
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hybrid acrylic/CeO2 nanocomposites using hydrophilic, spherical and high aspect ratio CeO2 nanoparticles†
Miren
Aguirre
a,
Eric
Johansson Salazar-Sandoval
bc,
Mats
Johansson
c,
Anwar
Ahniyaz
b,
Maria
Paulis
*a and
Jose Ramon
Leiza
*a
aPOLYMAT and Kimika Aplikatua saila, University of the Basque Country UPV/EHU, Joxe Mari Korta Zentroa, Tolosa Hiribidea 72, 20018 Donostia-San Sebastián, Spain. E-mail: jrleiza@ehu.es; maria.paulis@ehu.es
bSP Technical Research Institute of Sweden, Chemistry, Materials and Surfaces, Box 5607, SE-114 86, Stockholm, Sweden
cDepartment of Fibre and Polymer Technology, School of Chemical Science and Engineering, KTH Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 15th October 2014
Abstract
A dispersion of CeO2 nanoparticles and nanorods stabilized with nitrilotriacetic acid (NTA) and a 4,4′-azobis(4-cyanovaleric acid) (V-501) initiator has been used to initiate the emulsion polymerization of acrylic monomers, yielding stable hybrid CeO2 nanoparticle-nanorod/polyacrylate latexes for the first time. Films cast from these hybrid latexes are transparent due to the very homogenous distribution of the polymer compatibilized CeO2. Furthermore, it has been proven that the UV-Vis absorption capacity of the hybrid latexes is enhanced with the incorporation of the nanorods.
Introduction
It is well-known that inorganic nanomaterials have unique properties and they, when combined appropriately with polymers, produce hybrid materials with substantially better performance (mechanical, optical, electrical…).1–4 In this context, cerium oxide possesses interesting properties such as high UV-Vis absorbance capacity.5 Thus, hybrid latexes with cerium oxide nanoparticles are potential candidates for the outdoor coating industry and for hybrid organic/inorganic photoactive layers in solar cells. Several studies have already reported the synthesis of CeO2/polymer hybrids for UV protection purposes.6–8 However, all of them were produced with 3D CeO2 nanoparticles; namely, spherical nanoparticles. The use of CeO2 in photovoltaic applications has been scarce due to its wide band gap (≈3.19 eV), but recently doping or nanostructuration of this rare earth oxide has led to open the path to photoelectrochemical applications.9High aspect ratio materials attract more attention due to the fact that smaller amounts of these materials are enough to obtain similar characteristics.10 Those materials exhibit great optical, mechanical, catalytic, and electrical properties, and can be applied in a wide range of applications.11–13 In the last few years, 1-D ceria nanostructures with different morphologies, for example nanorods, nanowires and/or nanotubes, have been synthesized using a wide range of methods.14–18
Recently, it has been reported that with the combination of geometrically different nanofillers in a polymeric matrix, synergetic effects could be achieved, for instance, carbon nanotubes (CNTs) with carbon black,12 few-layered graphene with CNTs and nanodiamonds,19 graphene and CNTs,20 PbSxSe1−x quantum dots and nanorods21 and also two different high aspect ratio materials: clay and CNTs.22 For all of them a remarkable enhancement in the properties of the hybrids was observed.
Although many synthesis and surface modification methods for water dispersible ceria nanoparticles have been studied,23–25 there have been many challenges in the successful incorporation of ceria nanoparticles into the polymer structure;26,27 for example, in clear coatings, one of the key challenges is the stability of the paints formulated with ceria nanoparticles and in hybrid solar cells, the power conversion efficiency is limited by the large-scale aggregation of the nanoparticle phase.28 Therefore, the inherent instability of inorganic particles in the organic matrix makes it rather difficult to obtain stable dispersions from the physical mixture of inorganic nanoparticles and organic binders which often result in hazy films due to aggregation of nanoparticles.29 As a result, in situ incorporation of ceria nanoparticles and other inorganic nanoparticles into the polymer particles during the polymerization process has been pursued in the last decade in order to homogeneously distribute the inorganic nanoparticles in the polymeric film.30–36
Several approaches to produce waterborne hybrid CeO2/polymer coatings have been reported. We have recently shown the efficiency of a conventional seeded semibatch emulsion polymerization method for the encapsulation of CeO2 nanoparticles in waterborne polymer particles.7 Hydrophobic CeO2 nanoparticles were used and miniemulsion polymerization was employed to produce the seed hybrid particles. The monomer added under starved conditions buried the CeO2 nanoparticles incorporated into the seed particles, producing a high homogeneity of nanoparticle dispersion in the final polymeric film. Other authors used macro-RAFT modified CeO2 nanoparticle dispersions to start the polymerization.35–37 These modified particles were used as seeds in an emulsion polymerization reaction that yielded a homogeneous distribution of the nanofillers in the final latex. The advantage of the hybrid latexes with encapsulated morphology with respect to hybrids produced by physically blending the bare dispersions of the inorganic and organic materials is that the possible aggregation of ceria nanoparticles during the storage period of the paint or film formation is prevented.
In this work, a dispersion of semiconductor cerium oxide nanoparticles (NPs)/nanorods (NRs) has been synthesized. During the synthesis of the CeO2 nanoparticle dispersion in aqueous media, capping agents are usually added to stop the growth and hence, control the size of the nanoparticles. Generally, these capping agents (e.g. nitriloacetic acid, NTA; see the structure in Fig. 1)38 contain carboxylic groups that are adsorbed onto the surface of the growing inorganic nanoparticles stopping their growth and providing stability to the nanoparticle dispersion. In this work, in order to enhance the compatibility and dispersability of the inorganic material in the polymer matrix, the possibility of modifying the surface of CeO2 nanoparticles by adding carboxylic acids containing initiator moieties that can initiate the polymerization from the surface of CeO2 nanoparticles was considered. Particularly, the acidic initiator 4,4′-azobis(4-cyanovaleric acid) (V501) was used (see Fig. 1) together with the NTA capping agent.
