 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Paclitaxel-loaded polyphosphate nanoparticles: a potential strategy for bone cancer treatment†
Evandro M.
Alexandrino‡
a,
Sandra
Ritz‡
a,
Filippo
Marsico
a,
Grit
Baier
a,
Volker
Mailänder
ab,
Katharina
Landfester
a and
Frederik R.
Wurm
*a
aMax Planck Institute for Polymer Research, Ackermannweg 10, Mainz, 55128, Germany. E-mail: wurm@mpip-mainz.mpg.de
bIII. Medical Clinic (Hematology, Oncology and Pulmonology), University Medical Center of the Johannes-Gutenberg University, Langenbeckstraße 1, Mainz, 55131, Germany
First published on 7th January 2014
Abstract
While it has been shown that phosphates can target molecules and nanocarriers to bone we herein demonstrate the preparation of polyphosphate nanoparticles loaded with paclitaxel using a simple miniemulsion/solvent-evaporation technique as a model for chemotherapeutic delivery. Polyphosphates exhibit much higher structural versatility, relying on the pentavalence of the phosphorus center compared to conventional polyesters. This versatility allows for the development of new degradable polymeric carriers with inherent bone adhesion ability by the interaction of the nanoparticles with a calcium phosphate material used for bone regeneration. The novel polyphosphate nanoparticles were investigated in detail with respect to their size distribution, zeta-potential, thermal and morphological properties and were further proven to be efficiently loaded with a hydrophobic drug (up to 15 wt%). The in vitro cytotoxicity was assessed against human cancer cell lines (HeLa and Saos-2), and the paclitaxel-loaded nanoparticles showed a similar cytotoxicity profile similar to the commercially available formulation Taxomedac® and the pure paclitaxel for loading ratios of 10 wt% but additionally proved efficient adhesion on calcium phosphate granules allowing drug delivery to bone. This first report demonstrates that polyphosphate nanoparticles are promising materials for the development of systemic or local bone cancer treatment, even by direct application or by formation of composites with calcium phosphate cements.
Introduction
Bone is a site of cancer growth either of bone sarcomas, which include osteosarcoma and Ewing's sarcoma,1 or – more common – as a consequence of other tumors that spread by metastasis to the bone tissue.2,3 Among the types of cancer that spread by metastases to the bone tissue, breast and prostate cancer stand out, presenting metastases in approximately 70–80% in advanced stages of the disease.2–4 The most common treatment for primary bone tumors or bone metastases is a combination of different techniques, such as surgical removal, fixation and chemotherapy, treatment with radioisotopes or by radiation strategies.3 The chemotherapeutic treatment is normally performed after the surgical intervention, with the main purpose of slowing down the tumor growth and metastases outgrowth.5 Drugs are selected on the basis of the susceptibility of the primary tumor. For breast and ovarian tumors this would include doxorubicin6,7 and paclitaxel (PTX).5,8 PTX has high potency against many different types of cancer by stabilizing the tubulin system,9 which leads to a suppression of the microtubule dynamics, mitotic arrest, and finally to death by apoptosis.9–11Despite these interesting properties, PTX shows poor water solubility10 and does not have any bone surface targeting specificity. Higher solubility was achieved by the use of macrogolglycerol ricinoleate (Cremophor EL®) and ethanol to solubilize the drug.10,11 However, the toxicity of Cremophor EL created the necessity for the development of new carrier systems for the PTX application like nanoparticles or liposomes.10,12–19
Biodegradable polymer nanoparticles based on ‘conventional’ poly-(C)-esters were intensively studied with promising results concerning the encapsulation and activity of hydrophobic drugs.19 Recently, polyester,20–22 poly(benzyl-L-glutamate),23 poly[N-(2-hydroxypropyl) methacrylamide],24 poly(ethylene imine), and poly-L-lysine25 based micro- and nanoparticles for targeted delivery to bone have been reported. The targeting profile depends mainly on the nanoparticle surface-chemistry, which is typically achieved by the introduction of targeting groups at the nanoparticle surface,19,26 such as bisphosphonates and derivatives,27 or oligopeptides28 that can specifically target bone.29,30 Due to the high affinity to the bone mineral hydroxyapatite (HA), bisphosphonates have emerged as a great option for bone targeted therapy and have been studied intensively.3,31 However, side effects have been reported for bisphosphonates as osteonecrosis of the jaw (ONJ), atrial fibrillation, and renal insufficiency.31,32
Herein, we demonstrate a promising alternative for the production of biodegradable nanoparticles which have potential inherent bone targeting abilities based on polyphosphates. Polyphosphates belong to the major class of polymers called poly(phosphoester)s (PPEs), which are highly versatile biodegradable materials that allow easy modification of backbone, side chains, and end groups to tailor the functionality and solubility profile that stimulated the interest in these materials recently.33,34 No other polymeric material shows this high versatility which is mainly due to the ability of phosphorus to form triesters and, in addition, having the possibility of interacting with calcium. Further, the pendant ester results in usually less crystalline polymers compared to poly-(C)-esters. Even more, PPEs can be degraded both enzymatically and hydrolytically which makes them interesting also for long term use in vivo as no accumulation should occur. Interestingly, only a few publications have dealt with the synthesis and applications of PPEs to date.35 They have been successfully used in tissue engineering applications,36–38 and to generate liposomes with a good affinity to calcium deposits produced by the MC3T3-E1cell line.39
Herein, we present the formation of PPE-nanoparticles (PPE-NPs) prepared by the miniemulsion/solvent-evaporation technique, the loading of these nanoparticles with paclitaxel and the interaction with calcium phosphate cements.40 These PPE homopolymers were prepared by using a recently developed general synthetic protocol via olefin metathesis (via acyclic diene metathesis (ADMET) and ring opening metathesis (ROMP) polymerization).41–43 These novel hydrophobic PPEs were used for the preparation of potentially biodegradable and biocompatible nanoparticles that were efficiently loaded with PTX and that show a strong interaction with calcium phosphate surfaces.
