
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(II) nanocluster-grafted, Nb-doped TiO2 as an efficient visible-light-sensitive photocatalyst based on energy-level matching between surface and bulk states†
Min
Liu
b,
Xiaoqing
Qiu
b,
Kazuhito
Hashimoto
*bc and
Masahiro
Miyauchi
*ad
aDepartment of Metallurgy and Ceramics Science, Graduate School of Science and Engineering, Tokyo Institute of Technology, 2-12-1 Ookayama, Meguro-ku, Tokyo 152-8552, Japan. E-mail: mmiyauchi@ceram.titech.ac.jp
bResearch Center for Advanced Science and Technology, The University of Tokyo, 4-6-1 Komaba, Meguro-ku, Tokyo 153-8904, Japan. E-mail: hashimoto@light.t.u-tokyo.ac.jp
cGraduate School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
dJapan Science and Technology Agency (JST), ACT-C, 4-1-8 Honcho Kawaguchi, Saitama 332-0012, Japan
First published on 18th June 2014
Abstract
Although visible-light-sensitive photocatalysis has been reported for cupreous ion (Cu(II)) nanocluster-grafted titanium dioxide (TiO2), the visible-light absorption (α) of this system is relatively low because of its limited interfacial excitation. In the present study, we synthesized niobium (Nb)-doped TiO2 grafted with Cu(II) nanoclusters and examined its capacity for visible-light absorption and photocatalytic activity for decomposing organic molecules. We speculated that the matching of energy levels between the surface Cu(II) nanoclusters and bulk-doped Nb ions would markedly increase the visible-light activity of TiO2. The doped Nb ions produced partially occupied energy levels below the conduction band of TiO2 that closely matched the potential of the Cu2+/Cu+ redox couple in the surface-grafted Cu(II) nanoclusters. The well-matched energy levels induced the effective transfer of photogenerated electrons from the doped Nb states to the surface-grafted Cu(II) nanoclusters, which mediated the efficient multi-electron reduction of oxygen molecules. The prepared Cu(II)–NbxTi1−xO2 nanocomposites exhibited a high photocatalytic reaction rate for the decomposition of 2-propanol into CO2 under visible light. Our results demonstrate that efficient photocatalysts can be generated by matching the energy levels of bulk-doped ions and surface nanoclusters, which represents a strategic approach for the rational design and development of high-performance photocatalysts.
1. Introduction
Titanium dioxide (TiO2) has attracted considerable recent attention as an efficient photocatalyst for applications such as water splitting, organic decomposition, and solar cells.1 However, because TiO2 is a wide band-gap semiconductor, with band gap values of 3.2 and 3.0 eV for the anatase and rutile forms, respectively, it can only be activated under ultraviolet (UV) light irradiation, thereby limiting its practical applications.2 To increase the utilization of solar and indoor light sources, TiO2 has been doped with various transition metal cations, such as Cr, Mn, Fe, Pb, and Cu, and anions, including N, C, and S, in an attempt to extend the light absorption capacity of TiO2 into the visible light region.3 Despite extensive research effort being made to modify the properties of TiO2, most doped TiO2 systems remain unsuitable for practical use because their quantum efficiencies (QEs) under visible light are too low to support the efficient photocatalytic reactions.3,4 In the case of cationic doping, the increase in visible light sensitivity is mainly caused by impurity levels in the forbidden band, which act as recombination centers for photogenerated charge carriers.2a–c In contrast, anionic doping of TiO2 generally introduces isolated levels above the valence band (VB) that contain holes generated with lower oxidation power and mobility than those in the VB,4 resulting in low photocatalytic performance.Recently, the surface modification of TiO2 with Cu(II) or Fe(III) nanoclusters was shown to increase its visible-light sensitivity without inducing impurity levels in the band gap.5 Under visible-light irradiation, electrons in the VB of TiO2 are excited to these surface nanoclusters through an interfacial charge transfer (IFCT) process.6 Simultaneously, the excited electrons are consumed in the multi-electron reduction of oxygen mediated by the nanoclusters.7 Therefore, Cu(II) or Fe(III) nanocluster-grafted TiO2 exhibits a high QE under visible light. However, the capacity of this photocatalytic system for visible-light absorption is limited because IFCT only occurs at TiO2 particle/nanocluster interfaces.