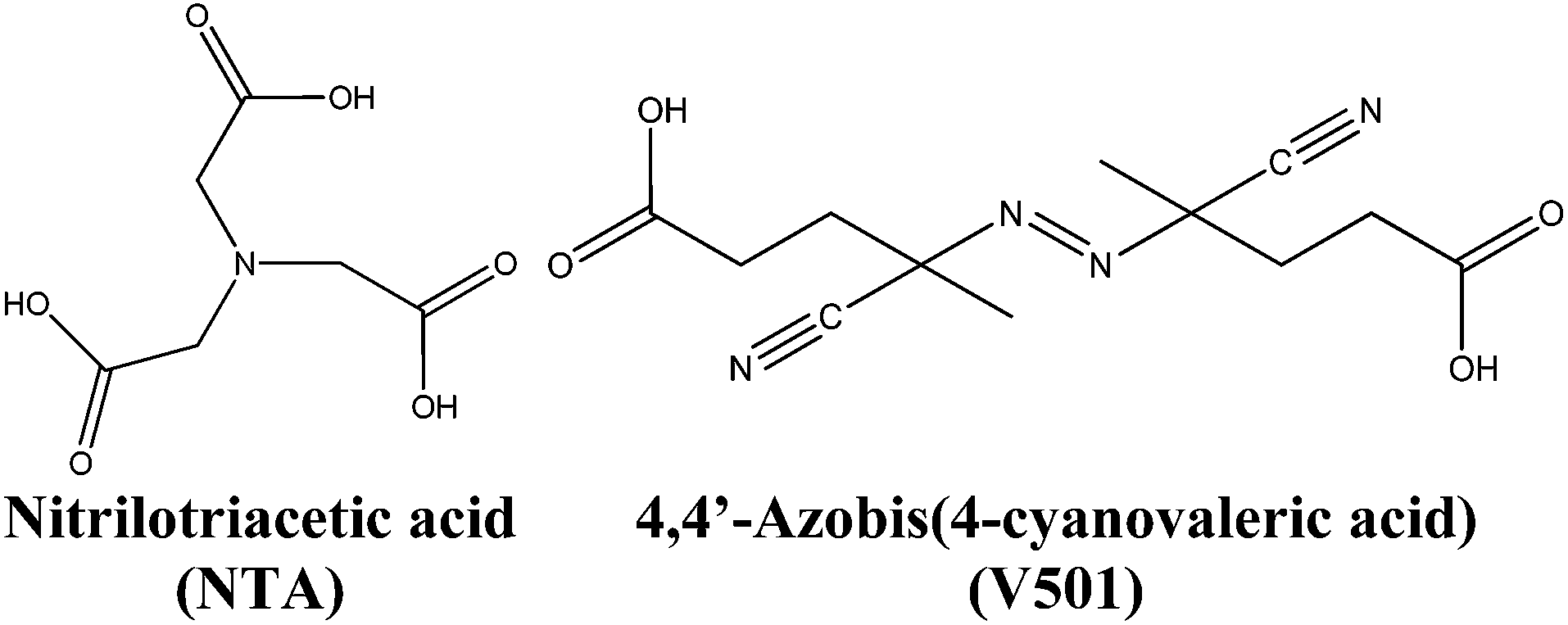 | ||
| Fig. 1 Chemical structures of NTA and V501 molecules. | ||
Then, the polymerization from the surface of CeO2 nanoparticles was carried out in the water phase in order to compatibilize the organic and the inorganic phases. Therefore the dispersion of the CeO2 NPs/NRs was used as the seed and initiator to produce hybrid acrylic latexes by emulsion polymerization. This is the first time that acrylic/CeO2 nanorod/nanoparticle hybrids are reported in the literature. The stability of the final hybrid latexes, together with their final morphology was analyzed. Furthermore, the morphology and UV absorption capacity of hybrid polymeric films containing CeO2 NPs/NRs were compared to those containing CeO2 nanoparticles alone.
Experimental
Materials
Cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O), ammonium hydroxide solution (28.0–30.0%), nitrilotriacetic acid (NTA, 98+% ACS reagent), sodium nitrilotriacetate (Na3NTA, Sigma grade) and 4,4′-azobis(4-cyanovaleric acid) (V501) were purchased from Sigma-Aldrich and together with hydrogen peroxide (30%, Merck) were used for the synthesis of the cerium oxide dispersion. Methyl methacrylate, MMA (Quimidroga) and n-butyl acrylate, BA (Quimidroga) were used as monomers. Dodecyl diphenyloxide disulfonate (Dowfax 2A1 45%, Dow Chemicals) was used as the anionic emulsifier. Double distilled water was used for the synthesis of cerium oxide dispersion whereas deionized water was used in the emulsion polymerization.Synthesis of CeO2 nanoparticle dispersions
The synthesis of hydrophilic nanoparticle/nanorod (NP/NR) dispersion was carried out as follows:30 g of Ce (NO3)3·6H2O were added to 300 mL of H2O and stirred until complete dissolution. Then, 24 g of NH4OH solution (28 wt%) were added to precipitate Ce(OH)x. After centrifuging this mixture and eliminating the supernatant, the cerium hydroxide cake was redispersed in 1.5 L of H2O previously heated at 85 °C using magnetic and Ultra-Turrax® stirring at high revolutions per minute (ca. 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm). The temperature was set to 88 °C while magnetic and Ultra-Turrax® stirring was continued. When the cake chunks were not seen any longer, 4 g of NTA and 2 g of Na3NTA were added to the reaction batch and it was left under stirring for 15 minutes. After obtaining a homogeneous system, 8 g of H2O2 solution (30 wt%) were added to oxidize cerium hydroxide (Ce(OH)x) to cerium dioxide (CeO2). 45 minutes after the addition of the peroxide, 3 g of Na3NTA were added to the dispersion. In this way the growth of CeO2 particles was stopped by the NTA capping agent. During the following 15 minutes, stirring and temperature were maintained. Then, both heating and Ultra-Turrax® stirring were turned off, while magnetic stirring was continued to help cooling down the system.
000 rpm). The temperature was set to 88 °C while magnetic and Ultra-Turrax® stirring was continued. When the cake chunks were not seen any longer, 4 g of NTA and 2 g of Na3NTA were added to the reaction batch and it was left under stirring for 15 minutes. After obtaining a homogeneous system, 8 g of H2O2 solution (30 wt%) were added to oxidize cerium hydroxide (Ce(OH)x) to cerium dioxide (CeO2). 45 minutes after the addition of the peroxide, 3 g of Na3NTA were added to the dispersion. In this way the growth of CeO2 particles was stopped by the NTA capping agent. During the following 15 minutes, stirring and temperature were maintained. Then, both heating and Ultra-Turrax® stirring were turned off, while magnetic stirring was continued to help cooling down the system.
When the temperature of the dispersion was below 30 °C (to avoid the decomposition of the thermal initiator), 1.2 g of V501 (acidic azo initiator) were added, followed by the addition of one more gram of Na3NTA. By the addition of V501, the exchange of some surface NTA units by V501 units was pursued. The final CeO2 content of the dispersion was 1.6 wt%. The sample was stored at 3–4 °C to prevent the decomposition of the thermal initiator V501. The synthesized dispersion of nanoparticles was stable for more than one month in a refrigerator.
