
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A conjugated porous poly-benzobisthiadiazole network for a visible light-driven photoredox reaction†
Zi Jun
Wang
,
Saman
Ghasimi
,
Katharina
Landfester
and
Kai A. I.
Zhang
*
Max Planck Institute for Polymer Research, Ackermannweg 10, D-55128 Mainz, Germany. E-mail: kai.zhang@mpip-mainz.mpg.de
First published on 16th September 2014
Abstract
A conjugated porous poly-benzobisthiadiazole network was synthesized via high internal phase emulsion polymerization as a highly active and stable heterogeneous photocatalyst. Under a household energy saving light bulb, dehalogenation of α-bromoacetophenones was performed in an almost quantitative manner at room temperature.
Photocatalysts, which absorb mainly in the visible range of light, have established notable prominence in applications such as water splitting, solar energy storage, and photovoltaics.1–3 Among these visible light-driven catalysts developed, rare metal complexes, especially ruthenium and iridium complexes have found large applications due to their commercial availability, excellent stability and photoredox properties.4–11 However, considering the high cost and toxicity of these rare metals, as well as their limited availability in nature, their sustainability still remains a huge challenge. There has been growing interest in developing metal-free visible light driven photocatalysts in recent years, where a vast number of organic chromophores and dyes were employed successfully in photoredox catalysis.12–14 In particular, fluorescein, eosin Y, nile red, and rhodamine B have been studied intensely.15 A common drawback associated with most single molecule organic dyes is that they easily suffer from the solvolytic attack in the reaction medium, the so-called photo-bleaching. In addition, homogeneous catalysts generally can be troublesome to remove after the reaction. Therefore, it is beneficial to develop a new class of organic heterogeneous visible light driven photocatalysts with high stability and easy processability.
Conjugated porous polymers, combining the photoactive π-electron backbone and porous surface properties, have been recently introduced as stable heterogeneous photocatalysts for organic synthesis under irradiation of visible light.16–21 High internal phase polymerization is a well-known technique previously employed to prepare highly porous materials. Polymers obtained by this technique (polyHIPEs) have found various applications such as enzyme immobilization,22–24 gas storage,25 tissue engineering scaffolds26 and separation media.27 Combining the interconnected porosity of polyHIPEs with the photoactivity of the conjugated polymer backbone, π-conjugated porous polyHIPEs have been recently employed as highly stable heterogeneous visible light-driven photocatalysts for singlet oxygen generation,28 highly selective oxidation of organic sulfides29 and effective visible light photoinitiation for free radical polymerization.30 Taking advantage of the insoluble nature and monolithic shape of these conjugated porous polymers, they can be readily incorporated into continuous flow systems, which offer facile automation, precise control over reaction parameters and predictable scale-up.28
In this work, we report on the design and preparation of a conjugated porous polymer via the Sonogashira cross-coupling reaction using a high internal phase emulsion polymerization technique. A strong electron acceptor, benzobisthiadiazole (BBT), was incorporated into the porous polymer backbone, gaining a broad absorption band and the desired energy level. The visible light-driven photocatalytic activity of the porous polymer was demonstrated by the reductive dehalogenaton reaction of α-bromoacetophenones under a 23 W household energy saving fluorescent light bulb.
In Scheme 1, the synthetic pathway of the conjugated porous polymer B2-FL2-BBT is displayed. As a comparison, another porous polymer only containing fluorene (B2-FL3) was synthesized for investigation of the photocatalytic performance of the dehalogenation reaction without the electron withdrawing BBT unit. The experimental synthetic details and polymer characterization data are described in the ESI.† It should be noted that, for the Sonogashira coupling reaction, K2CO3 seems to be one of the few bases which lead to stable high internal phase emulsion. By adding amines as the base, in comparison, only unstable emulsions were formed; rapid phase separation occurred during the polymerization. The formed monolithic cross-linked polymers were insoluble in all common organic solvents tested. Solid state 13C/MAS NMR spectroscopy showed chemical shifts between δ = 15 and 60 ppm that can be assigned to the alkyl groups of the fluorene units. The signals between 110 and 160 ppm can be assigned to the aromatic carbons in the polymer backbone (Fig. S1 in the ESI†).
