C and G, and G2-Trz-DAPMA (A4) in D and H. | ||
In addition, the cytotoxicity of the nanocarriers was investigated by the xCELLigence system (Fig. 6E–H) for continuous monitoring of cell viability and growth in real-time. The data from the xCELLigence system were normalized to the time before transfection (24 h after seeding) and were related to nT siRNA cells, demonstrated as a baseline (delta cell index = 0). Profiles from untreated cells (negative control, termed “Control”) and cells treated with Lipofectamine (positive control) served as references. Short-term rises (5 h period) of the cell index after amphiphile–siRNA polyplex treatment were observed for all four nanocarriers A1–A4 (E–H), while A2 (F) and A4 (H) led to stronger increases than the G1 analogs (A1, A3), potentially caused by nontoxic changes in cell adhesion and cell structure. Addition of Lipofectamine and the replacement of culture medium in the control group (Control) showed a minor effect on the cell index. Generally, it can be stated that all amphiphiles (A1–A4) induced no toxic long-term effects as observed by the xCELLigence system. Slight but non-significant decreases of the cell index compared to the controls (Control, Lipofectamine) (i.e., A3 N/P 10, 24–50 h post-treatment) may result from alteration of viability, adhesion, or cell structure during transfection. The differences between the outcomes of the two assay systems result from the varying methods, since the WST-1 assay generally gives information about the mitochondrial metabolism activity which signifies the endogenous redox potential of cells, whereas the xCELLigence system relates to changes of the cell impedance. In conclusion, although in several cases there is a drop of detectable mitochondrial activity at 48 h post-transfections (see above; WST assay), generally cell morphology and numbers do not differ significantly within the tested period of two days (xCELLigence system).
In vitro siRNA transfection
The in vitro transfection efficacy of amphiphile–siRNA complexes was evaluated using luciferase as a reporter gene in the human renal carcinoma cell line, 786-O-Luc. Due to the partial in vitro cytotoxicity of certain amphiphiles seen in HeLa cells, the effect on the cell viability of 786-O-Luc by all four constructs (A1–A4) was evaluated first. No reduction of cell viability was determined in A1 and A2 treated cells, as well as by the positive control Lipofectamine compared to the untreated control cells (Fig. 7A). However, the triazole-bridged compounds A3 and A4 revealed enhanced cytotoxicity vs. the ester-linked constructs (cf. ESI, Fig. S4A†), thereby confirming the effect seen with HeLa cells. Therefore, we focused on A1 and A2 for further studies. | ||
| Fig. 7 786-O Luc transgenic cells were transfected with luciferase specific and non-targeting siRNA (ON-TARGETplus Non-targeting siRNA, Dharmacon) complexed with nanocarriers A1 and A2 at N/P ratios of 10, 20, and 30 for 48 h. Lipofectamine was used as a positive control and untreated cells as the negative control. (A) Cell viability was measured by using the CellTiter-Glo Luminescent Cell Viability Assay (Promega). (B) Transfection efficacy was determined by using the commercial Luciferase Assay System (Promega). Results are shown as mean ± SD of triplicates. | ||
The transfection efficacy of the ester-linked amphiphiles A1 and A2 was largely influenced by the given N/P ratio of the amphiphiles (A1, A2)–anti-Luc-siRNA polyplexes, as shown in Fig. 7B. Both A1 and A2 showed the highest knockdown effect using N/P 30 (38% and 27%, respectively). N/P 20 caused a decrease of luciferase activity of 34% (A1) and 19% (A2) vs. control. However, knockdown was less than that obtained with Lipofectamine (positive control). Polyplexes with N/P 10 induced no reduction of luciferase expression. When using non-targeting siRNA, the luciferase activity of A1 at N/P 30 was also partially reduced, while the same applies to the A2 complexes (with all three tested N/P ratios). Due to the fact that these amphiphile–non-targeting siRNA complexes showed no cytotoxicity, an unspecific effect can be assumed in this case. In addition, polyplexes with an N/P ratio above 30 caused cytotoxicity (data not shown). An N/P ratio of 25 was therefore used for the following investigation (determination of pro-inflammatory cytokines). The triazole amphiphiles A3 and A4 showed, in addition to enhanced cytotoxicity, no specific transfection efficacy (cf. ESI, Fig. S4B†).