Polymerization of the acrylic/CeO2 hybrid latexes
The polymerizations were carried out in a 100 mL glass jacketed reactor fitted with a reflux condenser, a sampling device, a N2 inlet and a stirrer rotating at 200 rpm. In a typical experiment (see Run 1 in Table 1) the CeO2 NP/NR aqueous dispersion (24.75 g) and water (74.25 g) were placed in the reactor. The feeding of the monomer (using a syringe pump, Fisher Scientific, 78-11001 Series) and heating were started simultaneously. The temperature was set to 75 °C. The monomer was fed for 150 minutes at a flow rate of 0.004 g min−1 for Run 1. After 30 minutes the emulsifier was added in a shot dissolved in 2 mL of water. The total reaction time was 210 minutes, because after the monomer feeding, the polymerization was left to continue for one more hour. No extra initiator was added to the polymerization reaction, apart from the one present in the CeO2 NP/NR dispersion.In order to favor the initiation of the polymerization from the surface of the NPs/NRs in some experiments (not shown in Table 1) the emulsifier was not used or was used well below the critical micellar concentration (cmc). Unfortunately, these experiments yielded large amounts of coagulum and hence higher emulsifier concentration was used to obtain stable latexes.
Characterization techniques
The morphology of the nanoparticle/nanorod (NP/NR) dispersion as well as the hybrid latex dispersion was analyzed by using a transmission electron microscope (TEM), TECNAI G2 20 TWIN (FEI), operating at an accelerating voltage of 200 keV in a bright-field image mode. The samples were diluted to 0.5–1 wt% and dried in a refrigerator at 4 °C. Nanoceria particle and nanorod size distributions were obtained using Image Pro Plus 7.0 software on 500 particles of each kind.For the surface characterization of the CeO2 dispersion different steps were carried out: lyophilization, centrifugation and dialysis of the sample. Spectra/Por (MwCO: 12000–14000) was used as the dialysis membrane. The entire dialysate volume was changed for fresh water at least once a day. Dialysis was allowed to run until the conductivity of the dialysate was close to that of the DDI water (2 μS cm−1). The “clean” dispersion was kept in a refrigerator at 4 °C to avoid the decomposition of the thermal initiator V501.
The supernatant water of the centrifuged samples was characterized by 13C-NMR spectroscopy. Samples were dissolved in deuterated water (D2O) and analyzed using a Bruker AVANCE-400 spectrometer 400 MHz for 9 hours.
XPS composition data and spectra were acquired on a SPECS (Berlin, Germany) instrument equipped with a Phoibos 150 1D-DLD analyzer and a monochromatic Al Kα X-ray source. Compositional survey and detailed scans were acquired using a pass energy of 80 eV. High resolution spectra were acquired using a pass energy of 30 eV. The above data were taken at a 90° takeoff angle. Data analysis was performed with Casa XPS 2.3.16 Software to fit the signals to Gauss–Lorentzian curves, after removing the background (Shirley). Energy correction was performed by using the C1s peak at 284.6 eV as the reference. ATR-IR spectroscopy of the CeO2 dispersion was carried out using a FTIR-ATR Perkin-Elmer RX1 Spectrometer.
The UV-Vis absorption measurements were carried out using a Shimadzu spectrophotometer (model UV-2550 230 V). The measurements in the 250–600 nm range were carried out at room temperature on 50 μm films cast in Teflon panels at 23 °C and 55% humidity. The thermal properties of the hybrids were analysed by Differential Scanning Calorimetry (DSC, Q2000, TA Instruments) and Thermogravimetric Analysis (TGA, Q500 V6.5 build 196 TGA instrument) measurements.
Results and discussion
Morphology and surface characterization of the CeO2 NP/NR dispersion
In Fig. 2, TEM micrographs of the aqueous CeO2 nanoparticle dispersion are presented. It can be observed that both spherical CeO2 nanoparticles and nanorods were formed during the synthesis, and they were homogeneously dispersed in the aqueous phase. | ||
| Fig. 2 TEM micrographs of the CeO2 nanoparticle dispersion containing spherical nanoparticles and nanorods. | ||
The dimensions of 500 nanorods and nanoparticles were independently measured from the TEM images to compute the particle size distributions (PSDs) of each population (Fig. 2 and ESI† for the detailed PSDs). Nanorod dimensions were in the range of 25–275 nm in length and 1–15 nm in width, with an average aspect ratio of 16. Nanoparticle sizes between 1 and 15 nm were measured with a volume average diameter of 6 nm.
Fig. 3 describes the different steps carried out to characterize the surface of the NPs and NRs. In the first step, the CeO2 dispersion was lyophilized and analyzed by XPS. It should be mentioned, that this sample was used as a reference because it should contain all the V501 initiator. The powder was redispersed in water and it was seen that it initiated the polymerization of MMA (see details in the following sections).
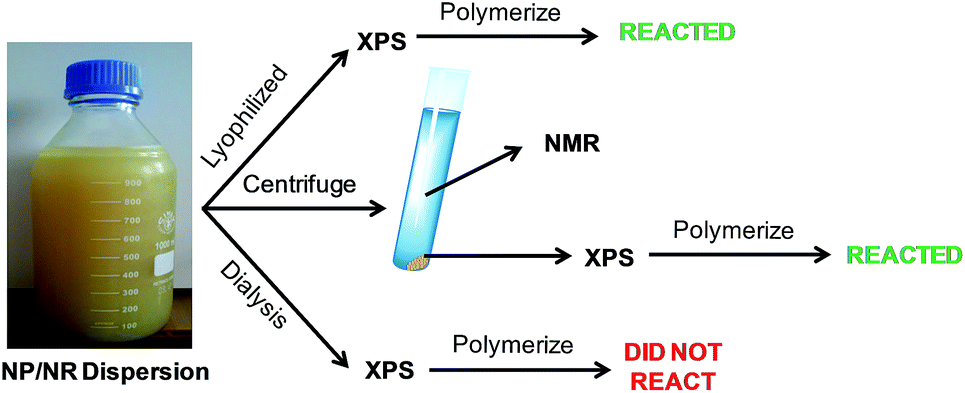 | ||
| Fig. 3 Experimental procedure followed to analyze the NP/NR dispersion. | ||
On the other hand, in order to check if the initiator was adsorbed on the surface of the NPs and NRs, a fraction of the NP/NR dispersion was centrifuged at 10 °C and 20000 rpm for 30 minutes. A fraction of the precipitated sample was analyzed by XPS and the supernatant of the centrifugation was analyzed by 13C-NMR.