Surface nanoclusters have also been demonstrated to increase the visible-light activities of doped semiconductors.5d,f,8 For example, Ti3+ self-doped TiO2, which is inactive even under UV light irradiation, is converted into an efficient visible-light-sensitive photocatalyst by the surface grafting of Cu(II) or Fe(III) nanoclusters.5d Although this finding indicates that photogenerated electrons are transferred from the doped levels to the surface Cu(II) or Fe(III) nanoclusters, the QEs of metal-doped TiO2 photocatalysts cannot exceed that of Cu(II)- or Fe(III)-grafted TiO2, because the charge transfer is limited from doped levels to surface nanoclusters. Thus, efficient charge transfer between the doped levels and surface nanoclusters is critical for the photocatalytic performance of these systems. To achieve this criterion, the concept of energy level matching between the doped ions and surface-grafted nanoclusters has been recently examined using Fe(III) ion-doped and -grafted TiO2 as a model system.5e The Fe(III)-doped and -grafted TiO2 system exhibits a QE of 47.3% and a reaction rate of 0.69 μmol h−1, suggesting the feasibility of this approach.
Compared with Fe(III)-based compounds, cupreous compounds have the added advantage of intrinsic anti-pathogenic properties under both dark and light conditions,9 as was demonstrated for a Cu-deposited thin film of TiO2.9a Despite this advantageous characteristic, the visible-light absorption and quantum efficiencies of Cu(II)-grafted TiO2 are less than those of Fe(III)-grafted TiO2.5a,b Thus, for the development of Cu(II)-based photocatalysts that are suitable for indoor applications aimed at reducing chemical irritation or spread of infectious disease, it is necessary to markedly increase the visible-light sensitivity and photocatalytic efficiency of these materials.9 Based on the concept of energy level matching, we hypothesized that this could be achieved by modifying TiO2 to have similar interband energy levels to the redox potential of Cu(II) nanoclusters.
Niobium (Nb)-doped TiO2 has attracted recent attention due to its intriguing electronic properties and potential applications in transparent conductive oxide, photovoltaic cells and sensors. Theoretical and experimental studies have shown that the doping of TiO2 with Nb ions generates an energy level at 0.1 to 0.2 eV below the conduction band (CB),10 a value that closely matches the redox potential of Cu2+/Cu+ (0.16 V vs. SHE, pH = 0).5a,b However, Nb-doped TiO2 exhibits low efficiency for photocatalytic reactions because pentavalent Nb ions are substituted for tetravalent Ti leading to the bulk formation of Ti3+ species that serve as recombination centers.5d,10 Instead, Cu(II) nanocluster-grafted (Cu(II)–TiO2) can increase its visible-light activity without introduction of impurity levels in the band gap. However, the visible-light absorption of Cu(II)–TiO2 is limited because IFCT proceeds only at the bulk/nanocluster interface.
In the present study, we investigated the coupling of the bulk doping of Nb ions and surface grafting of Cu(II) clusters on TiO2. We speculated that under visible light irradiation, excited electrons in the doped Nb levels would transfer to the Cu(II) nanoclusters owing to their similar energy levels (Fig. 1), thereby increasing the visible-light sensitivity and QE of TiO2 photocatalysts.
 | ||
| Fig. 1 Proposed photocatalytic processes for NbxTi1−xO2, Cu(II)–TiO2 and Cu(II)–NbxTi1−xO2, respectively. | ||
2. Experimental section
2.1 Synthesis of NbxTi1−xO2