This divergence in transfection efficiency can be attributed to the different incorporated linker groups at the core of the amphiphiles. The biodegradable and flexible nature of the ester linker unit (in A1 and A2) compared to a stiff, non-degradable triazole group present in A3 and A4 could be the reason for the low cytotoxicity and enhanced transfection activity for A1 and A2. It can be assumed that due to their high positive surface charges and consequential strong nucleic acid binding (as revealed by the conducted EthBr assay) which is clearly the result of the incorporated amine moiety DAPMA, the complexed siRNA presumably could not be liberated effectively in order to yield superior gene transfer activities. Similar observations are known from other gene carriers, as it can be difficult to find the delicate balance between initial protection and subsequent release of the genetic cargo.69,70
Determination of pro-inflammatory cytokines
It is known that nanoparticles can induce pro-inflammatory responses in vivo as an adverse reaction which can therefore constrict their administration.71 To evaluate if there are limitations regarding the biological application of the relevant amphiphiles A1 and A2 due to possible inflammatory side effects, the impact of their systemic injection in mice on pro-inflammatory cytokine levels in blood was determined (Fig. 8). | ||
| Fig. 8 Three BALB/c mice per group were treated intravenously with 8 mg kg−1 and 20 mg kg−1A1 (A) vs.A2 (B) complexed with non-targeting siRNA (ON-TARGETplus Non-targeting siRNA, Dharmacon) at an N/P ratio of 25, or Invivofectamine 2.0 Reagent, Invitrogen. HyPure water was used as the negative control (vehicle). Retrobulbar blood was taken 1 h after injection and serum was examined via a Meso Scale Discovery Multi-Spot Assay System, Mouse ProInflammatory 7-Plex Assay Ultra-Sensitive Kit. Results are shown as mean ± SD of triplicates. | ||
BALB/c mice were treated intravenously with A1– and A2–non-targeting siRNA complexes, respectively. Administration of Invivofectamine (Invitrogen) was used as a positive control, HyPure water as the negative control (vehicle). Blood was taken 1 h after administration and pro-inflammatory cytokines were determined in the serum. It was found that the secretion of the cytokines IL-6, IL-1β, and TNF-α was not affected by the treatment with A1 and A2. Furthermore, A1 treatment did not lead to a change in serum cytokine levels of IL-10 and KC. The treatment with 20 mg kg−1A2 N/P 25 caused a moderate increase of KC and IL-10 in the serum in comparison to the vehicle control. Followed by the administration of 20 mg kg−1, both amphiphiles showed an increased secretion of IFN-γ compared to the negative control. However, these measured average concentrations (A1: 5 pg mL−1, A2: 16 pg mL−1) do not reflect an acute inflammatory response.72,73
Interestingly, the positive control Invivofectamine induced higher levels of KC, IL-6, IL-10, and TNF-α compared to the tested amphiphiles (A1, A2). In conclusion, the administration of G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2) caused no induction of a pro-inflammatory response compared to the controls Invivofectamine and Hypure water. This lack of inflammatory side effects is promising for the application of this kind of delivery vehicle in vivo.
Conclusions
Within the scope of this work we have developed well-defined low-molecular-weight amphiphilic polyglycerol dendrons that display amine units in a multivalent fashion. The main distinctions that have been introduced relate to their dendron generation and the type of linker between the hydrophilic and hydrophobic domains, i.e., an ester functionality (A1, A2) and a triazole group (A3, A4). We have demonstrated that these molecules are able to self-assemble into small, spherical aggregates that exhibit high positive surface charges. By utilizing different physico-chemical techniques, such as DLS and zeta potential measurements, ethidium bromide displacement assay, and agarose gel electrophoresis, we showed that all four amphiphilic dendrimers (A1–A4) are able to effectively complex and condense DNA and RNA. Differences within this set of amphiphiles mainly emerged with regard to their biological performance. Although each amphiphilic dendrimer had the ability to bind siRNA effectively, only the ester-bridged constructs, G1-Ester-DAPMA (A1) and G2-Ester-DAPMA (A2), were able to result in a noticeable and target-specific reduction of luciferase activity in 786-O cells in vitro without affecting the cell viability. Notably, A1 as well as A2 revealed no inflammatory side effects after systemic administration in mice compared to the commercial transfection reagent “Invivofectamine” (Invitrogen). This is remarkable because such highly positively charged compounds often raise cytotoxicity issues,74–76 which renders these dendritic structures particularly appealing for in vivo gene delivery purposes.In conclusion, due to their valuable physico-chemical properties, and in vitro siRNA transfection efficiencies, as well as their safe in vivo profile, the ester-linked, DAPMA-functionalized dendritic amphiphiles (A1, A2) are promising candidates for the development of in vivo delivery systems in the future.
Acknowledgements
The authors acknowledge the support from the Biotransporter project of the Bundesministerium für Bildung und Forschung (BMBF).Notes and references
- Z. R. Yang, H. F. Wang, J. Zhao, Y. Y. Peng, J. Wang, B. A. Guinn and L. Q. Huang, Cancer Gene Ther., 2007, 14, 599–615 CrossRef CAS PubMed.
