
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Multiple linker half-squarylium dyes for dye-sensitized solar cells; are two linkers better than one?†
Arthur
Connell
a,
Peter J.
Holliman
*a,
Eurig W.
Jones
a,
Leo
Furnell
a,
Christopher
Kershaw
a,
Matthew L.
Davies
a,
Christopher D.
Gwenin
a,
Mateusz B.
Pitak
b,
Simon J.
Coles
b and
Graeme
Cooke
c
aSchool of Chemistry, Bangor University, Gwynedd LL57 2UW, UK. E-mail: p.j.holliman@bangor.ac.uk; Fax: +44 (0)1248 370528; Tel: +44 (0)1248 382375
bUK National Crystallography Service, Chemistry, Faculty of Natural and Environmental Sciences, University of Southampton, Highfield Campus, Southampton, SO17 1BJ, UK
cWestCHEM, School of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK
First published on 16th December 2014
Abstract
The synthesis and full characterization of new half-squaraine dyes (Hf-SQ) containing two or three carboxylate-based linker units is reported and these dyes tested in dye-sensitized solar cell (DSC) devices. The data show improved device efficiency for a Hf-SQ dye with two linkers (η = 5.5%) compared to the highest efficiency Hf-SQ previously reported which had only a single linker (η = 5.0%); this is mainly due to improved Voc. To understand the effects of using multiple dye linker groups, device I–V data have been correlated with single crystal X-ray structural analysis and dye electrical properties (both in solution and adsorbed to TiO2) using UV-visible and ATR-IR spectroscopy along with cyclic voltammetry, and also theoretical studies using density functional theory (DFT) calculations. These data show that positioning the linkers near the dye LUMO and so that this enables complete linker chemisorption are key factors for device performance.
Dye-sensitized solar cells (DSCs) are a promising form of 3rd generation photovoltaic technology. O'Regan and Grätzel's breakthrough in DSC devices used Ru bipyridyl dye N3 to sensitize nanoparticulate TiO2.1 Since 1991, Ru-bipy dyes have remained a widely used family of DSC dyes.2 However, these dyes are expensive which is partly due to raw material costs but also due to lengthy purification procedures which hinder scaling. They also have relatively low molar extinction coefficients (ε) and generally poor spectral response above 600 nm leading to DSC efficiencies of 11.1% for N719 (ref. 3) and 11.4% for C101.4 Modification of the organic ligands to enhance the extinction coefficient and broaden the spectral response of Ru based dyes has been reported in the literature.5–8 In addition, replacement of the thiocyanate ligands has been reported which can lead to improved dye properties. These improvements include extending the spectral response and enhancing the extinction coefficient8 as well as increasing open circuit voltage and greater stability.9–12 Furthermore, modification of the bipyridyl ligands has been reported to improve the hydrophobicity of Ru dyes.13 Finally, it has been reported that the purity of N3 and N719 has a profound influence on their performance as sensitizers in DSC devices.14 This has led to the development of modified bipyridyl ligands which have been used to prepare sensitizers which do not require such extensive purification during the dye synthesis.15 Whilst the aforementioned modifications of the organic ligands enable the properties of Ru dyes to be tuned, these dyes are still an expensive component of DSC devices because ruthenium is an expensive raw material, the synthetic procedures are multi-step and dye purification is complicated.
These issues have led to the development of “Ru-free” organic dyes (e.g. triarylamines,16–18 coumarins19 indolines,20,21 quinoxalines22 and natural dyes23,24). In general, all of these dyes have been designed with a donor–pi linker–acceptor (D–π–A) structural arrangement25 to maximize electron injection efficiency. These organic dyes often absorb in the same region as Ru-bipy dyes (450–600 nm) where AM1.5 solar intensity is highest but are generally simpler to purify than Ru-bipy complexes and have significantly higher ε. This allows thinner photo-electrodes to be used which can reduce recombination losses and improve Voc. High ε is also advantageous when co-sensitizing the TiO2 electrode to broaden spectral response because, if fewer dye sorption sites are utilized for the dye harvesting light at 400–650 nm, this leaves more space for near infrared (NIR) dyes which absorb at λ > 650 nm.26 Furthermore, organic dyes have surpassed Ru dyes with the highest η liquid DSC reported (η > 12%) for a combination of porphyrin and triphenylamine dyes27 or a push–pull porphyrin alone.28
A relatively new organic chromophore is the half-squarylium (Hf-SQ) dye. These dyes are both synthetically versatile and are also used as an intermediate during the synthesis of unsymmetrical squaraine dyes.29,30 Hf-SQ dyes are known to be fluorescent31 and early reports for ZnO-based DSC devices gave η = 0.27% (ref. 32) and 0.53% (ref. 33) with η = 3.54% (ref. 34) for TiO2 photo-electrodes. Recently, we have reported the highest efficiency Hf-SQ dyes to date (η = 5.0%).35 In this report, the highest Jsc was recorded for a Hf-SQ dye with a vinyl dicyano-modified squaric acid unit which desorbed when electrolyte was added so that the resulting device gave poor Voc and FF. In this paper, we report a study of multiple carboxylate linkers for Hf-SQ dyes and the effects these linkers have on DSC device performance when two or three linker groups positioned around the periphery of the Hf-SQ chromophore.
Results and discussion
Rationale for dye design