Nb-doped TiO2 (NbxTi1−xO2) nanocomposites were prepared using a simple impregnation method with commercial TiO2 (rutile phase, 15 nm grain size, 90 m2 g−1 specific surface area; MT-150A, Tayca Co.) as the starting material.5e In a typical synthesis, 1.5 g TiO2 powder was mixed with 10 mL ethanol to form a TiO2 suspension, to which niobium(V) chloride (NbCl5, Wako, 95%) was added as the source of Nb at a weight fraction to TiO2 of 0.1%. The resulting suspension was stirred for 0.5 h in a vial reactor and was then dried at room temperature. The obtained residue was calcined at 950 °C for 3 h to form NbxTi1−xO2 which was then further treated with a 6 M HCl aqueous solution at 90 °C for 3 h under stirring. The products were filtered through a 0.025 μm membrane filter (Millipore) and then washed with sufficient amounts of distilled water. NbxTi1−xO2 was obtained as a clear powder and was dried at 110 °C for 24 h before being ground to a fine powder using an agate mortar and pestle for the preparation of Cu(II) nanocluster-grafted NbxTi1−xO2 (Cu(II)–NbxTi1−xO2) nanocomposites. Pure TiO2 was obtained using the same annealing and acid treatment process without adding NbCl5 solution, and was used to prepare Cu(II)–TiO2 nanocomposites.2.2 Modification of NbxTi1−xO2 with Cu(II) nanoclusters
The grafting of Cu(II) nanoclusters onto NbxTi1−xO2 was performed using an impregnation method.5a,b Briefly, 1 g NbxTi1−xO2 powder was dispersed in 10 mL distilled water and CuCl2·2H2O (Wako, 99.9%) was then added to the TiO2 suspension as a source of Cu. The weight fraction of Cu relative to TiO2 was set to 0.1%. The resulting suspension was heated at 90 °C under stirring for 1 h in a vial reactor. The products were filtered through a 0.025 μm membrane filter (Millipore) and then washed with sufficient amounts of distilled water. The obtained residue was dried at 110 °C for 24 h and subsequently ground to a fine powder using an agate mortar and pestle. Cu(II)–TiO2 was also prepared by the same impregnation method.2.3 Sample characterization
The structural characteristics of the prepared nanocomposites were measured by powder X-ray diffraction (XRD) at room temperature on a Rigaku D/MAX25000 diffractometer with a copper target (λ = 1.54056 Å). Electron spin resonance (ESR) spectra were recorded on a Bruker ESP350E spectrometer. Elemental analyses of the samples were performed using an inductively coupled plasma-atomic emission spectrometer (ICP-AES; P-4010, Hitachi). UV-visible absorption spectra were obtained by the diffuse reflection method using a UV-2550 spectrometer (Shimadzu). The morphologies of the prepared NbxTi1−xO2 nanocomposites were investigated by scanning electron microscopy (SEM) using a Hitachi SU-8000 apparatus. Transmission electron microscopy (TEM) images were observed by the Hitachi HF-2000 instrument using acceleration voltage at 200 kV. The specific surface areas of the samples were determined from the nitrogen absorption data at liquid nitrogen temperature using the Barrett–Emmett–Teller (BET) technique. The samples were degassed at 200 °C and the pressure was kept below 100 mTorr for a minimum of 2 h prior to analysis using a Micromeritics VacPrep 061 instrument. Surface compositions and valence band X-ray photoelectron spectra were studied by X-ray photoelectron spectroscopy (XPS; model 5600, Perkin-Elmer). The binding energy data were calibrated with reference to the C 1s signal at 284.5 eV.2.4 Evaluation of photocatalytic properties