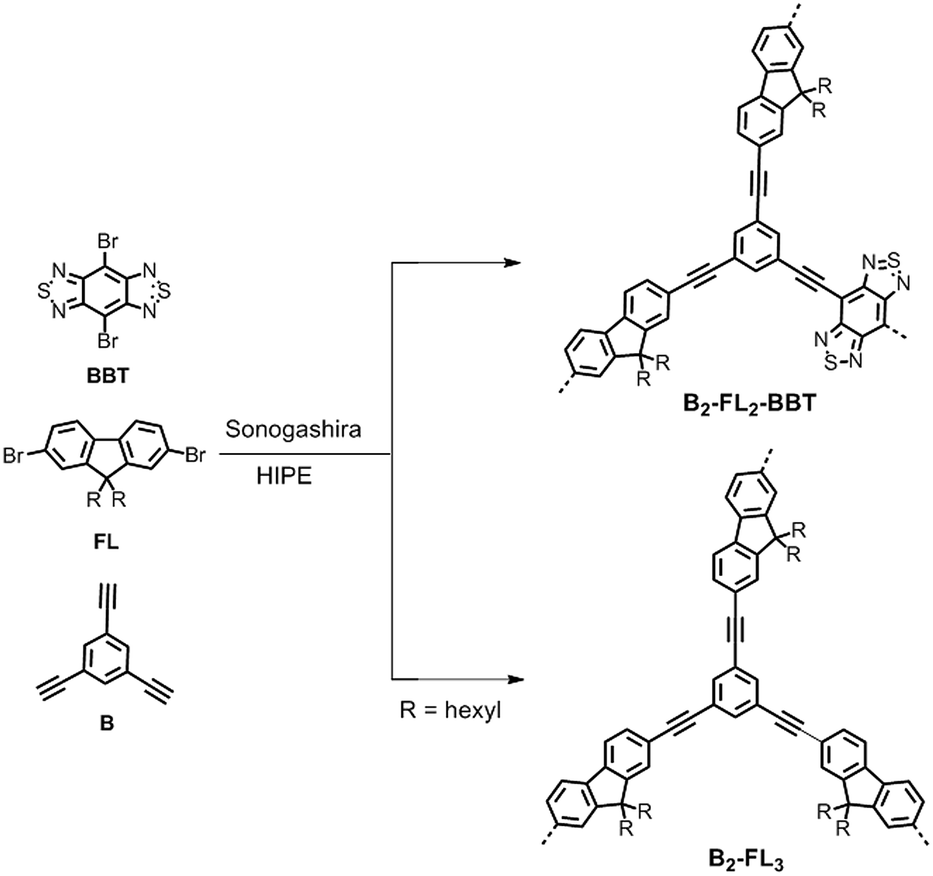 | ||
| Scheme 1 Synthetic route and idealized structure of B2-FL2-BBT and B2-FL3. Reaction conditions: (i) Pd(PPh3)4, CuI, K2CO3, span 80, toluene–H2O (v/v: 1/9), 80 °C, overnight. | ||
From the elemental analysis, a BBT/fluorene ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 inside the polymer B2-FL2-BBT was observed. This could be caused by the non-quantitative polymerization degree and different relativities of both building blocks during polymerization. Thermal gravimetric analysis (TGA) measurements showed that the conjugated network remained intact up to 450 °C and the weight loss at around 300 °C can be attributed to the alkyl groups (ESI, Fig. S2†). The Brunauer–Emmett–Teller (BET) surface areas of B2-FL2-BBT and B2-FL3 are around 23 and 17 m2 g−1 (ESI, Fig. S4†). Broad pore distributions from the meso to macropore range were observed for both polymers (ESI, Fig. S5†). This is similar to the previously reported surface area values and pore analyses of this class of conjugated porous polymers prepared using the HIPE technique.28,29 The broad pore size distribution could be caused by the alkyl side chains of the fluorene unit, which block the micropores and require larger space inside the porous polymers. As it was shown previously that micropores are too small to be effectively accessible by diffusion and no enhancement in catalytic efficiency was observed from higher surface area polymers with additional micropore volumes, the hierarchically porous structure of the polymers could be advantageous for a higher efficiency of the catalytic reactions.29
:3 inside the polymer B2-FL2-BBT was observed. This could be caused by the non-quantitative polymerization degree and different relativities of both building blocks during polymerization. Thermal gravimetric analysis (TGA) measurements showed that the conjugated network remained intact up to 450 °C and the weight loss at around 300 °C can be attributed to the alkyl groups (ESI, Fig. S2†). The Brunauer–Emmett–Teller (BET) surface areas of B2-FL2-BBT and B2-FL3 are around 23 and 17 m2 g−1 (ESI, Fig. S4†). Broad pore distributions from the meso to macropore range were observed for both polymers (ESI, Fig. S5†). This is similar to the previously reported surface area values and pore analyses of this class of conjugated porous polymers prepared using the HIPE technique.28,29 The broad pore size distribution could be caused by the alkyl side chains of the fluorene unit, which block the micropores and require larger space inside the porous polymers. As it was shown previously that micropores are too small to be effectively accessible by diffusion and no enhancement in catalytic efficiency was observed from higher surface area polymers with additional micropore volumes, the hierarchically porous structure of the polymers could be advantageous for a higher efficiency of the catalytic reactions.29
SEM images (Fig. 1) of two porous polymers illustrate the interconnected porous structures with cavities ranging from 10 to 20 μm in size, while the interconnected pores show diameters from 0.5 to 1 μm.