Results and discussion
Synthesis of PPE nanoparticles
It is well-known that charged phosphates exhibit a strong binding to bone surfaces, i.e. calcium phosphate. For cancer treatment it would be, however, favorable to generate hydrophobic polymers that can be loaded with a drug and that still possess bone targeting properties. In addition it would be desirable to avoid tedious chemical postmodification steps to attach targeting moieties or ionic groups that could further interfere with the drug loading process. We envisioned that PPEs would be the ideal candidates to exhibit an inherent bone-targeting motive due to the multiple phosphates along the polymer backbone. Further, due to the neutral triesters, efficient loading with hydrophobic drugs should be ensured. We prepared several PPEs with a variable number of methylene units (6, 10 or 20 carbon atoms) between the phosphate groups via the ADMET protocol (compared above). Fig. 1A presents exemplarily the synthesis of the polymer having 20 methylene spacers between the phosphate groups, i.e. PPE-C20. After the ADMET reaction, unsaturated PPEs (1) are obtained, which are amorphous for C6 and C10 or exhibit a rather low melting point (−7 °C for C20). These polymers were hydrogenated in order to remove the double bonds that act as defects during the crystallization and to generate saturated and crystalline PPEs (2). The saturated PPE-C20 shows a melting point of ca. 50 °C (see below). These polymers were used in the miniemulsion/solvent-evaporation method (see the Experimental section for details) to generate nanoparticles with mean diameters between approximately 80–300 nm (see ESI, Table S1†). As the polymers are soluble in hydrophobic polar solvents, they are ideal materials for the encapsulation of hydrophobic drugs or fluorescent dyes such as BODIPY-derivatives for further optical imaging (see below).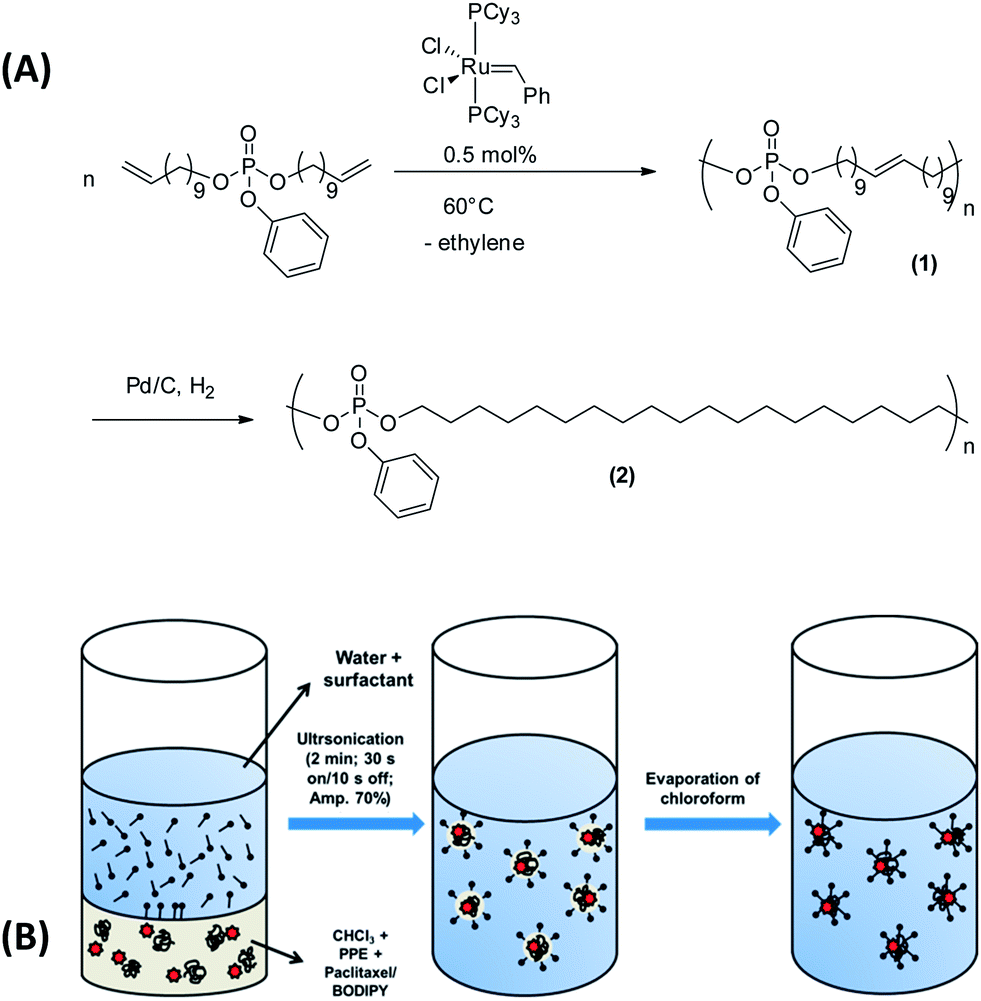 | ||
| Fig. 1 (a) Reaction scheme for the synthesis of the unsaturated (1) and saturated (2) poly(phosphoester)s (PPE-C20) via acyclic diene metathesis polymerization. (b) The miniemulsion/solvent-evaporation protocol used for the production of BODIPY or PTX loaded PPE nanoparticles. | ||
A GPC analysis of the colorless (without BODIPY) PPE dispersion after this process reveals that no molecular weight degradation has taken place (see ESI, Fig. S3–S5†) proving that the PPEs sustain the miniemulsion process. From the miniemulsion/solvent-evaporation process the formation of spherical particles is expected.40 The morphology of the PPE nanoparticles has been investigated by dynamic light scattering revealing a rather uniform scattering intensity at different angles, indicating the formation of spherical nanoparticles. Further, electron microscopy was used to visualize the shape of the nanoparticles. Due to the low melting point of PPE-C20, we expected rather soft materials (Fig. 2, PPE-C6 and C10 were detected as droplets due to their amorphous structures). Compared to other polyesters, like polylactide, which has a melting point of ca. 180 °C, the low melting point is another characteristic of the herein presented PPEs. The lower degree of crystallinity may further influence the degradation kinetics, which is currently under investigation. Fig. 2 shows exemplarily scanning and transmission electron micrographs of the nanoparticles.
 | ||
| Fig. 2 Morphological evaluation of the nanoparticles: (A) scanning electron micrograph PPE-C20-NP, scale bar 2 μm; (B) transmission electron micrograph PPE-C20-NP, scale bar 200 nm. | ||
The unloaded PPE nanoparticles are rather spherical particles, however – probably due to the low melting point of the polymer (and due to the carbon coating process) – their shape is a bit deformed. From scanning electron microscopy analysis (SEM), some of the nanoparticles suggest capsule-like morphologies, but transmission electron microscopy (TEM) analysis (Fig. 2b) confirms the formation of solid particles (see also ESI for further SEM images (Fig. S16 and S17†)).
Evaluation of bone adhesion
The PPE nanoparticle dispersions were then evaluated with respect to their capacity to interact with bone surfaces. MBCP+™ (Biomatlante) is a calcium phosphate material that has proven to be a good alternative to bone substitution for over 30 years,44,45 and is currently used in spinal, tumoral, orthopedic and periodontal applications.46,47 Biphasic calcium phosphate is usually composed of 20% hydroxyapatite (HA) and 80% β-tricalcium phosphate (β-TCP), a mixture that provides good bioactivity and osteoconduction properties.46 Due to these good bone substitution properties, MBCP+ was selected as a “model bone tissue” for the evaluation of the interaction with the PPE nanoparticles. The fluorescent and non-fluorescent PPE nanoparticles (PPE-NP-C6, -C10, -C20) were dispersed with the calcium phosphate granules and after extensive washing, the presence of the nanoparticles on the surface was evaluated by SEM and fluorescence microscopy (Fig. 3). The fluorescence intensity on the granules increased with increasing number of methylene units between the phosphate groups, indicating that there is a strong attachment of these nanoparticles to the calcium phosphate granules (note that the scaling between the SEM and the fluorescence images is not the same!). This first indication was confirmed by SEM analysis as the PPE-NP-C20s can be clearly visualized on top of the cement (Fig. 3). This behavior by the thermal properties of the three investigated polymers: as only PPE-NP-C20 is a crystalline solid at room temperature, the nanoparticles can attach