Fig. 4 presents the XPS analysis of the reference and the centrifuged sample, together with the pure V501. For the sake of clarity only the carbon and nitrogen component spectra are displayed in Fig. 4.
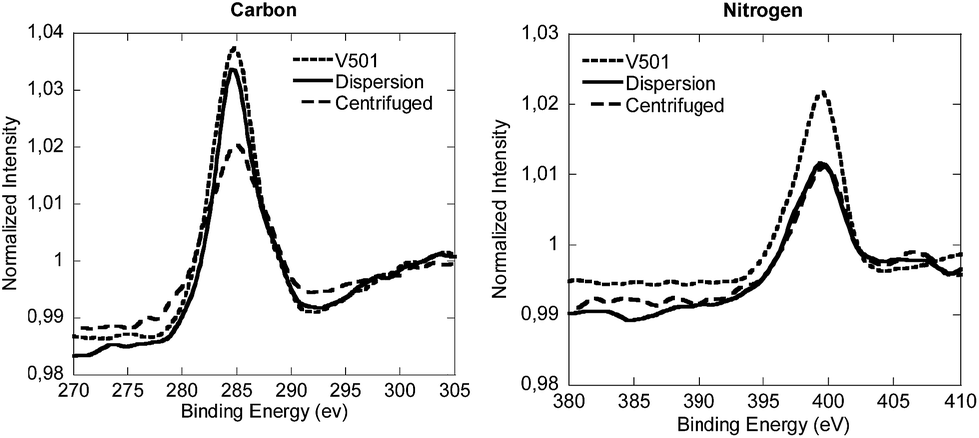 | ||
| Fig. 4 Normalized XPS spectra of the carbon and nitrogen components of the lyophilized CeO2 dispersion, the centrifuged sample and V501. | ||
As it can be seen in Fig. 4, the peaks of V501, original dispersion and centrifuged samples, for C and N signals, are very similar. Although the peaks of the centrifuged sample were slightly shifted, the components detected in V501 were in good agreement with the peaks of the original dispersion and centrifuged samples (the binding energies of each component together with the deconvoluted spectra are shown in the ESI†), which means that the initiator was at least partially associated with the NP/NR surfaces.
The precipitated sample was redispersed in water and used to initiate the polymerization of MMA. The sample was still able to polymerize MMA (19% of conversion was achieved). This further supports our XPS results and confirms that a fraction of the initiator was present in the NP/NR surface after centrifugation.
The composition of the supernatant obtained after the centrifugation was analyzed by NMR. Fig. 5 shows the 13C-NMR spectrum and the chemical structures of the NTA and V501 species. The assignments of the peaks are described in detail in the ESI.† The spectrum confirms the presence of both NTA and V501 in the aqueous phase after the centrifugation.
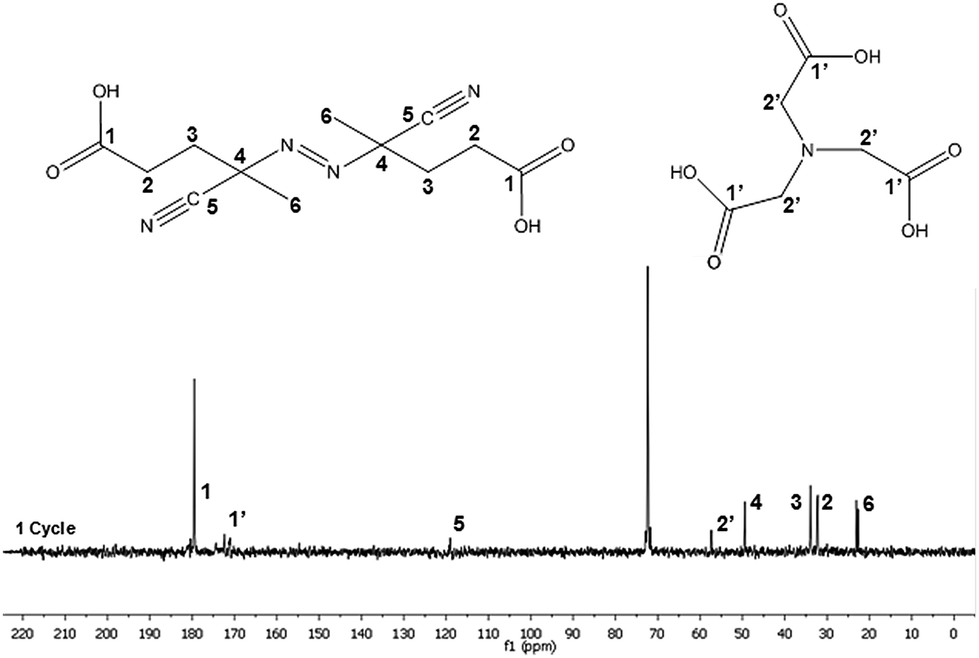 | ||
| Fig. 5 13C-NMR spectra corresponding to the supernatant water of the centrifuged CeO2 dispersion sample. | ||
These results indicate that the V501 initiator might be partitioned between the surface of the NPs/NRs and the aqueous phase since it was detected in both phases; the centrifuged solid (XPS, MMA initiation) as well as in the supernatant (NMR).
In order to check this partition, the equilibrium of V501 was shifted to the aqueous phase by dialysis (Fig. 3). The XPS spectra of the dialyzed and initial samples are presented in Fig. 6. It can be said that the V501 signals in the dialyzed sample were weaker and with more noise than those presented before. Moreover, it was confirmed that this sample was not able to initiate the polymerization of MMA. It can be concluded that there was no initiator left on the surface of the CeO2 NPs/NRs after the dialysis.
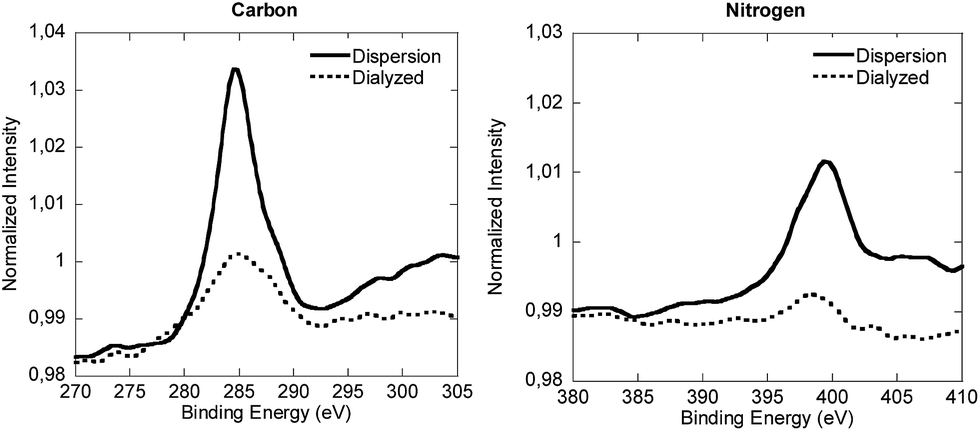 | ||
| Fig. 6 Normalized XPS spectra of the carbon and nitrogen components of the lyophilized CeO2 dispersion and the dialyzed sample. | ||
In order to better characterize the association of V501 with the CeO2 surface, Attenuated Total Reflectance Fourier Transform InfraRed spectroscopy (ATR-FTIR) was performed for the NP/NR dispersion and for pure NTA and V501, and the results are shown in Fig. 7. It can be seen that as already presented for the adsorption of NTA,38 the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bands of the carboxylic groups from NTA and V501 (1720, 1708 cm−1) are shifted to lower wavelengths (1565 cm−1), demonstrating their chelation to the CeO2 surface. Only a small fraction of carboxylic groups appear unshifted, which supports the amount of NTA and V501 found by NMR in the supernatant.
O bands of the carboxylic groups from NTA and V501 (1720, 1708 cm−1) are shifted to lower wavelengths (1565 cm−1), demonstrating their chelation to the CeO2 surface. Only a small fraction of carboxylic groups appear unshifted, which supports the amount of NTA and V501 found by NMR in the supernatant.
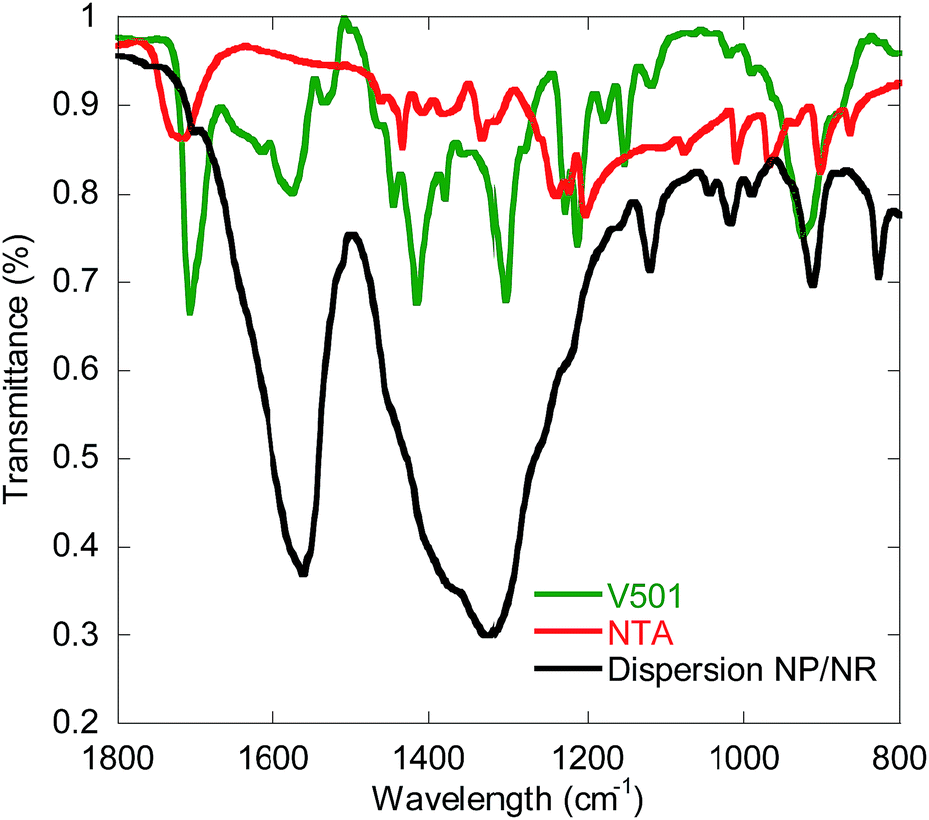 | ||
| Fig. 7 ATR-FTIR (Attenuated Total Reflectance-Fourier Transform InfraRed spectroscopy) measurements of pure V501 and NTA, and the NP/NR dispersion. | ||
Therefore the characterization of the NPs/NRs indicates that V501 was associated to the surface of the NPs/NRs and that it is sufficient to start the polymerization of MMA without adding further initiator. The results also confirm that there exists an equilibrium between the acidic species (NTA and V501) at the surface of the nanoparticles and in the aqueous phase.
Emulsion polymerization in the presence of the V501 modified CeO2 NP/NR dispersion
Semibatch emulsion polymerization reactions using the NP/NR dispersions and MMA or MMA/BA mixtures as monomers without further addition of initiator were carried out. The surfactant amount added in Run 1 was just below the cmc (critical micellar concentration, 70 mg L−1)39 to minimize the nucleation of particles and favor initiation from the surface of CeO2 NPs/NRs. However, it was observed that under this condition unstable latexes were obtained. Only when the amount of surfactant was twice the cmc, stable latexes could be obtained (see runs 2–4 in Table 2).In Run 2, (MMA) total conversion was achieved even if no extra initiator was added apart from the one present in the CeO2 dispersion, whereas in Run 3, (MMA/BA) 90% conversion was reached. On the other hand, in Run 4, 50% conversion was attained. The volume average polymer particle size measured by TEM was 31 nm for Run 2 and 83 nm for Run 4 (see ESI†). The size difference is related to the higher solid content of Run 4. Furthermore, the molecular weight distribution of Run 4 has also been measured, presenting a weight average molecular weight of 5.1 × 105 g mol−1 with a polydispersity of 4.1, which is in the typical range of polymers produced by emulsion polymerization. Therefore, stable hybrid acrylic/CeO2 NP/NR latexes with low solids content and CeO2 contents between 3.7 and 18.7 wt% with respect to the polymer were successfully produced by this approach.
Morphology of the hybrid latexes containing CeO2 NPs/NRs
Fig. 8 presents the TEM micrographs of Run 2 and Run 4 hybrid latexes containing CeO2 NPs/NRs. It can be clearly distinguished that the NP/NR concentration was higher in the hybrid latex of Run 2 (18.7% with respect to the polymer) than in the hybrid latex of Run 4 (3.7% with respect to the polymer).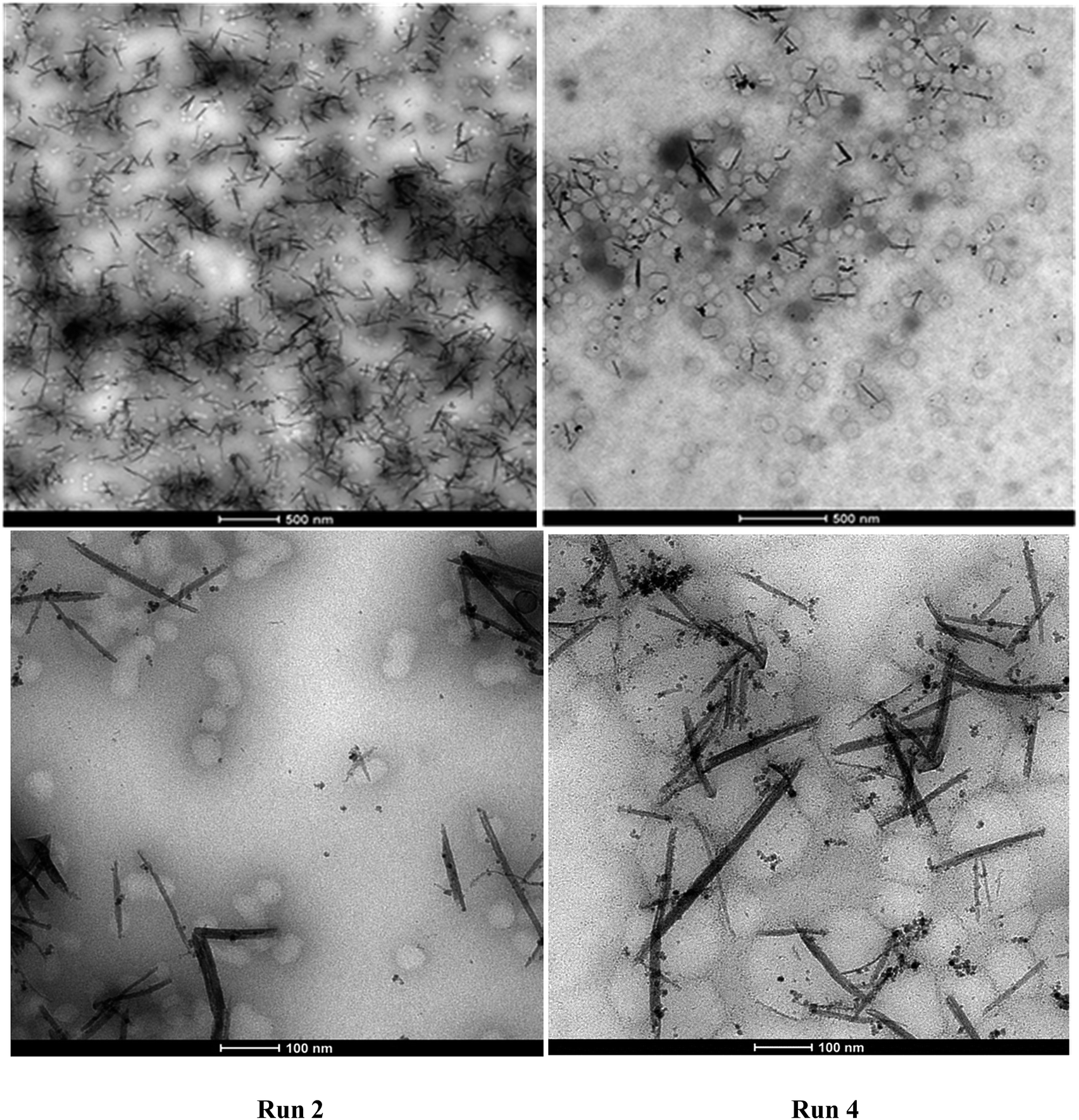 | ||
| Fig. 8 TEM micrographs of the hybrid latexes of Run 2 and Run 4. | ||
With regard to the location of the polymer particles, it seems that a large number of them are not associated with CeO2 NPs/NRs, while there are others that are closer or attached to CeO2 NPs/NRs. In the latter case, the images are not conclusive to elucidate whether the polymerization was initiated from the surface of the NPs/NRs or the attachment of the polymer particles and NPs/NRs occurred during the sample preparation for TEM.
Nonetheless, the TEM images are conclusive with regard to the occurrence of homogeneous or micellar nucleation, since a large number of free polymer particles (85%, counting 200 polymer particles), which apparently did not contain any CeO2 moieties were identified. It seems that the concentration of the emulsifier (above the cmc to ensure stability of the latex) and the partition of the V501 initiator between the CeO2 surface and the aqueous phase were significant enough to produce CeO2-free polymer particles. Note that the only initiator present was the one used in the synthesis of the CeO2 dispersion.
Film morphology and properties
In order to check the film forming properties of the hybrid latexes, films of Run 3 and Run 4 (MMA/BA, 50/50 wt%) were cast at room temperature for 24 hours (Fig. 9). Good quality films (crack and void-free) were formed despite the low solid content. The films obtained were yellowish but transparent and the color increased with the CeO2 content. | ||
| Fig. 9 Films of (a) Run 3 and (b) Run 4 both cast at room temperature. | ||
A TEM image of the hybrid film of Run 4 is presented in Fig. 10. It can be seen that after film formation the NPs/NRs are still well dispersed and they did not tend to form aggregates. This is one of the main reasons to obtain transparent films. Therefore the main objective of the work was fulfilled as by the modification of the surface of the CeO2 nanoparticles and nanorods, good compatibility between the inorganic and the polymeric phase was achieved, obtaining good dispersion of the inorganic material in the polymeric film. It must me pointed out that films of Runs 2–4 showed much higher stability than a physical blend of the CeO2 dispersion and a blank latex (see ESI†), indicating that even if the amount of polymer grown from the surface of the CeO2 nanoparticles was not large, it was enough to enhance the inorganic/organic compatibility.
 | ||
| Fig. 10 TEM micrograph of the film from Run 4. | ||
Due to the high UV absorbance capacity of CeO2, the UV-Vis absorption of these films could not be measured directly because at the minimum achievable thickness of the films the transmission of UV-Vis light was completely blocked in the spectrophotometer. Alternatively, in order to check if the CeO2 particle shape has any effect on the UV absorption capacity, the hybrid latex synthesized in this work, Run 4, was blended (by magnetically stirring for 1 h) with a blank latex to yield a hybrid latex with a final CeO2 content of 1 wt% (hybrid latex NPs/NRs). The absorption of this film was compared to that of the blank film (0% CeO2) and to that of a hybrid film containing only spherical CeO2 nanoparticles (1 wt% CeO2, hybrid latex NPs) prepared in a previous study.7
Fig. 11 presents the UV absorbance of the hybrids and the pristine latex films. It was observed that the films containing CeO2 performed considerably better than the pristine latex. However, in the dispersion that contained the nanorods, the UV absorption was enhanced at wavelengths above 350 nm, with respect to the dispersions that only contained spherical CeO2 nanoparticles. In both of them the CeO2 percentage was 1 wt% and therefore, the difference must come from the CeO2 rod-like structures prepared in this work.
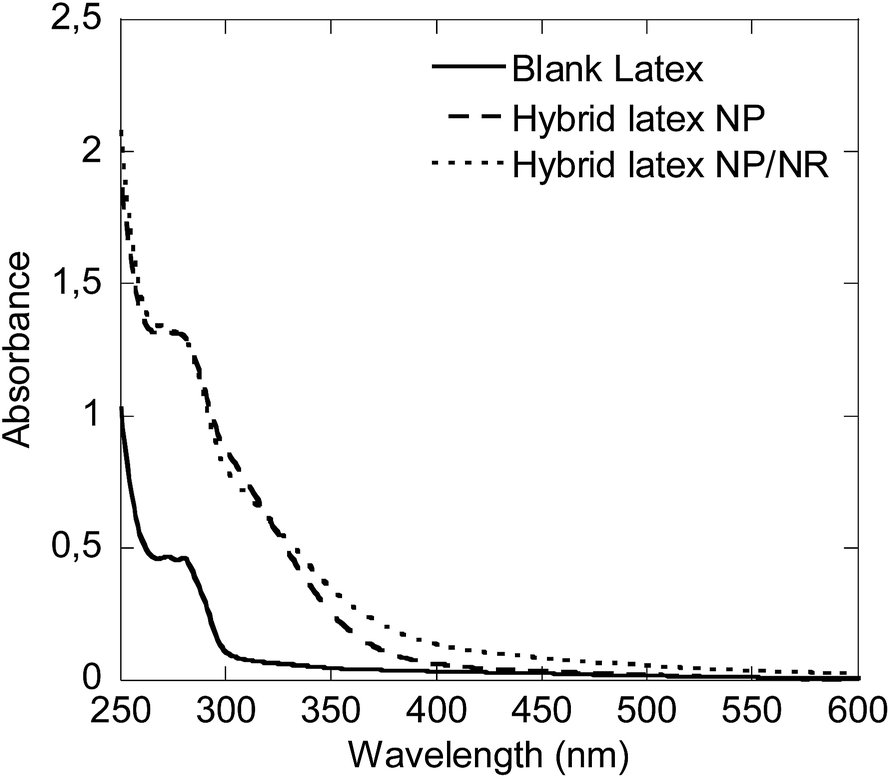 | ||
| Fig. 11 UV-Vis absorption capacity of the hybrid latexes prepared with 1 wt% of CeO2 NPs or NPs/NRs, compared to a blank latex. | ||
Furthermore the thermal properties of the films were also measured by DSC and TGA. Three different sample types have been analyzed in order to check the effect of the amount and aspect ratio of CeO2 in the hybrids: a blank polymer (obtained from a blank latex without CeO2), nanocomposites obtained by in situ polymerization with 1 and 2 wt% of spherical40 NPs and the nanocomposite from Run 4, which contains 3.7% of NPs/NRs. It was observed that the glass transition temperature (Tg) of the three samples was within the range of 14–16 °C, which suggests a negligible effect of the amount and aspect ratio of CeO2 in the range studied (see the ESI†). The TGA showed that the thermal stability decreased slightly with the CeO2 content. However, there were not big differences between the films containing NPs or the ones with NPs/NRs. The T50 (temperatures for 50% of degradation) of the three samples was 350 °C for the blank sample, 343 °C for the hybrid containing 1 wt% of CeO2 NPs, 341 °C for the hybrid containing 2 wt% of CeO2 NPs and 332 °C for the hybrid containing 3.7 wt% of CeO2 NPs/NRs (see the ESI†).
Conclusions
The emulsion polymerization of the dispersion of CeO2 NPs/NRs with acrylate monomers, without further addition of initiator, led to stable hybrid latexes when the amount of emulsifier was above the cmc. Therefore, a hypothetical nucleation mechanism was proposed in which the polymerization could be started from both the surface of the NPs/NRs and from the aqueous phase.This is the first time that high aspect CeO2 nanoparticles have been incorporated into polymer hybrids. Furthermore, they produced good quality and transparent films at room temperature, due to the enhanced compatibility between CeO2 and the polymer, which prevented the CeO2 aggregation during film formation. In addition, it has been proven that the UV-Vis absorption capacity was enhanced with the use of a mixture of different aspect ratio CeO2, with no significant changes in the thermal properties. Furthermore, this hybrid morphology opens also the way to new applications in the field of hybrid photoactive layers in solar cells, if the CeO2 band gap is accordingly engineered.
Acknowledgements
Financial support from the European Union (Woodlife project FP7-NMP-2009-SMALL-246434) is gratefully acknowledged. Miren Aguirre thanks the Basque Government for the scholarship “Ikertzaileak prestatzeko eta hobetzeko laguntzak” and the UPV/EHU for the scholarship “Doktore berriak kontratatzeko eta horiek doktorego ondoko prestakuntza programetan sartzeko laguntza”. The sGIKer UPV/EHU is acknowledged for the electron microscopy facilities of TEM and X-ray measurements (XPS).Notes and references
- J. Hu, M. Chen and L. Wu, Polym. Chem., 2011, 2, 760 RSC.
- K. Landfester, Angew. Chem., Int. Ed. Engl., 2009, 48, 4488–4507 CrossRef CAS PubMed.
- M. Paulis and J. R. Leiza, Advances in Polymer Nanocomposites Technologies, ed. V. Vittal, Nova Science, 2010 Search PubMed.
- C. K. Weiss and K. Landfester, Hybrid Latex Particles, 2010, 233, 185–236 CAS.
- R. Li, S. Yabe, M. Yamashita, S. Momose, S. Yoshida, S. Yin and T. Sato, Solid State Ionics, 2002, 151, 235–241 CrossRef CAS.
- T. Masui, M. Yamamoto, T. Sakata, H. Mori and G. Y. Adachi, R. Soc. Chem., 2000, 10, 353–357 CAS.
- M. Aguirre, M. Paulis and J. R. Leiza, J. Mater. Chem. A, 2013, 1, 3155–3162 CAS.
- M. Aguirre, M. Paulis, J. R. Leiza, T. Guraya, M. Iturrondobeitia, A. Okariz and J. Ibarretxe, Macromol. Chem. Phys., 2013, 214, 2157–2164 CAS.
- X. Lu, D. Zheng, P. Zhang, C. Liang, P. Liu and Y. Tong, Chem. Commun., 2010, 46, 7721–7723 RSC.
- P. C. Ma, M. Y. Liu, H. Zhang, S. Q. Wang, R. Wang, K. Wang, Y. K. Wong, B. Z. Tang, S. H. Hong, K. W. Paik and J. K. Kim, ACS Appl. Mater. Interfaces, 2009, 1, 1090–1096 CAS.
- E. Bourgeat-Lami and M. Lansalot, Adv. Polym. Sci., 2010, 233, 53–123 CrossRef CAS.
- E. D. Laird and C. Y. Li, Macromolecules, 2013, 46, 2877–2891 CrossRef CAS.
- B. R. Saunders and M. L. Turner, Adv. Colloid Interface Sci., 2008, 138, 1–23 CrossRef CAS PubMed.
- G. Chen, C. Xu, X. Song, W. Zhao, Y. Ding and S. Sun, Inorg. Chem., 2008, 47, 723–728 CrossRef CAS PubMed.
- K. S. Lin and S. Chowdhury, Int. J. Mol. Sci., 2010, 11, 3226–3251 CrossRef CAS PubMed.
- C. Sun, H. Li, H. Zhang, Z. Wang and L. Chen, Nanotechnology, 2005, 16, 1454–1463 CrossRef CAS.
- A. Vantomme, Z. Y. Yuan, G. Du and B. L. Su, Langmuir, 2005, 21, 1132–1135 CrossRef CAS PubMed.
- B. Tang, L. Zhuo, J. Ge, G. Wang, Z. Shi and J. Niu, Chem. Commun., 2005, 3565–3567 RSC.
- K. E. Prasad, B. Das, U. Maitra, U. Ramamurty and C. N. R. Rao, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 13186 CrossRef CAS PubMed.
- C. Zhang, S. Huang, W. W. Tjiu, W. Fan and T. Liu, J. Mater. Chem., 2012, 22, 2427–2434 RSC.
- M. Nam, S. Kim, S. W. Kim and K. Lee, Nanoscale, 2013, 5, 8202–8209 RSC.
- C. Tang, L. Xiang, J. Su, K. Wang, C. Yang, Q. Zhang and Q. Fu, J. Phys. Chem. B, 2008, 112, 3876–3881 CrossRef CAS PubMed.
- L. Qi, A. Sehgal, J. C. Castaing, J. P. Chapel, J. Fresnais, J. F. Berret and F. Cousin, ACS Nano, 2008, 2, 879–888 CrossRef CAS PubMed.
- J. Fresnais, C. Lavelle and J. F. Berret, J. Phys. Chem. C, 2009, 113, 16371–16379 CAS.
- B. Chanteau, J. Fresnais and J. F. Berret, Langmuir, 2009, 25, 9064–9070 CrossRef CAS PubMed.
- J. F. Berret, Macromolecules, 2007, 40, 4260–4266 CrossRef CAS.
- L. Qi, J. P. Chapel, J. C. Castaing, J. Fresnais and J. F. Berret, Langmuir, 2007, 23, 11996–11998 CrossRef CAS PubMed.
- R. Rhodes, M. Horie, H. Chen, Z. Wang, M. L. Turner and B. R. Saunders, J. Colloid Interface Sci., 2010, 344, 261–271 CrossRef CAS PubMed.
- S. Bardage, M. Henriksson, S. Olsson, P. Collins, D. Meng, A. Ahniyaz, E. Johansson, A. Rahier, M. Gasparini and N. Lamproye, Surf. Coat. Int., 2013, 96(2), 94–99 Search PubMed.
- B. Erdem, E. D. Sudol, V. L. Dimonie and M. S. El-Aasser, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 4441–4450 CrossRef CAS.
- H. Lu, B. Fei, J. H. Xin, R. Wang and L. Li, J. Colloid Interface Sci., 2006, 300, 111–116 CrossRef CAS PubMed.
- N. Bechthold, F. Tiarks, M. Willert, K. Landfester and M. Antonietti, Macromol. Symp., 2000, 151, 549–555 CrossRef CAS.
- J. Ramos and J. Forcada, Langmuir, 2011, 27, 7222–7230 CrossRef CAS PubMed.
- Y. Mori and H. Kawaguchi, Colloids Surf., B, 2007, 56, 246–254 CrossRef CAS PubMed.
- J. Garnier, J. Warnant, P. Lacroix-Desmazes, P. E. Dufils, J. Vinas, Y. Vanderveken and A. M. Van Herk, Macromol. Rapid Commun., 2012, 33, 1388–1392 CrossRef CAS PubMed.
- N. Zgheib, J. L. Putaux, A. Thill, E. Bourgeat-Lami, F. D'Agosto and M. Lansalot, Polym. Chem., 2013, 4, 607 RSC.
- N. Zgheib, J. L. Putaux, A. Thill, F. D'Agosto, M. Lansalot and E. Bourgeat-Lami, Langmuir, 2012, 28, 6163–6174 CrossRef CAS PubMed.
- E. Johansson Salazar-Sandoval, M. K. G. Johansson and A. Ahniyaz, RSC Adv., 2014, 4, 9048–9055 RSC.
- I. d. F. A. Mariz, High Solids Content Low Viscosity Latexes with Small Particle Size, PhD thesis, University of the Basque Country UPV/EHU, 2011 Search PubMed.
- M. Aguirre, M. Paulis and J. R. Leiza, Polymer, 2014, 55, 752–761 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Data on the particle size distribution of CeO2 nanoparticles and nanorods, 13C-NMR and XPS data assignments, particle size distribution of the polymer particles, stability tests and DSC and TGA measurements. See DOI: 10.1039/c4ta03620d |
| This journal is © The Royal Society of Chemistry 2014 |