 | ||
| Fig. 1 SEM images of B2-FL2-BBT (a) and B2-FL3 (b). | ||
The UV/Vis diffuse reflectance spectrum (Fig. 2) of B2-FL2-BBT showed a broad absorption band ranging from the visible and into the near infrared (NIR) region. An optical band gap of ca. 1.5 eV can be derived from the absorption edge. In comparison, the UV/Vis spectrum of B2-FL3 only containing fluorene showed a sharper absorption shoulder, exhibiting an optical band gap of ca. 2.40 eV. This is probably due to the strong electron withdrawing properties of the BBT unit, exhibiting lower HOMO–LUMO levels, and the resulting band gap of the polymer. It should be noted that the broad shoulder of the spectra could partially be due to the light scattering effect, giving a broad pore size range inside the porous polymers. Interestingly, by incorporating the electron withdrawing BBT group into the polymer backbone, the electron paramagnetic resonance (EPR) spectrum of B2-FL2-BBT exhibited significantly higher intensity than B2-FL3 (ESI, Fig. S4†).
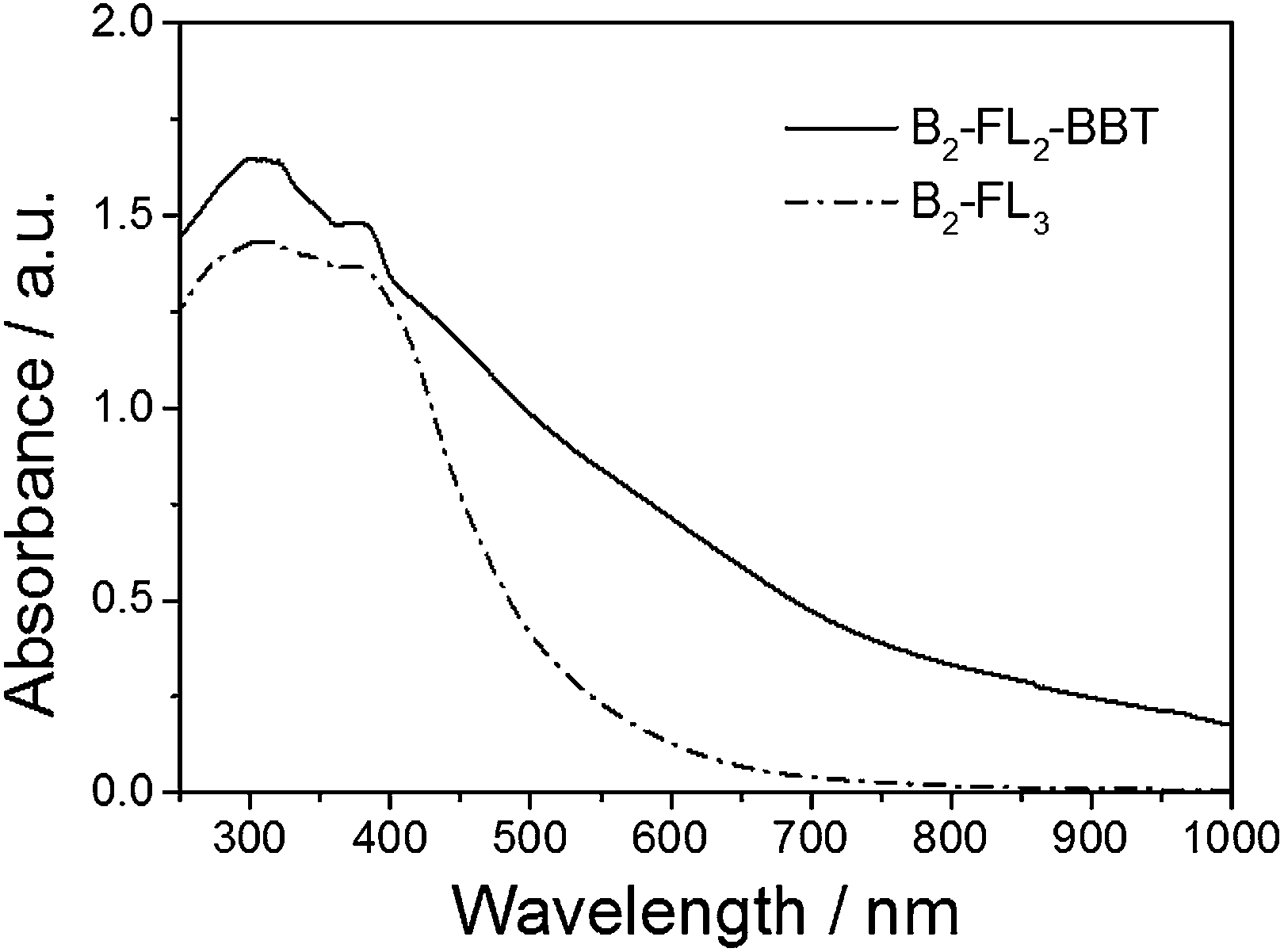 | ||
| Fig. 2 UV/Vis DRS spectra of B2-FL2-BBT and B2-FL3. | ||
The photocatalytic activity of the conjugated porous polymers as heterogeneous photocatalysts under visible light was investigated by the photoreductive dehalogenaton reaction of α-bromoacetophenones. A 23 W household energy saving light bulb was used as the light source. The simple illumination set-up is shown in Fig. S15 in the ESI.† Various α-bromoacetophenone derivatives were investigated (Table 1). B2-FL2-BBT showed notably higher photocatalytic activity than B2-FL3 (entry 1 and 2), which proved the importance of incorporation of the electron withdrawing BBT unit. We further tested different phenone bearing electron-withdrawing (entries 3–6) or electron-donating substituents (entries 7 and 8) on the phenyl ring as substrates for the reaction. As shown in Table 1, almost quantitative conversion of the dehalogenation reaction was achieved in every case, demonstrating the general applicability of the conjugated porous polymer B2-FL2-BBT as a highly efficient photocatalyst.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | Product | t (h) | Conversiona (%) | Yieldb (%) |
| a Conversion determined by 1H-NMR using trichloroethylene as the internal standard. b Isolated yield by chromatography. c Using B2-FL3 as the photocatalyst. d Blank experiment without B2-FL2-BBT, but under light. e Blank experiment with B2-FL2-BBT, but in the dark. Reaction conditions: 10 mg porous polymer, 0.2 mmol substrate, 0.4 mmol DIPEA, 0.22 mmol Hantzsch ester in 1 mL deuterated DMF under a 23 W household fluorescent light bulb at room temperature. | |||||
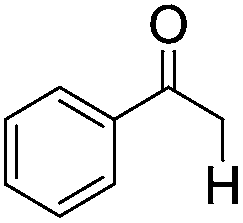
| 1 |

|
|
4 | >99 | 93 |
| 2c |
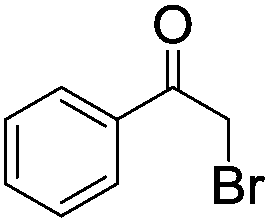
|
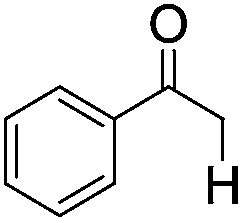
|
4 | 19 | 15 |
| 3 |
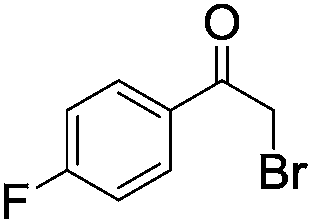
|
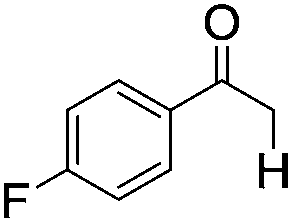
|
4 | 95 | 89 |
| 4 |
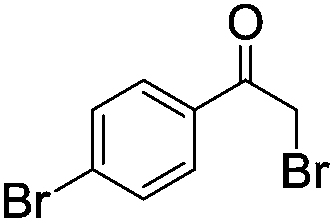
|
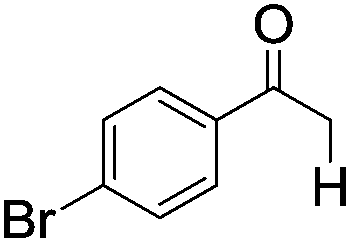
|
4 | >99 | 95 |
| 5 |
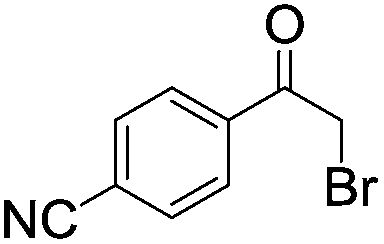
|
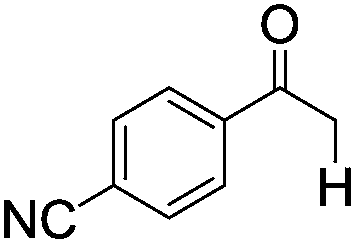
|
4 | >99 | 93 |
| 6 |
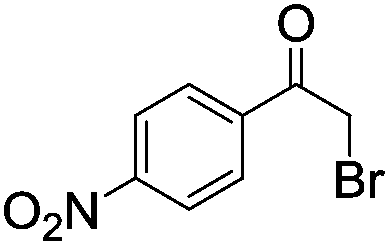
|
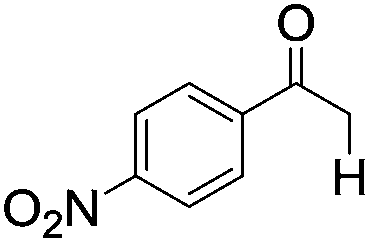
|
4 | >99 | 97 |
| 7 |

|
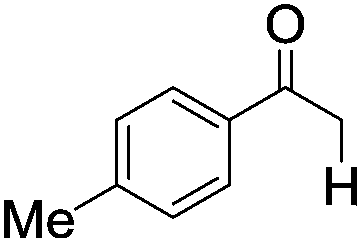
|
5 | >99 | 92 |
| 8 |
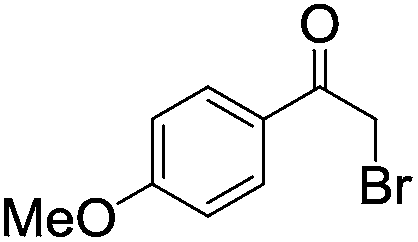
|
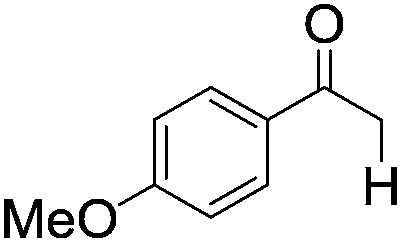
|
5 | >99 | 90 |
| 9d |
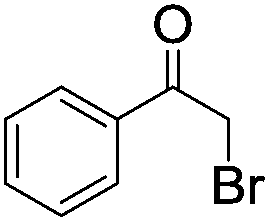
|
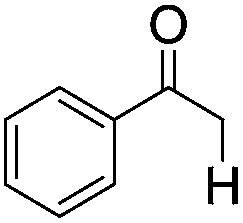
|
4 | <1 | — |
| 10e |
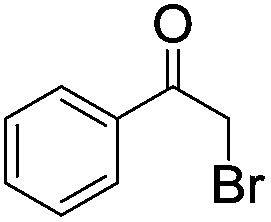
|
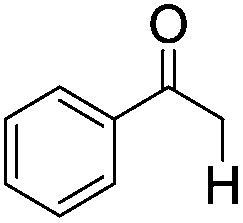
|
4 | <1 | — |

Five additional repeating experiments of the visible light induced dehalogenation of α-bromoacetophenone were carried out using B2-FL2-BBT under the same reaction conditions (Fig. S12 in the ESI†). The dehalogenated product was obtained in an almost quantitative manner, demonstrating the stability and reusability of B2-FL2-BBT as a photocatalyst. Blank experiments were also carried out as a control (entries 9 and 10). The same reaction with α-bromoacetophenone was carried out by using B2-FL2-BBT as a photocatalyst, but without light irradiation; or without B2-FL2-BBT but with light irradiation. The dehalogenated product, acetophenone, was barely obtained in either experiment, indicating that both components are indispensable.
