 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advanced dextran based nanogels for fighting Staphylococcus aureus infections by sustained zinc release
Kerstin
Malzahn
ab,
William D.
Jamieson
c,
Melanie
Dröge
a,
Volker
Mailänder
ad,
A. Toby A.
Jenkins
c,
Clemens K.
Weiss
*ae and
Katharina
Landfester
a
aMax Planck Institute for Polymer Research, Ackermannweg 10, 55128, Mainz, Germany
bGraduate School Materials Science in Mainz, Staudinger Weg 9, 55128, Mainz, Germany
cDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, United Kingdom
dIII. Medical Clinic, University Medical Center, Langenbeckstrassse 1, 55131 Mainz, Germany
eUniversity of Applied Sciences Bingen, Berlinstrasse 109, 55411 Bingen, Germany. E-mail: c.weiss@fh-bingen.de
First published on 3rd February 2014
Abstract
The growth in numbers and severity of hospital acquired infections has increased the need to target bacteria locally and specifically. Consequently, smart drug-delivery systems are being developed for local bactericidal action. The approach takes the concept of nanogels in drug delivery of small molecules to the next level by enclosing them in a shell. Versatile polysaccharide nanogels were loaded with zinc ions as antibacterial agents in a miniemulsion process, in order to target methicillin resistant strains of Staphylococcus aureus (MRSA). The encapsulation of drugs in nanogels is limited by the crosslinking density of the gel and the size of the drug. The characterization of the nanogels with inductively coupled plasma optical emission spectroscopy (ICP-OES) revealed that zinc ions cannot be retained within without an additional ‘shell’ layer. The nanogels were surrounded by a dextran–polyurethane shell, which can retain substances by reduction of water penetration. A delayed zinc release compared to the nanogels was confirmed by ICP-OES. Bacterial tests revealed an antibacterial effect of the shell enhanced nanogels against S. aureus. The studied nanogel system shows potential in locally addressing bacterial infections. The platform is extremely versatile and can be tailored to application as dextran and Zn(NO3)2 can be replaced by other polysaccharides (e.g. hyaluronic acid) and antibacterial agents, respectively.
Introduction
Hospital acquired infections by multi-drug-resistant bacteria have recently attracted a lot of attention. These infections are difficult to cure due to the resistance to common antibiotics and because infections are often noticed too late when a biofilm already has started to form. Biofilm formation is the second step in the biphasic bacterial attachment process.1,2 After the reversible attachment of bacteria within the first few hours the attachment becomes irreversible and a biofilm is formed. A biofilm typically reduces susceptibility to drugs through action as a physical and chemical barrier to antibiotic diffusion, and as a pool of genetic and phenotypic diversity allowing resistance to manifest rapidly.3 Efficient treatment of bacterial infections should thus be local and immediate.Smart drug-delivery systems, which can be applied locally (e.g. in wound dressings) and open up specifically in the presence of bacteria to release their antibacterial cargo have been developed to address the problem of bacterial attachment and colonization. Recent progress in this field was made by Baier et al.4 Delivered antibacterial agents at the wound site are, in principle, an alternative to the use of systemic antibiotics when treating wound infection.5,6 Metal ions and metal oxides7 have found applications in advanced antibacterial systems.8,9 To encapsulate and release metal salts or oxides, a hydrophilic environment like a hydrogel10,11 or a capsule with an aqueous core is more suitable than hydrophobic environments, which require the use of hydrophobic organo-metallic compounds.12,13 Additionally, the release into aqueous media requires accessibility by water. In drug-delivery applications nanogels are considered superior compared to common bulk hydrogels and were loaded with a variety of cargos.14,15 The confinement enables a local, specific release instead of a broad diffuse release. In contrast to bulk hydrogels, nanogels can be modified to target specific parts of the body leading to a cargo release at the desired location.16 In addition, phospholipid vesicles contained within a hydrogel matrix have been demonstrated as potential antimicrobial delivery vehicles.17
Potential opening mechanisms for the drug delivery systems include swelling of hydrogels,18,19 environmental pH changes,20 membrane lytic action of secretion toxins from the infecting bacteria, which open phospholipid vesicles,21–23 or polymer degradation by extracellular enzymes.24 Accordingly, biodegradable/compatible materials like polysaccharides seem very promising for such drug-delivery systems at the nanoscale as they can often be degraded specifically by bacterial enzymes.25,26 This is supported by the number of polysaccharide capsules, which are often based on diisocyanate chemistry for the shell forming reaction.27–29 The large variety of polysaccharides allows for interchange and fitting to the desired application. Furthermore, the resulting degradation products (saccharides) are typically non-toxic and can be easily excreted by the human body. The dense shell of the nanocapsules leads to good retention of small molecules whilst allowing for full payload release upon opening.
The combination of the benefits of capsules and nanogels in a hybrid gel capsule was realized in a one-pot synthesis utilizing a functionalized dextran for the crosslinking as well as the shell formation. In these hybrids the release can be controlled by the shell density and the swelling of the gel compared to common core–shell hydrogels which only rely on the latter.30,31 In this study, the nanogel forming procedure of Klinger et al.32 was modified to carry zinc ions as antimicrobial agents. Kobitskaya et al.33 described a complexation of zinc ions by polyacrylamide in polymer latexes, which is in agreement with the work of Gromov et al.,34 and should allow retention of zinc ions in the nanogels.
The zinc release from the nanogels was studied and the hydrogels were found to have a release pattern, which is too rapid for biomedical applications. Accordingly, the nanogels were enclosed in a shell, thus altering the release pattern and reducing the zinc release rate. Bacterial tests with a methicillin resistant strain of S. aureus were performed to study the antimicrobial effect of zinc ion loaded gel capsule hybrids on bacterial growth. Further the effect of the gel capsule hybrids on cell viability was studied to demonstrate applicability in a wound environment.
Experimental section
Materials
All chemicals were used without further purification. Dextran (MW ∼6000, 40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, 70000 g mol−1), dimethyl formamide (DMF), 2,4-toluene diisocyanate (TDI), D2O, cyclohexane (>99.9%, CH), SDS and acrylamide (AAm, ≥99%) were purchased from Sigma Aldrich. Methacrylic anhydride (94%) and zinc standard for plasma analysis were purchased from Alfa Aesar. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O) and triethylamine (TEA) were purchased from Fluka. Lubrizol U (poly(isobutylene-succinimide pentamine)) was kindly provided by Lubrizol France. The inhibitor 2,6 di-tert-butyl-4-methyl phenol (BHT) was purchased from Merck Schuchardt. The initiator V59 (2,2′-azobis(2-methyl-butyronitrile)) was a gift from Wako Chemicals. MilliQ water was used throughout the experiments.