effectively to the granules, while the nanoparticles from C6 and C10 probably form a polymer film on top of the granules, explaining that no distinct particles can be visualized via SEM. The weak fluorescence signal that is visible for amorphous materials (C6 and C10) is probably due to some remaining dye in the deposited film. The physical properties of the herein presented PPEs can be easily tuned by the ADMET protocol. These are the first aliphatic, crystalline PPEs reported to date and these results strongly indicate that a crystalline material (or a material with a high glass transition temperature) is needed to achieve an efficient attachment to the bone surface. The attachment of the investigated PPEs is thought to be caused by the interaction of the phosphate moieties along the backbone either in their neutral state or by partial hydrolysis of the pendant (and more labile) phenyl ester. PPE-C20 was incubated with 0.1 M HCl to induce hydrolysis and release of a low molecular weight compound (compare with the ESI Fig. S18,† at ca. 35 mL elution volume) was detected by the UV detector during the SEC experiment, while the apparent molecular weight of the polymer remained rather constant indicating that in this case the side chain is hydrolyzed first as reported previously by Baran and Penczek on similar PPEs.48 In order to further confirm the hypothesis of the degradation of the phenol group during the miniemulsion preparation, the polymer was dissolved in THF and ultrasonicated from 1 to 30 minutes and the samples taken in this interval were analyzed by GPC and HPLC (compare with ESI Fig. S24 and S25†), showing the presence of a new low molecular compound (with the same retention time as phenol) and simultaneously a decrease of the UV signal of the polymer proving the loss of the aromatic side chain.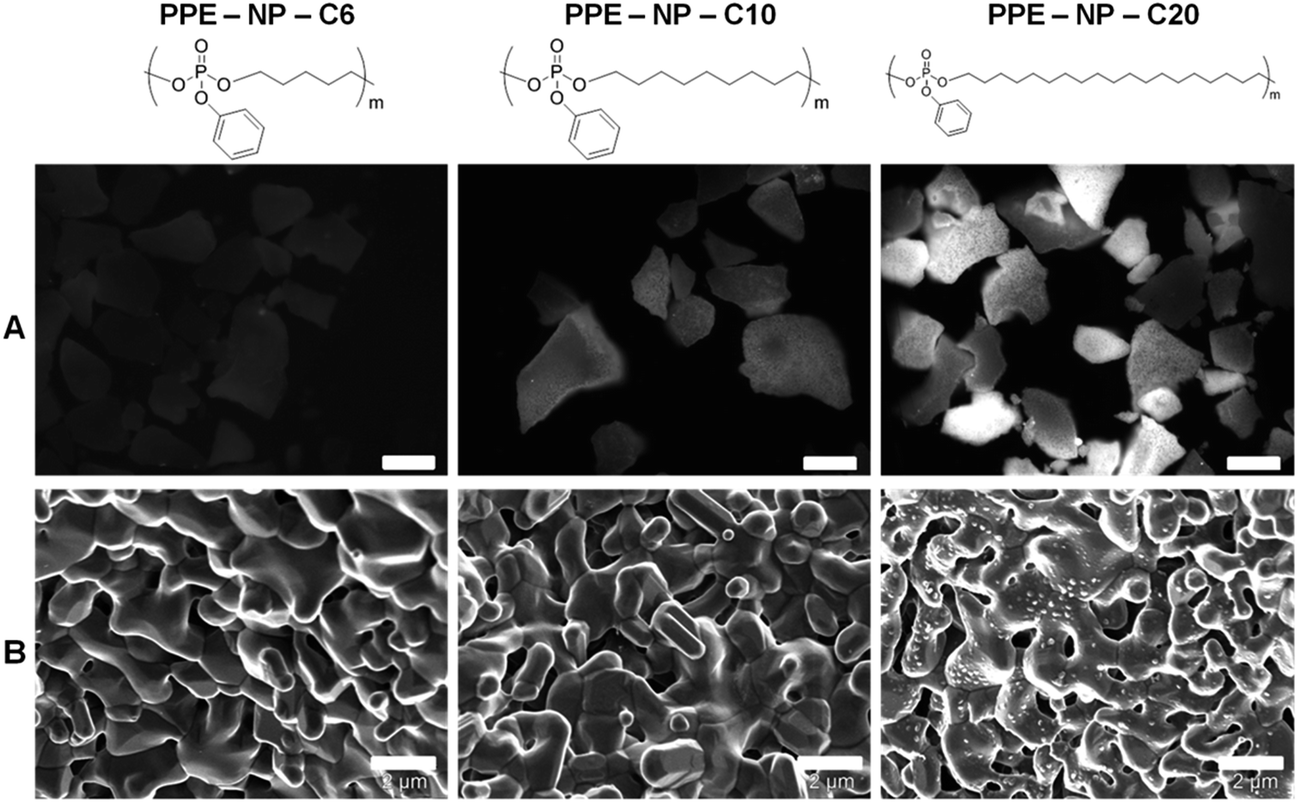 | ||
| Fig. 3 Fluorescence images (scale bar 50 μm) (A) and scanning electron images (scale bar 2 μm) (B) of the calcium phosphate cement MBCP+ surface after exposition to a dispersion of nanoparticles NP-PPE with increasing carbon linker length (C6, C10, and C20). MBCP+ is composed of 80% HA and 20% β-TCP. | ||
The stringency of the nanoparticle attachment on calcium phosphate granules was further supported by the investigation of the interaction of other hydrophobic nanoparticles with different detergents like poly(lactide), and poly(butyl cyanoacrylate) (PBCA) and modified polystyrene (ESI, Table S2 and Fig. S22†). None of these materials, except polystyrene nanoparticles modified with phosphonate surfmers (i.e. 12-methacrylamidododecylphosphonic acid), bound to the MBCP+.49,50 They were used as the positive control, because it was proven that the presence of the ionic phosphonate groups on the particle surface enables the crystallization of hydroxyapatite (HA),51 which might explain the high surface adsorption on the biphasic calcium phosphate granules (20% HA/80% β-TCP).
Paclitaxel loaded PPE nanoparticle synthesis
As shown above, the PPE nanoparticles effectively interact with the “model bone tissue”, this protocol should allow the development of a drug delivery system for local application in bone cancer therapies. With this goal, PTX, a strong anti-mitotic drug, was selected and the miniemulsion/solvent-evaporation methodology was used to produce PTX loaded-PPE nanoparticles (compare with Fig. 1b) by co-precipitation of both drug and polymer during solvent evaporation. Four PTX-loaded PPE nanoparticle formulations, with 1.5, 5, 10 and 15 wt% PTX (and a control without the addition of PTX) were produced. Table 1 summarizes the main physicochemical properties of these materials. The nanoparticle mean diameters were determined to be between 160 and 200 nm; it can be clearly seen that the size distribution (standard deviation) increased with the amount of PTX introduced initially to the particle preparation. The angle dependence of the hydrodynamic radius (see ESI, Fig. S2†) was also evaluated and revealed a similar angle dependence for all formulations, which indicates that, independent of the drug loading ratio, the shape of the particles in solution is approximately the same and isotropic. Two possible applications of the PTX-loaded NPs could be either their application on the bone cement before implantation or by injection over infected tissue (which is currently studied).| Sample | Paclitaxel loading ratioa (wt%) | Mean diameter ± S.D.b (nm) | Zeta potentialc (mV) | Encapsulation efficiencyd (%) | ΔHM ± 1e (J g−1) |
|---|---|---|---|---|---|
| a Theoretical weight percentage of paclitaxel. b Determined via dynamic light scattering. c Electrophoretic mobility determined using a Zetasizer. d Encapsulation efficiency determined by HPLC. e Melting enthalpy determined by differential scanning calorimetry. | |||||
| PPE-NP | 0 | 162 ± 38 | −47.6 ± 9.4 | — | 74.9 |
| PPE-NP-1 | 1.5 | 170 ± 33 | −44.3 ± 9.0 | 70 | 70.4 |
| PPE-NP-5 | 5 | 179 ± 73 | −49.0 ± 13.5 | 100 | 69.2 |
| PPE-NP-10 | 10 | 173 ± 98 | −48.7 ± 11.8 | 98 | 66.3 |
| PPE-NP-15 | 15 | 200 ± 100 | −29.0 ± 13.6 | 93 | 57.9 |
The zeta potential was measured after an exhaustive dialysis process. The zeta potential was, independent of the loading, in the range of −48 mV for all formulations, except for the formulation with the highest drug content (PPE-NP-15). The negatively charged stabilization field is related to the presence of sodium dodecyl sulfate (SDS) adsorbed at the surface of the particles, but could also be a result of partial hydrolysis of the phenyl ester (see above) of the PPE, generating negatively charged phosphate groups.