The photocatalytic activities of the prepared nanocomposites were evaluated by monitoring the decomposition of gaseous 2-propanol (IPA) under visible-light irradiation. A Xe lamp (LA-251Xe, Hayashi Tokei) equipped with L-42, B-47, and C-40C glass filters (Asahi Techno-Glass) was used as a source of visible light (420–530 nm, 1 mW cm−2). The light intensity was measured using a spectrum-radiometer (USR-45D, Ushio Co.) and was adjusted to 1 mW cm−2. A 500 mL cylindrical glass vessel was used as the photocatalysis reactor. To perform the photocatalytic experiments, 300 mg photocatalyst powder was evenly spread on the bottom of a circular glass dish (area of 5.5 cm2) that was mounted in the middle of the vessel reactor. The vessel was sealed with a rubber O-ring and a quartz cover, evacuated, and filled with fresh synthetic air. To eliminate organic contaminants from the sample surface, the vessel was illuminated with a Xe lamp (LA-251Xe) until the CO2 generation rate was less than 0.02 μmol per day. The vessel was then evacuated and refilled with fresh synthetic air. The pressure inside the vessel was kept at ∼1 atm. To begin the photocatalytic measurement, 300 ppmv (∼6 μmol) of gaseous IPA was injected into the vessel, which was then incubated in the dark for 12 h to achieve the absorption/desorption equilibrium of IPA on the photocatalyst surfaces. During this period, the IPA concentration first decreased and then remained constant, demonstrating that adsorption/desorption equilibrium had been reached. During the equilibration process, no acetone or CO2 was detected, demonstrating that the IPA molecules were not decomposed by the photocatalysts under dark conditions. The vessel was then irradiated with light, and 1 mL gaseous samples were periodically extracted from the reaction vessel to measure the concentrations of IPA, acetone, and CO2 using a gas chromatograph (model GC-8A; Shimadzu Co., Ltd.).3. Results and discussion
NbxTi1−xO2 nanocomposites were prepared by a simple impregnation method and heat treatment using commercial rutile TiO2 and NbCl5 as starting materials. The total amount of Nb in the prepared samples was measured by ICP-AES and was found to be nearly equal to the initial value used in the preparation process (Table S1†). The grafting of Cu(II) nanoclusters onto NbxTi1−xO2 was performed by a simple impregnation method.5 XRD analysis showed that the prepared samples maintained a pure rutile TiO2 crystalline structure (JCPDS card no. 21-1276) after Nb doping or surface grafting of Cu(II) nanoclusters (Fig. 2a and S1†), and an aggregated Nb2O5 phase was not detected at any of the examined doping concentrations (Fig. S1†). Notably, the diffraction peaks of the Nb-doped samples, NbxTi1−xO2 and Cu(II)–NbxTi1−xO2, were shifted towards smaller angles compared with that of the undoped sample (Fig. 2b). This finding indicates that Nb was substitutionally introduced into the TiO2 lattice at the Ti site, as the ionic radii of Nb5+ and Nb4+, which are 0.69 and 0.74 Å, respectively, are larger than the 0.68 Å radius of Ti4+.11,12 | ||
| Fig. 2 (a) XRD patterns of bare TiO2, Cu(II)–TiO2, NbxTi1−xO2, and Cu(II)–NbxTi1−xO2 nanocomposites at x = 0.1 wt%. (b) Extended XRD patterns of the samples in (a). | ||
The morphologies of the prepared nanocomposites were examined by SEM (Fig. 3 and S2†), which revealed that all samples were composed of uniformly distributed nanoparticles with an average grain size of approximately 200 nm. Introduction of Cu(II) nanoclusters on the TiO2 surface as well as the doping of Nb into the TiO2 lattice did not change the morphology or the particle size of the obtained Cu(II)–NbxTi1−xO2 samples. Therefore, the effects of morphology and particle size on the photocatalytic activity can be excluded in the present study.13 A TEM image clearly shows that the Cu(II) clusters, in the size of ∼2 nm, are well dispersed on the surface of NbxTi1−xO2 particles (Fig. S3†). The good attachment of Cu(II) clusters to the NbxTi1−xO2 surfaces was observed from the corresponding high-resolution TEM (HRTEM) image. Point analysis of energy dispersive X-ray spectroscopy (EDS, Fig. S3c†) and surface analysis of XPS (Fig. S4†) proved that these clusters consist of copper compound. Further, BET surface area analysis revealed that the introduction of Nb and Cu(II) ions did not alter the surface area of TiO2 (Table S2†).
 | ||
| Fig. 3 SEM images of (a) bare TiO2, (b) NbxTi1−xO2, and (c) Cu(II)–NbxTi1−xO2. | ||
The surface composition and elemental chemical states of the prepared nanocomposites were examined by XPS (Fig. 4, S4 and S5†). For bare TiO2, only Ti and O were detected. The additional peaks associated with Nb were clearly observed in the spectrum of NbxTi1−xO2, indicating that Nb ions were successfully introduced into the TiO2 lattice (Fig. S4†). For Cu(II)–NbxTi1−xO2, a signal attributable to Cu was clearly detected, confirming that Cu(II) nanoclusters were grafted onto the TiO2 surface (Fig. S4†).
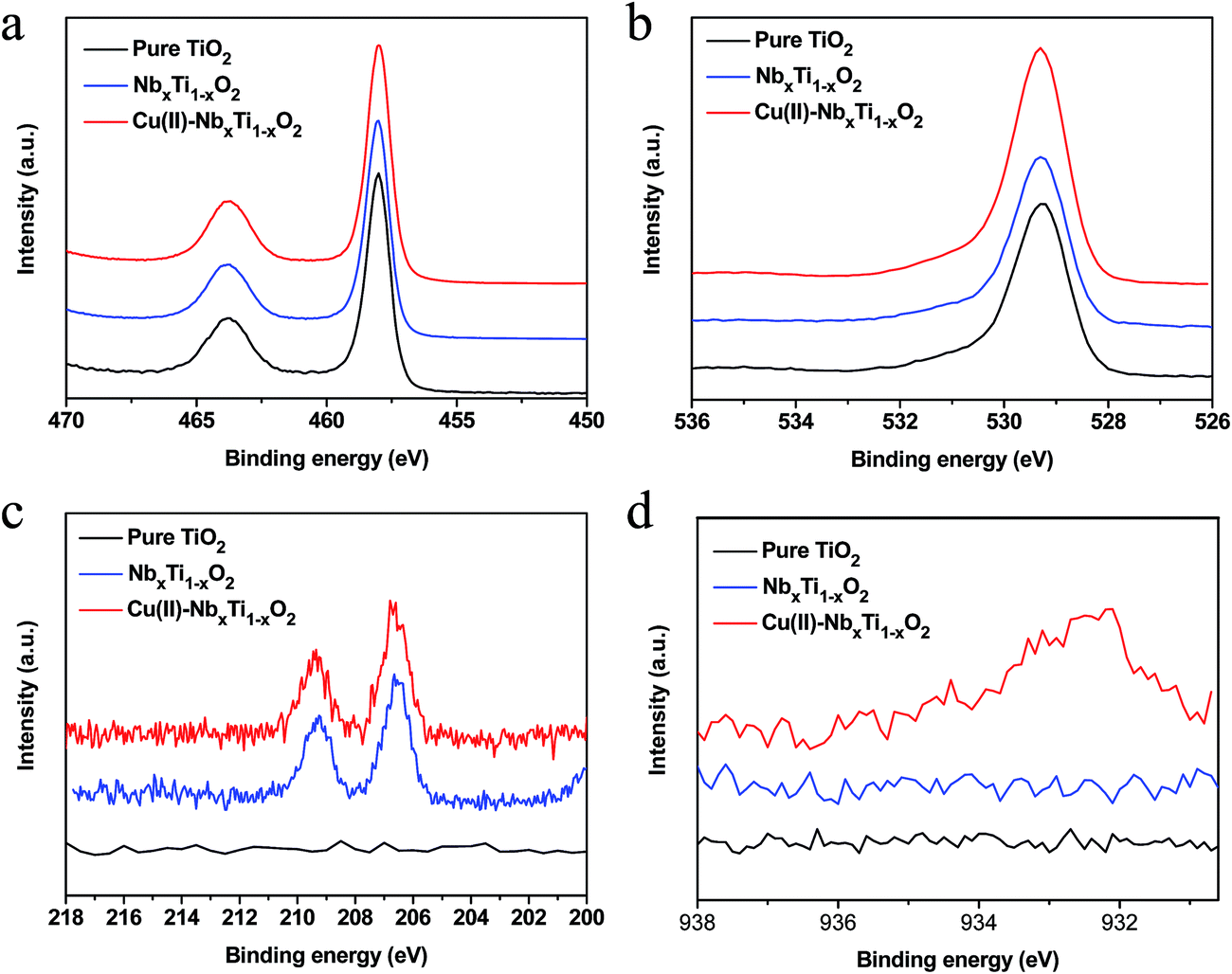 | ||
| Fig. 4 (a) Ti 2p, (b) O 1s, (c) Nb 3d and (d) Cu 2p core-level spectra of bare TiO2, NbxTi1−xO2, and Cu(II)–NbxTi1−xO2 nanocomposites at x = 0.1 wt%. | ||
Fig. 4a and b show the Ti 2p and O 1s core-level spectra, respectively, of the nanocomposite samples. No obvious differences between the chemical states of elemental Ti and O were observed,14 demonstrating that neither the grafted Cu(II) nanoclusters nor doped Nb ions affected the bonding structure between titanium and oxygen. Further, no shoulders associated with Ti3+ were observed in the Ti 2p core-level spectra (Fig. 4a and S5†), indicating that the density of Ti3+ was below the detection limit of the XPS analysis. The low density of Ti3+ in NbxTi1−xO2 was confirmed by ESR analysis (Fig. S6†). Hitosugi et al.15a studied the microstructure of Nb5+-doped TiO2 using XPS and found that the incorporation of Nb5+ into the TiO2 lattice resulted in the formation of minor Ti3+ components that maintain the charge balance. However, their XPS results also indicated that Nb5+ is reduced to Nb4+ at high annealing temperatures. Consistent with this finding, Khoviv et al.15b also showed that doped Nb ions in TiO2 mainly existed as Nb4+ after high temperature treatment.15b The formation of Nb4+ would not induce the generation of Ti3+ species.15,16
Fig. 4c shows the Nb 3d core-level XPS spectra of the nanocomposite samples. For NbxTi1−xO2 and Cu(II)–NbxTi1−xO2, well-defined photoelectron signals located at 206.5 and 209.3 eV were observed in the obtained spectra, whereas no feature was observed in the spectrum of the undoped sample, indicating that Nb was successfully doped into TiO2 and was presented in the Nb4+ oxidation state.15 In addition, the Nb signals in Nb2O5 and physically mixed TiO2 and Nb2O5 located at 207.5 and 210.4 eV (Fig. S7†), indicating that Nb5+ was not detected in NbxTi1−xO2 samples. Some previous reports indicated that the Nb doped TiO2 sample is possible to show Nb4+ in the bulk and Nb5+ on the surface because of the aggregation of Nb on the surface.15b,17 Our XRD data clearly demonstrated the single rutile phase of the obtained NbxTi1−xO2 samples, revealing the well distribution of Nb in the bulk TiO2. Thus, Nb5+ could not be detected in our NbxTi1−xO2 samples and the doped Nb existed as Nb4+ in the samples. On the other hand, previous reports demonstrated that the formation of Nb5+ and Ti3+ would result in the up-shift of the energy.11c–f However, our valence band (VB) XPS data clearly showed that the value of NbxTi1−xO2 was similar to that of pure TiO2 (Fig. S8†). Further, no shoulder associated with Ti3+ was observed in the Ti 2p core-level spectra (Fig. 4a and S5†), revealing that the density of Ti3+ was below the detection limit of the XPS analysis. Based on these results, it can be safely concluded that the doped Nb was well distributed in the sample and existed as the Nb4+ oxidation state.
After modification of NbxTi1−xO2 with Cu(II) nanoclusters, the Cu 2p3/2 core-level XPS signal was observed at 932.3 eV (Fig. 4d), a value that is consistent with the results from our previous studies that used a combination of X-ray absorption fine structure (XAFS) measurements, XPS analysis, and TEM observation to characterize the Cu(II) state in a Cu(II)–TiO2 system.5a,b Based on these analyses of the local crystal structure, we confirmed that Cu(II) nanoclusters were grafted onto the surface of TiO2 as distorted amorphous CuO-like structures with a five-coordinated square pyramidal form and a particle size of less than 3 nm.5d Our present results indicate that the chemical state and the environment of Cu(II) nanoclusters in the present Cu(II)–TiO2 and Cu(II)–NbxTi1−xO2 nanocomposites are identical to those of the previously characterized Cu(II)–TiO2 system.
The light absorption properties of the prepared samples were investigated by UV-visible spectroscopy (Fig. 5). The spectra presented in Fig. 5a clearly show that Nb doping into TiO2 increased the visible-light absorption in the range of 420 to 550 nm. In addition, the grafting of Cu(II) nanoclusters onto the surface of TiO2 enhanced the light absorption of the resulting nanocomposite in the 420–550 and 700–800 nm wavelength regions. The increase in the shorter wavelength region can be assigned to the IFCT of VB electrons to surface Cu(II) nanoclusters, and the longer wavelength region is attributable to the d–d transition of Cu(II).5a,b According to the band-gap estimation using the Kubelka–Munk function (Fig. S9†), introduction of Nb ions into the TiO2 lattice does not narrow the band gap of TiO2, as Nb ions are predicted to exist as isolated states in the forbidden gap. XPS (Fig. 4a) and ESR analyses (Fig. S6†) suggested that Ti3+ ions were not presented in the NbxTi1−xO2 nanocomposite. On the basis of these results, the visible-light absorption of the NbxTi1−xO2 nanocomposite is mainly attributable to the orbital of the doped Nb4+ ions.
 | ||
| Fig. 5 (a) UV-visible reflectance spectra of TiO2, Cu(II)–TiO2, NbxTi1−xO2 and Cu(II)–NbxTi1−xO2 nanocomposites. The inset shows the enlarged UV-visible reflectance spectra in the range of 400–520 nm. (b) Difference UV-vis spectra for Cu(II)–TiO2, NbxTi1−xO2 and Cu(II)–NbxTi1−xO2 nanocomposites at x = 0.1 wt% versus bare TiO2. | ||
A comparison of the difference absorption spectra of Cu(II)–TiO2, NbxTi1−xO2, and Cu(II)–NbxTi1−xO2versus bare TiO2 revealed that the grafted Cu(II) nanoclusters and doped Nb ions similarly increased the visible-light absorption of these systems between 420 and 550 nm (Fig. 5b). This finding indicates that doped Nb ions and grafted Cu(II) nanoclusters have similar energy levels, and that the enhanced light absorption of Cu(II)–NbxTi1−xO2 is mainly due to the doped Nb ions.
To specify similar energy levels between doped Nb ions and grafted Cu(II) nanoclusters, it is necessary to examine the detailed electronic structure of Nb-doped TiO2. It is generally considered that excess electrons are generated when pentavalent Nb ions are substituted for tetravalent Ti ions. The generated electrons could delocalize from Nb ions to the neighboring Ti ions, resulting in the formation of Ti3+ species, which introduce donor levels below the CB of TiO2.10,18 However, the localized states of Nb ions