- M. A. Mintzer and E. E. Simanek, Chem. Rev., 2009, 109, 259–302 CrossRef CAS PubMed.
- I. M. Verma and N. Somia, Nature, 1997, 389, 239–242 CrossRef CAS PubMed.
- F. Liu and L. Huang, J. Controlled Release, 2002, 78, 259–266 CrossRef CAS.
- A. G. Dalgleish, in Gene Therapy, Nature Publishing Group, 1997, vol. 4, pp. 629–630 Search PubMed.
- W. Filipowicz, L. Jaskiewicz, F. A. Kolb and R. S. Pillai, Curr. Opin. Struct. Biol., 2005, 15, 331–341 CrossRef CAS PubMed.
- J. Haensler and F. C. Szoka, Bioconjugate Chem., 1993, 4, 372–379 CrossRef CAS.
- W. Fischer, M. Calderoón, A. Schulz, I. Andreou, M. Weber and R. Haag, Bioconjugate Chem., 2010, 21, 1744–1752 CrossRef CAS PubMed.
- W. Fischer, B. Brissault, S. Prévost, M. Kopaczynska, I. Andreou, A. Janosch, M. Gradzielski and R. Haag, Macromol. Biosci., 2010, 10, 1073–1083 CrossRef CAS PubMed.
- P. Ofek, W. Fischer, M. Calderón, R. Haag and R. Satchi-Fainaro, FASEB J., 2010, 24, 3122–3134 CrossRef CAS PubMed.
- O. Germershaus, G. Pickaert, J. Konrad, U. Krüger, T. Kissel and R. Haag, Macromol. Biosci., 2010, 10, 1055–1062 CrossRef CAS PubMed.
- W. Fischer, M. A. Quadir, A. Barnard, D. K. Smith and R. Haag, Macromol. Biosci., 2011, 11, 1736–1746 CrossRef CAS PubMed.
- S. Prévost, S. Riemer, W. Fischer, R. Haag, C. Böttcher, J. Gummel, I. Grillo, M.-S. Appavou and M. Gradzielski, Biomacromolecules, 2011, 12, 4272–4282 CrossRef PubMed.
- A. Barnard, M. Calderon, A. Tschiche, R. Haag and D. K. Smith, Org. Biomol. Chem., 2012, 10, 8403–8409 CAS.
- A. Kwok, G. A. Eggimann, J.-L. Reymond, T. Darbre and F. Hollfelder, ACS Nano, 2013, 7, 4668–4682 CrossRef CAS PubMed.
- K. K. Hunt and S. A. Vorburger, Science, 2002, 297, 415–416 CrossRef CAS PubMed.
- S. P. Jones, N. P. Gabrielson, C.-H. Wong, H.-F. Chow, D. W. Pack, P. Posocco, M. Fermeglia, S. Pricl and D. K. Smith, Mol. Pharm., 2011, 8, 416–429 CrossRef CAS PubMed.
- M. L. Edelstein, M. R. Abedi and J. Wixon, J. Gene Med., 2007, 9, 833–842 CrossRef PubMed.
- D. W. Pack, A. S. Hoffman, S. Pun and P. S. Stayton, Nat. Rev. Drug Discovery, 2005, 4, 581–593 CrossRef CAS PubMed.
- C. Dufès, I. F. Uchegbu and A. G. Schätzlein, Adv. Drug Delivery Rev., 2005, 57, 2177–2202 CrossRef PubMed.
- D. K. Smith, Curr. Top. Med. Chem., 2008, 8, 1187–1203 CrossRef CAS.
- W. Fischer, M. Calderón and R. Haag, in Nucleic Acid Transfection, ed. W. Bielke and C. Erbacher, Springer, Berlin, Heidelberg, 2010, vol. 296, pp. 95–129 Search PubMed.
- F. Sheikhi Mehrabadi, W. Fischer and R. Haag, Curr. Opin. Solid State Mater. Sci., 2012, 16, 310–322 CrossRef CAS PubMed.
- S. Malhotra and R. Haag, in Encyclopedia of Polymeric Nanomaterials, ed. S. Kobayashi and K. Muellen, Springer, Berlin, Heidelberg, 2013 Search PubMed.
- S. D. Buckingham, B. Esmaeili, M. Wood and D. B. Sattelle, Hum. Mol. Genet., 2004, 13, R275–R288 CrossRef CAS PubMed.
- L. Aagaard and J. J. Rossi, Adv. Drug Delivery Rev., 2007, 59, 75–86 CrossRef CAS PubMed.