We propose a reaction mechanism as shown in Fig. 3. Under visible light irradiation, the excited state of the conjugated porous polymer generates a hole via charge separation, which oxidizes the amine DIPEA, leading to a radical anion inside the porous polymer. The radical anion reacts with α-bromoacetophenone, generating the phenone radical, as the Br leaving group cleaves off. The hydrogen radical is then subsequently donated by the sacrificial hydrogen donor Hantzsch ester to produce the final product. The dehydrogenated Hantzsch ester radical reacts with the DIPEA radical cation, and ammonium bromide and pyridine ester are obtained as side products.
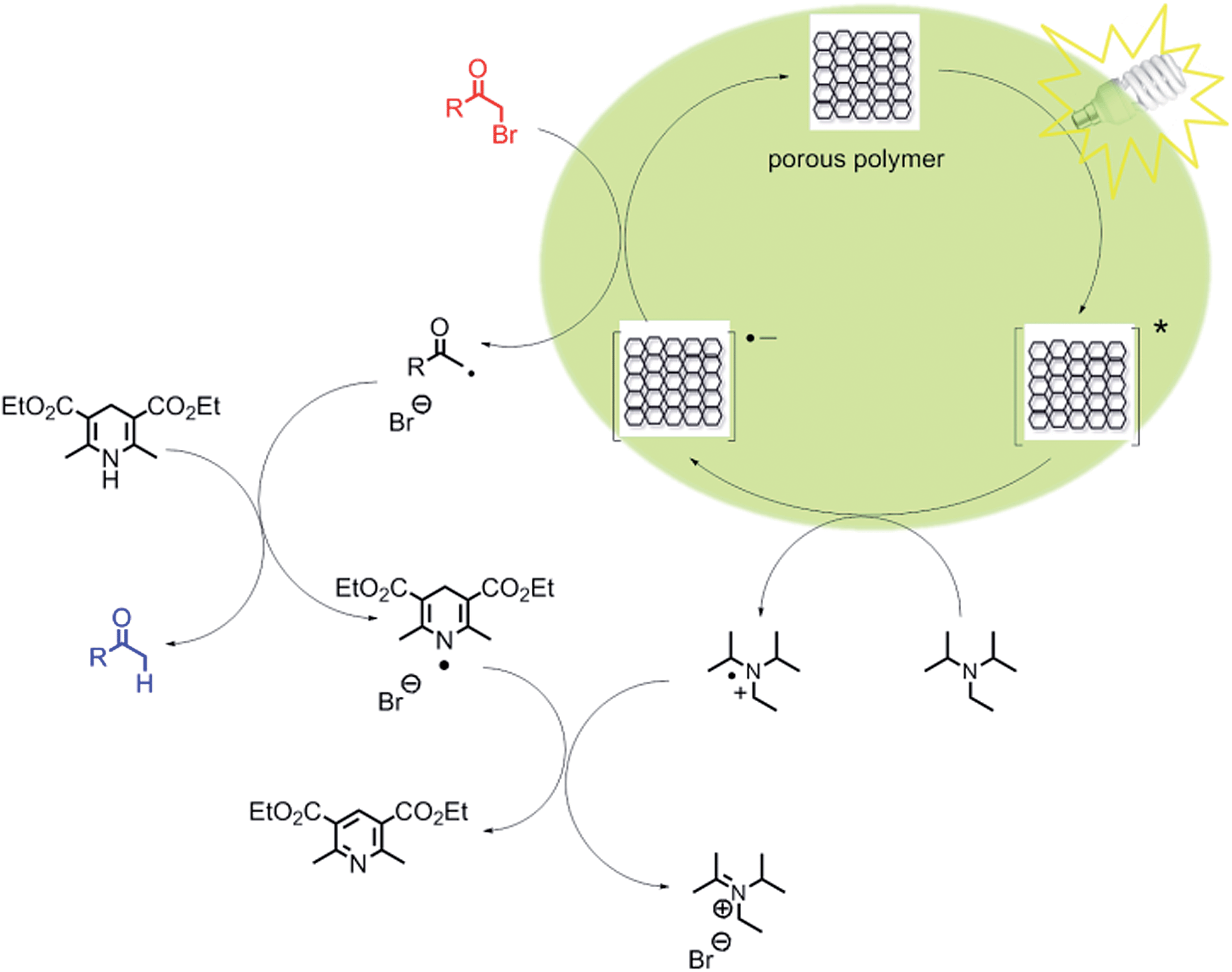 | ||
| Fig. 3 Proposed mechanism of the visible light-driven dehalogenation reaction using the conjugated porous polymers as heterogeneous photocatalysts. | ||
Inductively coupled plasma (ICP) spectroscopy was performed to determine the metal catalyst traces after the Sonogashira coupling reaction. For both B2-FL2-BBT and B2-FL3, traces of Pd (15 and 25 ppm) and Cu (∼20 ppm) were determined. To prove the origin of the photocatalytic activity, a short-chain linear polymer consisting of similar structure (see ESI†) was prepared and purified via chromatography for a homogeneous approach of the visible light-driven dehalogenation reaction. The ICP measurements showed very low Pd and Cu traces (<1 ppm). The dehalogenation reaction using the linear polymer as the homogeneous photocatalyst was carried out also in an almost quantitative manner. This indicates that the photocatalytic activity of the conjugated polymers does not stem from the minimal metal traces, but from the energetic level of the conjugated system. However, the drawback of using the soluble linear polymer is that photobleaching occurred during the dehalogenation reaction, shown by the decrease in the absorption maximum of the polymer by 27% (Fig. S12†). In comparison, the crosslinked porous polymer B2-FL2-BBT exhibited better stability.