Drug encapsulation efficiency
An important factor for drug delivery vehicles is the encapsulation efficiency of the drug of interest. As the PPEs used herein are hydrophobic materials, PTX was chosen as a model drug with low water solubility in the range of 1 μg mL−1.52 For all formulations, encapsulation values between 70 and 100% were obtained (see Table 1), proving that the miniemulsion/solvent-evaporation technique is a robust technique that can be also applied for obtaining drug loaded PPE nanoparticles.The drug loading and encapsulation efficiency of the PPE-NPs is comparable with other published nanoparticle systems. Feng and coworkers, for example, reported on several polyester-based nanoparticles for PTX delivery, reaching drug loading up to 12% and encapsulation efficiencies over 90%.15,53,54 Only very recently, Wooley and coworkers presented in an elegant work a very high PTX loading (up to 65 wt%) by covalent linkage of PTX to a PPE-block copolymer for micellar delivery.55
Thermal characterization
DSC was used to evaluate the dispersion of the drug within the nanoparticles. Fig. 4 shows the DSC scans of the PPE nanoparticles synthesized in this study versus pure PTX. Homogeneous incorporation of PTX in the nanoparticles can be detected if no crystallization peak of the drug can be observed in the different formulations. The appearance of a melting point would suggest inhomogeneous incorporation or the formation of crystals within the nanoparticles. PTX, as received, is a crystalline powder, with a melting point around 220–240 °C (variation depends on the water content52). At temperatures above the melting point, PTX decomposes; hence only one heating ramp was performed. In our setup, the DSC scan for PTX showed a melting transition at 228 °C. For all PPE-containing samples (control nanoparticles, a mixture of control nanoparticles and PTX (20 wt%), and the PTX-loaded nanoparticles) a melting transition at ca. 50 °C can be detected. For all the formulations with PTX content up to 10 wt%, only the melting transition of the PPE can be detected indicating a homogeneous dispersion without agglomerates inside the nanoparticles. For the sample with 15 wt% PTX incorporated, however, a slight endothermic response in the region of the melting point of the drug was observed, similar to the physical mixture of the nanoparticles and PTX indicating phase separation, i.e. the formation of drug crystals, for loading higher than 10 wt%. Another important result that can be determined from these experiments is the change in the melting enthalpy with the increasing amount of PTX incorporated in the nanoparticles (see Table 1). The comparison of the melting enthalpies reveals a lowering in the melting enthalpy with increasing drug content. This behavior can be related to a lowering in the degree of crystallinity of the polymer by the inclusion of an ‘impurity’ in the polymer matrix, leading to defects during the crystallization. Once again the effect was more evident for the sample PPE-NP-15, supporting also the formation of separated PTX domains. Since the polymer has a low glass transition temperature (around −50 °C), the mechanical stability of the nanoparticles is directly related to the degree of crystallinity of the polymer. Loading of the nanoparticles with the drug leads to a lowering of the degree of crystallinity and the nanoparticles become softer, which possibly explains the increase in the particle size and size distribution due to partial agglomeration and particle fusion during the evaporation process, especially for the sample PPE-NP-15.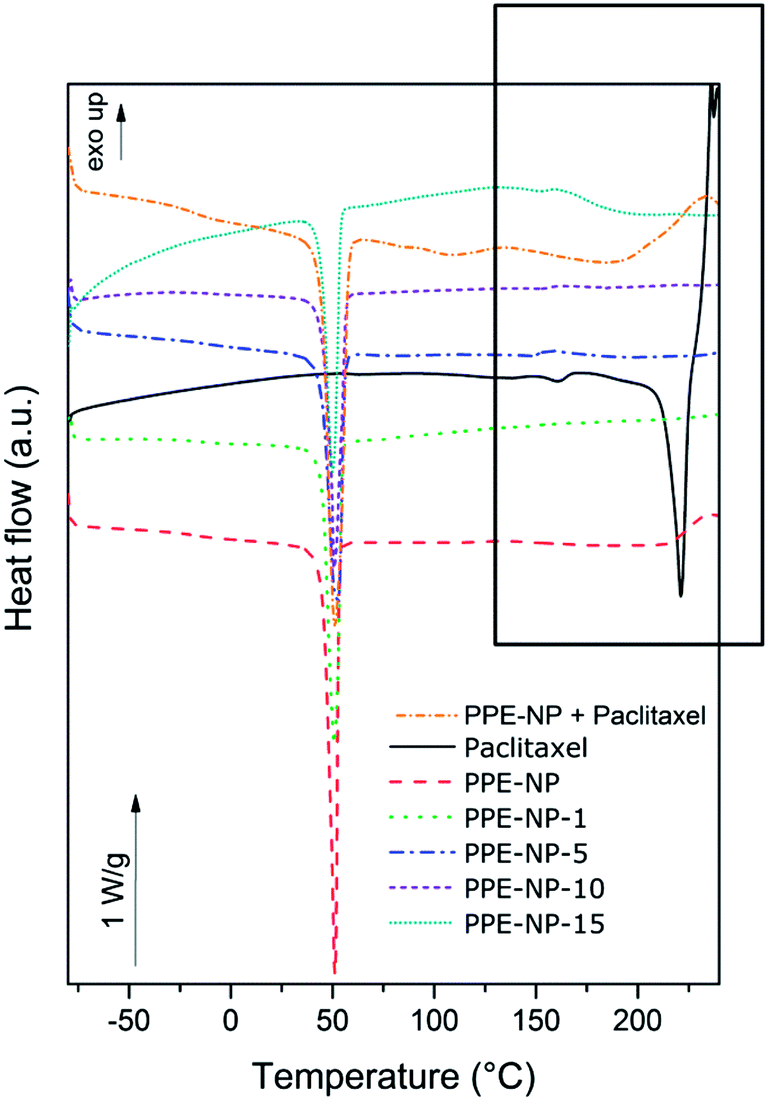 | ||
| Fig. 4 Differential scanning calorimetry obtained for PTX, the placebo nanoparticles NP-PPE, the mixture of paclitaxel and placebo nanoparticles and the formulations with up to 15 wt% paclitaxel. | ||
Effect of PPE-NPs and PPE-NPs loaded with paclitaxel on in vitro cell viability
For the investigation of the cytotoxicity of the PPE nanoparticles we first evaluated the effect without drug loading on the metabolic activity of HeLa cells. The cells were treated with 75 μg mL−1 to 600 μg mL−1 of nanoparticle dispersion for 24 h or 48 h and no cytotoxicity was detected as displayed in Fig. S21 & S26† (ESI) proving the biocompatibility under these conditions for the herein investigated PPEs.The cytotoxic effect of PTX-loaded PPE-NP-1 (1.5% wt) was analyzed on two different cancer cell lines (HeLa, Fig. 5A and Saos-2, Fig. 5B) and compared to PTX dissolved in DMSO and the chemotherapeutic compound Taxomedac®. The reference compounds, PTX/DMSO and Taxomedac, revealed generally a higher toxicity for osteoblastoma cells (Saos-2, EC50 ∼10 nM) for PTX when compared to the cervix cancer cells (HeLa, EC50 ∼25 nM) which is indicated by lower EC50 values (compare with Fig. 5A and B and S26†). The PPE-NPs with the low PTX loading (PPE-NP-1, 1.5 wt%) were less effective compared to the pure PTX/DMSO or Taxomedac which was reflected by an approximated EC50 value of 10–25 nM for Saos-2 cells (Fig. 5B) and >100 nM for the HeLa cells (Fig. 5A). The lower EC50 value of the drug loaded nanoparticles could probably be attributed to the loading efficiency (70% for the PPE-NP-1) or a different drug transfer mechanism.56 The actual drug availability is lower when PTX is encapsulated in comparison with the free drug, thus the EC50 depends in the first case also on the nanoparticle concentration (see ESI, Fig. S27†). Fig. 5C and D shows that the cytotoxic effect on Saos-2 cells can be improved by the application of PPE-NPs with increased compound loading. For example 1 μg mL−1 NP loaded with 10 wt% or 15 wt% of PTX showed a 75% reduction in cell viability, whereas the same concentration of nanoparticles loaded with 1 wt% PTX showed nearly no effect. Fig. 4 highlights this dependence of PPE-NP concentration, PTX loading and cytotoxicity for a fixed concentration of 30 nM PTX. The highest effect was obtained for 30 nM pure PTX/DMSO (0 μg mL−1 PPE-NP, 40% remaining cell viability), followed by the PPE-NPs with the highest PTX loading (15 wt%, 55% remaining cell viability) and further decreasing with lower PTX loading. These in vitro cell tests confirm that the drug can be loaded into the PPE nanoparticles and that it is still active, while a low load of PTX in wt% per polymer results in a lower effect. This would indicate that the release from a highly loaded nanocarrier is easier achieved compared to a lower loaded one. The delivery pathway is most likely a mixture of several ones. One delivery mechanism has been described recently which is especially attractive as it occurs rapidly for hydrophobic drugs at the cell membrane within minutes. As the nanoparticles only shortly touch and then detach, this mechanism has been termed “kiss-and-run” leaving behind the respective hydrophobic cargo.56 This appears to be reasonable when the supposed release from the nanoparticles may need release from the surface of the nanocarrier to a lipophilic surface like the cell membrane or an endosomal membrane. Therefore with a higher loading a higher concentration right at or below the surface would occur.