- S. Malhotra, H. Bauer, A. Tschiche, A. M. Staedtler, A. Mohr, M. Calderón, V. S. Parmar, L. Hoeke, S. Sharbati, R. Einspanier and R. Haag, Biomacromolecules, 2012, 13, 3087–3098 CrossRef CAS PubMed.
- T. Yu, X. Liu, A.-L. Bolcato-Bellemin, Y. Wang, C. Liu, P. Erbacher, F. Qu, P. Rocchi, J.-P. Behr and L. Peng, Angew. Chem., Int. Ed., 2012, 51, 8478–8484 CrossRef CAS PubMed.
- K. A. Whitehead, R. Langer and D. G. Anderson, Nat. Rev. Drug Discovery, 2009, 8, 129–138 CrossRef CAS PubMed.
- D. Castanotto and J. J. Rossi, Nature, 2009, 457, 426–433 CrossRef CAS PubMed.
- X. Liu, P. Rocchi and L. Peng, New J. Chem., 2012, 36, 256–263 RSC.
- J. F. Kukowska-Latallo, A. U. Bielinska, J. Johnson, R. Spindler, D. A. Tomalia and J. R. Baker, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4897–4902 CrossRef CAS.
- M. X. Tang, C. T. Redemann and F. C. Szoka, Bioconjugate Chem., 1996, 7, 703–714 CrossRef CAS PubMed.
- O. Taratula, O. B. Garbuzenko, P. Kirkpatrick, I. Pandya, R. Savla, V. P. Pozharov, H. He and T. Minko, J. Controlled Release, 2009, 140, 284–293 CrossRef CAS PubMed.
- F. Tack, A. Bakker, S. Maes, N. Dekeyser, M. Bruining, C. Elissen-Roman, M. Janicot, H. M. Janssen, B. F. M. De Waal, P. M. Fransen, X. Lou, E. W. Meijer, A. Arien and M. E. Brewster, J. Controlled Release, 2006, 116, e24–e26 CrossRef CAS PubMed.
- V. Russ, M. Günther, A. Halama, M. Ogris and E. Wagner, J. Controlled Release, 2008, 132, 131–140 CrossRef CAS PubMed.
- O. Boussif, F. Lezoualc'h, M. A. Zanta, M. D. Mergny, D. Scherman, B. Demeneix and J. P. Behr, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 7297–7301 CrossRef CAS.
- W. T. Godbey, K. K. Wu and A. G. Mikos, J. Controlled Release, 1999, 60, 149–160 CrossRef CAS.
- M. L. Forrest, J. T. Koerber and D. W. Pack, Bioconjugate Chem., 2003, 14, 934–940 CrossRef CAS PubMed.
- R. Duncan and L. Izzo, Adv. Drug Delivery Rev., 2005, 57, 2215–2237 CrossRef CAS PubMed.
- A. Barnard, P. Posocco, S. Pricl, M. Calderon, R. Haag, M. E. Hwang, V. W. T. Shum, D. W. Pack and D. K. Smith, J. Am. Chem. Soc., 2011, 133, 20288–20300 CrossRef CAS PubMed.
- I. Toth, T. Sakthivel, A. F. Wilderspin, H. Bayele, M. O'Donnell, D. J. Perry, K. J. Pasi, C. A. Lee and A. T. Florence, STP Pharma Sci., 1999, 9, 93–99 CAS.
- K. Ewert, A. Ahmad, H. M. Evans, H.-W. Schmidt and C. R. Safinya, J. Med. Chem., 2002, 45, 5023–5029 CrossRef CAS PubMed.
- D. Joester, M. Losson, R. Pugin, H. Heinzelmann, E. Walter, H. P. Merkle and F. Diederich, Angew. Chem., Int. Ed., 2003, 42, 1486–1490 CrossRef CAS PubMed.
- S. P. Jones, N. P. Gabrielson, D. W. Pack and D. K. Smith, Chem. Commun., 2008, 0, 4700–4702 RSC.
- T. Takahashi, C. Kojima, A. Harada and K. Kono, Bioconjugate Chem., 2007, 18, 1349–1354 CrossRef CAS PubMed.
- P. Posocco, S. Pricl, S. Jones, A. Barnard and D. K. Smith, Chem. Sci., 2010, 1, 393–404 RSC.
- C. l. Leal, K. K. Ewert, R. S. Shirazi, N. F. Bouxsein and C. R. Safinya, Langmuir, 2011, 27, 7691–7697 CrossRef CAS PubMed.
- S. P. Jones, G. M. Pavan, A. Danani, S. Pricl and D. K. Smith, Chem. – Eur. J., 2010, 16, 4519–4532 CrossRef CAS PubMed.
- M. S. Shim and Y. J. Kwon, Biomaterials, 2010, 31, 3404–3413 CrossRef CAS PubMed.
- C. He, X. Zhuang, Z. Tang, H. Tian and X. Chen, Adv. Healthcare Mater., 2012, 1, 48–78 CrossRef CAS PubMed.
- V. Bagnacani, V. Franceschi, M. Bassi, M. Lomazzi, G. Donofrio, F. Sansone, A. Casnati and R. Ungaro, Nat. Commun., 2013, 4, 1721 CrossRef PubMed.
- M. Wyszogrodzka and R. Haag, Chem. – Eur. J., 2008, 14, 9202–9214 CrossRef CAS PubMed.
- K. Kalyanasundaram and J. K. Thomas, J. Am. Chem. Soc., 1977, 99, 2039–2044 CrossRef CAS.
- M. A. Kostiainen, J. G. Hardy and D. K. Smith, Angew. Chem., Int. Ed., 2005, 44, 2556–2559 CrossRef CAS PubMed.
- M. A. Kostiainen, D. K. Smith and O. Ikkala, Angew. Chem., 2007, 119, 7744–7748 CrossRef.
- B. F. Cain, B. C. Baguley and W. A. Denny, J. Med. Chem., 1978, 21, 658–668 CrossRef CAS.
- H. Gershon, R. Ghirlando, S. B. Guttman and A. Minsky, Biochemistry, 1993, 32, 7143–7151 CrossRef CAS.
- M. Calderón, M. A. Quadir, S. K. Sharma and R. Haag, Adv. Mater., 2010, 22, 190–218 CrossRef PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- R. Haag and F. Kratz, Angew. Chem., Int. Ed., 2006, 45, 1198–1215 CrossRef CAS PubMed.
- V. A. Bloomfield, Biopolymers, 1997, 44, 269–282 CrossRef CAS.
- M. Morille, C. Passirani, A. Vonarbourg, A. Clavreul and J.-P. Benoit, Biomaterials, 2008, 29, 3477–3496 CrossRef CAS PubMed.
- C. Tanford, The Hydrophobic Effect: Formation of Micelles and Biological Membranes, John Wiley & Sons, New York, 1973 Search PubMed.
- M. Kellermann, W. Bauer, A. Hirsch, B. Schade, K. Ludwig and C. Böttcher, Angew. Chem., Int. Ed., 2004, 43, 2959–2962 CrossRef CAS PubMed.
- B. Trappmann, K. Ludwig, M. R. Radowski, A. Shukla, A. Mohr, H. Rehage, C. Böttcher and R. Haag, J. Am. Chem. Soc., 2010, 132, 11119–11124 CrossRef CAS PubMed.
- F. Sakurai, R. Inoue, Y. Nishino, A. Okuda, O. Matsumoto, T. Taga, F. Yamashita, Y. Takakura and M. Hashida, J. Controlled Release, 2000, 66, 255–269 CrossRef CAS.
- M. Guillot-Nieckowski, S. Eisler and F. Diederich, New J. Chem., 2007, 31, 1111–1127 RSC.
- C. L. Grigsby and K. W. Leong, J. R. Soc., Interface, 2010, 7, S67–S82 CrossRef CAS PubMed.
- L. Zhang, Z. Chen and Y. Li, Int. J. Nanomed., 2013, 8, 3689–3701 CrossRef PubMed.
- E.-J. Park and K. Park, Toxicol. Lett., 2009, 184, 18–25 CrossRef CAS PubMed.
- M. Bosmann, N. F. Russkamp and P. A. Ward, Exp. Mol. Pathol., 2012, 93, 319–323 CrossRef CAS PubMed.
- L. Huang, H. P. Lemos, L. Li, M. Li, P. R. Chandler, B. Baban, T. L. McGaha, B. Ravishankar, J. R. Lee, D. H. Munn and A. L. Mellor, J. Immunol., 2012, 188, 4913–4920 CrossRef CAS PubMed.
- A. E. Nel, L. Madler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef CAS PubMed.
- J. Khandare, M. Calderon, N. M. Dagia and R. Haag, Chem. Soc. Rev., 2012, 41, 2824–2848 RSC.
- C. L. Gebhart and A. V. Kabanov, J. Controlled Release, 2001, 73, 401–416 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and transfection results of A3 and A4, as well as CMC, DLS, and EthBr assay graphics. See DOI: 10.1039/c3tb21364a |
| ‡ These authors contributed equally to this study. |
| This journal is © The Royal Society of Chemistry 2014 |