Furthermore, the overall apparent quantum yield (AQY)31 of the reaction using B2-FL2-BBT as the photocatalyst was estimated to gain more information on its efficiency by using a single wavelength LED lamp (460 nm). The details are described in the ESI.† For this heterogeneous photocatalytic reaction, an overall AQY of 1.2% could be calculated according to the dehalogenation conversion.
Conclusions
In summary, we have developed a π-conjugated porous polymer via Sonogashira-coupling using the high internal phase emulsion technique as a heterogeneous, visible light-driven photocatalyst for the reductive dehalogenation of haloketones. The benzobisthiadiazole-based porous polymer offers a new class of robust visible light-driven heterogeneous catalyst systems, with strong and broad absorption from the visible to the near infra-red range. A household energy saving light bulb was used as the light source, which provides an environmentally sustainable and financially affordable solution for future catalytic processes. The heterogeneous photocatalyst can be easily recycled and reused without significant loss in photoactivity. Further studies of this highly versatile heterogeneous visible light driven photocatalyst system are expected to find more applications in more demanding catalytic processes.Acknowledgements
The Max Planck Society is acknowledged for financial support.Notes and references
- K. Kalyanasundaram, Coord. Chem. Rev., 1982, 46, 159–244 CrossRef CAS.
- A. Juris, V. Balzani, F. Barigelletti, S. Campagna, P. Belser and A. Vonzelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102–113 RSC.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- P. V. Pham, D. A. Nagib and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2011, 50, 6119–6122 CrossRef CAS PubMed.
- J. D. Nguyen, J. W. Tucker, M. D. Konieczynska and C. R. J. Stephenson, J. Am. Chem. Soc., 2011, 133, 4160–4163 CrossRef CAS PubMed.