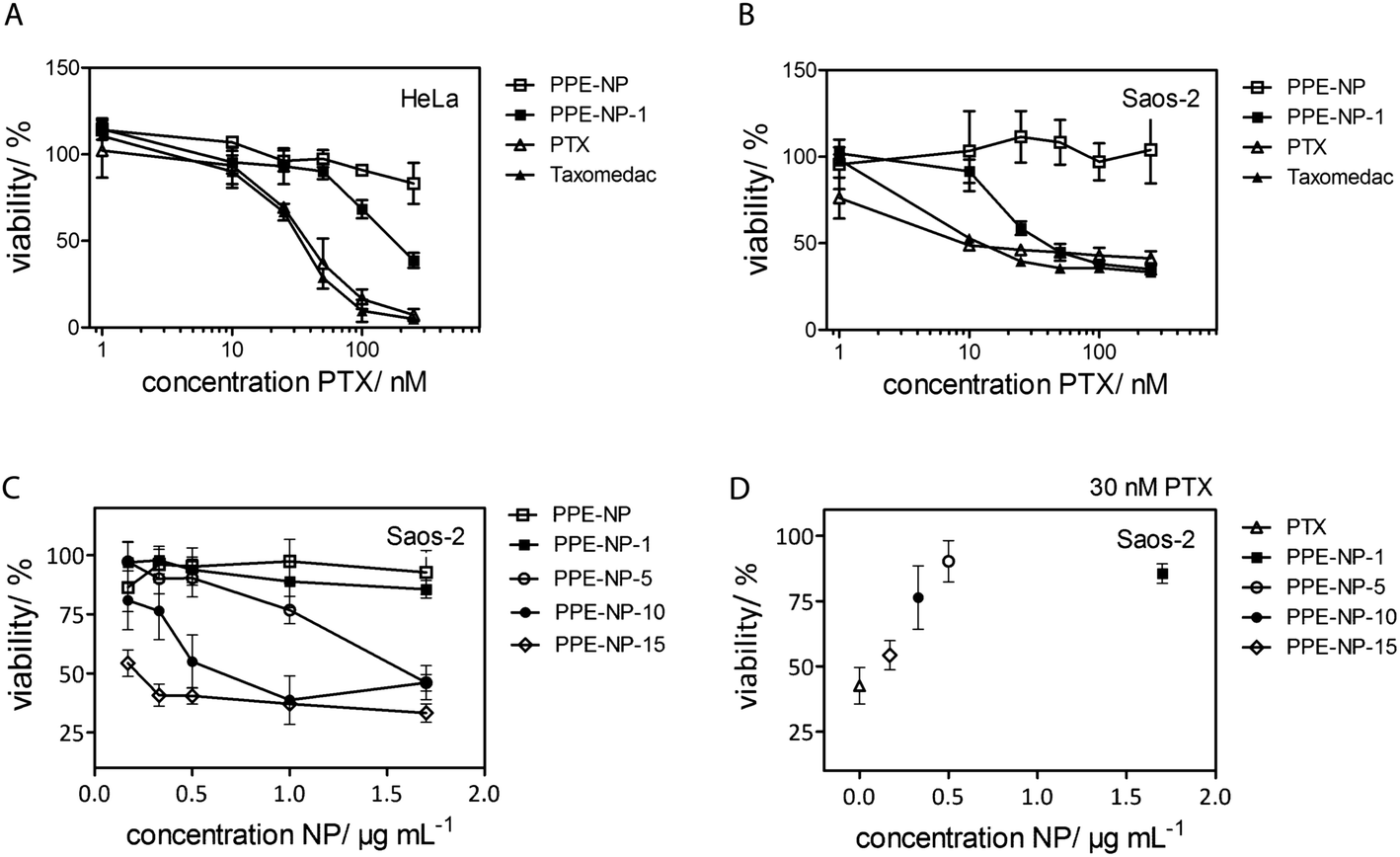 | ||
| Fig. 5 Effect of polyphosphate nanoparticles (PPE-NPs) and PPE-NPs loaded with paclitaxel (PTX) on in vitro cell viability. (A and B) Dose-response curves (1–250 nM) of paclitaxel loaded PPE-NP-1 (1.5 wt% PTX) in comparison to PPE-NP, PTX (0.1% DMSO) and Taxomedac in HeLa (A) and Saos-2 (B) cells. Cell viability was measured by PrestoBlue® staining after 48 h treatment. (C) Viability of Saos-2 cells depending on increased polymer concentration PPE-NP and PTX loading. PPE-NP-1, -5, -10, and -15 wt% correspond to 1.5, 5, 10, 15 wt% PTX loading. (D) Relation between cell viability, NP concentration and PTX loading for a fixed concentration of 30 nM PTX. Mean values ± standard deviation calculated from 4 replicates. | ||
Calcium phosphate attachment studies of drug-loaded nanoparticles
As a final proof that the PTX-loaded PPE-NP's are suitable for targeting bone tissue we investigated whether the drug loading affects the PPE-NP attachment to the calcium-phosphate cement.Fig. 6 shows the SEM pictures for the pure MBCP+ (Fig. 6A) and the granules after the attachment to the nanoparticles PPE-NP, PPE-NP-1 and PPE-NP-15 (Fig. 6B–D). From these images it can be clearly stated that the loaded PPE nanoparticles exhibit a high surface attachment with the calcium phosphate granules, even after vigorous washing steps. This proves that the phosphate groups are located at the surface of the PPE NPs and further indicates homogeneous drug dispersion within the nanoparticles. These attachment properties combined with the encapsulation efficiency and excellent toxicity data makes the herein presented PPE-NPs an ideal platform for further in vivo studies to generate a powerful anti-cancer drug delivery vehicle.