in the band gap differ between the anatase and rutile forms; in the former, doped Nb ions exist as Nb5+, because a relatively low temperature is required for the formation of the anatase structure. Thus, the excess electrons from Nb5+ ions could be transferred to neighboring Ti4+ ions, resulting in a shift of the Fermi level toward the CB and an enhancement of the metallic behavior of anatase TiO2.19 In other words, the transfer of excess electrons from Nb5+ ions to Ti4+ ions induces the generation of Ti3+ ions, which leads to the formation localized energy states at 0.3–0.8 eV below the CB.20 In contrast to the anatase form, Nb cations appear to substitute for Ti cations in rutile TiO2 and exist as Nb4+.15,16,21,22 The state of Nb in rutile single crystals and single crystal thin films, and rutile polymorph particles and polymorph particle thin films has been extensively analyzed using XRD, ESR, XPS, TEM, reflection high-energy and low-energy electron diffraction (RHEED and LEED), X-ray photoelectron diffraction (XPD), and scanning tunneling microscopy (STM).16,21 The results of these various analyses confirmed that Nb exists as Nb4+ in rutile TiO2.16,21 Investigation of the phase diagram of Ti–Nb–O also indicates that TiO2 and NbO2 may form a solid solution of NbxTi1−xO2 with 0 < x < 0.85 for the normal rutile phase and x > 0.85 in a deformed rutile phase at room temperature.12a The formation of Nb4+ is speculated to be because excess electrons remain on the Nb ions and form donor levels in the TiO2 lattice. Nb-doped rutile TiO2 exhibits semiconductor behavior, rather than the metallic behavior observed in the anatase form, and its Fermi level is located in the band gap.19 Theoretical and experimental studies have shown that Nb4+ (4d1) energy levels lie ∼0.12–0.22 eV below the bottom of the CB as a partially filled state.22 These findings are consistent with recent theoretical calculations that indicate Nb would form deep states in anatase and shallow states in rutile TiO2.23 Although doped Nb in rutile TiO2 has also been reported to remain as Nb5+ ions,24 whose 4d states would overlap with the O2p VB states of TiO2, our present UV-vis spectra, XPS, ESR, and VB XPS results (Fig. 4, 5, and S4–S9†) clearly indicate that doped Nb exists as Nb4+ in the band gap. Thus, we can conclude that the doped Nb existed as Nb4+ in our rutile TiO2 sample and produced a shallow energy level at ∼0.12–0.22 eV below the bottom of the CB, a value that matches the redox potential of Cu2+/Cu+ (0.16 V vs. SHE, pH = 0).5a,b
The photocatalytic performance of the nanocomposites prepared in the present study was evaluated by the visible-light induced decomposition of IPA, which was used as a representative gaseous volatile organic compound (VOC) and is a serious pollutant of indoor air.5 IPA can be completely decomposed to CO2 and water by photocatalytic oxidation.25 For the photocatalytic tests, the light intensity was set to 1 mW cm−2, which corresponds to an illuminance of 300 lux and is comparable to the intensity of white fluorescent light and LED light, the wavelength of the irradiation light ranged from 400 to 530 nm (Fig. S10†), and the initial IPA concentration was 300 ppmv (∼6 μmol). Under these conditions, the complete decomposition of IPA would result in a CO2 concentration of 900 ppmv (∼18 μmol), which is three times the