
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Decomposition synthesis of tuneable, macroporous carbon foams from crystalline precursors via in situ templating†
D.
Ressnig
*a,
T.
Corbiere
a,
T.
Lunkenbein
b,
U.
Braun
c,
M. G.
Willinger
b and
M.
Antonietti
a
aMax Planck Institute of Colloids and Interfaces, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: Debora.Ressnig@mpikg.mpg.de
bFritz Haber Institute of the Max Planck Society, Department of Inorganic Chemistry, Faradayweg 4-6, 14195 Berlin, Germany
cBundesanstalt für Materialforschung und Prüfung, Unter den Eichen 87, 12205 Berlin, Germany
First published on 29th September 2014
Abstract
A flexible, sustainable, one-step thermal decomposition route for the synthesis of hierarchical, heteroatom doped carbon foams is presented. Task-specific semi-organic crystals combine functions for three different purposes: the carbon and heteroatom source, a foaming agent (CO2) and an in situ generable template (NaCl). Insights to the decomposition pathway were gained through FTIR/MS coupled TGA and an ultrafast out-of-furnace heating procedure and the products were analysed with (HR)SEM/TEM, EELS, FTIR, and N2 sorption. The resulting macroporous carbon foams are excellent supports for metallic nanoparticles due to their hierarchical structure, high surface area and tuneable heteroatom contents. This was demonstrated for catalytically active copper or the magnetic CoNi alloy for water purification.
Introduction
Heteroatom doped (O, N, S) carbons are ubiquitous and display promising applications in different fields including hydrogen storage,1 catalysis,2 supercapacitors,1b,3 or sensors,4 owing to their high structural and chemical versatility. As macroporous materials, they are excellent supports for heterophase catalysts due to their tuneable surface acidity/basicity, wettability, electrical properties, unhindered reactant flow and in particular high thermal and chemical stability.5 The presence of heteroatoms in carbon based support materials is of crucial importance for the post-synthetic functionalization with metal/metal oxide nanoparticles through wet-chemical processes. Localized electron density and topological irregularities provide well-dispersed anchor sites for metal cations. Purely graphitic carbons in contrast need typically to be treated with harsh oxidants prior to functionalization with similar procedures.5The design of monolithic carbons is attractive from the perspectives of straightforward processing, device implementation and material recycling. Flexible and conceptually simple templating methods have been successfully established for the preparation of monolithic, well-ordered carbons during the past decades.6 However, practical drawbacks reduce the incentive to follow these routes, given the laborious template preparation and post-synthesis etching steps that bear the risk to destroy the materials's microstructure, which in particular holds for heteroatom doped carbons.
A simplified alternative profits from the in situ formation of hard templates, generated through the decomposition of metal organic precursor complexes. The metal rich templates such as MgO,7 also in combination with Mo,8 CaC2/SrC2,9 or Fe3C,10 come with the advantage that they act as both moulds and catalysts for graphitization of external carbon sources at elevated temperatures. Graphitization improves the electrical conductivity but typically reduces the amount of structural heteroatoms and often produces a mixture of different nanostructures including carbon cages and nanotubes.7 The graphitic shell and the inherently low reactivity of these templates against most etching agents moreover hinders their removal significantly and prolongs etching procedures, which likely damage the material and influence its physicochemical properties. Nevertheless, in situ templating is a remarkably simple approach and finding non-graphitizing, water soluble templates is a crucial next step. We here introduce a straightforward, flexible method for the synthesis of macroporous carbons that relies on the in situ precipitation of NaCl crystallites during the thermal decomposition of semi-organic, crystalline precursors. Alkali halide salts come with the clear advantages over other hard templates of being low-cost and easily removable by simple washing with water, hence ecologically and economically friendly. Furthermore, salt fluxes, pure or as eutectic mixtures, have recently emerged as powerful porogens in the synthesis of meso- and microporous carbons.11 Our method covers the macroporous range in this cascade.