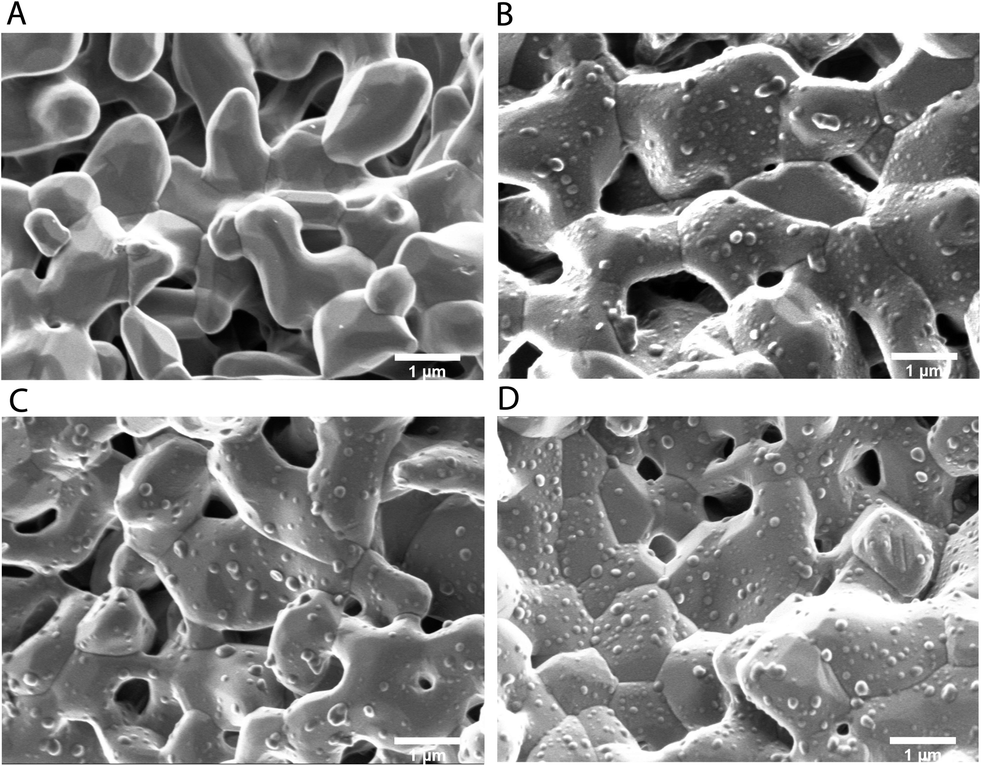 | ||
| Fig. 6 Scanning electron micrographs of the calcium phosphate attachment experiments: (A) MBCP+; (B) MBCP+/PPE-NP; (C) MBCP+/PPE-NP-1; (D) MBCP+/PPE-NP-15. Bar scales 1 μm. | ||
Conclusion
In conclusion we introduced a new class of nanoparticles based on potentially biodegradable and biocompatible polyphosphates with potential inherent bone adhesion abilities. The hydrophobic polyphosphates were prepared by using a metathesis protocol and the miniemulsion/solvent-evaporation method proportioned stable nanoparticle dispersions, with low size distribution and also an effective method for loading of a hydrophobic drug (up to 10 wt%). In vitro studies proved that the polyphosphate nanoparticles are non-toxic against HeLa or Saos-2 cells for concentrations up to 600 μg mL−1 or 300 μg mL−1, respectively, and that the polyphosphate nanoparticles loaded with paclitaxel exhibit a similar cytotoxicity to the commercially available Taxomedac® against osteosarcoma cells (Saos-2). The introduction of the hydrophobic drug did not influence the capacity of interaction of these nanoparticles with calcium phosphate, which creates the opportunity for the development of composite systems with bone cements that could also be applied for the delivery of paclitaxel. Therefore, the nanoparticles developed in this work showed a high potential for the development of systemic or local treatment. Further studies are in progress to understand the drug transfer mechanism, material degradation as well as in vivo validation are required to assess the potential for clinical application.Experimental section
Materials
The synthesis of the PPE (Fig. 1), with molecular weight Mwca. 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1, was previously described.43 The hydrogenation catalyst (Sigma-Aldrich 5% Pd/C) was used as received. Sodium dodecyl sulfate (SDS) was purchased from Sigma-Aldrich. Chloroform analytical grade used for nanoparticle synthesis was purchased from VWR and water was purified by reverse osmosis (Milli-Q, Millipore®). The BODIPY-dye was synthesized according to the reference method.57 Paclitaxel for research use was obtained from LC Laboratories and Taxomedac® (6 mg mL−1) was purchased from Medac (Wedel, Germany). A cellulose membrane from Carl Roth GmbH type 20/32 (molecular weight cut off 14000 g mol−1) was used for the dialysis purification process. The HPLC grade solvents THF, methanol and 0.1% trifluoroacetic aqueous solution were obtained from Sigma-Aldrich, VWR and Merck respectively. HCl solution (37%) was obtained from Sigma-Aldrich. A micro–macro porous biphasic calcium phosphate cement (MBCP+), composed of 80% hydroxyapatite and 20% β-tricalcium phosphate, was donated from Biomatlante (Vigneux de Bretagne, France).
000 g mol−1, was previously described.43 The hydrogenation catalyst (Sigma-Aldrich 5% Pd/C) was used as received. Sodium dodecyl sulfate (SDS) was purchased from Sigma-Aldrich. Chloroform analytical grade used for nanoparticle synthesis was purchased from VWR and water was purified by reverse osmosis (Milli-Q, Millipore®). The BODIPY-dye was synthesized according to the reference method.57 Paclitaxel for research use was obtained from LC Laboratories and Taxomedac® (6 mg mL−1) was purchased from Medac (Wedel, Germany). A cellulose membrane from Carl Roth GmbH type 20/32 (molecular weight cut off 14000 g mol−1) was used for the dialysis purification process. The HPLC grade solvents THF, methanol and 0.1% trifluoroacetic aqueous solution were obtained from Sigma-Aldrich, VWR and Merck respectively. HCl solution (37%) was obtained from Sigma-Aldrich. A micro–macro porous biphasic calcium phosphate cement (MBCP+), composed of 80% hydroxyapatite and 20% β-tricalcium phosphate, was donated from Biomatlante (Vigneux de Bretagne, France).
Nanoparticle preparation
PPE nanoparticles were synthesized using an adopted combination of the miniemulsion technique and the solvent-evaporation strategy.40 30 mg of the polymer were dissolved in 1.25 g of chloroform. To this solution, 0.05 mg of BODIPY or a given amount of paclitaxel was added. 5 g of Milli-Q water containing 10 mg of SDS were added to the chloroform solution and stirred over a period of 60 min for the formation of the pre-emulsion. Then, the pre-emulsion was submitted to a pulsed ultrasonication process in an ice bath during 120 s (30 s sonicated and 10 s paused) at 70% amplitude in a 1/4 tip Brason 450 W sonifier. The obtained miniemulsion was kept at 30 °C in an oil bath over a period of 8 h to completely evaporate the organic solvent. The obtained nanoparticle dispersion was further purified by exhaustive dialysis against water before being used for further studies.The average particle size and particle size distribution were obtained by dynamic light scattering (DLS) in a submicron particle sizer NICOMP® 380 equipped with a detector to measure the scattered light at 90°. The multi-angle dynamic light scattering measurement was performed in an ALV/CGS3 compact goniometer system with a He/Ne laser (632.8 nm), correlator ALV/LSE-5004 and evaluated with the ALV5000 software. The zeta-potentials of the solutions were obtained with a Zetasizer NanoZ using an aqueous 1 × 10−3 M KCl solution as the dispersive phase. The particle morphology was studied by scanning electron microscopy (SEM) using a Zeiss LEO Gemini 1530 microscope. Previous to the measurement a thin carbon coating layer was deposited using a vacuum coating system Leica EM MED020. Transmission electron microscopy (TEM) was performed in a JEOL 1400 microscope, after drop cast of the nanoparticle dispersion onto a carbon coated copper grid.
Thermal characterization
To investigate the crystallinity of the PPE nanoparticles and the influence of the dispersion of paclitaxel in the nanoparticles on the degree of crystallization, differential scanning calorimetry (DSC) of the samples was performed in a Perkin Elmer DSC 8500, with a single heating ramp from −80 °C to 240 °C, at a heating rate of 10 °C min−1. For the experiments, the nanoparticle samples were centrifuged at 18000 rpm at 4 °C over a period of 30 min, the supernatant was decanted, and the residue was lyophilized.