The series of dyes (4), (5), (7), (8), (10) and (11) have been synthesized following our recent report of the highest efficiency Hf-SQ DSC device (η = 5.0%).35 In the previous report, we studied the influence of the linker position on device performance. Interestingly, the highest Jsc was observed for a dye with vinyl dicyano-modified squaric acid unit; labelled compound (8) in paper;35 here this dye will be labelled (8′) (Fig. 1). However, the overall device efficiency was limited because (8′) appeared to desorb when electrolyte was added resulting in poor Voc and FF. Here, we have studied the desorption process further by pumping electrolyte through the cavity of DSC device dyed with (8′) and the data show that dye is desorbed by this (Fig. 2). The approach tested in this paper has been to synthesise Hf-SQ dyes which are related to the highly efficient dyes in35 but with more than one carboxylate linker to test if this enables the dyes to adsorb more strongly to TiO2 and to study how this affects device performance. For example, (4) is an analogous dye to (8′) but with a second carboxylate linker attached to the N of the indole. To test the effect of having 2 linkers on dye desorption, a TiO2 electrode was dyed with (4) and then electrolyte was pumped through the device cavity (Fig. 2b). This shows that dye desorption does not occur for (4). Instead the dye remains bonded to the surface and no discolouration is observed. By comparison, (5) is similar to (4) but without vinyl dicyano modification of the squaric acid unit. Hence, for both (4) and (5) the two linker groups are at the same end of the dye. To compare with this, dye (8) was designed with carboxylate linkers at opposite ends of the molecule and (11) with 3 carboxylate linkers; on the N atom and benzene of the indole and through the squaric acid unit. Dyes (7) and (10) are related to (8) and (11), respectively but without vinyl dicyano modification of the squaric acid unit.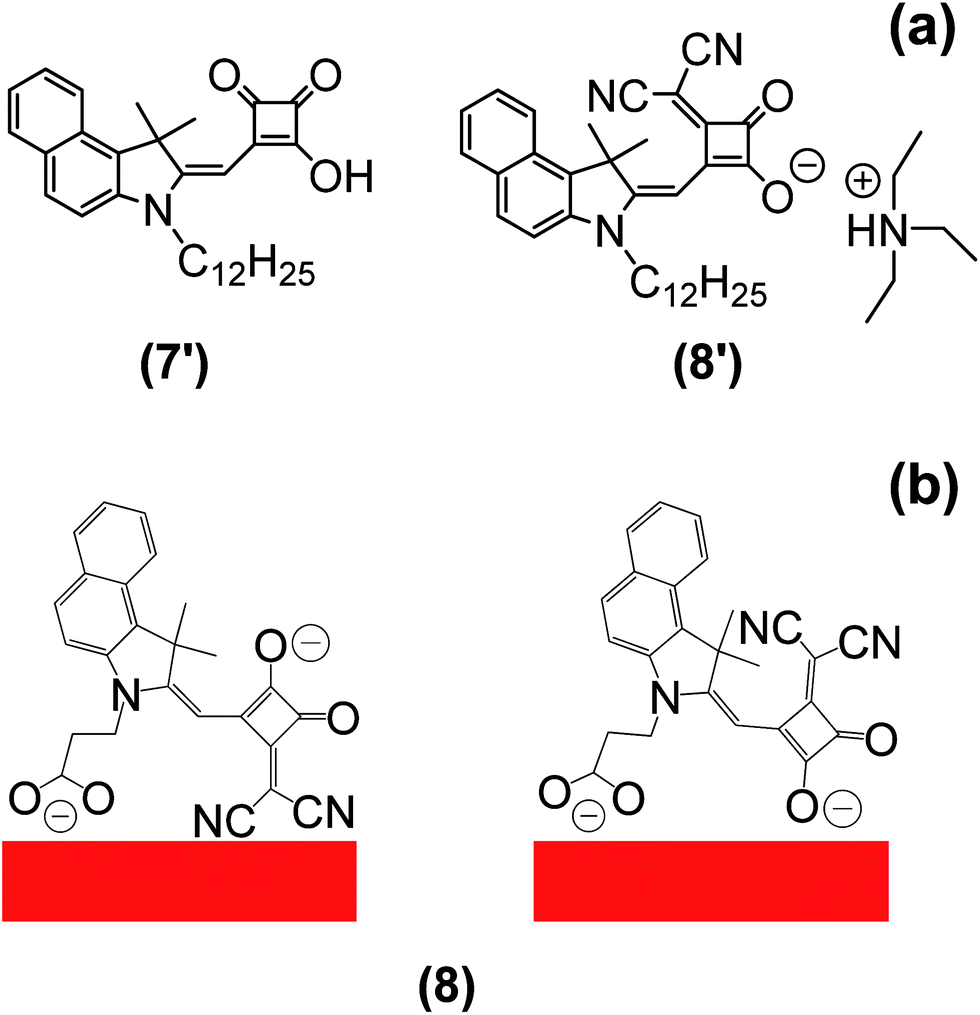 | ||
| Fig. 1 (a) Molecular structures of (7′) and (8′) from35 and (b) possible binding modes of (8) from this work. | ||
 | ||
| Fig. 2 (a) Desorption of (8′) from35 during acetonitrile electrolyte infusion showing (top) dyed film, (middle) during and (bottom) after electrolyte addition and (b) the equivalent data for (4) from this paper. | ||
Dye synthesis
The synthetic routes used to produce the Hf-SQ used in this work are shown in Scheme 1(a–c). The first step is the esterification of squaric acid in ethanol to give (1) which was prepared according to the method described by Terpetschnig et al. in good yield with spectroscopic data in line with the literature.36 Compounds (2) and (3) have been reported in our previous work.35 To the best of our knowledge (4–11) have not been reported in the literature and were identified using several analytical techniques. The resonances identified in the 1H NMR spectrum suggest that (4) has been isolated as the triethylamine salt. Six protons are identified in the aromatic region between 8.10 and 7.41 ppm with multiplicities including a doublet and multiplets. Resonances at 7.32, 4.25, 2.80 and 1.93 ppm have the correct multiplicities and integration to be identified as the methylene proton located near the squaric acid moiety, the ethyl group attached to the nitrogen of the indole and the methyl groups located at the 3 position of the indole unit, respectively. The quintet and triplet at 3.24 and 1.33 ppm, which integrate to six and nine protons respectively, are caused by the ethyl group of the triethylamine cation. Furthermore, the coupling constants calculated for the protons in each environment are similar to those reported for the previous half squaraine dyes.3513C NMR has a resonance at 112 ppm which is associated with the nitrile functional group. In addition, resonances for the carbonyl group of the squaric acid and carboxylic acid moiety are also identified between 191 and 173 ppm. The ethyl group of the triethylamine cation can be identified between 38.39 and 7.93 ppm. Analysis using high resolution mass spectrometry identifies the M+ ion at 424.1294 which corresponds to the target molecule in the non-salted form. In addition, an isotope profile is detected which fits the theoretical isotope profile expected for this molecule. Attenuated total reflectance infrared (ATR-IR) spectroscopy has been used to identify functional groups in the molecule including the nitrile and carbonyl groups at 2178, 1732 (s), 1626 (s), 1595 (s), 1537 (s), 1517 (s) cm−1, respectively. (4) was isolated in 75% yield and has a melting point between 175–179 °C, which is similar to a vinyl dicyano half squaraine reported in our previous work.35 Finally, the structure of (4) was confirmed from single crystal X-ray crystallography and is show in Fig. 3. The crystal structure of (4) shows the expected molecular configuration with a propionic acid linker attached to the indole N and a second carboxylate linker being the squaric acid moiety. The crystal structure also confirms that the squaric acid unit has been modified by the addition of a vinyl dicyano group but interestingly that this group lies on the same side of the molecule to the propionic acid linker. Finally, this crystal structure also shows the presence of a triethylamine counterion as suggested by the NMR data. As expected, this is located closest to the oxygen atoms of the modified squaric acid unit where the carboxylate anionic charge is found.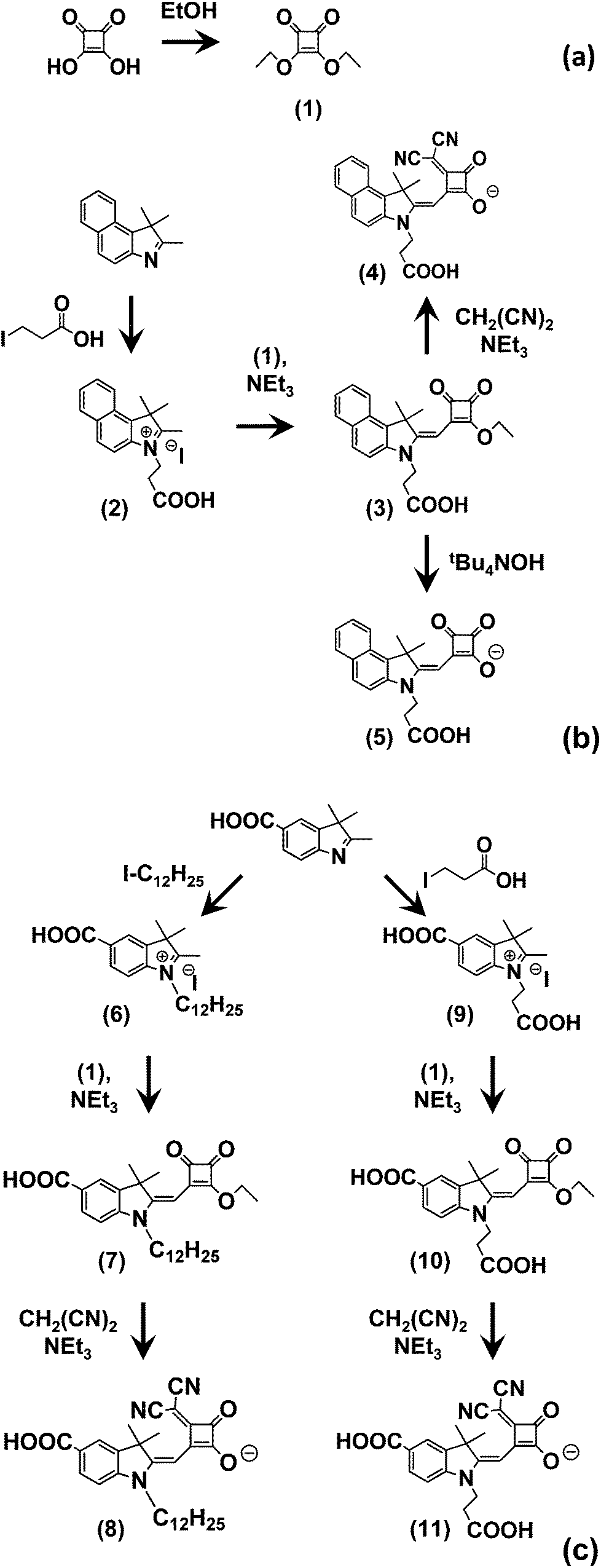 | ||
| Scheme 1 Synthetic pathways to multiple linker half-squaraine dyes. | ||
 | ||
| Fig. 3 X-ray crystal structures of (a) (4), (b) (5), (c) (7) and (d) (8) (polymorph 1). Displacement ellipsoids – 50% probability. For clarity counteranions as well as molecular disorder components have been omitted for (4), (5) and (8) (polymorph 1). Asymmetric part of the unit cell of (7) contains two independent molecules (Z′ = 2). Only one has been shown. | ||
Compounds (5–11) were isolated in good yield and analysed using the same techniques used to identify (4). Focussing on the major differences between these molecules, (5) is essentially the same as (4) but without a vinyl dicyano group on the squaric acid unit. The 13C NMR and ATR data confirm the absence of nitrile groups in (5) through the absence of signals at 112 ppm and 2178 cm−1, respectively. The other NMR and ATR data are similar to (4) as expected and mass spectrometry also confirms (5) with M+ at 270.1126. Single crystal X-ray crystallography also shows the expected molecular structure of (5) and it does mirror the structure of (4) as expected but without the vinyl dicyano modification to the squaric acid group.
The synthesis of (8) begins with alkylation of a carboxylate indole to produce (6) which is confirmed by mass spectrometry (M+ = 372.887). This was converted to the half squaraine dye (7) by reaction with (1) as evidenced by the additional peaks between 0.95 and 1.71 ppm for the alkyl chains on the indole nitrogen and in the 1H NMR and 3 signals for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O between 185 and 190 ppm. (7) was also confirmed by mass spectrometry (M+ = 494.2929). Single crystal X-ray crystallography (Fig. 3) verifies the molecular structure of (7) with a carboxylate linker attached to the benzene ring of the indole and an ester group attached to the squaraine moiety. In the context of this study, this is important because it means that (7) can only link to TiO2 through a single carboxylate linker.