The thermal decomposition of solid precursors currently emerges as a simple top-down pathway to access structured bulk materials.12 These transformations are mainly of topological nature, where significant amounts of the initial parent crystals are evolved as gaseous decomposition products leaving a porous bulk behind. Most of the prepared materials are oxide-ceramics that find widespread applications owing to their favourable, often also hierarchical, structuration.13 The solid state polymerizations of halogenated acrylates to polyesters build another class of thermally induced topological transformations. The polymerization goes in hand with intercalation of metal halide crystals. This however is an excellent starting point to synthesise macroporous carbons via in situ templating. To do so, we conceptually extend the thermal polymerization to a decomposition approach, to access tuneable, continuous, macroporous carbon foams. Heteroatom doping is easily achieved through the right choice of the precursor crystals. Their intrinsically good wettability further allows the direct functionalization with metal nanoparticles via wet-impregnation techniques.
Experimental
Synthetic procedures
For the fast, out-of-furnace heating, a heat gun setup as described previously was used.14 The powders were filled to a quartz tube and directly heated to 430 °C under nitrogen flow (0.4 L min−1) with a total reaction time of 10 min.
For the thermal post treatment, the washed carbons were heated at a rate of 5 K min−1 to 1200 °C under a nitrogen flow (15 L min−1) in a Nabertherm tube furnace for 3 h.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) for 1 h with magnetic stirring. The supernatant was removed by filtration and the solid was dried at 80 °C in air. The powders were subsequently heated to 600 °C (10 K min−1) under a nitrogen flow (15 L min−1) and directly cooled.
:1) for 1 h with magnetic stirring. The supernatant was removed by filtration and the solid was dried at 80 °C in air. The powders were subsequently heated to 600 °C (10 K min−1) under a nitrogen flow (15 L min−1) and directly cooled.
Characterization methods
FTIR spectra were collected in ATR geometry on a VARIAN 1000 spectrometer, Scimitar Series (FTS 1000). Elemental analysis was performed with an Elementar vario MICRO cube. Powder X-ray diffraction patterns were measured with copper Kα (1.54 Å) radiation on a Bruker D-8 device, equipped with a scintillation detector in reflection mode. Morphology observations and EDX measurements were performed on a JEOL (JSM – 7500 F) instrument, equipped with an X-MAX 80 mm2 detector from Oxford Instruments. Nitrogen sorption isotherms were recorded using a Quantachrome Quadrasorb SI porosimeter with N2 at 77 K, after degassing at 150 °C for 20 hours. Absorption branch points (p/p0 < 0.1) were applied in the BET model to calculate the apparent surface area.Thermogravimetric measurements were performed on a Netzsch thermal analysis device TG 209 F1 under a flow of nitrogen and with a heating rate of 10 K min−1. Transmission electron microscopy (TEM) and electron energy loss spectroscopy (EELS) were recorded on a Cs corrected FEI Titan 80-300 with an acceleration voltage of 80 kV and 300 kV. No beam induced damage of the sample was observed during measurements.