Encapsulation efficiency
The quantity of the encapsulated paclitaxel in the PPE nanoparticles was analyzed in triplicate on a HPLC (Hewlett Packard Series 1100). A reverse phase Spherisorb® ODS-2 column (250 × 4 mm i.d., pore size 5 μm, Lichrocart) was selected. A given amount of the nanoparticle dispersion was centrifuged at 18000 rpm at 4 °C over a period of 30 min, the supernatant was decanted, and the residue was lyophilized (this typically resulted in 1–3 mg of solid). The solid was dissolved in 1 mL of a mixture of THF:0.1% aqueous trifluoroacetic acid (70:30), ultrasonicated when necessary to help the dilution process, and filtered through a 0.45 μm PVDF filter before injection. A 10 μL injection was performed using an auto-sampler of the Agilent 1200 series. The mobile phase consisted of first an isocratic mixture of methanol:0.1% aqueous trifluoroacetic:THF (40:40:20) over a period of 8 min to elute the drug at 1 mL min−1 (its retention time under these conditions is approximately 6.6 min) and after a gradient until a pure THF mobile phase was performed in the next 5 min, followed by a 3 min isocratic flow of 100% THF for the elution of the polymer. Detection was accomplished with a UV-Vis detector (Soma S-3702) at a wavelength of 227 nm. To determine the amount of paclitaxel, the setup was calibrated with paclitaxel in the range of 50 ng mL−1 to 30000 ng mL−1. The encapsulation efficiency was determined as the ratio between the theoretical concentrations of paclitaxel expected for the starting amount of nanoparticles diluted to the concentration result obtained from the HPLC measurements, expressed in percentage.
Calcium phosphate attachment studies
The calcium phosphate particles (MBCP+, 80–200 μm, Biomatlante) were dispersed in ultrapure water (10 mg mL−1, Millipore) and washed for 30 min under horizontal agitation (200 rpm) before use. NP-PPE particles were dissolved in deionized water at an initial concentration of 1–3% and were diluted to application concentrations of 0.01% with deionized water in 1.5 mL Eppendorf centrifuge vials (1 mL nanoparticle solution volume). The calcium phosphate granules were left in the nanoparticle solution for 30 min immediately following deposition. The samples were placed on a shaker table at 180 rpm in order to expose all sides of the particles to the nanoparticles and stimulate attachment. After attachment the tubes were centrifuged at 1000 rpm for 5 min, the majority of the liquid was pipetted from the tube and removed, and the tube was refilled with deionized water and vortexed to remove the loose and weakly attached nanoparticles from the calcium-phosphate granules. This process of centrifuging, removing the liquid, replacing the liquid with fresh deionized water, and vortexing was repeated two additional times. After the three rinses were complete, the samples were centrifuged again and stored dry or in water before observation in the scanning electron microscope (SEM, Zeiss LEO Gemini 1530). Previous to the measurement a thin carbon coating layer was deposited using a vacuum coating system Balzer Union (BAE250).Cell viability assays
Human cervix carcinoma cells, HeLa cells (#ACC57, DMSZ, Germany), were cultured in DMEM medium (Invitrogen) and human osteosarcoma cells, SaOS-2 (#ACC 243, DSMZ, Germany) were cultured in RPMI (Invitrogen) medium in a humidified incubator at 37 °C/5% CO2. Both media were supplemented with 10% fetal calf serum (FCS, Gibco), 100 units penicillin and 100 mg mL−1 streptomycin (Life Technologies).The effect of PTX (10 mM stock solution in DMSO), NP-PPE, and Taxomedac® (6 mg mL−1 containing 527 mg mL−1 macrogolglycerol ricinoleate and 395 mg mL−1 ethanol) on cell viability was measured by CellTiter-Glo or PrestoBlue™ staining (Invitrogen) according to the manufacturer's protocol. Briefly, HeLa or Saos-2 cells (1.5 × 104 cells per well) were diluted in the indicated cell culture medium and seeded in 96 well-plates (black plate, clear bottom, corning, Amsterdam, Netherlands). The culture medium was replaced after ∼16 h by a compound supplemented medium (200 μL, DMEM, 10% FCS, 0.1% DMSO) or a medium without compound (DMEM, 10% FCS, 0.1% DMSO) as a specific control. After 24 h and 48 h, viable cells were stained with 20 μL PrestoBlue reagent per well and incubated for 20 min at 37 °C/5% CO2. Metabolically active cells reduce the cell permeable dye resazurin into fluorescent resorufin, which was measured with a fluorescence plate reader (excitation wavelength 560 nm, emission wavelength 590 nm, Tecan Infinite M1000, Austria).
Author contributions
The manuscript was written through contributions from all authors. All authors have approved the final version of the manuscript.Acknowledgements
E. M. A. thanks the International Max Planck Research School (IMPRS) for a fellowship. F. M. is a recipient of a fellowship through funding of the Excellence Initiative (DFG/GSC 266) in the context of the graduate school of excellence “MAINZ” (Materials Science in Mainz). F. R. W. thanks the Max Planck Graduate Center (MPGC) for support. We gratefully acknowledge the EU for financing. This study is supported by the 7th EU Framework program REBORNE (HEALTH-2009-1.4-2). The authors thank Dr Andrey Turshatov for experimental support and Melanie Stichelberger, Beate Stradmann-Bellinghausen and Carolin Hogl for skillful technical assistance. The authors thank Guy Daculsi (BIOMATLANE, France) for the kind donation of MBCP+.References
- V. L. Keedy, OncoTargets Ther., 2012, 5, 153–159 CrossRef PubMed.
- J. T. Buijs and G. van der Pluijm, Cancer Lett., 2009, 273, 177–193 CrossRef CAS PubMed.
- G. A. Clines and T. A. Guise, Expert Rev. Mol. Med., 2008, 10, 1–16 CrossRef PubMed.
- D. R. Clohisy and P. W. Mantyh, Cancer, 2003, 97, 866–873 CrossRef PubMed.
- M. A. Lopez-Heredia, G. J. B. Kamphuis, P. C. Thune, E. C. Oner, J. A. Jansen and X. F. Walboomers, Biomaterials, 2011, 32, 5411–5416 CrossRef CAS PubMed.
- T. Tani, K. Okada, S. Takahashi, N. Suzuki, Y. Shimada and E. Itoi, In Vivo, 2006, 20, 55–60 CAS.
- A. Spina, L. Sorvillo, F. Di Maiolo, A. Esposito, R. D'Auria, D. Di Gesto, E. Chiosi and S. Naviglio, J. Cell. Physiol., 2013, 228, 198–206 CrossRef CAS PubMed.
- S. Magnetto, S. Boissier, P. D. Delmas and P. Clezardin, Int. J. Cancer, 1999, 83, 263–269 CrossRef CAS.
- T. H. Wang, H. S. Wang and Y. K. Soong, Cancer, 2000, 88, 2619–2628 CrossRef CAS.
- C. Luo, Y. Wang, Q. Chen, X. Han, X. Liu, J. Sun and Z. He, Mini-Rev. Med. Chem., 2012, 12, 434–444 CrossRef CAS.
- H. Gelderblom, J. Verweij, K. Nooter and A. Sparreboom, Eur. J. Cancer, 2001, 37, 1590–1598 CrossRef CAS.
- A. Jager, D. Gromadzki, E. Jager, F. C. Giacomelli, A. Kozlowska, L. Kobera, J. Brus, B. Rihova, M. El Fray, K. Ulbrich and P. Stepanek, Soft Matter, 2012, 8, 4343–4354 RSC.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jerome, J. Marchand-Brynaert, O. Feron and V. Preat, J. Controlled Release, 2009, 133, 11–17 CrossRef CAS PubMed.
- C. Fonseca, S. Simoes and R. Gaspar, J. Controlled Release, 2002, 83, 273–286 CrossRef CAS.
- L. Mu and S. S. Feng, Pharm. Res., 2003, 20, 1864–1872 CrossRef CAS.
- X. Jiang, H. Xin, J. Gu, X. Xu, W. Xia, S. Chen, Y. Xie, L. Chen, Y. Chen, X. Sha and X. Fang, Biomaterials, 2013, 34, 1739–1746 CrossRef CAS PubMed.
- S. S. Feng, L. Mu, K. Y. Win and G. F. Huang, Curr. Med. Chem., 2004, 11, 413–424 CrossRef CAS.