O between 185 and 190 ppm. (7) was also confirmed by mass spectrometry (M+ = 494.2929). Single crystal X-ray crystallography (Fig. 3) verifies the molecular structure of (7) with a carboxylate linker attached to the benzene ring of the indole and an ester group attached to the squaraine moiety. In the context of this study, this is important because it means that (7) can only link to TiO2 through a single carboxylate linker.
The squaraine ester of (7) is then de-esterified and a vinyl dicyano group added by reaction with CH2(CN)2 to produce (8). This is confirmed by additional signals for nitrile at 117–118 ppm in the 13C NMR and at 2182 cm−1 in the infrared data. Single crystal X-ray crystallography again proves the expected structure (Fig. 3d). Importantly, the data show the presence of two potential linker groups for this dye. These are the carboxylate linker on the benzene ring of the indole and the vinyl dicyano-modified squaric acid unit. These are oriented on one side of the molecule in the solid state which suggests it might be possible for both linkers to adsorb to TiO2 at the same time. Interestingly, we have found that (8) also crystallises into a second polymorph; (8) (polymorph 2) (see ESI†). This varies from (8) (polymorph 1) shown in Fig. 3 mainly through the H-bonding which influences the both the crystal packing and the relative positions of the Hf-SQ anions and triethylamine cations.
In the first step towards the synthesis of (11), a propionic group is first added to the N atom of a carboxy indole to make (9) which is confirmed by mass spectrometry (M+ = 276.1227). This is then converted to the half-squaraine (10) which is evidenced by the 1H NMR signals at between 3.2 and 4.4 ppm for the C–H of the propionic acid and squaraine ester groups, the CO signals at 190–200 ppm in the 13C NMR and the mass spectrometry (M+ = 398.1235). This molecular structure of (10) is important for this study because the molecule contains two potential carboxylate linkers attached to the benzene ring and the N atom of the indole, respectively. The final dye (11) is produced from (10) by simultaneous modification and de-esterification of the squaraine ester to produce the vinyl dicyano modified half-squaraine. The production of (11) is confirmed by the additional nitrile signals in the 13C NMR between 117–119 ppm and in the IR at 2198 cm−1 and the mass spectrometry (M+ = 418.1036). This molecule is important for this study because it contains 3 potential carboxylate linkers; the squaric acid moiety in addition to carboxylates on the benzene ring and N atom of the indole.
Spectroscopic analysis
Fig. 4 shows UV-vis spectra of dyes in solution and after sorption onto TiO2 photo-electrodes. The spectra in solution (Fig. 4a) show that modification of the central squaric acid moiety by replacing a carbonyl group with a vinyl dicyano group causes red shifts in the spectra of (4), (8) and (11) by comparison to the unmodified dyes (5), (7) and (10), respectively. In addition, the highest molar extinction coefficient (ε) is observed for the esterified squaraine (7) with generally higher ε for vinyl dicyano modified dyes apart from (11) which has the lowest ε of all the dyes synthesized. The spectra obtained for (4) and (5) are similar to dyes (7b′) and (8′) reported in our previous work.35 This is interesting because the main structural difference associated with the new dyes reported in this paper is the addition of a propionic acid linker group to the N of the indole. This suggests that this new group plays little role in the HOMO or LUMO of these dyes in solution. In turn, this suggests that the main role of these additional groups is predominantly as linkers onto TiO2. | ||
| Fig. 4 UV-vis spectra of selected dyes (a) in solution (1 μM) and (b) adsorbed to TiO2 films; (4) solid line, (5) dotted line, (7) long dashes, (8) open circles, (10) crosses, (11) filled circles. | ||
Fig. 4b shows the transmission UV-vis data when the dyes are adsorbed onto transparent, mesoporous titania films. The data show that the absorption is broadened as might be expected for molecules which are chemisorbed on a surface. Interestingly, as for the solution data, the spectra for adsorbed (4) and (5) are similar to (7b′) and (8′) reported previously.35 This supports the notion that, because the additional propionic linkers of (4) and (5) are attached to the indole N atom, they only act as additional linkers to the TiO2 surface and that they do not contribute to the HOMO or LUMO of the adsorbed dyes. This is important because (7b′) from35 can only chemisorb to TiO2 through covalent ester bonds between the squaric acid moiety and surface hydroxyls whilst the adsorption mode of and (8′) is less clear. By comparison, (4) and (5) can chemisorb both through the squaric acid and/or from the propionic acid linker. Fig. 2 illustrates our observations that (5) adsorbs much more strongly to TiO2 than (8′) producing stable devices that can easily be measured over periods of days. By comparison, devices made from (8′) last minutes at most. Hence, we can assume that (4) does attach to the TiO2 surface via the propionic linker group because the dye is not desorbed upon infusion of electrolyte into the cell. In addition, because the UV-vis spectra is similar to (8′),35 this suggests that the vinyl dicyano-modified squaric acid moiety may also interact with the TiO2. For (5), one could argue that this can attach to the surface via either the central squaric acid moiety and/or from the carboxylic acid linker attached to the nitrogen of the indole. Given that our previous observations35 are that unmodified squaric acid interacts strongly with TiO2, it seems most likely that (5) may bonds to TiO2 in a similar orientation to (4); i.e. that both the squaric and propionic acid units are involved. In further support of this assertion, the UV-vis spectrum for (5) is similar to those of (7b′) and (10′) reported in our previous work35 because (7) can only attach through the squaric acid unit and (10′) can only attach through the indole propionate because it's squaraine unit is esterified.
To further study how Hf-SQ dyes adsorb on TiO2, new Hf-SQ dyes were prepared with an esterified squaraine moiety to prevent this bonding to the surface. In addition, the carboxylate linker was positioned on the benzene ring of the indole so that the squaraine ester should be orientated away from the TiO2 surface. For the first example, (7), the UV-vis spectrum of TiO2-sorbed (7) exhibits a smaller bathochromic shift relative to solvated (7) than that observed for dyes where it is believed that the squaric acid unit can interact with the surface. These dyes include (7b′), (8′) and (10′) from,35 and (4) and (5) reported here. By comparison, when (7) is modified to form the vinyl dicyano analogue (8), this also de-esterifies the squaric acid unit which would be expected to enable dye-TiO2 chemisorption through the squaraine moiety. In line with this assertion, a larger bathochromic shift is observed for (8) which is in line with other vinyl dicyano modified dyes (60 to 80 nm). Whilst the UV-vis data cannot prove the orientation of the dyes on the TiO2 surface, these data do suggest that larger bathochromic shifts are observed for sorbed dyes when adsorbed through the squaraine moiety and that (8) may bind preferentially through the vinyl dicyano-modified squaraine compared to the indole benzene carboxylate. The peak broadening and red-shifts observed when the dyes chemisorb to TiO2 are common in DSC dyes and has been observed previously for Hf-SQ dyes.33,35 To rationalise this, Cicero et al. used DFT and related the phenomena to the formation of ester bonds between squaric acid oxygen and surface metal atoms which lowers the HOMO–LUMO gap. Interestingly, whilst these workers found that the HOMO changes little on sorption, they reported that the LUMO of the TiO2-dye system does shift. They also suggested that electron excitation into the new LUMO, which includes character from the O and C 2p orbitals of the squaric acid moiety and surface Ti 3d orbitals, should enhance electron injection from the dye into the TiO2.
The next dye (10) was designed with an esterified squaraine and two carboxylate linkers; on the benzene ring and the N of the indole. The UV-vis spectrum of solvated (10) shows three absorption bands; at 420 nm and weaker bands at 480 and 515 nm. For TiO2-adsorbed (10), these bands all broaden considerably. Whilst this makes precise assignment of peak positions difficult, there is little evidence of any major shifts on these peaks and certainly not to the extent observed for the squaraine-bonding dyes. In line with our previous data, modification of (10) to form the de-esterified vinyl dicyano squaraine analogue (11) does show a bathochromic shift for the adsorbed versus solvated data. This is interesting because (11) has three potential carboxylate linkers; on the benzene ring and N atom of the indole and through the modified squaraine unit. Whilst these data do not rule out some interaction through the first two linkers, they do suggest that that the modified squaraine moiety does interact with the TiO2 surface.
To further investigate dye–TiO2 interactions, ATR-infrared spectroscopy has been used to compare adsorbed dye and dye powders (see ESI†). Firstly, the molecular structure of dyes (4) and (5) should make it possible for these dyes to covalently link to TiO2 through two carboxylate linker groups; the squaraine moiety and the propionic acid on the indole N. For (4), the sharp, intense carbonyl (1650 cm−1) and nitrile (2225 cm−1) peaks in the powder broaden and shift in frequency (to 2000–2100 cm−1 for nitrile). The shift in the nitrile peak suggests an interaction with the TiO2 surface which, although it cannot clarify any surface interaction from the propionic acid, does suggest that (4) may interact with TiO2 through the nitrile groups of the vinyl dicyano-modified squaraine unit. For (5), which has no nitrile groups, the data for the dyed film show a broadening and downward shift of the sharp carbonyl signal of the powder. This dye can only chemisorb through carboxylate linkers but the broadness of the sorbed carbonyl signals does not allow any differentiation between sorption modes. This may be due to the poor resolution of the peaks or it may reflect a mixture of surface binding modes (e.g. mono- and bi-dentate). Interestingly, the spectra for (4) and (5) show much greater carbonyl peak shifts between neat powder and TiO2-sorbed dyes than the single linker dyes reported previously35 (see ESI†); thus, (7b′) or (8′) can only bond to the surface from the squaraine unit and (10′) which can only link to TiO2via an indole N linker. For instance, the carbonyl signals of (7b′) and nitrile and carbonyl peaks of (8′) do broaden on sorption to TiO2 but only shift ca. 10 cm−1 in frequency. These data suggest that, for dyes (4) and (5) which contain both propionic and squaric acid groups, both are involved in surface binding to TiO2.
By comparison with (4) and (5), dye (7) possesses only one carboxylate linker which is located on the benzene ring of the indole. The ATR-IR data show that, although there is a broadening of the peaks for the TiO2-sorbed (7) compared to the neat powder (ESI Fig. 4†), the carbonyl peaks do not shift although there is a significant reduction in the O–H signal (>3000 cm−1) on sorption. This presumably reflects the formation of an ester linkage between the carboxylate group and the TiO2 surface. A similar scenario is observed for (8) with a reduction in the O–H signal, and a slight broadening but very little shift on the nitrile and carbonyl peaks. This suggests that bigger peak shifts are observed when the dye sorbs to TiO2 through linker groups which are more associated with increased electron density of the LUMO.35 In support of this assertion, dye (10) shows two carbonyl signals in the powder which corresponds to the two linkers in the structure. These broaden and shift very slightly to lower frequency on sorption which is in line with the molecule sorbing to TiO2 at the opposite end to the squaraine moiety. By comparison, (11) is interesting because it possesses 3 linker groups and the position of these groups around the outside of the molecule suggest it would be difficult for all 3 linkers to chemisorb to TiO2 simultaneously (Scheme 1). In this context, the absence of a significant shift in the nitrile signal (ESI Fig. 7†) suggests that this dye does not appear to sorb strongly through the squaraine unit but rather through the other carboxylate linkers.
Device testing
Table 1 shows I–V data for Hf-SQ devices with and without 5 mM chenodeoxycholic acid (CDCA). As stated above, one aim when designing these Hf-SQ dyes was to retain the optical properties of (8′) from our previous work35 but to modify the molecular structure to enable the new dye to bind more strongly to a TiO2 electrode so that it might remain bonded after the addition of electrolyte solution.| Dye | η/% | FF | V oc/V | J sc/mA cm−2 |
|---|---|---|---|---|
| (4) | 4.5 | 0.69 | 0.67 | 9.80 |
| (4) + CDCA | 4.7 | 0.69 | 0.68 | 10.00 |
| (5) | 5.4 | 0.69 | 0.72 | 10.80 |
| (5) + CDCA | 5.5 | 0.66 | 0.79 | 10.50 |
| (7) | 2.1 | 0.66 | 0.64 | 5.20 |
| (7) + CDCA | 2.9 | 0.64 | 0.71 | 6.12 |
| (8) | 3.1 | 0.78 | 0.69 | 5.68 |
| (8) + CDCA | 3.1 | 0.76 | 0.68 | 6.10 |
| (10) | 2.8 | 0.71 | 0.66 | 5.39 |
| (10) + CDCA | 2.9 | 0.69 | 0.63 | 6.76 |
| (11) | 2.8 | 0.68 | 0.59 | 7.10 |
| (11) + CDCA | 3.0 | 0.71 | 0.64 | 6.57 |
| N719 only | 6.4 | 0.60 | 0.73 | 14.69 |
Thus, dye (4) was designed and synthesized with a vinyl dicyano modified squaric acid unit but with a second carboxylate linker attached the indole N. The power conversion efficiency (η) of devices made using (4) is 4.7% and 4.5%, with and without CDCA, respectively. By comparison, the previous dye (8′) gave η = 2.8% as a result of a lower fill factor (FF) and open circuit voltage (Voc) which was ascribed to rapid dye desorption in liquid electrolyte during measurement. The data for (4) show much higher Voc and FF and the stability of the device measurements confirm that the two linker approach has been successful.
However, in this study, the highest performing dye was actually (5) which gives η = 5.5%. This dye actually performs slightly better than (7b′) from our recent report35 which gave η = 5.0% and, at that time, was the most efficient Hf-SQ dye. Thus, (5) is currently the most efficient Hf-SQ dye mainly due to an increase in Voc which here is 0.79 V compared to 0.71 V in the presence of CDCA. This is further evidence that attaching the Hf-SQ dyes to the surface from two anchoring points is advantageous to device performance. However, for dyes which can link to the surface from a carboxylic acid attached to the benzene ring of the indole an increase of Voc was not observed when an additional linker group was added to the nitrogen of the indole. For example, dyes (7) and (8) gave devices with Voc of 0.71 and 0.68 V, respectively. By comparison, (10) and (11) also have a carboxylic acid linker which is attached to the nitrogen of the indole but the Voc is lower i.e. 0.63 and 0.64 V, respectively. In addition, modification of the central unit to a vinyl dicyano group results in a lower Voc in analogous dyes and a comparable Jsc, despite a broadening of the spectral response. This, allied to the ATR-IR data, suggests that the close proximity of the vinyl dicyano unit to the squaric acid linker may have a negative influence on electron injection and/or dye regeneration processes.
The spectral response of the dyes has also been tested (Fig. 5) and the data show that dye (4) responds most strongly (EQE ca. 60%) between 500 and 575 nm which is where solar intensity is highest. This correlates with this dye having good efficiency (η = 4.7%) and also the vinyl dicyano modification of the squaraine unit which re-shifts the light harvesting relative to (5). Thus, by comparison, the spectral response of (5) is highest between 430 and 540 nm. However, the EQE of (5) is significantly higher ca. 80% which is reflected in the higher efficiency of (5) with η = 5.5%. The spectral response data are very much in line with the DRUV data of the adsorbed dyes (Fig. 4b) and also with the ε measured for dissolved dyes which are 47![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1 for (4) and 18400 M−1 cm−1 for (5) suggesting that, for devices with similar TiO2 films and electrolyte, light harvesting most strongly influences EQE and device performance. A similar pattern is observed for the other dyes tested in that the EQE of (7), (8), (10) and (11) all strongly resemble the DRUV of the adsorbed dyes (Fig. 4b). Thus, as expected, the EQE of dyes (8) and (11) are red-shifted relative to (7) or (10) due to the presence of the vinyl dicyano modifications of the squaraine unit. However, the solution ε of these dyes seems less important as all four dyes have very similar device efficiencies (η is ca. 3.0%) whilst ε varies from 2500 to 78000 M−1 cm−1.
000 M−1 cm−1 for (4) and 18400 M−1 cm−1 for (5) suggesting that, for devices with similar TiO2 films and electrolyte, light harvesting most strongly influences EQE and device performance. A similar pattern is observed for the other dyes tested in that the EQE of (7), (8), (10) and (11) all strongly resemble the DRUV of the adsorbed dyes (Fig. 4b). Thus, as expected, the EQE of dyes (8) and (11) are red-shifted relative to (7) or (10) due to the presence of the vinyl dicyano modifications of the squaraine unit. However, the solution ε of these dyes seems less important as all four dyes have very similar device efficiencies (η is ca. 3.0%) whilst ε varies from 2500 to 78000 M−1 cm−1.
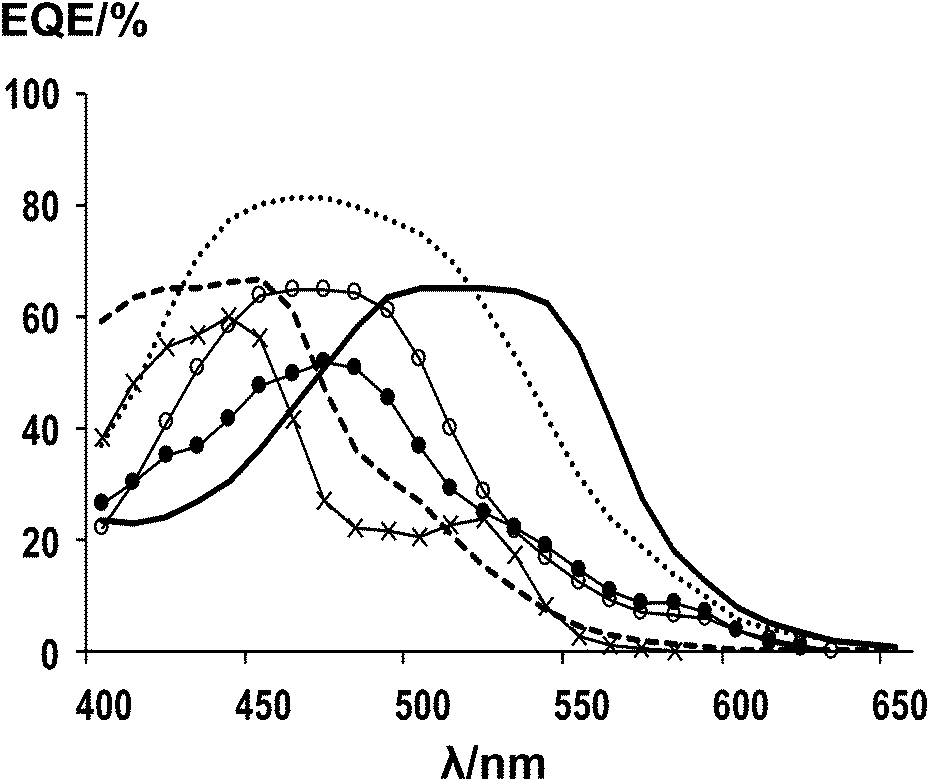 | ||
| Fig. 5 EQE data of DSC devices using; (4) solid line, (5) dotted line, (7) long dashes, (8) open circles, (10) crosses, (11) filled circles. | ||
HOMO–LUMO characteristics
Cyclic voltammetry (CV) measurements of selected dyes (10 mM) have been carried out in degassed DMF to compare dye oxidation and reduction processes with spectral and DSC device data along with theoretical DFT calculations to try to further examine any structure–activity relationships arising from changes to the dye linker position.As an example, the CV data for (10) are shown in Fig. 6 with the onsets of the oxidation and reduction peaks which have been used to calculate the dye band gap labelled. Furthermore, the CV data show no changes following ten cycles scanned at a rate of 50 mV s−1. However, the reduction signals for all the dyes tested are very broad (see ESI Fig. 11–19†). To clarify the reduction voltages, each dye was held at 2.0 V for 60 s to oxidise all the dye molecules in solution. By doing this, the shape of the reduction peak changed and a more defined peak was obtained. By using these peaks to calculate the band gap of the dyes, good correlations with the bang gaps calculated using the UV-vis onsets of the dyes in solution were obtained (Table 2). In addition, the UV-vis data (Fig. 4) have shown that modification of the central squaric acid moiety to a vinyl dicyano group resulted in a bathochromic shift in solution when the dye attaches to a titania surface compared to in solution (e.g. for dyes (4), (8) and (11)). This resulted in a decrease of the dye band gap when calculated from the absorption onset which was also observed in the CV data (Table 2). For example, shifts in the anodic and cathodic waves were observed for (4), (8) and (11) by comparison to (5), (7) and (10), respectively. In addition, both the onset and mid-wave CV data have been used to calculate HOMO–LUMO potentials and band gaps versus the NHE. As expected, the band gap energies obtained from onset CV data correlate with the onset of the absorption measured from the solution UV-visible spectroscopy whilst the mid-wave CV values correlate well with the DFT calculations. Furthermore, the CV data (Fig. 6 and Table 2) suggest that the LUMO levels of the dyes reported here should be above the conduction band of TiO2 to facilitate successful electron injection from the dye into the TiO2.
 | ||
| Fig. 6 Cyclic Voltammetry of (10) 10 mM in degassed DMF vs. Ag/AgCl. | ||
| Dye | Onset values | Half-wave values | |||
|---|---|---|---|---|---|
| λ/nm | λ | From CV | From CV | From DFT | |
| (4) | 526 | 2.35 | 2.30 | 2.70 | 2.70 |
| (5) | 476 | 2.60 | 2.55 | 2.80 | 2.40 |
| (7) | 464 | 2.67 | 2.57 | — | — |
| (8) | 508 | 2.44 | 2.37 | 2.75 | 2.80 |
| (10) | 544 | 2.28 | 2.30 | 2.40 | 2.50 |
| (11) | 580 | 2.14 | 2.15 | 2.80 | 2.80 |
In order to compare the band gaps calculated from the HOMO–LUMO levels (Table 2) with device Voc values measured from the I–V data (Table 1), the Voc data from CDCA devices have been used based on the assumption that CDCA treatment should mitigate dye aggregation and recombination losses. The data show the highest Voc values measured are for (5) and (7) and these dyes have the highest dye band gaps (2.60 and 2.67 eV). By comparison, the lowest Voc is recorded is for (11) which also has the lowest band bap; Voc = 0.64 V and EB = 2.14 eV. This suggests that higher dye band gaps do result in higher Voc. However, the dye energy levels will set the maximum Voc and, as shown with the data from the CDCA treatment, voltage losses must be minimised to achieve this.
DFT molecular modelling studies of (4), (5), (8), (10) and (11) have also been carried out to study both the optimized molecular geometry and HOMO and LUMO maps of these dyes (Fig. 7). Whilst it is important to note that these are effectively gas phase structures (i.e. solvent free), for vinyl dicyano modified dyes ((4), (8) and (11)) the nitrile groups are consistently located on the same side of the molecule as the propionic acid linker. This implies that, if these dyes chemisorb through the indole propionate, the vinyl dicyano group is located closer to the TiO2 surface than the acid unit of the squaraine moiety. This is supported by the ATR-IR data for these dyes which show significant nitrile shifts on sorption to TiO2. In addition, the HOMO–LUMO calculations show electron density spread through the π-framework of each dye but not on the propionic acid linker. This suggests that the latter group acts mainly as a dye–TiO2 anchoring point and does not play a major role in electron injection from the dye excited state. This correlates well with the UV-vis data which suggests that the indole propionate is not strongly involved with the dye HOMO or LUMO levels. Analysis of the HOMO–LUMO energy level calculations shows that the band gaps are in good agreement with the mid-wave CV data apart from for (5) where there is closer agreement to the UV-vis onset value. This may reflect that the DFT relates to gas phase structures. Interestingly, when considering the relative energies of the HOMO and LUMO levels, the dyes are divided into 2 groups with the vinyl dicyano modified dyes much higher in energy. If these differences occurred for sorbed dyes, this might result in poorer overlap between dye and TiO2 orbitals which might result in poorer electron injection. This might explain why these dyes performed less well than expected in DSC devices. This is further supported when comparing the HOMO and LUMO maps of (4) and (5). For (4), the LUMO map shows little electron density near the vinyl dicyano-modified squaraine or propionic acid groups which are expected to be closest to TiO2. By comparison, (5) is the best performing dye and the LUMO map shows electron density on the squaraine moiety which would be expected to enhance electron injection for this dye.
 | ||
| Fig. 7 Optimized geometries and HOMO–LUMO maps of selected dyes using CH3-indoline derivatives to simplify DFT calculations. | ||
Experimental
Instrumentation and chemicals
All chemicals were purchased from Aldrich and used as supplied unless otherwise stated. Anhydrous solvents were used as supplied except tetrahydrofuran (THF) which was dried using Na wire. NMR spectra were recorded on a Bruker AC500 at 500 MHz for 1H and 125 MHz for 13C. Chemical shifts (δ) are given in ppm relative to (CH3)4Si and J values (in Hz) refer to JH,H unless otherwise stated. Mass spectra were recorded at the EPSRC National Mass Spectrometry Service at the University of Swansea. Infrared spectra were recorded on a PE1600 series FTIR spectrometer using an ATR attachment. UV-visible spectroscopy was measured on a Perkin Elmer spectrometer. Cyclic voltammetry was measured on 10 mM dyes in N2-saturated DMF at RT using 0.1 M Bu4NPF6 as supporting electrolyte on an Autolab PGSTAT 30 computer-controlled electrochemical measurement system (Eco Chemie, Holland). Sweep rate was 50 mV s−1 in a 3-electrode cell using a glassy carbon (1 cm2) working electrode, a Ag/AgCl reference electrode and a Pt counter electrode (1 cm2). All voltammetric potentials were re-calculated and are reported versus NHE.X-ray crystallography
Single-crystal X-ray diffraction data of (4), (5), (7), (8) (polymorph 1) and (8) (polymorph 2) were collected at 100 K on Rigaku AFC12 goniometer equipped with an enhanced sensitivity (HG) Saturn 724 + detector mounted at the window of an FR-E+ Superbright MoK rotating anode generator with HF Varimax optics.37Unit cell parameters were refined against all data. An empirical absorption correction was carried out using CrystalClear software.38 All crystal structures were solved by charge flipping methods39 and refined on Fo2 by full-matrix least-squares refinements using programs of the SHELX-2013 software.40 All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were added at calculated positions and refined using a riding model with isotropic displacement parameters based on the equivalent isotropic displacement parameter (Ueq) of the parent atom.
In the crystal structure of (4), the HNEt3+ cation is disordered and modelled over two sites with a 78:22 ratio, whereas in (7) disordered ethoxy group on one of two independent molecules present in the asymmetric unit was modelled over two sites with 68:32 ratio. In (8) (polymorph 2), the aliphatic chain is badly disordered and modelled and constrained over two sites with a 72:28 ratio.
Vibrational restraints (SIMU/DELU) as well as distance/angle restraints DFIX/DANG were used to maintain sensible molecular geometry and atomic displacement ellipsoids. Some disordered atoms required EADP and ISOR restraints to be used.
Due to the poor crystal quality, data and highly disordered model obtained for (8) (polymorph 2), the solution for these data are not ideal. However, it is still sufficient to prove structural connectivity.†
549 measured reflections, 6256 unique reflections [Rint = 0.0682], 4361 with F2 > 2σ, R(F, F2 > 2σ) = 0.0477; Rw(F2, all data) = 0.1168; CCDC: 1030510.
283 measured reflections, 8162 unique reflections [Rint = 0.0596], 4856 with F2 > 2σ, R(F, F2 > 2σ) = 0.0592; Rw(F2, all data) = 0.1351; CCDC: 1030511.
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z = 4, ρcalc = 1.189 Mg m−3; μ = 0.080 mm−1; T = 100(2) K; θmax = 27.528°, 37607 measured reflections, 12648 unique reflections [Rint = 0.1004], 7291 with F2 > 2σ, R(F, F2 > 2σ) = 0.0646; Rw(F2, all data) = 0.1746; CCDC: 1030512.
, Z = 2, ρcalc = 1.145 Mg m−3; μ = 0.074 mm−1; T = 100(2) K; θmax = 27.485°, 31053 measured reflections, 8194 unique reflections [Rint = 0.0882], 5922 with F2 > 2σ, R(F, F2 > 2σ) = 0.0617; Rw(F2, all data) = 0.0836; CCDC: 1030513.
, Z = 2, calc = 1.165 Mg m−3; μ = 0.076 mm−1; T = 100(2) K; θmax = 25.026°, 18323 measured reflections, 6166 unique reflections [Rint = 0.0845], 3532 with F2 > 2σ, R(F, F2 > 2σ) = 0.1184; Rw(F2, all data) = 0.3751; CCDC: 1030514.
, Z = 4, ρcalc = 1.189 Mg m−3; μ = 0.080 mm−1; T = 100(2) K; θmax = 27.528°, 37607 measured reflections, 12648 unique reflections [Rint = 0.1004], 7291 with F2 > 2σ, R(F, F2 > 2σ) = 0.0646; Rw(F2, all data) = 0.1746; CCDC: 1030512.
, Z = 2, ρcalc = 1.145 Mg m−3; μ = 0.074 mm−1; T = 100(2) K; θmax = 27.485°, 31053 measured reflections, 8194 unique reflections [Rint = 0.0882], 5922 with F2 > 2σ, R(F, F2 > 2σ) = 0.0617; Rw(F2, all data) = 0.0836; CCDC: 1030513.
, Z = 2, calc = 1.165 Mg m−3; μ = 0.076 mm−1; T = 100(2) K; θmax = 25.026°, 18323 measured reflections, 6166 unique reflections [Rint = 0.0845], 3532 with F2 > 2σ, R(F, F2 > 2σ) = 0.1184; Rw(F2, all data) = 0.3751; CCDC: 1030514.
Preparation of triethylammonium-2-[(2-carboxyethyl-3,3-dimethyl-1,3-dihydro-2H-indol-2-ylidene)]-3-(dicyanomethylidene)-4-oxocyclobut-1-en-1-olate (4)
Triethylamine (0.33 ml, 2.38 mmol) was added dropwise to a mixture of (3) (0.81 g, 2 mmol) and malononitrile (145 mg, 2.22 mmol) in ethanol (15 ml) and stirred for 2 h at room temperature. The solvent was removed in vacuo and the product was obtained as an orange solid after purification by column chromatography (SiO2) with chloroform and methanol as eluent.(Yield 1 g, 75%) m.p. 175–179 °C. δH (400 MHz, MeOD) 8.11 (1H, d, J 8.4), 7.89–7.80 (3H, m), 7.53–7.41 (2H, m), 7.32 (1H, t, J 7.4), 4.25 (2H, bs), 3.24 (6H, q, J 7.3), 2.80 (2H, t, J 6.4), 1.93 (6H, s), 1.33 (9H, t, J 7.3).
δ C (101 MHz, MeOD) 191.78, 191.28, 185.69, 181.53, 173.25, 138.03, 130.69, 130.12, 129.36, 128.99, 128.65, 127.93, 126.43, 125.99, 122.82, 122.40, 121.73, 112.87, 109.97, 38.39, 30.99, 26.07, 7.93.
MS (FTMS+) M+ calculated = 424, M+ observed = 424, m/z accurate mass (FTMS+), reference compound: NH4OAc, M+ calculated = 424.1292, M+ observed = 424.1294. FT-IR (ATR) ν/cm−1 3141 (s), 2982 (s), 2685 (s), 2515 (s), 2197 (s), 2178 (s), 1732 (s), 1626 (s), 1595 (s), 1537 (s), 1517 (s). UV-visible λmax 467 nm (47000 M−1 cm−1 ± 500 M−1 cm−1) in ethanol.
Preparation of 4-[(1-carboxy-1,3-dihydro-3,3-dimethyl-2H-benzo[e]indol-2-ylidene)methyl]-3-cyclobutene-1,2-dione (5)
3-Ethoxy-[(1-carboxyethyl)-1,1,2-trimethyl-1H-benzo[e]indolium iodide-2-ylidene)methyl]-3-cyclobutene-1,2-dione (1.07 g, 2.7 mmol) was dissolved in ethanol (10 ml) and heated under reflux. Tetrabutyl ammonium hydroxide (1 ml, 40%) was added and the solution was refluxed for 10 min. The mixture was concentrated and 15 ml of cold ethanol was added. The yellow product crystallized and was washed with ethanol three times.Yield 0.5 g (50%) m.p. 185–191 °C.
δ H (400 MHz, MeOD) 8.12 (1H, d, J 8.6), 7.83 (2H, dd, J 11.4, 8.6), 7.51–7.43 (2H, m), 7.28 (1H, t, J 7.5), 5.77 (1H, s), 4.29–4.21 (2H, m), 3.29–3.19 (10H, m), 2.68–2.60 (2H, m), 1.95 (6H, s), 1.67 (10H, dt, J 15.9, 7.9), 1.50–1.35 (8H, m), 1.03 (8H, t, J 7.3).
δ C (101 MHz, MeOD) 204.15, 196.50, 179.97, 177.48, 164.56, 140.82, 130.72, 130.34, 129.33, 128.88, 126.09, 122.19, 121.66, 109.93, 82.58, 58.13, 58.11, 58.08, 39.72, 34.14, 26.23, 23.39, 19.32, 19.30, 12.55.
MS (FTMS+) M+ calculated = 270, M+ observed = 270, m/z accurate mass (FTMS+), reference compound: NH4OAc, M+ calculated = 270.1125, M+ observed = 270.1126.
FT-IR (ATR) ν/cm−1 3512 (w), 2960 (s), 2937 (s), 2876 (s), 1751 (m), 1666 (w), 1623 (m), 1592 (s), 1548 (s), 1516 (s). UV-visible λmax 443 nm (18400 M−1 cm−1 ± 200 M−1 cm−1) in ethanol.
Preparation of 5-carboxy-2,3,3-trimethyl-1-dodecyl-3H-indolium iodide (6)
A mixture of 5-carboxy-2,3,3-trimethyl-3H-indolium iodide (2 g, 6 mmol) and iodododecane (4.1 g, 24 mmol) was heated under reflux overnight under nitrogen in anhydrous acetonitrile (80 ml). After cooling, the solvent was removed in vacuo and the product was re-crystallized from methanol to give a yellow solid. (Yield 2.42 g, 72%).MS (FTMS+) M+ calculated = 372, M+ observed = 372. m/z accurate mass (FTMS+), reference compound: NH4OAc, M+ calculated = 372.2897, M+ observed = 372.887.
Preparation of 2-((2-ethoxy-3,4-dioxocyclobut-1-enyl)methylene)-3,3-dimethyl-1-dodecylindoline-5-carboxylic acid (7)
Synthetic procedures followed to (3).(2 g, 65%) m.p. 180–184 °C.
δ H (400 MHz, THF) 8.04–8.02 (doublet and singlet overlapping 3H), 7.10 (1H, d, J 8.2), 5.58 (1H, s), 4.92 (2H, q, J 7.1), 3.98 (2H, t, J 7.4), 3.65 (1H, s), 1.71 (6H, s), 1.56 (3H, t, J 7.1), 1.48 (4H, m), 1.35 (14H, s), 0.95 (3H, t, J 6.6).
δ C (101 MHz, THF) 189.65, 187.42, 185.97, 171.22, 164.78, 164.58, 144.90, 138.61, 128.58, 122.74, 121.28, 105.69, 80.66, 67.84, 45.19, 40.59, 30.02, 27.73, 27.67, 27.62, 27.45, 27.44, 24.93, 24.51, 24.29, 20.71, 13.33, 11.61.
MS (FTMS−) [M − H]− calculated = 494, [M − H]− observed = 494. m/z accurate mass (FTMS−), [M − H]− calculated = 494.2912, [M − H]− observed = 494.2919.
FT-IR (ATR) ν/cm−1 3065 (b), 2925 (s), 2856 (s), 2530 (b), 1773 (s), 1716 (s), 1678 (s), 1603 (s), 1539 (s). UV-visible λmax 428 nm (78000 M−1 cm−1 ± 700 M−1 cm−1) in ethanol.
Preparation of 2-((2-ethoxy)methylene)-3,3-dimethyl-1-dodecylindoline-3-(dicyanomethylidene)-5-carboxylic acid (8)
Synthetic procedures followed to (4).Yield 0.8 g (70%) m.p. 170–175 °C.
δ H (400 MHz, MeOD) 7.98 (1H, d, J 8.3), 7.90 (1H, s), 6.99 (1H, d, J 8.3), 6.14 (1H, s), 3.86 (2H, t, J 7.0), 3.24 (6H, m), 1.75 (2H, d, J 6.6), 1.68 (6H, s), 1.32 (24H, m), 0.91 (3H, t, J 6.3).
δ C (101 MHz, MeOD) 192.35, 185.79, 178.53, 168.78, 167.78, 163.87, 147.41, 140.60, 130.48, 123.27, 122.81, 118.66, 117.84, 107.01, 86.06, 42.47, 41.36, 37.18, 35.58, 31.67, 29.32, 29.08, 29.04, 28.97, 27.91, 26.47, 26.21, 26.14, 23.01, 22.33, 17.98, 13.06, 8.46, 7.85.
MS (FTMS−) [M]− calculated = 514, [M]− observed = 514 m/z accurate mass (FTMS−), [M]− calculated = 514.2711, [M]− observed = 514.2699.
FT-IR (ATR) ν/cm−1 3158 (bs), 2925 (s), 2854 (s), 2639 (bs), 2201 (s), 2182 (s), 1749 (s), 1705 (s), 1666 (s), 1618 (m), 1604 (m), 1533 (s). UV-visible λmax 466 nm (55000 M−1 cm−1 ± 600 M−1 cm−1) in ethanol.
Preparation of 5-carboxy-2,3,3-trimethyl-1-carboxyethyl-3H-indolium iodide (9)
A mixture of 5-carboxy-2,3,3-trimethyl-3H-indolium iodide (2 g, 6 mmol) and 3-iodopropionic acid (2 g, 7.5 mmol) was heated under reflux overnight under nitrogen in anhydrous 1,2 dichlorobenzene (30 ml). After cooling, the solvent was removed in vacuo and the product was re-crystallized from methanol to give a yellow solid. (Yield 1.67 g, 70%).MS (FTMS+) M+ calculated = 276, M+ observed = 276, m/z accurate mass (FTMS+), reference compound: NH4OAc, M+ calculated = 276.1230, M+ observed = 276.1227.
Preparation of 2-((2-ethoxy-3,4-dioxocyclobut-1-enyl)methylene)-3,3-dimethyl-1-carboxyethylindoline-5-carboxylic acid (10)
Synthetic procedures followed (3).Yield 0.5 g (80%) m.p. 180–185 °C.
δ H (400 MHz, MeOD) 8.04–7.97 (doublet and singlet overlapping 3H), 7.20 (1H, d, J 8.3), 6.02 (1H, s), 4.44 (3H, t, J 6.7), 4.27–4.20 (two overlapping triplets 4H), 4.17 (3H, t, J 7.4), 3.20 (2H, q, J 7.3), 1.66 (6H, s).
δ C (101 MHz, MeOD) 196.95, 191.89, 179.38, 177.80, 170.28, 167.43, 161.47, 147.39, 145.20, 140.18, 129.77, 122.63, 108.18, 106.88, 84.29, 69.90, 39.13, 32.26, 28.79, 25.94, 14.62.
MS (FTMS−) [M − H]− calculated = 398, [M − H]− observed = 398. m/z accurate mass (FTMS−), [M − H]− calculated = 398.1245, [M − H]− observed = 398.1235.
FT-IR (ATR) ν/cm−1 3543–3060 (bs), 2978 (w), 2965 (m), 2866 (s), 1738 (s), 1707 (s), 1618 (s), 1554 (s). UV-visible λmax 425 nm (16000 M−1 cm−1 ± 400 M−1 cm−1) in ethanol.
Preparation of 2-((2-ethoxy)methylene)-3,3-dimethyl-1-carboxyethylindoline-3-(dicyanomethylidene)-5-carboxylic acid (11)
Synthetic procedures followed (4).Yield 0.3 g (60%) m.p. 170–175 °C.
δ H (400 MHz, MeOD) 7.96 (1H, d, J 8.3), 7.89 (1H, d, J 1.0), 7.10 (1H, d, J 8.4), 6.10 (1H, s), 4.17 (2H, t, J 6.8), 3.23 (6H, q, J 7.3), 2.75 (2H, t, J 6.8), 1.68 (6H, s), 1.33 (12H, t, J 7.3).
δ C (101 MHz, MeOD) 192.58, 185.94, 179.98, 173.74, 168.71, 163.04, 147.08, 140.16, 130.42, 123.55, 118.47, 117.57, 111.91, 106.87, 86.20, 68.02, 40.39, 38.80, 30.98, 26.60, 22.17, 18.40, 9.98, 7.40.
MS (FTMS−) [M]− calculated = 418, [M]− observed = 418. m/z accurate mass (FTMS−), [M]− calculated = 418.1045, [M]− observed = 418.1036.
FT-IR (ATR) ν/cm−1 3420 (b), 2988 (w), 2974 (w), 2924 (w), 2500 (b), 2198 (s), 1744 (s), 1681 (s), 1603 (s), 1544 (s). UV-visible λmax 459 nm (2500 M−1 cm−1 ± 60 M−1 cm−1) in ethanol.
Device manufacture and testing
DSC devices were prepared using 1.0 cm2 (2 × 0.5 cm) TiO2 working electrodes by doctor blading two layers of transparent paste (DSL18NR-T, Dyesol) onto TEC15™ FTO glass (NSG) followed by one layer of scattering paste (WER4-O, Dyesol). Each layer was sintered (450 °C, 30 min) before TiCl4:THF2(aq) treatment (50 mM, 70 °C, 30 min), rinsing with de-I H2O and re-sintering (450 °C, 30 min). To prepare counter electrodes, after washing (iPrOH) and air drying TEC8™ glass (NSG) Pt colloid (Pt-1, Dyesol) was deposited and then sintered (400 °C, 30 min). After sealing the counter and working electrodes together with Surlyn™ (DuPont) at 120 °C, the devices were fast dyed according to the method described previously41 after which acetonitrile-based electrolyte was added (50 mM I2, 50 mM LiI, 50 mM guanidinium thiocyanate, 800 mM 1-methyl-3-propyl-imidazolium iodide, 300 mM benzimidazole). Conductive Ag paint (Agar) was applied to ensure good electrical contact.
Device performance was measured using I–V data and an ABET Solar Simmulator with Xe arc lamp and a Keithley 2400 at 100 mW cm−2 or 1 Sun between 0 and 1 V. Spectral response was measured in DC mode using a QEX10 Quantum Efficiency Measurement System (resolution 10 nm). Lamp calibration to 1 Sun (100 mW cm−2) used a certified (Oriel 91150V) monocrystalline silicon reference cell traceable to the National Renewable Energy Laboratory (NREL).
Molecular modeling
DFT calculations to predict the geometries and properties of the dyes were performed using a 64 bit version of Spartan'14.42 The structures were first optimized semi-empirically (AM1) and then re-optimized using B3LYP (6-31G*). Frequency calculations confirmed the optimizations gave rise to local minima. To facilitate geometry optimization, some of the longer side-chains were shortened to methyl groups.Conclusions
This study shows that increasing the number of linker groups for Hf-SQ dyes improves DSC device efficiency leading to the highest efficiency reported for this class of sensitizer. Furthermore, increasing the number of linker groups enables the favourable optical properties of a vinyl dicyano modified squaric acid dye to be retained whilst also ensuring that the dye does not desorb from the electrode surface during electrolyte infusion. However, the data support previous studies35 which suggest that the position of linker groups in the dye molecule is important and that they need to be positioned to enable the LUMO of the dye to be in close proximity to the TiO2 surface.Our data also show that infrared spectroscopy can be used to study dye sorption from the different linker groups. These data suggest that, to optimize DSC device performance, it is necessary to place the second linker closer to the squaraine unit. If this happens, it appears that the squaraine moiety can orient itself closer to the TiO2 which is believed to be a better position to enable electron injection from the dye excited state into the TiO2. However, DFT calculations and ATR-IR data suggest that, for vinyl dicyano modified squaraines, the nitrile groups orient themselves closer to the TiO2 resulting in poorer than expected electron injection from this group of dyes. This explains the poor binding and device performance of (8′) reported in.35 The DFT data also support our previous assertion35 that, for the best performing dyes, the LUMO is located near to the squaraine linker moiety. We are currently studying the long-term stability of these dyes but our initial data suggest that they are stable in DSC devices under ambient conditions in the short term (i.e. weeks).
Acknowledgements
We gratefully acknowledge Welsh Government funding for Sêr Cymru (AC, EWJ, CK, PJH), EPSRC/TSB funding for SPECIFIC (EP/I019278/1) for MLD, EPSRC CASE and Tata support for LF and the EPSRC UK National Mass Spectrometry Facility at Swansea University. We thank EPSRC for funding the National Crystallography Service (Southampton and Newcastle), and Diamond Light Source for access to synchrotron facilities.Notes and references
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737 CrossRef.
- A. Hagfeldt, G. Boschloo, L. C. Sun, L. Kloo and H. Pettersson, Chem. Rev., 2010, 110, 6595 CrossRef CAS PubMed.
- M. K. Nazeeruddin, F. De Angelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835 CrossRef CAS PubMed.
- Y. Cao, Y. Bai, Q. Yu, Y. Cheng, S. Liu, D. Shi, F. Gao and P. Wang, J. Phys. Chem. C, 2009, 113, 6290 CAS.
- K.-L. Wu, H.-C. Hsu, K. Chen, Y. Chi, M.-W. Chung, W.-H. Liu and P.-T. Chou, Chem. Commun., 2010, 46, 5124 RSC.
- K. Cao, J. Lu, J. Cui, Y. Shen, W. Chen, G. Alemu, Z. Wang, H. Yuan, J. Xu, M. Wang and Y. Cheng, J. Mater. Chem. A, 2014, 2, 4945 CAS.
- C.-Y. Chen, J.-G. Chen, S.-J. Wu, J.-Y. Li, C.-G. Wu and K.-C. Ho, Angew. Chem., Int. Ed., 2008, 47, 7342 CrossRef CAS PubMed.
- T. Funaki, N. O. Komatsuzaki, K. Kasuga, K. Sayama and H. Sugihara, Inorg. Chem. Commun., 2013, 35, 281 CrossRef CAS PubMed.
- T. Bessho, E. Yoneda, J.-H. Yum, M. Guglielmi, I. Tavernelli, H. Imai, U. Rothlisberger, M. K. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2009, 131, 5930 CrossRef CAS PubMed.
- K.-L. Wu, W.-P. Ku, S.-W. Wang, A. Yella, Y. Chi, S.-H. Liu, P.-T. Chou, M. K. Nazeeruddin and M. Grätzel, Adv. Funct. Mater., 2013, 23, 2285 CrossRef CAS.
- K.-L. Wu, W.-P. Ku, J. N. Clifford, E. Palomares, S.-T. Ho, Y. Chi, S.-H. Liu, P.-T. Chou, M. K. Nazeeruddin and M. Grätzel, Energy Environ. Sci., 2013, 6, 859 CAS.
- G. C. Vougioukalakis, A. I. Philippopoulos, T. Stergiopoulos and P. Falaras, Coord. Chem. Rev., 2011, 255, 2602 CrossRef CAS PubMed.
- M. Wang, S.-J. Moon, D. Zhou, F. L. Formal, N. Lê, C. Ha, R. H. Baker, C. Grätzel, P. Wang, S. M. Zakeeruddin and M. Grätzel, Adv. Funct. Mater., 2010, 20, 1821 CrossRef CAS.
- K. Fantai, A. I. Songyuan and W. Kongj, Plasma Sci. Technol., 2006, 8, 5 CrossRef.
- A. J. Hallett and J. E. Jones, Dalton Trans., 2011, 40, 3871 RSC.
- H. Im, S. Kim, C. Park, S.-H. Jang, C.-J. Kim, K. Kim, N.-G. Park and C. Kim, Chem. Commun., 2010, 46, 1335 RSC.
- M. Liang and J. Chen, Chem. Soc. Rev., 2013, 42, 3453 RSC.
- N. Mohammad, P. J. Mahon and F. Wang, J. Mol. Graphics Modell., 2013, 40, 64 CrossRef PubMed.
- K. Hara, Z. S. Wang, T. Sato, A. Furube, R. Katoh, H. Sugihara, Y. D. Oh, C. Kasada, A. Shinpo and S. Suga, J. Phys. Chem. B, 2005, 109, 15476 CrossRef CAS PubMed.
- S. Ito, S. M. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. K. Nazeeruddin, P. Pechy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202 CrossRef CAS.
- Y. Wu, M. Marszalek, S. M. Zakeeruddin, Q. Zhang, H. Tian, M. Grätzel and W. Zhu, Energy Environ. Sci., 2012, 5, 8261 CAS.
- X. Lu, Q. Feng, T. Lan, G. Zhou and Z.-S. Wang, Chem. Mater., 2012, 24, 3179 CrossRef CAS.
- H. Hug, M. Bader, P. Mair and T. Glatzel, Appl. Energy, 2014, 115, 216 CrossRef CAS PubMed.
- N. A. Ludin, A. M. A. Mahmoud, A. B. Mohamad, A. A. H. Kadhum, K. Sopian and N. S. A. Karim, Renewable Sustainable Energy Rev., 2014, 31, 386 CrossRef CAS PubMed.
- Y.-S. Yen, H.-H. Chou, Y.-C. Chen, C.-Y. Hsu and J. T. Lin, J. Mater. Chem., 2012, 22, 8734 RSC.
- P. J. Holliman, K. J. Al-Salihi, A. Connell, M. L. Davies, E. W. Jones and D. A. Worsley, RSC Adv., 2014, 4(5), 2515 RSC.
- A. Yella, H.-W. Lee, H. N. Tsao, C. Yi, A. K. Chandiran, M. K. Nazeeruddin, E. W.-G. Diau, C.-Y. Yeh, S. M. Zakeeruddin and M. Grätzel, Science, 2011, 334, 629 CrossRef CAS PubMed.
- A. Yella, C.-L. Mai, S. M. Zakeeruddin, S.-N. Chang, C.-H. Hsieh, C.-Y. Yeh and M. Grätzel, Angew. Chem., Int. Ed., 2014, 53(11), 2973 CrossRef CAS PubMed.
- A. L. Tatarets, I. A. Fedyunyaeva, E. Terpetschnig and L. D. Patsenker, Dyes Pigm., 2005, 64, 125 CrossRef CAS PubMed.
- L. Beverina and P. Salice, Eur. J. Org. Chem., 2010, 1207 CrossRef CAS.
- M. Matsui, T. Shibata, M. Fukushima, Y. Kubota and K. Funabiki, Tetrahedron, 2012, 68, 9936 CrossRef CAS PubMed.
- M. Matsui, H. Mase, J.-Y. Jin, K. Funabiki, T. Yoshida and H. Minoura, Dyes Pigm., 2006, 70, 48 CrossRef CAS PubMed.
- N. Shahzad, F. Risplendi, D. Pugilese, S. Bianco, A. Sacco, A. Lamberti, R. Gazzia, E. Tresso and G. Cicero, J. Phys. Chem. C, 2013, 117, 22778 CAS.
- G. Cicero, G. Musso, A. Lambertini, B. Camino, S. Bianco, D. Pugilese, F. Risplendi, A. Sacco, N. Shahzad, A. M. Ferrari, B. Ballarin, C. Barolo, E. Tresso and G. Caputo, Phys. Chem. Chem. Phys., 2013, 15, 7198 RSC.
- A. Connell, P. J. Holliman, M. L. Davies, C. D. Gwenin, S. Weiss, M. B. Pitak, P. N. Horton, S. J. Coles and G. Cooke, J. Mater. Chem. A, 2014, 2(11), 4055 CAS.
- E. Terpetschnig and J. R. Lakowicz, Dyes Pigm., 1993, 21(3), 227 CrossRef CAS.
- S. J. Coles and P. A. Gale, Chem. Sci., 2012, 3, 683 RSC.
- CrystalClear-SM Expert 3.1ba16, Rigaku, 2012.
- L. Palatinus and G. Chapuis, J. Appl. Crystallogr., 2007, 40, 786 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- P. J. Holliman, M. L. Davies, A. Connell, B. Vaca Velasco and T. M. Watson, Chem. Commun., 2010, 46, 7256 RSC.
- Wavefunction, Inc. 18401 Von Karman Ave., Suite 370, Irvine, CA 92612.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1030511–1030514. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ta06896c |
| This journal is © The Royal Society of Chemistry 2015 |