Results and discussion
For the one-step preparation of a porous carbon from an ionic crystal, the precursor must generally include a metal cation and an organic, carbonizable residue that is functionalized with the anion of the target porogen. Sodium monochloroacetate (NaAc-Cl) is one of the simplest of these systems as it consists of sodium and chloride as template cation and anion respectively, separated by an acetate residue as the minimal organic subunit (Fig. 1). Our other precursors, sodium-2-chloropyridine-3-carboxylate (NaPyr-Cl) and chloramine-T (CT) are in contrast rich in sp2 carbon and provide heteroatoms (S, N) for direct in situ doping (Fig. 1). NaAc-Cl and CT are low cost and commercially available, while NaPyr-Cl was prepared by the deprotonation of acetic acid with NaOH in a water/acetone mixture, followed by slow crystallization (cf. experimental section). | ||
| Fig. 1 TGA curves, recorded with 10 K min−1 under a flow of N2, track the thermal decomposition of sodium-2-monochloroacetate (1 NaAc-Cl), sodium-2-chloropyridine-3-carboxylate (2 NaPyr-Cl) and N-chloro-p-toluenesulfonamide sodium salt (3 CT). | ||
The finely-crushed precursor powders were heated at a rate of 10 K min−1 to 550 °C for 30 min under an inert nitrogen atmosphere for the decomposition synthesis. The three corresponding thermogravimetric analysis (TGA) curves are compared in Fig. 1. For NaAc-Cl a single, but pronounced weight loss sets in at around 240 °C (46 wt%). For NaPyr-Cl, a pre-step starts at 55 °C (6 wt%), which corresponds to evaporating acetone (b.p. 58 °C) that remains from the deprotonation/crystallization reaction. A pronounced weight loss sets in at 274 °C (35 wt%), while CT starts to decompose at 185 °C (41 wt%). The mass remaining at 550 °C (i.e. after subtraction of NaCl) is higher for NaPyr-Cl (28 wt%) and CT (34 wt%) products compared to NaAc-Cl (4 wt%), consistent with the molecular structure of the precursors.
The general synthetic procedure is scalable and the inset in Fig. 2a shows the product of 20 g NaAc-Cl precursor. The initial mass was reduced by 95% to 0.79 g after decomposition and removal of NaCl by repeated washing with water. All three precursors yield stable, easily processable monoliths, which are truncated with pores in the milli- micrometre ranges. Scanning electron microscopy (SEM) images show that the monoliths possesses hierarchical properties as removal of NaCl generates cubic shaped pores in the micro- nanometre ranges. This implies that the in situ generated NaCl crystals are replicated as hard endo-templates and excludes significant intermediate formation processes in a salt melt, which would lead to the creation percolation networks with smaller, less defined porosity.11 The typical edge length of the cavities depend on the starting compounds and range from 200 nm up to approximately 2 μm for NaAc-Cl, from 80 nm to 250 nm for NaPyr-Cl and from 100 nm to 350 nm for CT. Faster decomposition – as described below – prohibits the diffusion and growth of NaCl crystallites by immediate inclusion to the carbon matrix and hence, leads to formation of smaller more uniform cubic voids of 0.5 μm to 1 μm for NaAc-Cl (Fig. S1†). The thickness of carbon scaffold is defined by the ratio of template to organic residue in the precursor compounds. The finest features are therefore obtained from NaAc-Cl with a width of 10 nm to 25 nm, as it consists of just one available carbon atom per NaCl unit. The decomposition of NaAc-Cl was additionally executed by applying an ultrafast (10 min) out-of-furnace heating technique that can be visually followed (cf. experimental section). It was observed that the precursor passes a liquid stage during the transformation, which may occur just locally along the reaction front or within the entire reactant volume depending on the amount of material. The subsequent carbonization goes in hand with a drastic volume expansion due to the evolution of volatile products (Fig. 2e).
 | ||
| Fig. 2 SEM images of thermally decomposed (a and b) NaAc-Cl, (c) NaPyr-Cl and (d) CT. The inset in (a) shows a macroscopic foam synthesized from 20 g NaAc-Cl. (e) Photograph of NaAc-Cl powders before and after the thermal decomposition with the heat-gun setup illustrate the drastic volume expansion. In addition, functions of different precursor moieties are described at the example of NaAc-Cl. | ||
The volatile decomposition products of NaAc-Cl were analysed with FTIR or MS coupled TGA (Fig. S2 and S3†). The mass change from the decomposition step at around 300 °C can be attributed to the loss of mainly carbon dioxide and water according to MS. FTIR further indicates formation of acetic acid and other hydrocarbon components, likely small condensed molecular units. The above data suggest that the nucleation of NaCl, which is thermodynamically favoured, occurs quickly after reaching a certain temperature threshold and in a direct solid–solid fashion. The precipitation of NaCl forces the remaining organic fragments to a liquid state, thus allowing the formation of continuous monoliths. These highly reactive organic fragments undergo polymerization, dehydration and decarboxylation reactions to form the resulting monolithic carbons within just seconds. The evolution of volatile by-products has an additional foaming effect to generate hierarchic structures. The majority of the evolved gas is CO2, which is a well-known industrial foaming agent.15 A carbon/template/foaming agent ratio of (1/1/1) appears to be favourable to the formation of light weight monoliths with an ultrathin scaffold structure.