- P. Lopez-Gasco, I. Iglesias, J. Benedi, R. Lozano, J. M. Teijon and M. D. Blanco, J. Microencapsulation, 2011, 28, 417–429 CrossRef CAS PubMed.
- L. Brannon-Peppas and J. O. Blanchette, Adv. Drug Delivery Rev., 2004, 56, 1649–1659 CrossRef CAS PubMed.
- F. C. J. van de Watering, P. Laverman, V. M. Cuijpers, M. Gotthardt, E. M. Bronkhorst, O. C. Boerman, J. A. Jansen and J. van den Beucken, Biomed. Mater., 2013, 8, 035012 CrossRef PubMed.
- M. Salerno, E. Cenni, C. Fotia, S. Avnet, D. Granchi, F. Castelli, D. Micieli, R. Pignatello, M. Capulli, N. Rucci, A. Angelucci, A. Del Fattore, A. Teti, N. Zini, A. Giunti and N. Baldini, Curr. Cancer Drug Targets, 2010, 10, 649–659 CrossRef CAS.
- S. W. Choi and J. H. Kim, J. Controlled Release, 2007, 122, 24–30 CrossRef CAS PubMed.
- I. Ozcan, K. Bouchemal, F. Segura-Sanchez, O. Ozer, T. Guneri and G. Ponchel, J. Pharm. Sci., 2011, 100, 4877–4887 CrossRef CAS PubMed.
- D. Wang, S. Miller, M. Sima, P. Kopeckova and J. Kopecek, Bioconjugate Chem., 2003, 14, 853–859 CrossRef CAS PubMed.
- S. Zhang, J. E. Wright, N. Ozber and H. Uludag, Macromol. Biosci., 2007, 7, 656–670 CrossRef CAS PubMed.
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed.
- J. N. Yewle, D. A. Puleo and L. G. Bachas, Bioconjugate Chem., 2011, 22, 2496–2506 CrossRef CAS PubMed.
- K. Aoki, N. Alles, N. Soysa and K. Ohya, Adv. Drug Delivery Rev., 2012, 64, 1220–1238 CrossRef CAS PubMed.
- A. Tautzenberger, A. Kovtun and A. Ignatius, Int. J. Nanomed., 2012, 7, 4545–4557 CrossRef CAS PubMed.
- E. Cenni, S. Avnet, D. Granchi, C. Fotia, M. Salerno, D. Micieli, M. G. Sarpietro, R. Pignatello, F. Castelli and N. Baldini, J. Biomater. Sci., Polym. Ed., 2012, 23, 1285–1300 CAS.
- P. Anderson, L. Kopp, N. Anderson, K. Cornelius, C. Herzog, D. Hughes and W. Htth, Expert Opin. Invest. Drugs, 2008, 17, 1703–1715 CrossRef CAS PubMed.
- A. Bamias, E. Kastritis, C. Bamia, L. A. Moulopoulos, L. Melakopoulos, G. Bozas, V. Koutsoukou, D. Gika, A. Anagnostopoulos, C. Papadimitriou, E. Terpos and M. A. Dimopoulos, J. Clin. Oncol., 2005, 23, 8580–8587 CrossRef PubMed.
- S. Zhang, H. Wang, Y. Shen, F. Zhang, K. Seetho, J. Zou, J.-S. A. Taylor, A. P. Dove and K. L. Wooley, Macromolecules, 2013, 46, 5141–5149 CrossRef CAS PubMed.
- S. Y. Zhang, A. Li, J. Zou, L. Y. Lin and K. L. Wooley, ACS Macro Lett., 2012, 1, 328–333 CrossRef CAS PubMed.
- Y. C. Wang, Y. Y. Yuan, J. Z. Du, X. Z. Yang and J. Wang, Macromol. Biosci., 2009, 9, 1154–1164 CrossRef CAS PubMed.
- Q. Li, J. Wang, S. Shahani, D. D. N. Sun, B. Sharma, J. H. Elisseeff and K. W. Leong, Biomaterials, 2006, 27, 1027–1034 CrossRef CAS PubMed.
- J. J. Qiu, Z. X. He, C. M. Liu, X. D. Guo and Q. X. Zheng, Biomed. Mater., 2008, 3, 044107 CrossRef PubMed.
- Z. X. Zhang, X. L. Feng, J. Mao, J. Z. Xiao, C. M. Liu and J. J. Qiu, Biochem. Biophys. Res. Commun., 2009, 379, 557–561 CrossRef CAS PubMed.
- R. Ikeuchi and Y. Iwasaki, J. Biomed. Mater. Res., Part A, 2013, 101, 318–325 CrossRef PubMed.
- A. Musyanovych, J. Schmitz-Wienke, V. Mailander, P. Walther and K. Landfester, Macromol. Biosci., 2008, 8, 127–139 CrossRef CAS PubMed.
- H. Mutlu, L. M. de Espinosa and M. A. R. Meier, Chem. Soc. Rev., 2011, 40, 1404–1445 RSC.
- T. Steinbach, E. Alexandrino and F. Wurm, Polym. Chem., 2013, 4, 3800–3806 RSC.
- F. Marsico, M. Wagner, K. Landfester and F. R. Wurm, Macromolecules, 2012, 45, 8511–8518 CrossRef CAS.
- M. P. Ginebra, C. Canal, M. Espanol, D. Pastorino and E. B. Montufar, Adv. Drug Delivery Rev., 2012, 64, 1090–1110 CrossRef CAS PubMed.
- G. Daculsi, F. Jegoux and P. Layrolle, in Advanced Biomaterials, John Wiley & Sons, Inc., 2010, pp. 101–141 Search PubMed.
- O. Gauthier, E. Goyenvalle, J. M. Bouler, J. Guicheux, P. Pilet, P. Weiss and G. Daculsi, J. Mater. Sci.: Mater. Med., 2001, 12, 385–390 CrossRef CAS.
- O. Gauthier, J. M. Bouler, E. Aguado, P. Pilet and G. Daculsi, Biomaterials, 1998, 19, 133–139 CrossRef CAS.
- J. Baran and S. Penczek, Macromolecules, 1995, 28, 5167–5176 CrossRef CAS.
- G. Baier, A. Cavallaro, K. Friedemann, B. Müller, G. Glasser, K. Vasilev and K. Landfester, Nanomedicine: Nanotechnology, Biology and Medicine, 2014, 10, 131–139 CrossRef CAS PubMed.
- G. Baier, C. Costa, A. Zeller, D. Baumann, C. Sayer, P. H. H. Araujo, V. Mailänder, A. Musyanovych and K. Landfester, Macromol. Biosci., 2011, 11, 628–638 CrossRef CAS PubMed.
- R. Sauer, P. Froimowicz, K. Scholler, J. M. Cramer, S. Ritz, V. Mailander and K. Landfester, Chem. – Eur. J., 2012, 18, 5201–5212 CrossRef CAS PubMed.
- R. T. Liggins, W. L. Hunter and H. M. Burt, J. Pharm. Sci., 1997, 86, 1458–1463 CrossRef CAS PubMed.
- Z. P. Zhang, L. Mei and S. S. Feng, Expert Opin. Drug Delivery, 2013, 10, 325–340 CrossRef CAS PubMed.
- L. Mu and S. S. Feng, J. Controlled Release, 2003, 86, 33–48 CrossRef CAS.
- S. Zhang, J. Zou, M. Elsabahy, A. Karwa, D. A. Moore, R. B. Dorshow and K. Wooley, Chem. Sci., 2013, 4, 2122–2126 RSC.
- D. Hofmann, C. Messerschmidt, M. Bannwarth, K. Landfester and V. Mailander, Chem. Commun., 2014, 50, 1369–1371 RSC.
- R. W. Wagner and J. S. Lindsey, Pure Appl. Chem., 1996, 68, 1373–1380 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3tb21295e |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |