
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Molecular weight and end capping effects on the optoelectronic properties of structurally related ‘heavy atom’ donor–acceptor polymers†
Gregory L.
Gibson
,
Dong
Gao
,
Ashlee A.
Jahnke
,
Jing
Sun
,
Andrew J.
Tilley
and
Dwight S.
Seferos
*
Lash Miller Chemical Laboratories, Department of Chemistry, University of Toronto, 80 St. George Street, Toronto, Ontario, M5S 3H6 Canada. E-mail: dseferos@chem.utoronto.ca
First published on 17th July 2014
Abstract
Two donor–acceptor polymers containing either Si or Ge in the donor and Se in the acceptor, poly[(4,4′-bis(2-ethylhexyl)dithieno[3,2-b:2′,3′-d]silole)-2,6-diyl-alt-(2,1,3-benzoselenadiazole)-4,7-diyl] and poly[(4,4′-bis(2-ethylhexyl)dithieno[3,2-b:2′,3′-d]germole)-2,6-diyl-alt-(2,1,3-benzoselena diazole)-4,7-diyl], were synthesized by microwave assisted polymerization. These polymer structures are attractive because they combine the red light absorption characteristics of the Se acceptor, with high charge carrier mobility inherent to the Si- or Ge-containing donor. Here we study the effects of molecular weight and end capping on the photophysical, morphological, and photovoltaic properties. The solution and film absorption profiles and solution onset are dictated by molecular weight, whereas the subtler heteroatom effect dictates the absorption onset in the polymer films. Molecular weight appears to affect polymer absorption to the greatest degree in a medium molecular weight regime and these effects have a significant aggregation component. Highlighting the red-light absorption of the Se-acceptor monomer, both Si-donor and Ge-donor polymer devices display improved photon harvesting beyond 850 nm relative to their S-acceptor analogues. Higher hole mobility relative to the C-donor/Se-acceptor polymer analogue indicates successful integration of heavy atom donor properties with the 2,1,3-benzoselenadiazole acceptor. Molecular weight invokes the greatest change on polymer/fullerene blend morphology, followed by phenyl end capping, and finally by the Si or Ge heteroatom.
Introduction
As donor–acceptor (D–A) conjugated polymers have gained attention as photovoltaic active layer components, much effort has been invested in appropriately tuning their optical properties.1–8 In addition to solid state structure, it is well known that the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels are critical for device performance.9–13 Extensive monomer modification has been undertaken in order to tune energy levels and solid state structures, including: extending the conjugation of the polymer repeat unit,14,15 single atom substitution in one or both of the donor and acceptor monomers,16–22 and monomer side chain substitution.23,24Recently, single atom substitution has gained increasing attention because polymer properties may be predictably tuned through modular chemistry. Two donor monomer examples are silicon25 and germanium16,26 substitution in fluorene and cyclopentadithiophene27–29 parent structures. Motivation for this work was inspired by high silole charge transport mobility in transistors.30–32 Silicon incorporation in place of carbon in the well-known PCPDTBT structure results in a more crystalline material with greater charge carrier mobility than its carbon donor counterpart.33,34 Further, germanium substitution has produced materials with similar properties to their silicon analogues but with enhanced stability.35,36 This has led to recent work designing new donor co-monomers containing germanium as well as analogous small molecule structures.37–39 While less work has been conducted on acceptor monomers, fluorination has become an increasingly popular method of positioning D–A polymer HOMO and LUMO energy levels.40–46 Selenium and tellurium substitutions into 2,1,3-benzothiadiazole have shed significant light on the D–A absorption origin and mechanism.47 Replacing sulfur in conjugated heterocycles with selenium and tellurium consistently resulted in red-shifted absorption spectra in the resulting materials.9,11,48 With respect to organic photovoltaic cells, greater red light absorption should increase device short circuit current by utilizing a larger portion of the visible spectrum.49
Despite the many structural variables that have been investigated in the D–A polymer context, molecular weight remains relatively understudied. It has been observed empirically that given two polymers of the same repeat unit structure, generally the larger one will perform better in an organic photovoltaic device.50 One obstacle to systematic molecular weight study has been synthetic control over step growth polymer molecular weight. While chain growth polymerization methods produce polythiophenes and polyselenophenes with controlled molecular weight,51,52 similar methodology is not currently available for step growth polymerizations. It has been demonstrated, however, that general molecular weight targets can be achieved by changing the monomer ratio within the polymerization reaction, assuming the extent of reaction goes to unity.16
Herein we report new optical, electrochemical, and photovoltaic data for D–A polymers containing either silicon or germanium and selenium in the repeat unit. Ideally these atom combinations will provide materials with high charge carrier mobility, due to the silicon or germanium-containing donor, and enhanced red-light collection due to the 2,1,3-benzoselenadiazole acceptor. For each polymer structure a molecular weight series and a phenyl end-capped derivative were synthesized. This allows us to draw conclusions about how molecular weight and end groups affect the optical, electrochemical, and photovoltaic properties of polymers containing multiple heavy atoms in the repeat unit, and more broadly, to D–A polymers in general.
Results and discussion
Synthesis
Silicon and germanium cyclopentadithiophene-based monomers were functionalized with trimethyl tin groups by treatment with n-butyllithium followed by trimethyltin chloride, according to literature procedures.32,35 In our prior study on these polymer structures, stannylated monomer purity limited heavy atom polymer molecular weight.50,53 Purification is difficult due to the carbon–tin bond instability and decomposition is observed when purifying these monomers using conventional column chromatography techniques. Alternative purification procedures include reverse phase HPLC and recycling GPC, both of which require expensive instruments and chromatography columns dedicated to this purpose. Reverse-phase column chromatography is a more convenient and widely accessible purification method, and has been previously demonstrated to effectively purify tin-functionalized conjugated monomers.18 Therefore in this study monomers 2 and 4 were purified using C18-end capped silica with 30% v/v ethyl acetate in acetonitrile as the mobile phase. Both compounds display 1H and 13C NMR resonances that are consistent with previous reports.D–A polymer series incorporating either 2 or 4 and 2,1,3-benzoselenadiazole were synthesized using step growth polymerization (Scheme 1 and Table 1). In contrast to our previous report where we made one polymer sample of each repeat unit configuration, here a 60 minute microwave assisted Stille polymerization was carried out at 160 °C to make a molecular weight series and phenyl end-capped derivative of each repeat unit structure. Each reaction mixture was precipitated into methanol, and the solid was extracted in a Soxhlet apparatus successively with methanol, hexanes, dichloromethane, and chloroform. In our naming scheme the number preceding the ‘P’ indicates the GPC determined number average molecular weight (Mn) in kg mol−1 rounded to the nearest one and the subscript indicates the identity of the D–A fragment. In the case of silicon-donor polymer 33PSiSe, a further extraction with chlorobenzene was performed to collect the chloroform-insoluble material. All polymers were passed through a short silica column eluting with chloroform, except 33PSiSe, which was eluted with chlorobenzene, and the solvent was removed to afford the target compound. Two phenyl end-capped polymers were also synthesized and are abbreviated, ‘Ph’ in place of a number (Scheme 1). All polymer 1H NMR spectra, except 11PSiSe, 8PSiSe and 5PGeSe, were collected at 130 °C in 1,1,2,2-tetrachloroethane-d2 solvent due to limited polymer solubility at room temperature in traditional NMR solvents (ESI, Fig. S1–S9†). 11PSiSe and 5PGeSe1H NMR spectra were collected in CDCl3 at ambient temperature. All polymers have two broad signals in the 1H NMR aromatic region and broad alkyl peaks characteristic of branched 2-ethylhexyl alkyl chains, consistent with previously reported spectra at ambient temperature in chloroform. The two end-capped polymers, PhPSiSe and PhPGeSe have additional signals in the 1H NMR aromatic region, consistent with phenyl end groups at either end of the polymer chain. Considering that each end group can be located next to either a bithiophene monomer or a selenadiazole monomer, and that these should exist in roughly equal amounts, we expect six additional aromatic signals, which are observed.
 | ||
| Scheme 1 Synthesis of heavy atom monomers and corresponding 2,1,3-benzoselenadiazole-containing polymers PSiSe (D = Si), PGeSe (D = Ge); MW = microwave. | ||
| M n/kg mol−1 | Đ | λ max, abs nm−1 | ||||||
|---|---|---|---|---|---|---|---|---|
| Solutionb | Film | |||||||
| a Calibrated with polystyrene standards using 1,2,4-trichlorobenzene as eluent at 130 °C. b Approximately 10−6 M in chlorobenzene. c Shoulder. | ||||||||
| 8PSiSe | 8.0 | 2.2 | — | 708 | 427 | 803 | 739 | 438 |
| 11PSiSe | 10.6 | 1.84 | — | 717 | 429 | 809 | 740 | 447 |
| 18PSiSe | 17.6 | 1.85 | 793 | 736 | 441 | 814 | 758c | 444 |
| 33PSiSe | 33.0 | 1.83 | 829 | 759 | 452 | 830 | 775c | 455 |
| 5PGeSe | 4.9 | 2.6 | — | 695 | 422 | 833 | 750 | 441 |
| 16PGeSe | 15.6 | 1.99 | 796 | 740 | 439 | 822 | 762c | 446 |
| 24PGeSe | 23.6 | 1.61 | 813 | 749 | 443 | 836 | 779c | 455 |
| PhPSiSe | 11.0 | 2.60 | 807c | 724 | 434 | 825 | 751c | 451 |
| PhPGeSe | 10.7 | 2.17 | 809c | 736 | 437 | 827 | 756c | 449 |
Because of the step growth mechanism by which D–A copolymers are synthesized, molecular weight control has been difficult to achieve. While molecular weight effects on D–A polymer properties have been observed anecdotally,50 during the course of this project a publication by You and co-workers demonstrated that pseudo-control over D–A polymer molecular weight can be achieved by invoking the Carothers equation.16 The general degree of polymerization can be controlled by systematically varying the monomer ratio after optimizing the reaction conditions such that the extent of reaction is close to unity. In our case 1.00, 1.02, and 1.05 donor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :acceptor monomer ratios produced high, medium, and low molecular weight polymers of each repeat unit structure (Scheme 1, Table 1). One additional example in the silicon series, 8PSiSe, was synthesized using a 1.07:1 monomer ratio in order to complete the photovoltaic studies (see below). Analogous monomer ratios did not produce equal degrees of polymerization for each polymer structure, which would be expected assuming strict adherence to the Carothers equation. Thus, we do not observe a strictly linear relationship between monomer ratio and molecular weight. We attribute these results to an extent of reaction below unity for each polymerization. Even though we are not able to strictly control the polymer degree of polymerization, we were successful in synthesizing a molecular weight series for each polymer structure, which will allow us to study the molecular weight effects on materials properties. Two polymers, 18PSiSe and 16PGeSe, have roughly equal degree of polymerization, which will allow us to draw molecular weight-independent conclusions about the donor heteroatom effect. Additionally, two phenyl-end capped polymers allow us to study the end group effect on D–A polymer properties. Despite similar reaction stoichiometry to their high molecular weight analogues, the end-capped derivatives were produced in lower molecular weight. This is likely due to a combination of unobservable monomer impurities and the batch-to-batch variability currently inherent in step growth polymerization reactions. Conveniently, the end-capped polymers are almost identical molecular weight to each other and fall within the molecular weight series created by their structural analogues.
:acceptor monomer ratios produced high, medium, and low molecular weight polymers of each repeat unit structure (Scheme 1, Table 1). One additional example in the silicon series, 8PSiSe, was synthesized using a 1.07:1 monomer ratio in order to complete the photovoltaic studies (see below). Analogous monomer ratios did not produce equal degrees of polymerization for each polymer structure, which would be expected assuming strict adherence to the Carothers equation. Thus, we do not observe a strictly linear relationship between monomer ratio and molecular weight. We attribute these results to an extent of reaction below unity for each polymerization. Even though we are not able to strictly control the polymer degree of polymerization, we were successful in synthesizing a molecular weight series for each polymer structure, which will allow us to study the molecular weight effects on materials properties. Two polymers, 18PSiSe and 16PGeSe, have roughly equal degree of polymerization, which will allow us to draw molecular weight-independent conclusions about the donor heteroatom effect. Additionally, two phenyl-end capped polymers allow us to study the end group effect on D–A polymer properties. Despite similar reaction stoichiometry to their high molecular weight analogues, the end-capped derivatives were produced in lower molecular weight. This is likely due to a combination of unobservable monomer impurities and the batch-to-batch variability currently inherent in step growth polymerization reactions. Conveniently, the end-capped polymers are almost identical molecular weight to each other and fall within the molecular weight series created by their structural analogues.
Optical properties
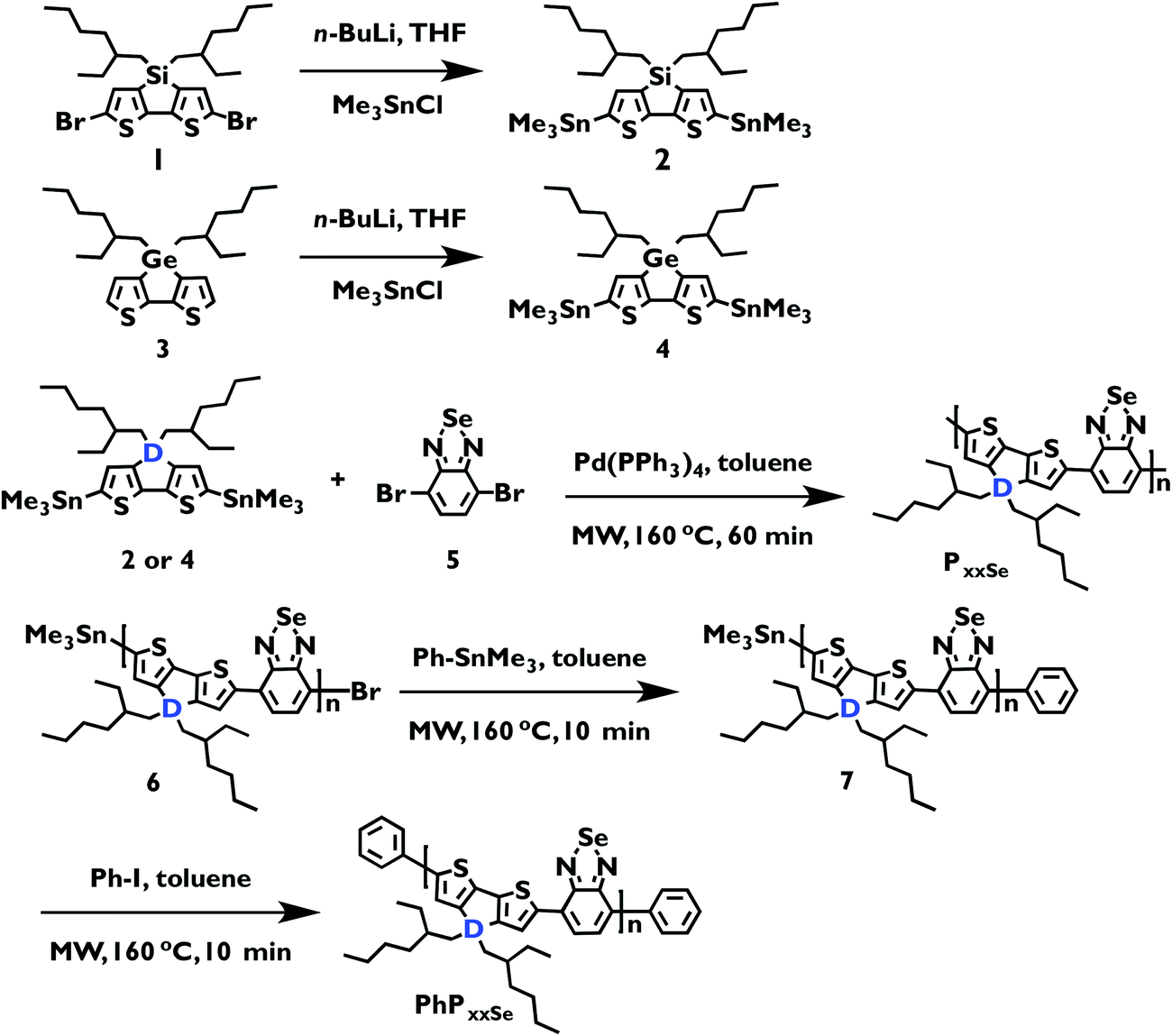
Polymer absorption spectra were collected at room temperature in chlorobenzene and as thin films deposited by spin coating onto a glass slide (Fig. 1, Table 1, ESI Fig. S10†). Both solution and film spectra demonstrate the typical D–A polymer dual-band absorption.27 The longer wavelength band is brought about by a charge transfer (CT) HOMO–LUMO transition that localizes excited state electron density on the acceptor units, while the shorter wavelength absorption is a neutral π–π* transition that results in excited state electron density that is delocalized across both donor and acceptor units.10,12 More specifically, the solution spectra at higher molecular weights have a bimodal CT absorption, resulting in three λmax values for each spectrum (Table 1). Film spectra, on the other hand, have a main CT λmax value with a shoulder peak where the second λmax occurs in the solution spectra. This change in spectral profiles from solution to film can be attributed to greater polymer π-stacking that is known to occur in the solid state with polythiophene-type polymers.14,54 This suggests that polymer aggregation in solution increases with molecular weight regardless of which heteroatom is in the donor bridge position. Solution and film spectral profiles also reveal that molecular weight has less effect on the solid-state absorption than in solution. This is consistent with the greater order in the polymer films. Further donor heteroatom effects are observed when comparing the relative spectral onset positions in solution with those in the film. In solution 33PSiSe has the most red-shifted peak onset, while 24PGeSe has the most red-shifted film onset (Table 2). The solution onset positions agree with prior density functional theory (DFT) calculations where replacing Si with Ge slightly widens the HOMO–LUMO gap by lowering the HOMO energy level.17,53 The film onset positions indicate, however, that 24PGeSe has the narrowest solid-state HOMO–LUMO gap. We attribute the relatively narrow 24PGeSe film HOMO–LUMO gap to greater π-stacking in the Ge-donor polymer due to its slightly larger Ge–C bond length, which allows closer proximity of the polymer π–electron systems.14,23,35 Thus the solution and film absorption profiles and solution onsets are dictated by molecular weight, whereas the subtler heteroatom effect dictates the absorption onset in the polymer films. This is likely due to organization in the polymer films, which lessens molecular weight effects. Further, it is notable that the 33PSiSe solution and film absorption profiles are nearly identical, indicating that there is similar structural order in the solution and solid state. If one considers the film spectrum to represent the upper limit of aggregation-induced order in solution, this result suggests that there is a threshold molecular weight above which the solution absorption becomes molecular weight-independent because it has reached its maximum degree of order in solution while maintaining solubility. The increasing intensity of the π-stacking shoulder with molecular weight in the film spectra suggests this molecular-weight independent absorption threshold may exist in the solid state also. Taken with our previous experiments that show identical CT λmax values for Mn = 3000 g mol−1 and Mn = 6000 g mol−1 D–A polymers in solution,47 the similarity in absorption profiles for 8PSiSe and 11PSiSe suggest that molecular weight has the greatest impact on polymer absorption in a medium molecular weight regime. This range extends from the long edge of the chromophore conjugation length to a polymer-specific molecular weight in solution and film where order is maximized.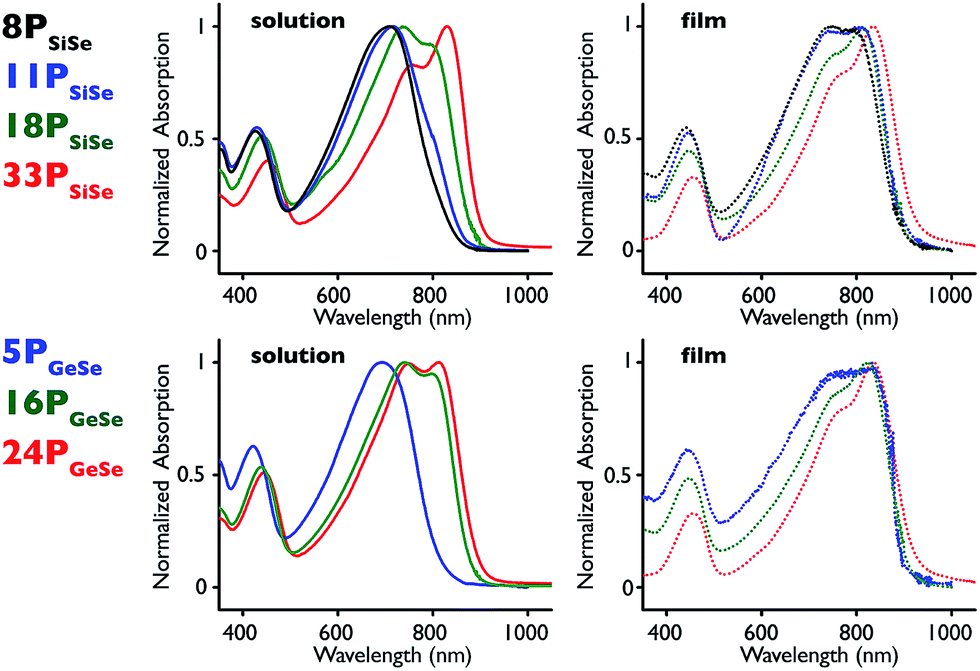 | ||
| Fig. 1 PSiSe series absorption spectra (top row) in ∼10−6 M chlorobenzene solution and film cast from chlorobenzene solution; PGeSe series absorption spectra (bottom row) in ∼10−6 M chlorobenzene solution and film cast from chlorobenzene solution. | ||
| λ onset nm−1 | Energy levels/eV | X-ray spacing/Å | ||||||
|---|---|---|---|---|---|---|---|---|
| HOMOb | LUMOelecb | E elecg | E optg | LUMOoptc | Lamella | π–π | ||
| a Measurement taken from film absorption spectrum. b HOMO and LUMOelec energy levels are approximated using the negative IP and EA values, respectively, which were calculated from cyclic voltammetry and were referenced to ferrocene (∼0.4 V vs. Ag/Ag+ −4.8 eV vs. vacuum). c LUMOopt was calculated from IP + Eoptg. | ||||||||
| 11PSiSe | 903 | −5.08 | −3.62 | 1.46 | 1.37 | −3.71 | 17.4 | — |
| 18PSiSe | 904 | −5.05 | −3.34 | 1.71 | 1.37 | −3.68 | 17.8 | 3.60 |
| 33PSiSe | 919 | −4.96 | −3.20 | 1.76 | 1.35 | −3.61 | 17.5 | — |
| 5PGeSe | 900 | −4.99 | −3.33 | 1.66 | 1.38 | −3.61 | 17.2 | — |
| 16PGeSe | 909 | −5.00 | −3.28 | 1.72 | 1.36 | −3.64 | 17.8 | — |
| 24PGeSe | 939 | −4.90 | −3.25 | 1.65 | 1.32 | −3.58 | 17.7 | 3.56 |
To further understand molecular weight and aggregation influence on optical properties, we carried out variable temperature absorption and emission experiments (Fig. 2 and S11†).14 Polymers 33PSiSe and 24PGeSe were dissolved in chlorobenzene at approximately 10−6 M and heated from 20 °C to 100 °C at 20 °C intervals. The absorption spectra display a consistent decrease in their π-stacking absorption peak intensity with increasing temperature, consistent with disrupting polymer aggregates.55 We observe concurrent blue shifts in the CT absorption and neutral transition λmax values as well as slightly increased peak intensity in the short-wavelength absorption. This shift suggests that a polymer aggregation effect contributes to the CT absorption position and intensity, in addition to the π-stacking peak present at lower temperatures. Further, the increasing short-wavelength peak intensity indicates that as fewer electrons are excited through the CT transition the probability of excitation through the neutral transition is increased. At 100 °C the 33PSiSe π-stacking shoulder remains visible, whereas it is absent in the 24PGeSe spectrum, indicating that the higher molecular weight polymer requires greater energy input to disrupt aggregation.
 | ||
| Fig. 2 (left) 33PSiSe and (right) 24PGeSe absorption spectra in chlorobenzene (∼10 −6 M) collected at 20 °C (black), 40 °C (blue), 60 °C (green), 80 °C (orange) and 100 °C (red). | ||
Having observed significant changes to the polymer absorption spectra as a function of temperature, we were interested in comparing elevated temperature absorption spectra of the highest molecular weight polymers against the ambient temperature spectra of their smaller analogues (Fig. S12†). Interestingly, the 80 °C 33PSiSe spectrum nearly superimposes on the 18PSiSe spectrum at ambient temperature and trends towards the ambient temperature 11PSiSe CT band position and peak shape. While the result is not as striking for 24PGeSe, the elevated temperature spectrum CT band peak shape and position fall within its molecular weight series limits. Thus, a high molecular weight polymer absorption band can be converted to that resembling its lower molecular weight analogue by elevating the temperature. This suggests that polymer aggregation red-shifts the CT chromophore band position as well as inducing the growth of an additional shoulder related to π-stacking.14,25 If the CT band position were solely determined by conjugation length at these molecular weights then disappearance of the CT π-stacking peak would not be observed together with a blue shift in the CT band λmax value. In the context of our prior work with low molecular weight D–A polymers, we therefore hypothesize that spectral changes in different size D–A polymers above roughly 6000 g mol−1 are induced by aggregation and not conjugation length.
Partial 33PSiSe and 24PGeSe variable temperature photoluminescence spectra (Fig. S11†) show increased photoluminescence intensity with increasing temperature. This is counter to the general observation of increased photoluminescence at lower temperatures and suggests a self-quenching effect as the degree of polymer aggregation increases. Attempts to collect film photoluminescence spectra were unsuccessful. The heavy atom combination in both polymer repeat units enhances intersystem crossing, populating the first triplet excited state.56,57 This effect likely causes the fairly weak photoluminescence observed in both polymer samples, as they contain two second- and third-row atoms in their respective repeat units. The position and low photoluminescence signal make spectra collection difficult and influence the spectral shapes. Whereas the photoluminescence blue-edge onset values match the expected transitions and we are confident that the signal originates from the polymer, the visible detector limit likely truncates the signal at approximately 850 nm, where detector roll off is observed. Based on the spectral shape, the absorption λmax values, and the expected Stokes shift, the emission λmax values probably occur at wavelengths beyond 850 nm. Attempts to collect this data with a near-IR photomultiplier tube were unsuccessful due to sensitivity limitations.
Electrochemistry
Polymer oxidation and reduction potentials were determined with cyclic voltammetry (Table 2 and Fig. S13†). The electrochemical experiments were conducted in 0.1 M tetrabutylammonium hexafluorophosphate (TBAPF6) on gold working electrodes with silver reference, and platinum counter electrodes. The voltammograms were referenced to the ferrocene redox couple, measured at 0.4 V vs. Ag/Ag+. The negative ionization potential (IP) and electron affinity (EA) are used to approximate the polymer HOMO and LUMO energy levels, respectively, and were calculated assuming −4.8 V ionization potential for ferrocene vs. vacuum.26,58 Polymer HOMO energy level values range from −5.08 eV to −4.90 eV with those of the Si-donor series being lower. These generally high-lying energy level values are a natural consequence of the low polymer HOMO–LUMO optical gaps, which are around 1.35 eV. 33PSiSe has quasi-reversible oxidation and reduction waves. Interestingly the voltammogram exhibits a sharp irreversible oxidation onset peak. Continued reversible oxidation occurs at larger potentials. 18PSiSe, 16PGeSe, and 24PGeSe have the most reversible oxidation waves. All polymers exhibit greater oxidation current relative to reduction, characteristic of p-type donor materials. 24PGeSe exhibits a similar oxidation shoulder to 33PSiSe, albeit with less current, while this shoulder is absent in the other samples. These oxidation shoulders indicate at least two distinct oxidations occur in 33PSiSe and 24PGeSe and result in relatively high HOMO energy levels relative to their lower molecular weight analogues. The shoulder intensity corresponds to the degree of polymer aggregation, as seen in the optical experiments, and could be a function of increased π-stacking in the solid state. Polymer LUMO energy level values range from −3.62 ev to −3.20 eV. Interestingly, reduction reversibility is greatest for 33PSiSe and 24PSiSe. This may result from higher quality films and suggests that the ability to form a stable radical anion increases with molecular weight.X-ray diffraction
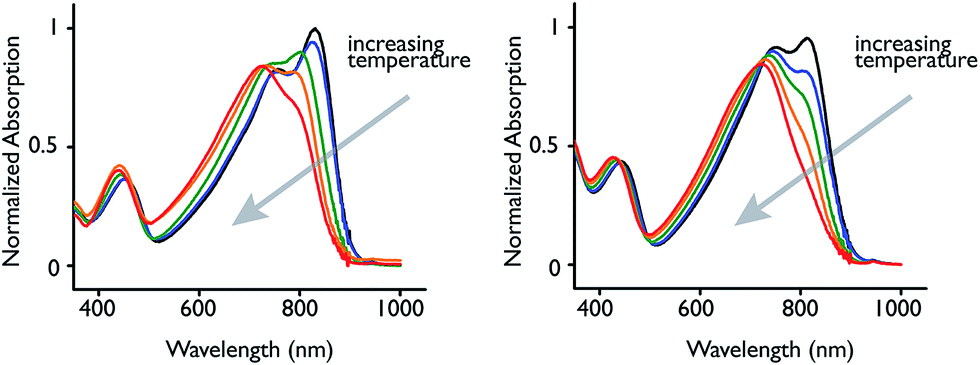
The ordering in the polymer thin films was investigated with two-dimensional wide-angle X-ray scattering (2D-WAXS). Films were cast onto silicon wafers from chlorobenzene solution before annealing at 130 °C for 30 min. Because the films are highly disordered, the scattering patterns (Fig. S14†) were radially averaged to produce one-dimensional traces (Fig. 3). The one-dimensional scattering patterns show lamellar crystalline peaks ranging from 17.2 Å for 5PGeSe to 17.8 Å in both 18PSiSe and 16PGeSe. There appears to be no obvious trend in lamellar spacing distance with respect to molecular weight or donor heteroatom. The π–π stacking distances are 3.56 Å and 3.60 Å for 24PGeSe and 18PSiSe, respectively. These represent the most crystalline polymers in each series with respect to π–π stacking, and the films on which a reliable measurement could be made. The similar lamellar π–π stacking distances in these polymers are not surprising given their very similar molecular geometries. The slightly smaller π–π stacking distances in the PGeSe series could result from its longer Ge–C bond length, which allows closer contact between neighboring chains. Overall the 2D scattering patterns show little directional orientation for either lamellar spacing or π–π stacking. Attempts were made to cast films from solutions of equal concentration; however, it is possible that some undissolved 33PSiSe resulted in thinner films, and less intense X-ray diffraction signal. Equal casting solution concentration for the other polymers indicates generally greater crystalline character in the Ge-donor polymers relative to their Si-donor analogues. This result is consistent with X-ray experiments conducted on the benzothiadiazole-acceptor analogues where carbon substitution with silicon in the cyclopentadithiophene donor results in a dramatic increase in polymer crystallinity.33 In this context we conclude that heavy group 14-atom substitution in the donor produces more crystalline polymers. Molecular weight effects, on the other hand, are less straightforward. For the silicon-donor series the low molecular weight 11PSiSe has the greatest lamellar crystallinity. The lamellar spacing signal decreases with increasing molecular weight and coincides with growth in the π–π stacking signal, indicating that as the molecular weight increases some lamellar order is exchanged for π–π stacking. This result is consistent with a recent study by You and co-workers.16 In the germanium-donor case, however, 24PGeSe exhibits the most crystalline character with respect to lamellar spacing and π–π stacking. This difference in crystallinity trend with molecular weight is possibly another result of the change in Si/Ge–C bond distance, as this is the only major structural difference between these two polymers. It is possible that the inter-chain distance created by this longer heteroatom-carbon bond allows neighboring Se–N interactions to contribute to polymer crystallinity. | ||
| Fig. 3 (top left) Radially averaged PSiSe traces from film 2-D WAXS plots; (bottom left) radially averaged PGeSe traces from film 2-D WAXS plots; (top right) 11PSiSe 2-D WAXS plot as a representative example; (bottom right) 24PGeSe 2-D WAXS plot as a representative example; all films drop cast onto silicon substrates from chlorobenzene solution and annealed for 30 min at 130 °C. | ||
Solar cell performance
Organic photovoltaic cells with the configuration ITO/PEDOT:PSS/polymer:PC71BM/LiF/aluminum were constructed to investigate the polymer photovoltaic properties (Fig. 4 and Table 3). Briefly, polymer:PC71BM 1:1 ratio solutions were spin coated onto PEDOT:PSS coated indium tin oxide (ITO) substrates before a LiF layer was deposited, followed by an aluminum anode. The optimized device processing conditions differed for each polymer repeat unit structure. The PSiSe series polymers were spin coated from a 1,2-dichlorobenzene solution with 2% v/v 2-chloronapthalene as the solvent additive whereas PGeSe polymers were cast from chlorobenzene solution with 10% v/v 1,2,4-trichlorobenzene and 0.5% v/v 1,8-diiodooctane as solvent additives, except in the 24PGeSe case where no 1,8-diiodooctane was used. Devices containing 33PSiSe blend films could not be fabricated due to the extremely limited solubility of this polymer, even after heating at 100 °C for 24 h. The limited solution concentration did not produce a thick enough film to construct a working device. Thus, devices were fabricated using a fourth polymer with Mn = 8000 g mol−1 (8PSiSe) in order to establish a molecular weight trend in the PSiSe series.
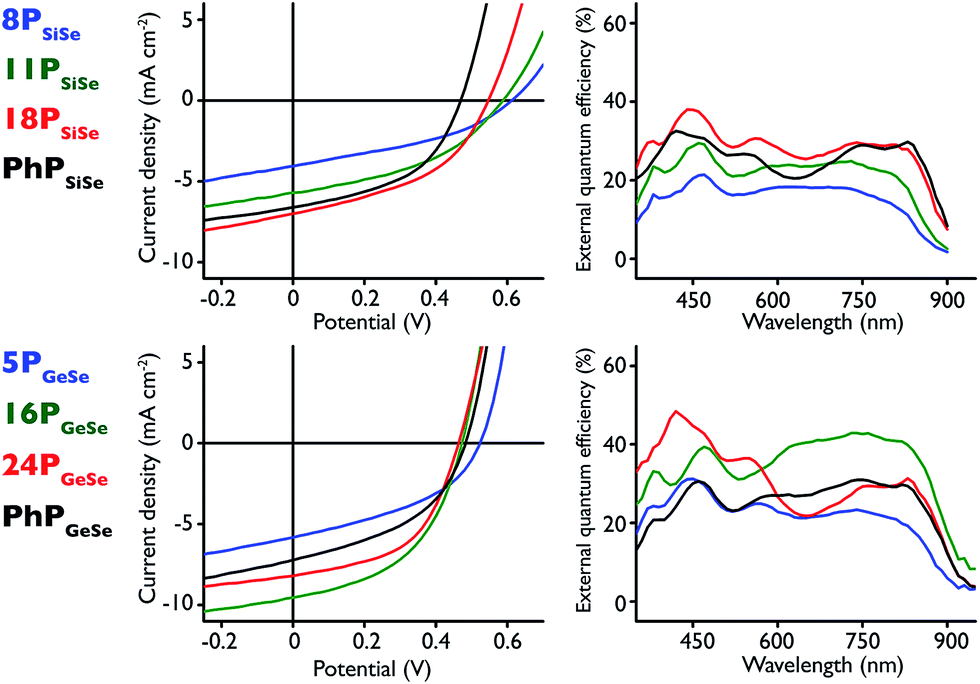 | ||
| Fig. 4 (top left) PSiSe series J–V curves; (top right) corresponding PSiSe series EQE curves; (bottom left) PGeSeJ–V curves; (bottom right) corresponding PGeSe EQE curves. | ||
| Polymer:PC71BM |
V OC/V | J SC/mA cm−2 | FF/% | η/% | Mobility/cm2 V−1 s−1 | |
|---|---|---|---|---|---|---|
| a Active layer cast from dichlorobenzene with 2% v/v chloronaphthalene additive. b Active layer cast from chlorobenzene with 10% v/v trichlorobenzene and 0.5% v/v 1,8-diiodooctane additives. c Active layer cast from chlorobenzene with 20% v/v trichlorobenzene additive. | ||||||
| 8PSiSe | 1:1a |
0.62 ± 0.01 | 4.27 ± 0.11 | 39.0 ± 1.1 | 1.03 ± 0.04 | — |
| 11PSiSe | 1:1a |
0.58 ± 0.01 | 5.57 ± 0.13 | 42.3 ± 0.9 | 1.38 ± 0.02 | — |
| 18PSiSe | 1:1a |
0.55 ± 0.01 | 6.72 ± 0.30 | 44.7 ± 1.6 | 1.64 ± 0.06 | 1.53 × 10−4 |
| 33PSiSe | 1:1a |
— | — | — | — | — |
| 5PGeSe | 1:1b |
0.51 ± 0.01 | 5.78 ± 0.03 | 41.0 ± 1.8 | 1.21 ± 0.07 | — |
| 16PGeSe | 1:1b |
0.48 ± 0.01 | 9.48 ± 0.28 | 44.3 ± 2.6 | 2.02 ± 0.12 | 4.55 × 10−5 |
| 24PGeSe | 1:1c |
0.45 ± 0.01 | 7.8 ± 1.0 | 51.7 ± 1.6 | 1.82 ± 0.22 | — |
| PhPSiSe | 1:1b |
0.46 ± 0.01 | 6.17 ± 0.30 | 47.7 ± 1.1 | 1.35 ± 0.10 | — |
| PhPGeSe | 1:1b |
0.46 ± 0.01 | 6.60 ± 0.19 | 47.8 ± 0.6 | 1.45 ± 0.06 | — |
In the PSiSe polymer series there is a clear improvement in device performance with increasing molecular weight, consistent with previous observations on conjugated polymer-based photovoltaic devices.59 As polymer size increases, the short circuit current (JSC) values increase along with the fill factor (FF), while there is a concomitant decrease in open circuit voltage (VOC). 18PSiSe produced the best performing Si-donor polymer-based devices with VOC = 0.55 V, JSC = 6.72 mA cm−2, FF = 44.7% and a power conversion efficiency (η) of 1.64%. This represents an increased power conversion efficiency compared to devices utilizing the carbon-donor analogue polymer in the active layer (PCSe),28,29,60 but decreased relative to those containing the silicon-donor and sulfur-acceptor (PSiS)30–32,34,61 owing largely to changes in JSC and FF. The 18PSiSe external quantum efficiency (EQE) spectrum reaches nearly 40% at roughly 440 nm and maintains around 30% at ∼850 nm. The EQE spectral profiles maintain the same shape, and trend to increased efficiency, at higher molecular weights, consistent with the observed JSC values. Interestingly, EQE increases consistently with molecular weight across the entire spectrum, indicating that increased photon harvesting could be the result of improved charge transport. The broader EQE spectra represent an improvement in light harvesting at lower energy compared to both PCSe and PSiS structural analogues. The 18PSiSe device demonstrates a 1.53 × 10−4 cm2 V−1 s−1 mobility value, which is an order of magnitude larger than its carbon-donor analogue (PCSe). We attribute this improvement to the silicon atom present in the donor because of its increased polarizability relative to carbon.
PGeSe-based device performance peaks at an intermediate molecular weight. Similar to PSiSe polymers, the PGeSe device FF increases with increasing the molecular weight, while the VOC is decreased. 16PGeSe produced the best performing cells in the germanium series, and in this study overall, with VOC = 0.48 V, JSC = 9.48 mA cm−2, FF = 44.3%, and η = 2.02%. It should be noted that 24PGeSe cells demonstrate 1.82% power conversion efficiency, which is close to the 16PGeSe value. The 16PGeSe cells benefit from a high JSC value whereas the 24PGeSe devices maintain a higher FF. This effect is likely the result of film morphology, and differs from the PSiSe results where the current consistently improves with increasing molecular weight. Indeed the 16PGeSe EQE spectrum shows remarkably greater photon harvesting at wavelengths beyond 500 nm. Compared to its best performing silicon-donor analogue, 16PGeSe maintains an EQE around 40% between 600 nm and 850 nm and roughly 20% at 900 nm. Like its silicon-donor analogue, these 16PGeSe device results show improvement over the lighter PCSe-based devices but show roughly half the overall power conversion efficiency of devices containing the germanium-donor and sulfur-acceptor polymer analogue (PGeS).36,62 This decrease in performance is a direct result of lower device JSC. One possible explanation is that the 2,1,3-benzothiadiazole acceptor polymers have generally greater absorption coefficients across the longer wavelengths than their Se analogues.35,36,53 Nonetheless, 16PGeSe devices display improved EQE at wavelengths beyond 850 nm, highlighting the 2,1,3-benzoselenadiazole acceptor red–absorption effect. The 16PGeSe device demonstrates a 4.55 × 10−5 cm2 V−1 s−1 mobility value, roughly half that of the 18PSiSe devices and a two-fold improvement over devices containing its carbon-donor analogue.
To better study heavy atom substitution effects and the role of polymer end groups, devices of phenyl-end capped polymers with nearly identical molecular weights were fabricated as well. The PhPSiSe and PhPGeSe devices present nearly identical VOC and FF, with very similar JSC values. This is not surprising given that the polymer molecular weights are nearly identical and the polymer end groups are likely all the same. The PhPGeSeJSC, FF, and η fall between those measured for 5PGeSe and 16PGeSe-based devices. Remarkably, the power conversion results for PhPSiSe and its equal-sized counterpart 11PSiSe are similar, whereas PhPSiSe maintains higher FF and JSC values and a lower VOC. Overall the molecular weight series results suggest that there is an intermediate molecular weight at which ideal device conditions are achieved, and that this molecular weight is likely different for each polymer structure. Similar PhPSiSe and PhPGeSe device results indicate that, in this case, heavy donor atom substitution is less of a factor in device performance than polymer molecular weight. As well, end capping appears to have little effect on device performance in this study. Overall we observe red light harvesting attributable to the 2,13-benzoselenadiazole and improved hole mobility resulting from the silicon- or germanium-containing donor monomers. The combination of separate monomer attributes into observed polymer properties further demonstrates the utility of D–A polymers to be engineered to a specific function through judicious choice of donor and acceptor units. The polymers presented here may have utility in the red light absorbing layer in a tandem device,46,63 or as long wavelength photosensitizers in a single bulk-heterojunction photovoltaic.
Morphology characterization
In order to gain insight into molecular weight and end capping effects on materials properties, tapping-mode atomic force microscopy (AFM), transmission electron microscopy (TEM), and dark field scanning TEM (STEM) images of 1:1 polymer:PC71BM blend films were collected (Fig. 5, 6 and S15†). AFM images were collected directly on the device films between the top electrodes. Films for TEM were cast from chlorobenzene at 5 mg mL−1 of polymer and PC71BM onto PSS coated glass substrates, producing thinner films than those used in the devices. These films were imaged with greater contrast than the thicker device films. The films were delaminated from the glass substrate in water, collected on a TEM grid, and dried before imaging. All polymers produced relatively smooth films, which increased in quality with molecular weight. Pinholes are visible in the TEM images of the low molecular weight samples. These indicate fast aggregation during drying and are consistent with lower quality films, which may contribute to the moderate device performance. Generally the polymer domain shapes shift from sphere-like to fiber-like as molecular weight increases. Focusing on the silicon-donor series, the 33PSiSe films display the greatest degree of order with a distinct morphology. A similar, but less obvious, morphology is present in the 18PSiSe films. The less-ordered 18PSiSe films produce the best device results in the series, despite less structural order than the 33PSiSe film. While this may suggest that an intermediate degree of order is ideal, it should be noted that the low 33PSiSe solubility affected our ability to produce sufficiently thick films for working photovoltaic devices. Thus we cannot say for certain that the morphology is directly responsible for the device results, which highlights the importance of balance between polymer properties when pursuing high performance devices.
 | ||
| Fig. 5 (top row) PSiSe/PC71BM blend film AFM height images; (middle row) AFM phase images; (bottom row) TEM images at 10000× magnification; columns are labeled with the specific polymer blended with PC71BM in the top panel; AFM data were collected directly on the device film between surface electrodes; TEM films were cast from 5 mg mL−1 polymer/PC71BM solutions in chlorobenzene onto PSS coated glass slides, delaminated in water and collected onto a TEM grid. | ||
 | ||
| Fig. 6 (top row) PGeSe/PC71BM blend film AFM height images; (middle row) AFM phase images; (bottom row) TEM images at 10000× magnification; columns are labeled with the specific polymer blended with PC71BM in the top panel; AFM data were collected directly on the device film between surface electrodes; TEM films were cast from 5 mg mL−1 polymer/PC71BM solutions in chlorobenzene onto PSS coated glass slides, delaminated in water and collected onto a TEM grid. | ||
TEM images show the germanium-donor series produces less ordered films with PC71BM than its silicon-donor analogues, which is consistent with its lower hole mobility, and less planar repeat unit structure.37,53 Taken with the 2D-WAXS data which show generally greater crystalline character in the germanium-donor polymer series, we postulate that PSiSe polymers produce larger ordered domains, as seen in the microscope images, whereas PGeSe films contain a greater area of smaller crystalline domains, resulting in more X-ray scattering in the 2D-WAXS data. The morphology differences among polymers with different molecular weights are less obvious in the PGeSe films than in the PSiSe films, thus the molecular weight effect is less pronounced. It is interesting to note that the relatively disordered 16PGeSe films produced the highest performing devices in this study. This suggests that an intermediate molecular weight polymer is ideal for photovoltaic device performance.
The PhPGeSe film TEM image shows domain sizes most closely resembling those in the 5PGeSe film image. Taken together with the PhPGeSe device data we observe that in this case molecular weight, and not end-groups, appears to most greatly affect the film morphology and subsequent device performance. In the PhPSiSe film the domain size is similar but somewhat more ordered than in the PhPGeSe film, consistent with the generally more structured films in the PSiSe series. The PhPSiSe film contains more structural order than its equal-molecular weight 11PSiSe analogue. It is interesting to note that these two polymers of equal molecular weight and different film morphologies produce very similar device data. This suggests that while the phenyl end groups may contribute to more order in the film, this is not sufficient to produce greater-than-expected device performance based on the polymer molecular weight. Finally, it should be noted that polymer dispersity likely affects the polymer:fullerene blend morphology and solar cell results to some degree. Presently there is no methodology for controlling its batch-to-batch variation in a step growth polymerization. As a result it is difficult to determine exactly how polymer dispersity affects the data presented here.
Conclusions
The effects of molecular weight and phenyl end capping were studied on two narrow band-gap (<1.4 eV) donor–acceptor polymer structures containing selenium and either germanium or silicon in their repeat unit. A molecular weight series and one phenyl-end capped derivative were synthesized using a microwave assisted Stille coupling polymerization. The solution and film absorption profiles and solution onset change as a function of molecular weight, whereas the subtler Si or Ge heteroatom changes the absorption onset in the polymer films. Molecular weight affects polymer absorption to the greatest degree in a medium molecular weight regime and the charge-transfer band has a significant aggregation component. Two-dimensional small angle X-ray scattering data indicate that generally a heavier atom in the donor produces more crystalline polymers, and polymer crystallinity increases with molecular weight. Both silicon-donor and germanium-donor polymer devices display improved photon conversion at 850 nm relative to their sulfur-acceptor analogues, highlighting the red-light absorption contributed by the 2,1,3-benzoselenadiazole monomer. Improved hole mobility is also observed relative to a structurally analogous donor–acceptor polymer with carbon in the donor bridge position. Additionally, the device results reveal that there is an intermediate molecular weight range at which the best device performance is achieved. Phenyl end capping appears to have little effect on overall device performance in this case. Molecular weight invokes the greatest change on polymer/fullerene blend morphology, followed by phenyl end capping, and finally by donor heteroatom. The largest silicon-donor polymer displays the most ordered blend morphology but is not sufficiently solution-processable to construct a working photovoltaic device. This highlights the importance of balance between polymer properties when pursuing high performance photovoltaic devices. The polymers presented here should have further utility as red light harvesters in a tandem device, or as photosensitizers in a single bulk-heterojunction photovoltaic.Experimental section
General considerations
Unless stated otherwise, starting materials were purchased and used as received. 4,4′-Bis(2-ethyl-hexyl)-5,5′-dibromo-dithieno[3,2-b:2′,3′-d]silole was purchased from Solarmer Materials, Inc. 4,4′-Bis(2-ethyl-hexyl)-dithieno[3,2-b:2′,3′-d]germole was purchased from 1-Material, Inc. 4,4′-bis(2-ethyl-hexyl)-5,5′-bis(trimethyltin)-dithieno[3,2-b:2′,3′-d]silole, and 4,4′-bis(2-ethyl-hexyl)-5,5′-bis(trimethyltin)-dithieno[3,2-b:2′,3′-d]germole were prepared by literature procedures and purified by reverse phase chromatography in 30% v/v ethyl acetate in acetonitrile. Deuterated 1,1,2,2-tetrachloroethane-d2 was purchased from Sigma-Aldrich and used as received. Deuterated chloroform was purchased from Cambridge Isotope Laboratories and used as received. All other chemicals were purchased from Sigma-Aldrich and used as received. Unless otherwise noted, all manipulations involving air- or water-sensitive reagents were performed under a dry argon atmosphere using standard Schlenk techniques or under dry nitrogen in a glovebox. Solvents were degassed with argon for 25 minutes and dried using an Innovative Technologies solvent purification system. Microwave reactions were carried out with a Biotage Initiator Classic microwave reactor.Instrumentation
NMR spectra were recorded on an Agilent DD2 600 spectrometer operating at 600 MHz for 1H at 130 °C or on a Varian Mercury 400 spectrometer operating at 400 MHz for 1H, as noted. Chemical shifts are reported in ppm at 130 °C or ambient temperature, as noted. 1H chemical shifts are referenced to the residual 1,1,2,2-tetrachloroethane proton peak at 6.00 ppm or the residual chloroform peak at 7.26 ppm, as noted. Absorption spectra were recorded on a Varian Cary 5000 UV-Vis-NIR spectrophotometer. Solution spectra were recorded in chlorobenzene; films for absorption were spin-coated onto glass substrates at 1000 rpm from chlorobenzene. Emission spectra were recorded on a Photon Technology International QuantaMaster 40-F spectrofluorometer in chlorobenzene. Polymer molecular weights were determined with a Viscotek HT-GPC (1,2,4-trichlorobenzene, 140 °C) using Tosoh Bioscience LLC TSK-GEL GMHHR-HT mixed-bed columns and narrow molecular weight distribution polystyrene standards. Cyclic voltammetry was conducted on Au button electrodes with a Ag/AgCl reference electrode, and Pt counter electrode in 0.1 M TBAPF6 in dry acetonitrile at 50 mV s−1 scan rate. Two-dimensional wide angle X-ray (2D-WAXS) scattering experiments were conducted at McMaster University on a Bruker D8 Discover with Davinci. Design diffractometer equipped with a cobalt sealed tube source and a Vantac 500 area detector. AFM images were obtained with a Vecco Dimension 300 microscope. TEM images were obtained on a Hitachi H-7000 microscope at an accelerating voltage of 100 kV. STEM images were obtained on a FEI Quanta FEG ESEM at 30 kV.Photovoltaic device fabrication and testing
PC71BM (American Dye Source) was purchased and used as received. Devices were fabricated on commercial indium tin oxide (ITO) substrates. These substrates were cleaned in aqueous detergent, deionized (DI) water, acetone, and methanol, and subsequently treated in an air-plasma cleaner for 5 min. Next, poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate) (PEDOT:PSS) (Clevios P VP AI 4083) was coated onto the substrates at 3000 rpm and annealed at 130 °C in air for 15 min, after which the substrate was transferred into a nitrogen-filled glove box. Polymer:PC71BM films were spin-coated from chlorobenzene (PGeSe) or 1,2-dichlorobenzene (PSiSe) solutions. For PSiSe polymers 2% v/v 1-chloronaphthalene was used as a processing additive, while 0.5% v/v 1,8-diiodooctane was used as the additive for PGeSe polymers. Solutions were stirred at 50 °C (80 °C for 33PSiSe) overnight before spin-coating onto the PEDOT:PSS coated substrate. To finish the device, a 0.8 nm LiF layer and 100 nm Al anode were thermally deposited through a shadow mask at ∼10−6 torr using an Angstrom Engineering Covap II. All device areas were 0.07 cm2 as defined by the area of the circular Al anode. J–V characteristics were measured using a Keithley 2400 source meter under simulated AM 1.5 G conditions. The mismatch of the simulator spectrum was calibrated using a Si diode with a KG-5 filter. EQE spectra were recorded and compared with a Si reference cell traceable to the National Institute of Standards and Technology (NIST).
Synthesis and characterization
An analogous procedure with a 1.02:1.00 4,7-dibromo-2,1,3-benzoselenadiazole:2 monomer ratio gave a chloroform recovered polymer (18PSiSe, 66%) with the same NMR data as above and the following GPC data: Mn = 17.7 kg mol−1, Mw = 32.7 kg mol−1, Đ = 1.85.
An analogous procedure with a 1.05:1.00 4,7-dibromo-2,1,3-benzoselenadiazole:2 monomer ratio gave a chloroform recovered polymer (11PSiSe, 45%) with the same NMR data as above and the following GPC data: Mn = 10.6 kg mol−1, Mw = 19.5 kg mol−1, Đ = 1.84.
An analogous procedure with a 1.07:1.00 4,7-dibromo-2,1,3-benzoselenadiazole:2 monomer ratio gave a chloroform recovered polymer (8PSiSe, 34%) with the same NMR data as above and the following GPC data: Mn = 8.1 kg mol−1, Mw = 17.0 kg mol−1, Đ = 2.2.
An analogous procedure with a 1.02:1.00 4,7-dibromo-2,1,3-benzoselenadiazole:4 monomer ratio gave a polymer (16PGeSe, 55%) with the same NMR data as above and the following GPC data: Mn = 15.6 kg mol−1, Mw = 31.0 kg mol−1, Đ = 1.99.
An analogous procedure with a 1.05:1.00 4,7-dibromo-2,1,3-benzoselenadiazole:4 monomer ratio gave a polymer (5PGeSe, 62%) with the same NMR data as above and the following GPC data: Mn = 4.6 kg mol−1, Mw = 12.9 kg mol−1, Đ = 2.6.
Acknowledgements
The authors would like to thank Victoria Jarvis at McMaster University for conducting 2D-WAXS experiments. This work was supported by the University of Toronto, NSERC, the CFI, and the Ontario Research Fund. D.S.S. is grateful to the Connaught Foundation for an Innovation Award and DuPont for a Young Professor Grant. G.L.G. is grateful for the F.E. Beamish Graduate Scholarship in Science and Technology.References
- A. K. Bakhshi, Y. Yamaguchi, H. Ago and T. Yamabe, Synth. Met., 1996, 79, 115–120 CrossRef CAS.
- A. Bakhshi, Y. Yamaguchi, H. Ago and T. Yamabe, J. Mol. Struct.: THEOCHEM, 1998, 427, 211–219 CrossRef CAS.
- Q. T. Zhang and J. M. Tour, J. Am. Chem. Soc., 1998, 120, 5355–5362 CrossRef CAS.
- R. S. Kularatne, H. D. Magurudeniya, P. Sista, M. C. Biewer and M. C. Stefan, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 743–768 CrossRef CAS.
- A. Pron, P. Berrouard and M. Leclerc, Macromol. Chem. Phys., 2012, 214, 7–16 CrossRef.
- L. Pandey, C. Risko, J. E. Norton and J.-L. Brédas, Macromolecules, 2012, 45, 6405–6414 CrossRef CAS.
- Z. Zhu, D. Waller, R. Gaudiana, M. Morana, D. Mühlbacher, M. Scharber and C. Brabec, Macromolecules, 2007, 40, 1981–1986 CrossRef CAS.
- H. Zhou, L. Yang and W. You, Macromolecules, 2012, 45, 607–632 CrossRef CAS.
- A. A. Jahnke, B. Djukic, T. M. McCormick, E. Buchaca Domingo, C. Hellmann, Y. Lee and D. S. Seferos, J. Am. Chem. Soc., 2013, 135, 951–954 CrossRef CAS PubMed.
- J.-L. Brédas, J. E. Norton, J. Cornil and V. Coropceanu, Acc. Chem. Res., 2009, 42, 1691–1699 CrossRef PubMed.
- J. Hollinger, A. A. Jahnke, N. Coombs and D. S. Seferos, J. Am. Chem. Soc., 2010, 132, 8546–8547 CrossRef CAS PubMed.
- H. Zhou, L. Yang, S. Stoneking and W. You, ACS Appl. Mater. Interfaces, 2010, 2, 1377–1383 CAS.
- S. Shoaee, T. M. Clarke, C. Huang, S. Barlow, S. R. Marder, M. Heeney, I. McCulloch and J. R. Durrant, J. Am. Chem. Soc., 2010, 132, 12919–12926 CrossRef CAS PubMed.
- C.-Y. Kuo, W. Nie, H. Tsai, H.-J. Yen, A. D. Mohite, G. Gupta, A. M. Dattelbaum, D. J. William, K. C. Cha, Y. Yang, L. Wang and H.-L. Wang, Macromolecules, 2014, 47, 1008–1020 CrossRef CAS.
- J. L. Jellison, C.-H. Lee, X. Zhu, J. D. Wood and K. N. Plunkett, Angew. Chem., Int. Ed., 2012, 51, 12321–12324 CrossRef CAS PubMed.
- W. Li, L. Yang, J. R. Tumbleston, L. Yan, H. Ade and W. You, Adv. Mater., 2014, 26, 4456–4462 CrossRef CAS PubMed.
- G. L. Gibson and D. S. Seferos, Macromol. Chem. Phys., 2014, 215, 811–823 CrossRef CAS.
- J. D. Azoulay, Z. A. Koretz, B. M. Wong and G. C. Bazan, Macromolecules, 2013, 46, 1337–1342 CrossRef CAS.
- J. S. Kim, Z. Fei, D. T. James, M. Heeney and J.-S. Kim, J. Mater. Chem., 2012, 22, 9975–9982 RSC.
- B. J. A. Caputo, G. C. Welch, D. A. Kamkar, Z. B. Henson, T.-Q. Nguyen and G. C. Bazan, Small, 2011, 7, 1422–1426 CrossRef CAS PubMed.
- A. A. B. Alghamdi, D. C. Watters, H. Yi, S. Al-Faifi, M. S. Almeataq, D. Coles, J. Kingsley, D. G. Lidzey and A. Iraqi, J. Mater. Chem. A, 2013, 1, 5165–5171 CAS.
- D. R. Levine, M. A. Siegler and J. D. Tovar, J. Am. Chem. Soc., 2014, 136, 7132–7139 CrossRef CAS PubMed.
- H. Zhong, Z. Li, E. Buchaca-Domingo, S. Rossbauer, S. E. Watkins, N. Stingelin, T. D. Anthopoulos and M. Heeney, J. Mater. Chem. A, 2013, 1, 14973–14981 CAS.
- Y. Li, J. Zou, H.-L. Yip, C.-Z. Li, Y. Zhang, C.-C. Chueh, J. Intemann, Y. Xu, P.-W. Liang and Y. Chen, Macromolecules, 2013, 46, 5497–5503 CrossRef CAS.
- R. Yang, R. Tian, J. Yan, Y. Zhang, J. Yang, Q. Hou, W. Yang, C. Zhang and Y. Cao, Macromolecules, 2005, 38, 244–253 CrossRef CAS.
- N. Allard, R. B. Aïch, D. Gendron, P.-L. T. Boudreault, C. Tessier, S. Alem, S.-C. Tse, Y. Tao and M. Leclerc, Macromolecules, 2010, 43, 2328–2333 CrossRef CAS.
- P. M. Beaujuge, C. M. Amb and J. R. Reynolds, Acc. Chem. Res., 2010, 43, 1396–1407 CrossRef CAS PubMed.
- L. Huo, H.-Y. Chen, J. Hou, T. L. Chen and Y. Yang, Chem. Commun., 2009, 5570–5572 RSC.
- Y.-M. Hwang, J. Ohshita, Y. Harima, T. Mizumo, Y. Ooyama, Y. Morihara, T. Izawa, T. Sugioka and A. Fujita, Polymer, 2011, 52, 3912–3916 CrossRef CAS PubMed.
- F. Wang, J. Luo, K. Yang, J. Chen, F. Huang and Y. Cao, Macromolecules, 2005, 38, 2253–2260 CrossRef CAS.
- P. M. Beaujuge, W. Pisula, H. N. Tsao, S. Ellinger, K. Müllen and J. R. Reynolds, J. Am. Chem. Soc., 2009, 131, 7514–7515 CrossRef CAS PubMed.
- G. Lu, H. Usta, C. Risko, L. Wang, A. Facchetti, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2008, 130, 7670–7685 CrossRef CAS PubMed.
- H.-Y. Chen, J. Hou, A. E. Hayden, H. Yang, K. N. Houk and Y. Yang, Adv. Mater., 2010, 22, 371–375 CrossRef CAS PubMed.
- M. C. Scharber, M. Koppe, J. Gao, F. Cordella, M. A. Loi, P. Denk, M. Morana, H.-J. Egelhaaf, K. Forberich, G. Dennler, R. Gaudiana, D. Waller, Z. Zhu, X. Shi and C. J. Brabec, Adv. Mater., 2010, 22, 367–370 CrossRef CAS PubMed.
- C. M. Amb, S. Chen, K. R. Graham, J. Subbiah, C. E. Small, F. So and J. R. Reynolds, J. Am. Chem. Soc., 2011, 133, 10062–10065 CrossRef CAS PubMed.
- D. Gendron, P.-O. Morin, P. Berrouard, N. Allard, B. R. Aïch, C. N. Garon, Y. Tao and M. Leclerc, Macromolecules, 2011, 44, 7188–7193 CrossRef CAS.
- H. Zhong, Z. Li, F. Deledalle, E. C. Fregoso, M. Shahid, Z. Fei, C. B. Nielsen, N. Yaacobi-Gross, S. Rossbauer, T. D. Anthopoulos, J. R. Durrant and M. Heeney, J. Am. Chem. Soc., 2013, 135, 2040–2043 CrossRef CAS PubMed.
- Z. Fei, R. S. Ashraf, Z. Huang, J. Smith, R. J. Kline, P. D'Angelo, T. D. Anthopoulos, J. R. Durrant, I. McCulloch and M. Heeney, Chem. Commun., 2012, 48, 2955–2957 RSC.
- G. C. Welch, R. C. Bakus II, S. J. Teat and G. C. Bazan, J. Am. Chem. Soc., 2013, 135, 2298–2305 CrossRef CAS PubMed.
- H. Zhou, L. Yang, A. C. Stuart, S. C. Price, S. Liu and W. You, Angew. Chem., Int. Ed., 2011, 50, 2995–2998 CrossRef CAS PubMed.
- S. Albrecht, S. Janietz, W. Schindler, J. Frisch, J. Kurpiers, J. Kniepert, S. Inal, P. Pingel, K. Fostiropoulos, N. Koch and D. Neher, J. Am. Chem. Soc., 2012, 134, 14932–14944 CrossRef CAS PubMed.
- Y. Zhang, J. Zou, C.-C. Cheuh, H.-L. Yip and A. K. Y. Jen, Macromolecules, 2012, 45, 5427–5435 CrossRef CAS.
- S. A. Shin, J.-H. Kim, J. B. Park, I.-N. Kang, M.-J. Park and D.-H. Hwang, Macromol. Chem. Phys., 2013, 214, 1780–1788 CrossRef CAS.
- N. Wang, Z. Chen, W. Wei and Z. Jiang, J. Am. Chem. Soc., 2013, 135, 17060–17068 CrossRef CAS PubMed.
- P. Shen, H. Bin, Y. Zhang and Y. Li, Polym. Chem., 2014, 5, 567–577 RSC.
- J.-H. Kim, S. A. Shin, J. B. Park, C. E. Song, W. S. Shin, H. Yang, Y. Li and D.-H. Hwang, Macromolecules, 2014, 47, 1613–1622 CrossRef CAS.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Am. Chem. Soc., 2012, 134, 539–547 CrossRef CAS PubMed.
- A. M. Ballantyne, L. Chen, J. Nelson, D. D. C. Bradley, Y. Astuti, A. Maurano, C. G. Shuttle, J. R. Durrant, M. Heeney, W. Duffy and I. McCulloch, Adv. Mater., 2007, 19, 4544–4547 CrossRef CAS.
- S. Günes, H. Neugebauer and N. S. Sariciftci, Chem. Rev., 2007, 107, 1324–1338 CrossRef PubMed.
- R. C. Coffin, J. Peet, J. Rogers and G. C. Bazan, Nat. Chem, 2009, 1, 657–661 CrossRef CAS PubMed.
- R. S. Loewe, S. M. Khersonsky and R. D. McCullough, Adv. Mater., 1999, 11, 250–253 CrossRef CAS.
- I. Osaka and R. D. McCullough, Acc. Chem. Res., 2008, 41, 1202–1214 CrossRef CAS PubMed.
- G. L. Gibson, T. M. McCormick and D. S. Seferos, J. Phys. Chem. C, 2013, 117, 16606–16615 CAS.
- J. Hollinger, P. M. DiCarmine, D. Karl and D. S. Seferos, Macromolecules, 2012, 45, 3772–3778 CrossRef CAS.
- J. Peet, N. S. Cho, S. K. Lee and G. C. Bazan, Macromolecules, 2008, 41, 8655–8659 CrossRef CAS.
- P. Serguievski and M. R. Detty, Organometallics, 1997, 16, 4386–4391 CrossRef CAS.
- G. He, W. Torres Delgado, D. J. Schatz, C. Merten, A. Mohammadpour, L. Mayr, M. J. Ferguson, R. McDonald, A. Brown, K. Shankar and E. Rivard, Angew. Chem., Int. Ed., 2014, 53, 4587–4591 CrossRef CAS PubMed.
- J. Pommerehne, H. Vestweber, W. Guss, R. F. Mahrt, H. Bässler, M. Porsch and J. Daub, Adv. Mater., 1995, 7, 551–554 CrossRef CAS.
- J. J. Intemann, K. Yao, H.-L. Yip, Y.-X. Xu, Y.-X. Li, P.-W. Liang, F.-Z. Ding, X. Li and A. K. Y. Jen, Chem. Mater., 2013, 25, 3188–3195 CrossRef CAS.
- J. Hou, T. L. Chen, S. Zhang and Y. Yang, J. Phys. Chem. C, 2009, 113, 1601–1605 CAS.
- J. Hou, H.-Y. Chen, S. Zhang, G. Li and Y. Yang, J. Am. Chem. Soc., 2008, 130, 16144–16145 CrossRef CAS PubMed.
- Z. Fei, J. S. Kim, J. Smith, E. B. Domingo, T. D. Anthopoulos, N. Stingelin, S. E. Watkins, J.-S. Kim and M. Heeney, J. Mater. Chem., 2011, 21, 16257–16263 RSC.
- L. Dou, J. You, J. Yang, C.-C. Chen, Y. He, S. Murase, T. Moriarty, K. Emery, G. Li and Y. Yang, Nat. Photonics, 2012, 6, 180–185 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4ta02820a |
| This journal is © The Royal Society of Chemistry 2014 |