The carbon foam derived from NaAc-Cl features an apparent surface area SBET of 483 m2 g−1 with a total pore volume of 0.57 cm2 g−1, which can be attributed to the contribution of micropores and mesopores with a broad size distribution (Fig. S3†). This surface area is twice as high as for example carbon aerogels synthesized by hydrothermal routes.16 The apparent SBET surface areas of NaPyr-Cl (15 m2 g−1) and CT (8 m2 g−1) are significantly lower.
The FTIR spectra give insight to remaining functional groups after carbonization (Fig. 3). The absence of aromatic or aliphatic C–H stretches in the region of 3000 cm−1 points to a high degree of aromatization. The band at 1702 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) is relatively weak for NaAc-Cl, and even absent for NaPyr-Cl, indicating that most of the carbonyl functionalities were eliminated in the course of the reaction. The pronounced band at 1590–1580 cm−1 is present in all three spectra and typical for aromatic (CC) valences. The regions in-between 1500 cm−1 and 1050 cm−1 correspond to an overlap of vibrations from (hetero-)aromatic systems with alkoxy (C–O–C) groups, and the sharp bands <1000 cm−1 are characteristic for the fingerprint region of aromatics.17 The band at 1146 cm−1 in the CT spectrum corresponds to sulfonamide groups (–SO2N) and consequently the absorptions between 770 cm−1 and 618 cm−1 to (S–N) and (S–C) vibrations.18
O stretch) is relatively weak for NaAc-Cl, and even absent for NaPyr-Cl, indicating that most of the carbonyl functionalities were eliminated in the course of the reaction. The pronounced band at 1590–1580 cm−1 is present in all three spectra and typical for aromatic (CC) valences. The regions in-between 1500 cm−1 and 1050 cm−1 correspond to an overlap of vibrations from (hetero-)aromatic systems with alkoxy (C–O–C) groups, and the sharp bands <1000 cm−1 are characteristic for the fingerprint region of aromatics.17 The band at 1146 cm−1 in the CT spectrum corresponds to sulfonamide groups (–SO2N) and consequently the absorptions between 770 cm−1 and 618 cm−1 to (S–N) and (S–C) vibrations.18
 | ||
| Fig. 3 FTIR spectra of carbons derived from different semi-organic precursor crystals. | ||
The composition of the different foams was deduced by elemental analysis (Table 1) and identifies carbon as major component. Negligible amounts of hydrogen are present in NaAc-Cl foams and 3 wt% remain in NaPyr-Cl or CT products. The contents of oxygen heteroatoms range above 10 wt%. Nitrogen and sulphur could also be successfully incorporated to the foams. The nitrogen/carbon ratio in NaPyr-Cl is 0.2, which is equal to the value of pure pyridine pointing towards high stability of the aromatic ring structure during the decomposition. Also the (–SO2–) unit appears to remain intact according to the constant atomic ratio of oxygen to sulfur and FTIR results.