000, 70000 g mol−1), dimethyl formamide (DMF), 2,4-toluene diisocyanate (TDI), D2O, cyclohexane (>99.9%, CH), SDS and acrylamide (AAm, ≥99%) were purchased from Sigma Aldrich. Methacrylic anhydride (94%) and zinc standard for plasma analysis were purchased from Alfa Aesar. Zinc nitrate hexahydrate (Zn(NO3)2·6H2O) and triethylamine (TEA) were purchased from Fluka. Lubrizol U (poly(isobutylene-succinimide pentamine)) was kindly provided by Lubrizol France. The inhibitor 2,6 di-tert-butyl-4-methyl phenol (BHT) was purchased from Merck Schuchardt. The initiator V59 (2,2′-azobis(2-methyl-butyronitrile)) was a gift from Wako Chemicals. MilliQ water was used throughout the experiments.
Synthesis of methacrylated dextran
The synthesis of methacrylated dextran (DexMa) was adapted from Kim et al.35 In brief, 1 g of dextran (MW 6000–70000 g mol−1) was dissolved in 60 mL 6 wt% LiCl solution (3.4 g, in DMF) at 95 °C under an Ar atmosphere. Subsequently, the temperature was reduced to 60 °C and 1 mL of a 0.1 wt% solution of BHT in DMF was added dropwise, followed by 145 μL TEA, which was previously dissolved in a small amount of DMF. Subsequent to the addition of the amine, 156 μL methacrylic anhydride dissolved in a small amount of DMF was added dropwise to the solution. Afterwards, the reaction was allowed to proceed for two hours. To stop the reaction and precipitate the modified dextran, the solution was transferred into 2-propanol and the precipitate was isolated by centrifugation (Sigma 3K30, 10000 rpm, 5 min). The supernatant was discarded and the pellet was resuspended in MilliQ water. This procedure was repeated 3 times, always using approx. a water/DMF to isopropanol ratio of 1:4. The resulting DexMa solution was dialyzed (Roth Spectra Por MWCO 3500, Roth Visking MWCO 14000) against water for several days, changing the water daily. The dialyzed product was freeze dried and stored until use.
The experimental conditions are given for an exemplarily degree of substitution (DS) of 0.1. The DS was evaluated by 1H-NMR spectroscopy using a Bruker spectrometer (300 MHz) following the method of Aschenbrenner.36 2 mg of the dialyzed DexMa were dissolved in D2O for the analysis.
Synthesis of DexMa–PAAm nanogels
The miniemulsion process for the synthesis of the DexMa–PAAm nanogels (PAAm:polyacrylamide) follows the procedure of Klinger et al.32 The dispersed phase was prepared with a 1:1 ratio (wt) for the liquid and solid. 5.8 mmol acrylamide and 0.47 mmol DexMa were dissolved in 500 mg of an aqueous 0.5 M NaCl solution. The resulting solution was added dropwise to 10 g of cyclohexane with 1 wt% Lubrizol U. Afterwards, the mixture (pre-emulsion) was allowed to stir for 2 h at 1750 rpm. Subsequently, the mixture was subjected to sonification under ice-cooling with a 1/2′′ tip for 2 min at 90% amplitude (Branson sonifier 450W-Digital). The vessel was sealed with parafilm and purged with argon to remove oxygen that could affect the radical polymerization. Before increasing the temperature to 60 °C 10 mg of the initiator (V-59) were dissolved in cyclohexane and added dropwise to the emulsion. When the desired temperature was reached the reaction was allowed to proceed for 15 h. The steps from the two phase system through sonication to hydrogels are shown in Fig. 1(I)–(III). The magnified section for step two depicts the ongoing reactions during polymerization. The multiple methacrylate moieties per DexMa molecule will react with AAm and thus serve as a crosslinker. Further steps in the figure correspond to the formation of a gel hybrid and will be discussed in the following section.
 | ||
| Fig. 1 Schematic of the modified miniemulsion process, the steps I–V depict the relevant steps to obtain nanogels with a TDI crosslinked dextran shell. The process of polymerization (II) and shell formation (V) is presented in more detail in the enlarged sections. For clarity Zn2+ is not shown in the magnified illustrations. | ||
To remove excess surfactant the nanogels were centrifuged at 5000 rpm for 10 min and washed repetitively. The pellet was suspended in cyclohexane and the procedure was repeated two times until the supernatant appeared transparent after centrifugation.
Shell formation around DexMa–PAAm nanogels
In order to form a shell surrounding the DexMa–PAAm nanogels, TDI was added to crosslink pendant nucleophilic groups of the gel. The process of shell formation directly followed the synthesis for preparation of the nanogels as depicted in Fig. 1(IV) and (V).After 15 h of radical polymerization, as indicated above, TDI dissolved in cyclohexane was added dropwise to the emulsion. The required amounts of TDI were chosen with respect to the free hydroxyl moieties of the DexMa molecules, ratios of 1:1 and 1:2 for TDI and hydroxyl groups were used, respectively.
Characterization
The size and size distribution of the nanogels and hybrids were studied by dynamic light scattering (DLS) at 90° scattering angle (20 °C) using a PSS Nicomp Particle Sizer 380 (Nicomp Particle Sizing Systems). After dilution of the samples with the respective solvent the nanocapsules were measured to obtain the hydrodynamic radius (rh).The capsule morphology of the nanogel and gel–shell hybrids was investigated by scanning electron microscopy (SEM) using a field emission microscope (LEO (Zeiss), 1530 Gemini, Oberkochen, Germany) with an acceleration voltage of 200 V. Prior to measurement the sample was diluted with cyclohexane to about 0.01% solid content and dropcast onto a silicon wafer.
Zinc release experiments
In order to evaluate the zinc content of the nanogels, two important parameters had to be studied, namely the zinc loading of the nanogels and the zinc leakage/release from the nanogels over time. In Fig. 2(A) the sampling steps for the respective processes from loading to release are shown. All steps were investigated by inductively coupled plasma optical emission spectroscopy (ICP-OES), which is a sensitive technique for elemental analysis in solutions or dispersions.37 | ||
| Fig. 2 (A) Scheme showing the three steps of sample characterization by ICP-OES. The steps include the investigation of zinc loading in the gel, zinc leakage during the redispersion process and the release of zinc during consecutive water immersion. (B) Sample collection for assessment of zinc loading. (C) Sample collection for assessment of zinc release and additional sampling after 72 h for nanogel–shell hybrids. | ||
ICP-OES measurements were performed using an ACTIVA M spectrometer (Horiba Jobin Yvon, Bernsheim, Germany) equipped with a Meinhardt-type nebulizer, a cyclone chamber, and controlled by using ACTIVAnalyst 5.4 software. The following conditions were applied: 1250 W forward plasma power, 12 L min−1 Ar flow and 15 rpm pump flow. The argon emission line at 404.442 nm was used as the reference line. Measurements were performed using four different standard concentrations, two different elemental emission lines and 5 s integration time. As a baseline correction, a dynamic underground correction provided by the software was used. The emission lines chosen for the quantification of the Zn were 206.201 nm and 213.857 nm.
To assess the total zinc loading of the dispersions in cyclohexane the dispersions were freeze dried and the solid was degraded with nitric acid before being diluted in water for ICP-OES measurements (Fig. 2(B)). The zinc leakage during the redispersion process as well as the zinc release in the following 24 h was studied (Fig. 2(C)). To ensure that the redispersion process was finished, the first point in time for determination of the zinc leakage was set to 24 h from the beginning of the redispersion process. The total zinc content was assessed by a measurement of a diluted dispersion aliquot. Another aliquot was collected and centrifuged for 10 min at 4000 rpm, and the supernatant was removed and measured by ICP-OES. To study the further zinc release the supernatant was discarded after centrifugation and the pellet was redispersed in fresh water for 24 h; afterwards, the sample was centrifuged again and the zinc concentration in the supernatant was measured.
Bacterial testing
The antimicrobial activity of the zinc nanogel–shell hybrids was tested as it is highly relevant to the biomedical applications of this project. For this purpose two methicillin resistant Staphylococcus aureus strains (ST239 μ2, TW20) were exposed to the nanostructures and their behavior were studied.Tryptic Soy Broth (TSB) was prepared with MilliQ water according to the manufacturer's instructions and autoclaved before use in bacterial culture. The bacterial cultures were grown overnight at 37 °C using a shaking incubator. The optical density (OD) was measured at 600 nm and the bacteria were diluted for bacterial testing to an OD of 0.0001. The resulting initial number of colony forming units (CFU mL−1) was determined by conventional plating and colony counting. The adjusted OD corresponded to an initial bacterial concentration of 0.7 × 105 CFU mL−1 and 1.3 × 105 CFU mL−1 for S. aureus ST239 μ2 and TW20, respectively.
To test the antibacterial activity, the bacteria were inoculated in 96 well plates with the nanogel samples (20 μL) at a total volume of 200 μL per well. The resulting zinc concentration was 0.5 mM Zn(NO3)2, which corresponds to 1000 μg mL−1 nanogels. The change in absorbance at 600 nm was monitored over a duration of 18 h in 5 min increments. Each sample was tested in triplicate.
Cytotoxicity study
To ensure the usability of the nanogels in biomedical applications like wound treatment the cytotoxicity of the drug delivery system is studied on human keratinocytes (HaCaT) as a relevant model cell line for keratinocytes. Furthermore toxicity tests were performed on HeLa cells. The viability of the cells in the presence of the nanogel shell hybrids was studied using the CellTiter-Glo® luminescence assay (Promega).The HaCaT cells were received from CLS Cell Service GmbH while the HeLa cells were from DSMZ GmbH. Both were precultured in Dulbecco's modified eagle medium (DMEM) with an addition of 10% FCS and 1% penicillin–streptomycin stock solution. To test cell viability in the presence of the nanogel shell hybrids the cells were seeded in a 96 well plate at a cell density of 20000 cells per cm2. The nanogels with and without zinc were diluted in cell medium to the desired concentration and 100 μL were added to each well. Nanogels and cells were incubated together for 24 h before 100 μL of CellTiter-Glo® solution were added. The well plate was shaken well for two minutes and the solution was allowed to incubate for 10 min prior to luminescence readout using a plate reader (Tecan) All experiments were performed in triplicate.
Results and discussion
As described above, the aim of the project was to develop a nanogel system based on polysaccharides, such as dextran. The nanogels shall incorporate zinc nitrate as an active antimicrobial agent, which is to be released upon exposure to bacteria. Accordingly, nanogels had to be prepared and the loading and release of zinc from the nanogels had to be studied. Dextran methacrylate served as an enzyme degradable crosslinker32 for PAAm hydrogels. The molecular weight of dextran (Mw(Dex) 6000–70000 g mol−1) and its crosslinking ability (depending on modification with methacrylate units as characterized by the degree of substitution (DS 0.05–0.2)) were varied.
In order to prolong the release of Zn2+ for sustained antibacterial action a shell was introduced to enclose the nanogel and control the zinc elution. The shell was generated by crosslinking the pendant groups with toluene diisocyanate.
Hydrogels
The zinc loaded hydrogels were obtained from polymerization of AAm and crosslinking of DexMa in inverse miniemulsion. The nanogels were characterized with scanning electron microscopy (SEM) as well as dynamic light scattering (DLS). They can be redispersed in H2O without the addition of surfactant due to the stabilizing effect of dangling dextran chains.38When dispersing the nanogels in water, swelling is expected, the extent of which is inversely proportional to the degree of crosslinking of the nanogels. In Table 1 results from DLS measurements in cyclohexane and H2O are shown exemplarily for dextran methacrylate with a molecular weight of 6000 g mol−1. First of all, it can be noted that all nanogels show a similar size in cyclohexane (around 205 nm), which was expected as the same sonication conditions and surfactant concentrations have been used in the miniemulsion process. This is in good agreement with the theory of miniemulsions which states that ideally the droplets, which are created during sonication, turn into single particles without coalescence.39
| d Cyclohexane | d H2O | |
|---|---|---|
| DS 0.05 | 210 nm ± 90 nm | 790 nm ± 120 nm |
| DS 0.1 | 205 nm ± 65 nm | 610 nm ± 90 nm |
| DS 0.2 | 205 nm ± 55 nm | 390 nm ± 65 nm |
Additionally, the size of the nanogels was assessed after redispersion in H2O to investigate the full extent of swelling of the nanogels. It was expected that the swelling capability of the nanogels decreases when an increased DS (of dextran with Ma) leads to an increased degree of crosslinking. While nanogels with a low cross-linking density (DS 0.05) exhibit nearly a 4-fold increase of their initial hydrodynamic diameters an increased crosslinking density (DS 0.2) only leads to a doubled hydrodynamic diameter of 390 nm. A linear relationship between the degree of crosslinking and swelling of the nanogels was found for the various Mw(Dex), which were utilized.
The micrographs in Fig. 3 give an overview of the nanogels which were prepared by the variation of Mw(Dex) and DS and dried from cyclohexane onto a silicon wafer. All structures show a spherical morphology and a diameter ranging between 100 and 250 nm.
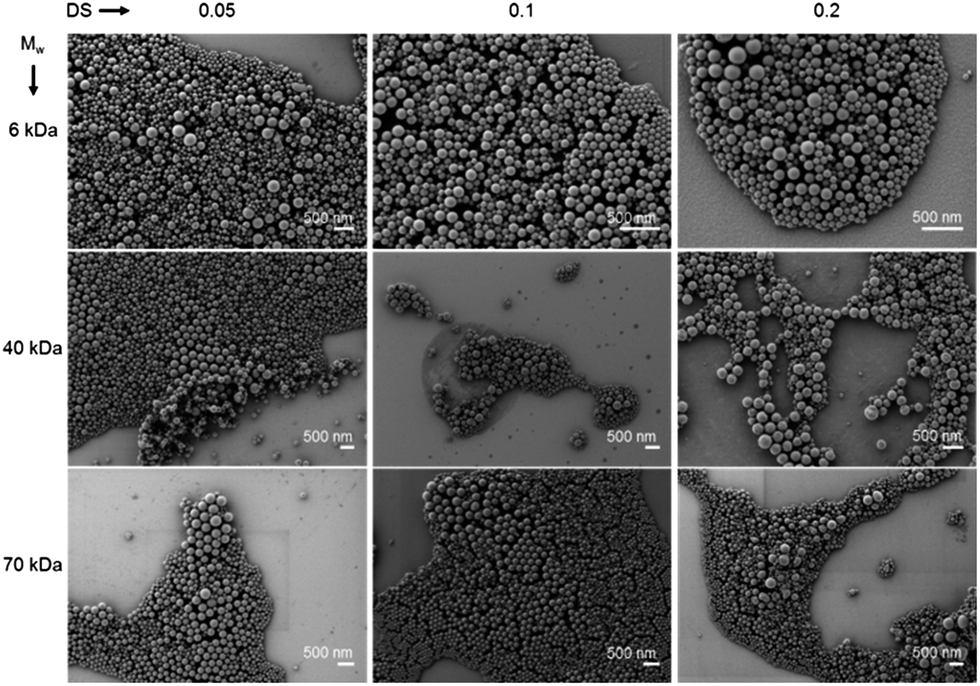 | ||
| Fig. 3 Scanning electron micrographs showing zinc loaded dextran–polyacrylamide nanogels dried from cyclohexane. | ||
The size estimated from the SEM evaluation of the nanogels is slightly smaller than the values measured by dynamic light scattering from cyclohexane. While the nanogels are completely dried under SEM conditions in CH dispersion the gels are swollen to some extent limited by the droplet size of the miniemulsion.
Zinc content/release
A theoretical Zn2+ content of 2.4 wt% was calculated from the initial amount of zinc nitrate used in the preparation of the nanogels. The total amount of Zn2+ in the gels was assessed to determine the encapsulation efficiency. To be able to assess the zinc content in the nanogels while they were dispersed in cyclohexane the samples were freeze dried and degraded with nitric acid prior to ICP-OES measurements. All combinations of Mw(Dex) and DS were assessed. Fig. 4 shows the zinc content as a wt% of dry gel in the respective sample vs. the DS for the types of dextran. It can be seen that the zinc content in all samples ranges between 2 and 2.5 wt% with respect to the solid content of the aqueous phase. | ||
| Fig. 4 Weight percent of zinc with respect to the dry gel is plotted vs. the degree of substitution of dextran (Mw: 6, 40 and 70 kDa). The average zinc loading is noted in the graph. | ||
The values, which were calculated from the zinc loading experiment, support the assumption of nearly quantitative incorporation of zinc nitrate, which was drawn from the observation that no precipitate appeared during preparation. Additionally, it can be concluded that the incorporation of zinc nitrate is independent of the type of dextran (molecular weight and degree of substitution) and the resulting crosslinking density. This is in good agreement with the principles of miniemulsion40 and the potential complexation of Zn2+ by PAAm.41,42
Retention and sustained release of Zn2+ in the gels are highly desired in order to allow redispersion in an aqueous phase and a long shelf-life time of the gels in an aqueous environment. However, the swelling of the nanogels in water suggested that water can penetrate the nanogels, accordingly a leaching can only be avoided by complexation of encapsulated ions.
The relative Zn2+ content in the nanogels after redispersion was determined by first measuring the dispersion (Fig. 5, black bars) and then the supernatant subsequent to centrifugation (red bars). Most of the Zn2+, which was found in the dispersion, had already been released from the nanogels into the supernatant. This was further confirmed when the supernatant which was collected after 24 h from a redispersed nanogel pellet only contained low amounts of zinc (blue bars).
 | ||
| Fig. 5 Zn2+ release from the nanogels with respect to the combination of Mw(Dex) and DS. The Zn2+ content in the dispersion and in the respective supernatant obtained after centrifugation at 4000 rpm is displayed in black and red, respectively. The blue bars represent the zinc released from the gels after additional 24 h incubation in fresh water. | ||
This implies that during the redispersion process the majority of zinc is already released and only small quantities are retained within the gel. It can be concluded that complexation by PAAm does not provide sufficient retention of Zn2+, which is leached out by a diffusive process during swelling of the hydrogels and water penetration. To guarantee the desired sustained release, the nanogels were modified with a shell, thus creating a unique gel–shell hybrid system. The shell was expected to enhance the Zn2+ retention by limiting the water penetration and hindering the swelling of the nanogel.
Formation of a dextran shell
Following the preparation and purification process the gel–shell hybrid dispersions in cyclohexane were stable for several weeks. For the nanogels steric self-stabilization through the dangling dextran or PAAm chains was reported, while for the dispersion of the hybrids a surfactant like SDS (0.3 wt%) was required to avoid aggregation and precipitation. The impact of the shell on the swelling behavior of the nanostructures was studied by DLS measurements in cyclohexane and water after TDI addition. In cyclohexane the gel–shell hybrids show a slightly larger diameter (250 nm) than that of the pure gels (Table 1, 205 nm). After dispersion in water the hydrodynamic diameter (dh) of the gel–shell hybrids is increased to 355 nm. The increased dh is caused by the formation of a hydration layer of the surfactant, and a swelling of the nanogel, which is limited by the surrounding shell. The slight swelling further indicates that a certain penetration of the hybrid structure by water is still possible.
The micrographs in Fig. 6(A) and (B) depict the nanogels/hybrids before and after addition of TDI, respectively. The nanogels appear particle-like in SEM imaging, which differed significantly from the results observed after shell formation. The small dried spheres (∼100 nm) reflect what was observed in Fig. 3, while the structures in Fig. 6(B) rather resemble partially deflated capsules. The observed crumpled morphology is likely to be caused by the drying process and the vacuum conditions during SEM measurement. The inner gel network hinders the shell from complete deflation or rupture as reported by Baier et al.4
 | ||
| Fig. 6 Micrographs of dried DexMa–PAAm nanogels (A) and (C) before TDI addition show a spherical morphology, (B) and (D) Nanogels after shell formation with TDI, nanogels show a crumpled morphology caused by water evaporation during the drying process in SEM and TEM, respectively. | ||
Fig. 6(C) and (D) show the images of the respective nanostructures as obtained by transmission electron microscopy. The nanogels appear homogenous throughout the object (Fig. 6(C) magnification), which is expected from homogenous polymeric particles. After the shell was formed, the nanostructures show a dark core (gel) which is surrounded by a brighter irregular rim. The dark core corresponds to the collapsed hydrogel inside the structure which is surrounded by a shell which appears as the bright rim. Additionally, the crumpled morphology observed in SEM, indicating a partial deflation, seems to be represented by a slightly inhomogeneous contrast of the particles. Bearing in mind that the nanogel–shell hybrids have been prepared in subsequent steps through a miniemulsion process the observations fit well to the expectations.
During the miniemulsion process water droplets were formed in the continuous phase and were filled out completely with the swollen nanogel network after polymerization. The dangling chains of the gel can participate in the shell forming reaction, thus being hindered from collapsing during the drying process.
The experiments were conducted as described above for the nanogels. The results are plotted in Fig. 7 as the relative amount of zinc versus the ratio of TDI to hydroxyl groups of dextran. The black bars indicate the zinc content in the dispersion after the redispersion process into aqueous solution and are regarded as 100%. An aliquot of the dispersion was centrifuged and the Zn2+ content in the supernatant is shown as red bars relative to the content in the full dispersion. The pellet obtained by centrifugation was resuspended in MilliQ water and the process was repeated twice in 24 h intervals after the start of the redispersion (blue and green bars).
 | ||
| Fig. 7 Relative zinc release from DexMa–PAAm nanogels with a TDI shell is plotted with respect to the amount of TDI used for the shell formation (expressed as the ratio of TDI units to the amount of OH groups available from the dextran). The Zn2+ content in the supernatant after redispersion (24 h) and in the following two days is displayed relative to the redispersed samples (‘Dispersion’) which is set to 100%. | ||
The zinc release into the supernatant during the redispersion process as well as in the following 48 h was found to be in the range between 5 and 15% of the total zinc content. This represents a 9-fold reduction in zinc release compared to the bare nanogels without the surrounding shell. The deviation in the zinc release over the three days is thought to be caused by deviations in sample handling as implied by a similar release pattern of both TDI to OH ratios. A similar release pattern of both TDI to OH/amide ratios implies that both samples have a comparable shell structure, which is in good agreement with the literature where no impact of the TDI amount on the shell thickness was observed.43
In conclusion, the nanogel–shell hybrids seem to exhibit a slow and rather steady release which could be described as continuous leakage rather than a boost release as seen for the nanogels. Hereby, it was shown that the hybrid structures allow the encapsulation of small drugs, which makes them superior to common nanogels.
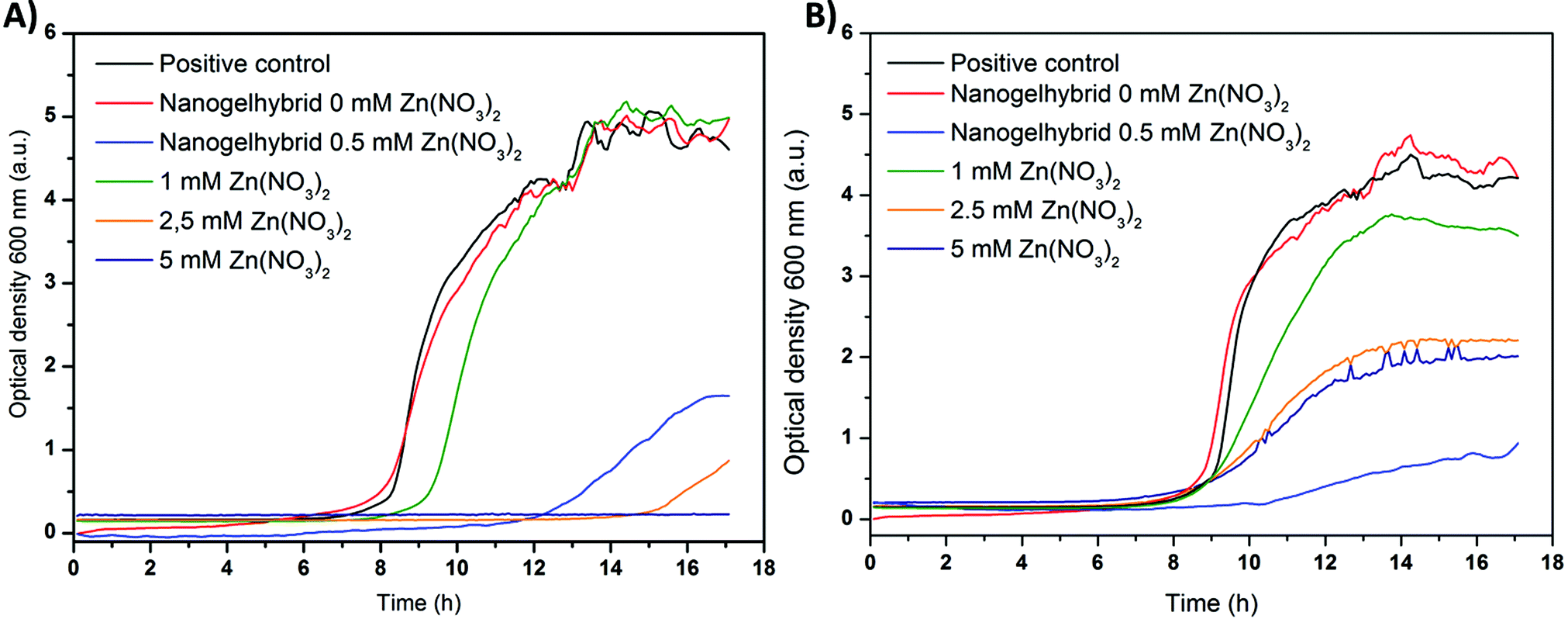 | ||
| Fig. 8 Bacterial growth of MRSA ST239 μ2 (A) and MRSA TW20 (B) in the presence of zinc nitrate and dextran nanogels with and without zinc nitrate loading. | ||
To better classify the impact of the zinc loaded gel–shell hybrids bacteria were inoculated with Zn(NO3)2 containing medium (1, 2.5, 5 mM). Low concentrations (1 mM) of zinc nitrate (green) caused a delay of ST239 μ2 growth indicated by the shifted growth curve (Fig. 8(A)). The TW20 strain did not only show a delayed growth but also a lower maximum absorbance (Fig. 8(B)). Higher concentrations of zinc (2.5 mM) further delayed bacterial growth of both strains and the maximum absorbance of the positive control was not reached. Thus the growth of bacteria is limited, while for concentrations of 5 mM (dark blue) no change in absorbance is recorded for ST239 μ2.
Accordingly the MIC100, where bacterial growth has been completely inhibited, is estimated to be about 5 mM Zn(NO3)2. The value is much higher than that reported in the literature for zinc acetate (S. aureus MSSA 476, 0.7 mM).44 This deviation may be due to the utilization of a potentially more resistant strain of S. aureus. This is further supported by the fact that for the strain TW20 no complete inhibition of growth could be observed within the concentration range tested, thus showing an even lower susceptibility to zinc than that of ST239 μ2.
Hybrid gel–shell samples with the resulting 0.5 mM Zn2+ concentration were used during the bacterial test. This corresponds to 1 mg mL−1 dried nanogels. The change of absorbance over time (light blue) shows a strong shift of the growth curve for both bacterial strains, which indicates a strong delay of bacterial growth. Furthermore, a plateau is reached within 18 h at a comparatively low absorbance, which implies that the bacterial growth is retarded. Hence, the MIC100 for the gel–shell-hybrids seems to be slightly higher than the 0.5 mM Zn2+. Comparing the growth curves of Zn(NO3)2 and the gel–shell hybrids, the latter appears to be more effective than expected compared with the same concentration of non-encapsulated Zn2+. One explanation of this effect is that the encapsulation of Zn(NO3)2 and the subsequent release leads to a high local concentration of Zn2+ in the vicinity of the bacteria. While for chitosan nanoparticles an interaction with S. aureus bacteria was observed due to the positive zeta potential,45 no such effect is expected for dextran, a neutral polysaccharide. The interaction of bacteria and polysaccharides in biofilms is not understood,46 and an alteration of bacterial metabolism in the presence of the presented dextran based hybrid nanogel shell structures cannot be excluded. Nevertheless, the reason for the enhanced antibacterial activity remains unresolved.
Possible cytotoxic effects of the pure nanogel material on the cell viability have been assessed by incubating the cells with zinc-free nanogel shell hybrids in various concentrations. Subsequently, the cell viability was monitored in the presence of zinc loaded nanogel shell hybrids. To be able to relate the outcome to the results of the bacterial testing similar nanogel contents were studied.
Fig. 9 shows the relative viability of the cells vs. the concentration of nanogel shell hybrids. The viability of cells which have been grown in medium only was set to 100%. The black line shows the viability for increasing concentrations of zinc free nanogels. Up to the concentration used in bacterial testing (1000 μg mL−1, dashed line) the viability remains close to 100%. When further doubling the concentration (2000 μg mL−1) the viability of the cells starts to be affected. The LD50 is approximated to be around 3000 μg mL−1. The effect of the nanogel shell hybrids on the cell viability at high nanocapsules could be attributed to several reasons. These could include e.g. effect of residual surfactant.
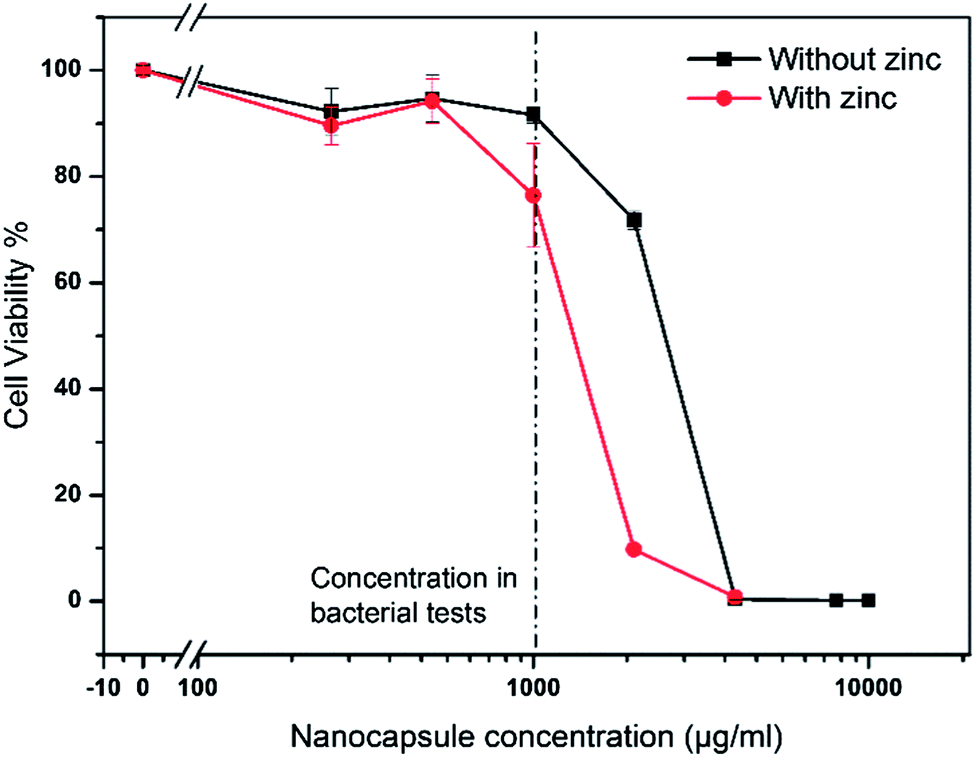 | ||
| Fig. 9 Cell viability assay for HaCaT cells. Relative cell viability with respect to untreated cells is plotted against increasing nanogel concentrations. Cell viability in the presence of zinc free (black) and zinc loaded (red) nanogels. | ||
The viability of the cells in the presence of zinc loaded nanogels was reduced to 75% at the concentration used in bacterial tests (1000 μg mL−1). When increasing the nanogel concentration to 2000 μg mL−1 the viability drops to about 10%. This is probably a cumulative effect of the zinc and nanogel concentration (cells with zinc free gels already showed reduced viability). Therefore the LD50 is between 1000 and 2000 μg mL−1. Additionally, the effect of the nanostructures on HeLa cells was investigated and found to be slightly less severe than the effect on HaCaT cells. For 1000 μg mL−1 pure nanogels and zinc loaded nanogels nearly 100% and 90% viability were observed, respectively (results not shown). A potential application would include the immobilization of the nanogel shell hybrids and thus enhance the viability of the cells, as internalization of the nanogels can thus be avoided.
Conclusions
Dextran-crosslinked polyacrylamide nanogels were transferred successfully into a nanocarrier system. Zinc nitrate was incorporated into the nanogels to serve as an antimicrobial agent. When performing zinc release studies deficient zinc retention of the nanogels was observed. Polyacrylamide did not provide sufficient complexation of the zinc ions. The release system was modified to retain the Zn2+ over a longer period and be better suited for biomedical applications.For this purpose a gel–shell hybrid nanostructure was prepared in consecutive steps based on radical polymerization and polycondensation in inverse miniemulsion. The resulting nanostructures showed an altered morphology as well as a different release pattern compared to the gels. The zinc release was changed from a fast boost release into a reduced, but continuous release from the gels, which is more beneficial in biomedical applications.
Polysaccharides are a very versatile group of polymers, accordingly, the nanocarrier concept, here presented with dextran as a model compound, is transferable to other polysaccharides to fit the desired application. Currently, the preparation of gels based on methacrylated hyaluronic acid as a cross linker is under investigation.
In order to verify the antimicrobial activity of the nanocarriers, bacterial experiments were conducted with a relevant methicillin resistant strain of S. aureus. Growth was delayed and reduced in the presence of Zn2+ loaded gel–shell hybrids. To further enhance the antibacterial activity Zn2+ can easily be replaced by other metals which are known for their antibacterial effect (e.g. silver).
The nanogel shell hybrids show no relevant cytotoxicity on HaCaT cells in the concentration regime required for bacterial inhibition. At the same concentration the zinc loaded nanogel shell hybrids exhibit only a slight effect on cell viability. To summarize, a drug release system with high potential for applications in wound environments was developed, combining strong inhibition of bacterial growth with high biocompatibility.
Acknowledgements
K.M. is a recipient of a fellowship through the excellence initiative (DFG/GSC 266). The authors kindly acknowledge funding of this work through the European Commission, (BacterioSafe) grant#245500 and Katrin Kirchhoff (MPI-P Mainz) for TEM measurements.Notes and references
- P. Tenke, C. R. Riedl, G. L. Jones, G. J. Williams, D. Stickler and E. Nagy, Int. J. Antimicrob. Agents, 2004, 23, S67–S74 CrossRef CAS PubMed.
- M. Katsikogianni and Y. F. Missirlis, Eur. Cells Mater., 2004, 8, 37–57 CAS.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34–40 CAS.
- G. Baier, A. Cavallaro, K. Vasilev, V. Mailänder, A. Musyanovych and K. Landfester, Biomacromolecules, 2013, 14, 1103–1112 CAS.
- A. Dubin, P. Mak, G. Dubin, M. Rzychon, J. Stec-Niemczyk, B. Wladyka, K. Maziarka and D. Chmiel, Acta Biochim. Pol., 2005, 52, 633–638 CAS.
- T. Koburger, N.-O. Hübner, M. Braun, J. Siebert and A. Kramer, J. Antimicrob. Chemother., 2010, 65, 1712–1719 CAS.
- M. L. Kääriäinen, C. K. Weiss, S. Ritz, S. Pütz, D. C. Cameron, V. Mailänder and K. Landfester, Appl. Surf. Sci., 2013, 287, 375–380 Search PubMed.
- C. James, A. L. Johnson and A. T. A. Jenkins, Chem. Commun., 2011, 47, 12777–12779 CAS.
- C. James, T. Pugh, A. L. Johnson and A. T. A. Jenkins, Eur. Polym. J., 2011, 47, 1338–1345 CAS.
- V. Thomas, M. Namdeo, Y. Murali Mohan, S. K. Bajpai and M. Bajpai, J. Macromol. Sci., Part A: Pure Appl.Chem., 2007, 45, 107–119 Search PubMed.
- V. B. Schwartz, F. Thétiot, S. Ritz, S. Pütz, L. Choritz, A. Lappas, R. Förch, K. Landfester and U. Jonas, Adv. Funct. Mater., 2012, 22, 2376–2386 CAS.