- D. A. Nagib and D. W. C. MacMillan, Nature, 2011, 480, 224–228 CrossRef CAS PubMed.
- L. Furst, J. M. R. Narayanam and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2011, 50, 9655–9659 CrossRef CAS PubMed.
- T. P. Yoon, M. A. Ischay and J. Du, Nat. Chem., 2010, 2, 527–532 CrossRef CAS PubMed.
- J. W. Tucker, J. D. Nguyen, J. M. R. Narayanam, S. W. Krabbe and C. R. J. Stephenson, Chem. Commun., 2010, 46, 4985–4987 RSC.
- J. Du and T. P. Yoon, J. Am. Chem. Soc., 2009, 131, 14604 CrossRef CAS PubMed.
- M. L. Marin, L. Santos-Juanes, A. Arques, A. M. Amat and M. A. Miranda, Chem. Rev., 2012, 112, 1710–1750 CrossRef CAS PubMed.
- M. Neumann, S. Fuldner, B. Konig and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- H. J. Liu, W. Feng, C. W. Kee, Y. J. Zhao, D. Leow, Y. H. Pan and C. H. Tan, Green Chem., 2010, 12, 953–956 RSC.
- M. Neumann, S. Füldner, B. König and K. Zeitler, Angew. Chem., Int. Ed., 2011, 50, 951–954 CrossRef CAS PubMed.
- L. Chen, Y. Honsho, S. Seki and D. Jiang, J. Am. Chem. Soc., 2010, 132, 6742–6748 CrossRef CAS PubMed.
- A. I. Cooper, Adv. Mater., 2009, 21, 1291–1295 CrossRef CAS.
- Y. Chen, J. Zhang, M. Zhang and X. Wang, Chem. Sci., 2013, 4, 3244–3248 RSC.
- X. C. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76 CrossRef CAS PubMed.
- F. Vilela, K. Zhang and M. Antonietti, Energy Environ. Sci., 2012, 5, 7819–7832 CAS.
- K. Zhang, D. Kopetzki, P. H. Seeberger, M. Antonietti and F. Vilela, Angew. Chem., Int. Ed., 2013, 52, 1432–1436 CrossRef CAS PubMed.
- S. J. Pierre, J. C. Thies, A. Dureault, N. R. Cameron, J. C. M. van Hest, N. Carette, T. Michon and R. Weberskirch, Adv. Mater., 2006, 18, 1822–1826 CrossRef CAS.
- I. Pulko, J. Wall, P. Krajnc and N. R. Cameron, Chem.–Eur. J., 2010, 16, 2350–2354 CrossRef CAS PubMed.
- P. Krajnc, N. Leber, J. F. Brown and N. R. Cameron, React. Funct. Polym., 2006, 66, 81–91 CrossRef CAS PubMed.
- F. Su, C. L. Bray, B. Tan and A. I. Cooper, Adv. Mater., 2008, 20, 2663–2666 CrossRef CAS PubMed.
- M. Bokhari, R. J. Carnachan, N. R. Cameron and S. A. Przyborski, Biochem. Biophys. Res. Commun., 2007, 354, 1095–1100 CrossRef CAS PubMed.
- E. H. Mert, H. Yıldırım, A. T. Üzümcü and H. Kavas, React. Funct. Polym., 2013, 73, 175–181 CrossRef CAS PubMed.
- K. Zhang, Z. Vobecka, K. Tauer, M. Antonietti and F. Vilela, Chem. Commun., 2013, 49, 11158–11160 RSC.
- Z. J. Wang, S. Ghasimi, K. Landfester and K. A. I. Zhang, Chem. Commun., 2014, 50, 8177–8180 RSC.
- Z. J. Wang, K. Landfester and K. A. I. Zhang, Polym. Chem., 2014, 5, 3559–3562 RSC.
- T. Maschmeyer and M. Che, Angew. Chem., Int. Ed., 2010, 49, 1536–1539 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Monomer synthesis, FT-IR, fluorescence spectra, TGA, 1H, 13C, solid state 13C CP/MAS NMR and EPR spectra, N2 sorption isotherms and pore size distribution. See DOI: 10.1039/c4ta03887h |
| This journal is © The Royal Society of Chemistry 2014 |