| [Wt%] | C | N | H | S | O |
|---|---|---|---|---|---|
| NaAc-Cl 550 | 79 | <1 | 1 | <1 | 15 |
| NaAc-Cl 1200 | 95 | <1 | <1 | <1 | 5 |
| NaPyr-Cl | 70 | 14 | 3 | <1 | 13 |
| CT | 63 | 7.5 | 3.5 | 11 | 14 |
Further modifications will be discussed in depth for the for NaAc-Cl derived foams as representative system. The surface polarity of the carbon monoliths was switched from hydrophilic to hydrophobic through elimination of the oxygen groups by thermal post-treatment (Fig. 4a). The oxygen content was thereby reduced from 15 wt% to <5 wt% (Table 1). Restructuration through loss of oxygen affects the texture of the material and reduces the apparent surface area from 483 m2 g−1 to 296 m2 g−1, whilst maintaining the cubic porous structure (Fig. 4b and S5†). The long and short-range connectivity motifs of carbon are essential for defining the electronic properties, which becomes more relevant for the thermally reduced carbons. The atomic binding state was evaluated by electron energy loss spectroscopy (EELS) (Fig. 4c). The EELS spectra display energy loss peaks at 286 eV and 294 eV that are characteristic of the 1s to π* and σ* transitions of sp2 hybridized carbon.19 The relative strength of the π* and the high π* to σ* ratio further verifies the predominance of graphitic bound carbon.20 No long-range atomic order can be determined from HRTEM analyses, as expected from fast polymerizing fragments (Fig. 4d). Nevertheless, a pattern of prolated structures is indicative for the formation of small graphitic platelets. Moreover, the selected area diffraction pattern (SAED) shows two diffuse cycles that reflect the basal and edge plain stacking distances of graphite, pointing towards minimal short range atomic order.
 | ||
| Fig. 4 (a) The surface polarity is switched from polar to apolar by thermal post treatment at 1200 °C. (b) TEM analysis indicates that the cubic pore shape is retained in the thermal post treatment. (c) Two representative EELS spectra of post-treated foams depict the predominance of sp2 configured carbon. (d) HRTEM analysis shows atomic orientation into small graphitic platelets with no significant long range order. The inset shows the corresponding SAED pattern. | ||