- C. P. Hauser, D. T. Thielemann, M. Adlung, C. Wickleder, P. W. Roesky, C. K. Weiss and K. Landfester, Macromol. Chem. Phys., 2011, 212, 286–296 CrossRef CAS.
- A. Manzke, N. Vogel, C. K. Weiss, U. Ziener, A. Plettl, K. Landfester and P. Ziemann, Nanoscale, 2011, 3, 2523–2528 RSC.
- L. Messager, N. Portecop, E. Hachet, V. Lapeyre, I. Pignot-Paintrand, B. Catargi, R. Auzely-Velty and V. Ravaine, J. Mater. Chem. B, 2013, 1, 3369–3379 RSC.
- R. T. Chacko, J. Ventura, J. Zhuang and S. Thayumanavan, Adv. Drug Delivery Rev., 2012, 64, 836–851 CrossRef CAS PubMed.
- S. V. Vinogradov, T. K. Bronich and A. V. Kabanov, Adv. Drug Delivery Rev., 2002, 54, 135–147 CrossRef CAS.
- S. E. Marshall, S.-H. Hong, N. T. Thet and A. T. A. Jenkins, Langmuir, 2013, 29, 6989–6995 CrossRef CAS PubMed.
- J. K. Oh, R. Drumright, D. J. Siegwart and K. Matyjaszewski, Prog. Polym. Sci., 2008, 33, 448–477 CrossRef CAS PubMed.
- M. Karg and T. Hellweg, Curr. Opin. Colloid Interface Sci., 2009, 14, 438–450 CrossRef CAS PubMed.
- L. Zhang, D. Pornpattananangkul, C. M. J. Hu and C. M. Huang, Curr. Med. Chem., 2010, 17, 585–594 CrossRef CAS.
- S. Tamilvanan, N. Venkateshan and A. Ludwig, J. Controlled Release, 2008, 128, 2–22 CrossRef CAS PubMed.
- J. Zhou, A. L. Loftus, G. Mulley and A. T. A. Jenkins, J. Am. Chem. Soc., 2010, 132, 6566–6570 CrossRef CAS PubMed.
- M. Laabei, A. Young and A. T. A. Jenkins, Pediatr. Infect. Dis. J., 2012, 31, e73–e77, DOI:10.1097/inf.1090b1013e3182493b3182421.
- J. Andrieu, N. Kotman, M. Maier, V. Mailänder, W. S. L. Strauss, C. K. Weiss and K. Landfester, Macromol. Rapid Commun., 2012, 33, 248–253 CrossRef CAS PubMed.
- K. Raemdonck, J. Demeester and S. De Smedt, Soft Matter, 2009, 5, 707–715 RSC.
- V. R. Sinha and R. Kumria, Int. J. Pharm., 2001, 224, 19–38 CrossRef CAS.
- N. Jagielski, S. Sharma, V. Hombach, V. Mailänder, V. Rasche and K. Landfester, Macromol. Chem. Phys., 2007, 208, 2229–2241 CrossRef CAS.
- G. Baier, A. Musyanovych, M. Dass, S. Theisinger and K. Landfester, Biomacromolecules, 2010, 11, 960–968 CrossRef CAS PubMed.
- G. Baier, D. Baumann, J. M. Siebert, A. Musyanovych, V. Mailänder and K. Landfester, Biomacromolecules, 2012, 13, 2704–2715 CrossRef CAS PubMed.
- C. D. Jones and L. A. Lyon, Macromolecules, 2000, 33, 8301–8306 CrossRef CAS.
- D. Suzuki, T. Yamagata and M. Murai, Langmuir, 2013, 29, 10579–10585 CrossRef CAS PubMed.
- D. Klinger, E. M. Aschenbrenner, C. K. Weiss and K. Landfester, Polym. Chem., 2012, 3, 204–216 RSC.
- E. Kobitskaya, D. Ekinci, A. Manzke, A. Plettl, U. Wiedwald, P. Ziemann, J. Biskupek, U. Kaiser, U. Ziener and K. Landfester, Macromolecules, 2010, 43, 3294–3305 CrossRef CAS.
- V. F. Gromov, Usp. Khim., 1995, 64, 93–104 CAS.
- S.-H. Kim and C.-C. Chu, J. Biomed. Mater. Res., 2000, 49, 517–527 CrossRef CAS.
- E. Aschenbrenner, K. Bley, K. Koynov, M. Makowski, M. Kappl, K. Landfester and C. K. Weiss, Langmuir, 2013, 29, 8845–8855 CrossRef CAS PubMed.
- N. Vogel, C. P. Hauser, K. Schuller, K. Landfester and C. K. Weiss, Macromol. Chem. Phys., 2010, 211, 1355–1368 CrossRef CAS.
- D. Klinger and K. Landfester, Polymer, 2012, 53, 5209–5231 CrossRef CAS PubMed.
- F. J. Schork, Y. Luo, W. Smulders, J. Russum, A. Butté and K. Fontenot, in Polymer Particles, ed. M. Okubo, Springer, Berlin, Heidelberg, 2005, pp. 129–255 Search PubMed.
- K. Landfester and C. Weiss, in Modern Techniques for Nano- and Microreactors/-reactions, ed. F. Caruso, Springer, Berlin, Heidelberg, 2010, pp. 1–49 Search PubMed.
- G. Dzhardimalieva, A. Pomogailo and V. Volpert, J. Inorg. Organomet. Polym., 2002, 12, 1–21 CrossRef CAS.
- K. B. Girma, V. Lorenz, S. Blaurock and F. T. Edelmann, Coord. Chem. Rev., 2005, 249, 1283–1293 CrossRef CAS PubMed.
- D. Crespy, M. Stark, C. Hoffmann-Richter, U. Ziener and K. Landfester, Macromolecules, 2007, 40, 3122–3135 CrossRef CAS.
- N. Poulter, M. Donaldson, G. Mulley, L. Duque, N. Waterfield, A. G. Shard, S. Spencer, A. T. A. Jenkins and A. L. Johnson, New J. Chem., 2011, 35, 1477–1484 RSC.
- L. Qi, Z. Xu, X. Jiang, C. Hu and X. Zou, Carbohydr. Res., 2004, 339, 2693–2700 CrossRef CAS PubMed.
- I. W. Sutherland, Microbiology, 2001, 147, 3–9 CAS.
| This journal is © The Royal Society of Chemistry 2014 |