The high sensitivity of the thin carbon layers unfortunately does not allow for the collection of meaningful Raman spectra for further structure elucidation. The electronic conductivity of carbon foams is a function of a complex interplay of structural and density sensitive factors and typically affected by meso- and macroporosity.21 The conductivity was evaluated with impedance spectroscopy (1–1000 Hz) using a two electrode setup and accounts for the thermally post-treated samples 0.85 S m−1 (graphitic powder, experimental: 2.66 S m−1).
Functionalization
The carbons were functionalized by a wet-chemical procedure through impregnation with aqueous solutions of Cu(NO3)2 or Co/Ni(NO3)2 (0.3 M). The presence of oxygen heteroatoms is essential to the wettability and cation adsorption, hence defining the resultant degree of dispersion of the metal nanoparticles. Reduction of the metal cations was achieved by thermal treatment at 600 °C. Carbon materials feature the advantage over other supports (e.g. zeolites or metal oxides), that they act as both reductant and stabilizers for the metal nanoparticles.22 HRSEM and TEM analyses confirm that metallic particles are homogeneously dispersed over the entire carbon foam (Fig. 5). The CoNi alloy forms particles of approximately 20 nm in diameter. The powder X-ray diffraction (PXRD) reflections are shifted versus the references of pristine Co or Ni to intermediate 2θ positions, indicative for the adaption of the crystal lattice in accordance to formation of an alloy (Fig. S6†). The deposited copper particles show a larger size variation between approximately 50 nm and 100 nm (Fig. 5b). Also here, PXRD reflections can be clearly assigned to metallic copper (Fig. S6†).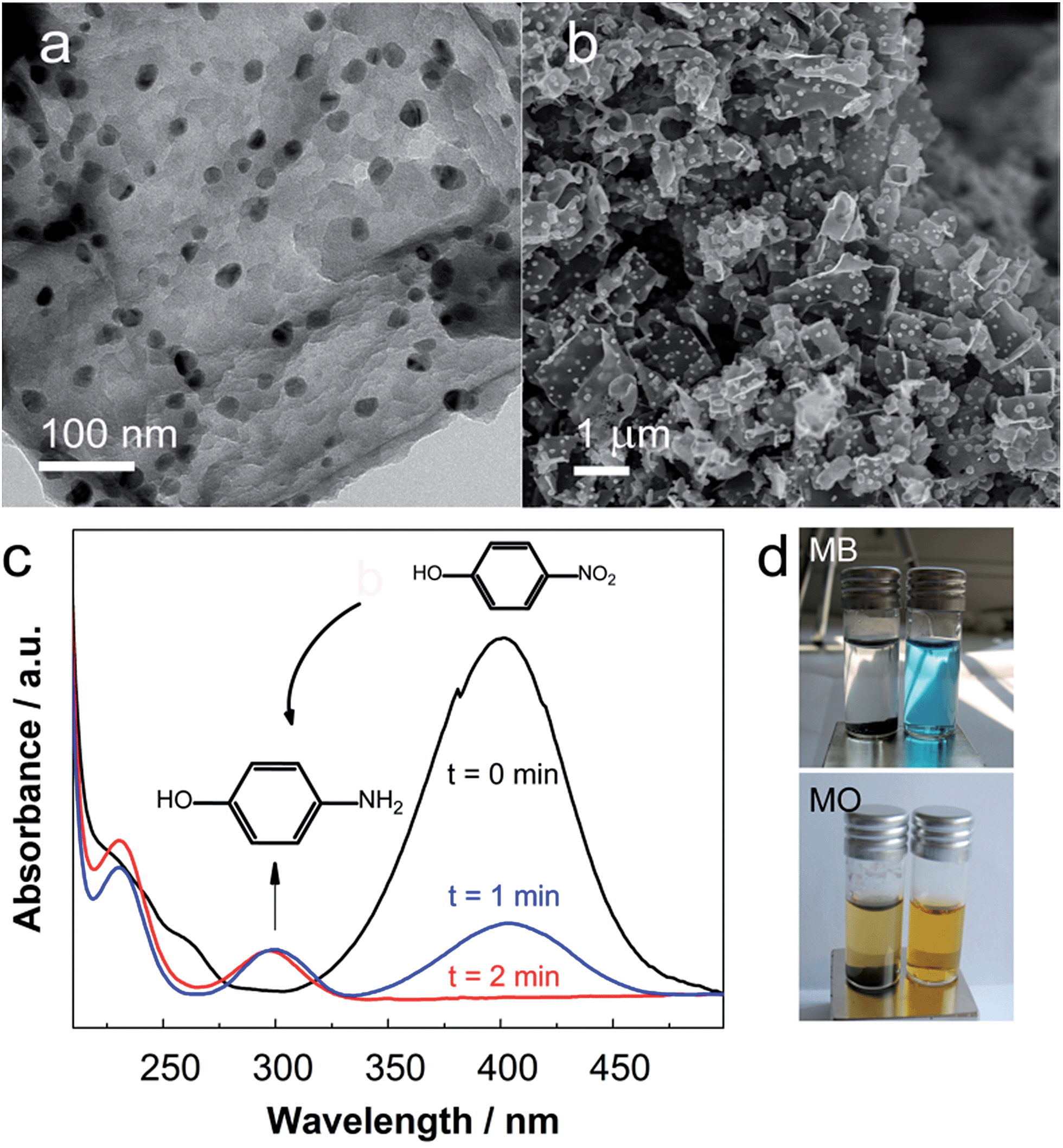 | ||
| Fig. 5 (a) TEM image of a CoNi/C composite. (b) SEM image of a Cu/C composite. (c) The UV-vis spectra track the reduction of p-nitrophenolate catalysed by Cu/C. (d) Adsorption of methylene blue (MB) or methyl orange (MO) to CoNi/C. The photographs compare pristine solutions (right) with solution after dye adsorption and magnetic separation of the adsorbent (left). | ||
The catalytic activity and accessibility of the deposited copper nanoparticles was approved with the catalytic reduction of p-nitrophenolate in the presence of NaBH4.23 The transformation can be easily tracked with UV-vis spectroscopy by following the disappearance of the absorption band at 400 nm and appearance of the band at 300 nm corresponding to p-aminophenolate (Fig. 4c). The hydrophilic character of the oxygen functionalized carbon is here not only an advantage but rather essential, to disperse the composite in water.
Magnetic carbon composites have great potential for application in different fields, e.g. as magnetic toners, as ferrofluids, magnetic resonance imaging or for environmental issues.24 Water purification is a major concern due to the increased pharmaceutical waste that accumulates in the water cycle and drastically disturbs ecological systems.25
In a proof of principle we demonstrate adsorption of organic dyes as waste model systems. The CoNi modified carbons adsorb pollutants of different polarities, with predominance for cationic species (Fig. 4d). Knowing that macropores are dominant, which are characteristically not molecule traps, it can be assumed that surface defects and composition are relevant for the adsorption of the dyes. The NiCo/C monoliths can be easily recovered through magnetic separation.
Conclusions
We present a versatile, one-step route to synthesize heteroatom rich, hierarchical macroporous carbon foams via the thermal decomposition of semi-organic crystals without the need for external templates. The key is that the precursor crystals are functionalized for three main purposes: (1) with the carbon and heteroatom source, (2) with carboxylate that generates CO2 as foaming agent, (3) with a chloride group and a sodium cation as in situ generable template (NaCl). Phase separation, i.e. nucleation of NaCl, is thermodynamically favoured and allows carbonization at comparably low temperatures, which offers optimal conditions to maintain structural heteroatoms. The amounts of heteroatoms can therefore be adjusted through thermal post-treatments. We further demonstrate straightforward functionalization with metal nanoparticles and achieve high dispersion due to the presence of oxygen in the pristine carbon foam. The metal/carbon composites may be used for different purposes from which catalysis and waste water treatments are just two options.The absolute porosity of the foams as well as mechanical properties remain to be elucidated with regards to specific applications. Moreover, there is still much more room to investigate other crystalline precursors e.g. through variation of the alkali halide or organic unit to obtain products with different structures, morphologies and properties.
Notes and references
- (a) Y. Xia, Z. Yang and Y. Zhu, J. Mater. Chem. A, 2013, 1, 9365 RSC; (b) M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250 RSC.
- (a) X.-K. Kong, C.-L. Chen and Q.-W. Chen, Chem. Soc. Rev., 2014, 43, 2841 RSC; (b) T.-P. Fellinger, F. Hasché, P. Strasser and M. Antonietti, J. Am. Chem. Soc., 2012, 134, 4072 CrossRef CAS PubMed.
- G. Wang, L. Zhang and J. Zhang, Chem. Soc. Rev., 2012, 41, 797 RSC.
- (a) S. Peng and K. Cho, Nano Lett., 2003, 3, 513 CrossRef CAS; (b) F. Villalpando-Páez, A. H. Romero, E. Muñoz-Sandoval, L. M. Martınez, H. Terrones and M. Terrones, Chem. Phys. Lett., 2004, 386, 137 CrossRef PubMed.
- J. Zhu, A. Holmen and D. Chen, ChemCatChem, 2013, 5, 378 CrossRef CAS.
- (a) H. Nishihara and T. Kyotani, Adv. Mater., 2012, 24, 4473 CrossRef CAS PubMed; (b) A. H. Lu and F. Schüth, Adv. Mater., 2006, 18, 1793 CrossRef CAS; (c) A. Thomas, F. Goettmann and M. Antonietti, Chem. Mater., 2008, 20, 738 CrossRef CAS.
- (a) K. Xie, X. Qin, X. Wang, Y. Wang, H. Tao, Q. Wu, L. Yang and Z. Hu, Adv. Mater., 2012, 24, 347 CrossRef CAS PubMed; (b) S. Chen, J. Bi, Y. Zhao, L. Yang, C. Zhang, Y. Ma, Q. Wu, X. Wang and Z. Hu, Adv. Mater., 2012, 24, 5593 CrossRef CAS PubMed.
- M. Motiei, J. Calderon-Moreno and A. Gedanken, Adv. Mater., 2002, 14, 1169 CrossRef CAS.
- Y. Saito and T. Matsumoto, Nature, 1998, 392, 237 CrossRef CAS PubMed.
- J. N. Wang, L. Zhang, J. J. Niu, F. Yu, Z. M. Sheng, Y. Z. Zhao, H. Chang and C. Pak, Chem. Mater., 2007, 19, 453 CrossRef CAS.
- (a) N. Fechler, T.-P. Fellinger and M. Antonietti, Adv. Mater., 2013, 25, 75 CrossRef CAS PubMed; (b) X. Liu, N. Fechler and M. Antonietti, Chem. Soc. Rev., 2013, 42, 8237 RSC.
- H.-D. Yu, M. D. Regulacio, E. Ye and M.-Y. Han, Chem. Soc. Rev., 2013, 42, 6006 RSC.
- (a) O. Herzberg and M. Epple, Eur. J. Inorg. Chem., 2001, 1395 CrossRef CAS; (b) M. Epple and O. Herzberg, J. Mater. Chem., 1997, 7, 1037 RSC.
- D. Ressnig, G. Clavel, N. Scharnagl and M. Antonietti, Part. Part. Syst. Charact., 2014, 31, 557 CrossRef CAS.
- L. J. M. Jacobs, M. F. Kemmere and J. T. F. Keurentjes, Green Chem., 2008, 10, 731 RSC.
- S.-A. Wohlgemuth, R. J. White, M.-G. Willinger, M.-M. Titirici and M. Antonietti, Green Chem., 2012, 14, 1515 RSC.
- T. Hartono, S. Wang, Q. Ma and Z. Zhu, J. Colloid Interface Sci., 2009, 333, 114 CrossRef CAS PubMed.
- C.-O. Argelia, M.-R. Christian, O.-S. Fabián, M.-H. Sheila, B.-G. Virgilio and R. Gildardo, Quím. Nova, 2011, 34, 787 Search PubMed.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893 RSC.
- K. A. Katrinak, P. Rez and P. R. Buseck, Environ. Sci. Technol., 1992, 26, 1967 CrossRef CAS.
- N. Amini, PhD Thesis, University of London, 2010 Search PubMed.
- (a) J. Zhu, A. Holmen and D. Chen, ChemCatChem, 2013, 5, 378 CrossRef CAS; (b) G. Chieffi, C. Giordano, M. Antonietti and D. Esposito, J. Mater. Chem. A, 2014, 2, 11591 RSC; (c) J. M. Campelo, D. Luna, R. Luque, J. M. Marinas and A. A. Romero, ChemSusChem, 2009, 2, 18 CrossRef CAS PubMed.
- P. Herves, M. Perez-Lorenzo, L. M. Liz-Marzan, J. Dzubiella, Y. Lu and M. Ballauff, Chem. Soc. Rev., 2012, 51, 5577 RSC.
- M. Zhu and G. Diao, Nanoscale, 2011, 3, 2748 RSC.
- (a) D. G. J. Larsson, Upsala J. Med. Sci., 2014, 119, 108 CrossRef PubMed; (b) S. Kaplan, Crit. Rev. Environ. Sci. Technol., 2013, 43, 1074 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: SEM images for NaAc-Cl derived pristine and functionalized foams and nitrogen sorption isotherms. FTIR/MS coupled TGA analyses of the thermal decomposition of NaAc-Cl. See DOI: 10.1039/c4ta03646h |
| This journal is © The Royal Society of Chemistry 2014 |