initial IPA concentration (CH3CHOHCH3 + 9/2O2 → 3CO2 + 4H2O). A representative curve of the change in gas concentration during the decomposition of IPA by the Cu(II)–NbxTi1−xO2 sample is shown in Fig. 6a. Under dark conditions, the IPA concentration initially decreased and then remained constant, demonstrating that adsorption equilibrium had been established. In addition, acetone and CO2 were not detected, indicating that IPA was not decomposed by Cu(II)–NbxTi1−xO2 under these conditions. With the onset of light irradiation, the IPA concentration decreased rapidly and the amount of acetone increased sharply until reaching a peak at approximately 300 ppmv, after which, the acetone concentration started to decrease. Accompanying the decrease of acetone, the concentration of CO2 increased quickly. The observed reaction profile is consistent with the photocatalytic decomposition of IPA proceeding via the formation of acetone as an intermediate, followed by the decomposition of acetone to the final products CO2 and H2O.25 After 200 h of irradiation, the concentration of CO2 in the reaction vessel reached approximately 900 ppmv (∼18 μmol), which was nearly 3 times the amount of the initially injected IPA (300 ppmv), indicating that IPA was completely decomposed.
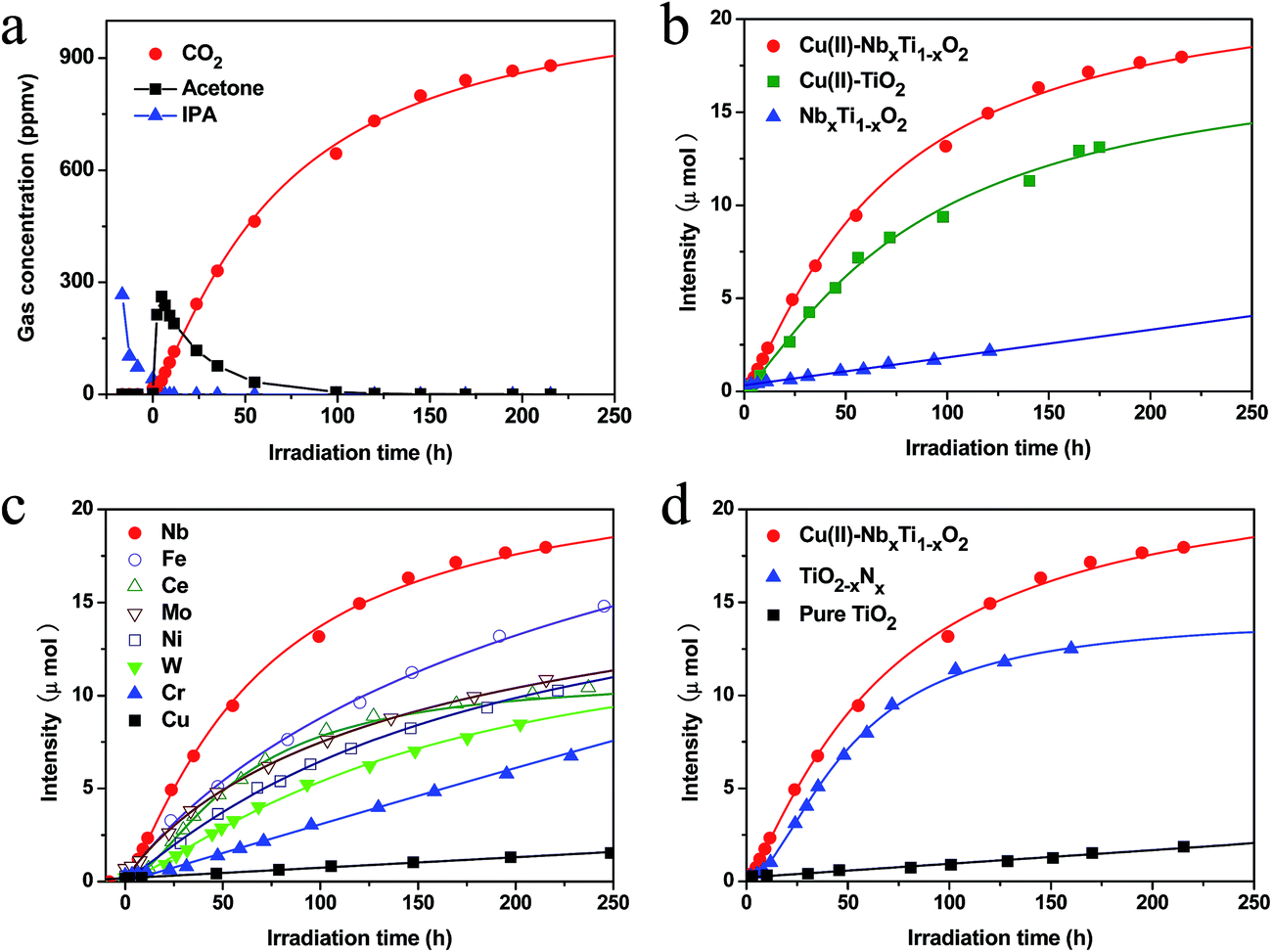 | ||
| Fig. 6 (a) Representative time-dependent gas concentrations during IPA decomposition by Cu(II)–NbxTi1−xO2. (b) Comparative studies of CO2 generation by bare NbxTi1−xO2, Cu(II)–TiO2, and Cu(II)–NbxTi1−xO2 at x = 0.1 wt%. (c) CO2 generation curves for Cu(II)–MxTi1−xO2 (M = Nb, Fe, Ce, Mo, Ni, W, Cr, and Cu, x = 0.1 wt%) samples under visible-light irradiation. (d) CO2 generation over bare TiO2, TiO2−xNx, and Cu(II)–NbxTi1−xO2 at x = 0.1 wt%. | ||
Comparative studies of the photocatalytic activities of NbxTi1−xO2, Cu(II)–TiO2 and Cu(II)–NbxTi1−xO2 samples under the same visible light source are shown in Fig. 6b. After the doping of Nb ions into TiO2, the generated NbxTi1−xO2 nanocomposite exhibited visible-light sensitivity (Fig. 5), but had relatively low photocatalytic performance, because the photogenerated charge carriers were not efficiently separated and transferred to the surface.3 The Cu(II)–TiO2 nanocomposites exhibited high visible-light activity, owing to the IFCT from the VB of TiO2 to the surface Cu(II) nanoclusters and the efficient multi-electron reduction of oxygen by these nanoclusters.5–7 Irie et al.5b investigated the role of Cu(II) nanoclusters in electron-trapping by performing in situ XAFS analysis under visible light in the presence of IPA and absence of oxygen and found that Cu(I) was generated under these conditions, but was converted back to Cu(II) upon exposure to oxygen. However, theoretical and experimental studies have revealed that the doping of Nb into TiO2 induces the formation of an energy level approximately 0.12–0.22 eV below the CB,22 which is within the range of the reported redox potential of Cu2+/Cu+, 0.16 V (vs. SHE, pH = 0).5a,b These results indicate that grafted Cu(II) nanoclusters and doped Nb ions have closely matched energy levels. The enhanced photocatalytic performance of Cu(II)–NbxTi1−xO2 compared with those of Cu(II)–TiO2 and NbxTi1−xO2 demonstrates that efficient energy level matching is achieved between the grafted Cu(II) nanoclusters and doped Nb ions. Based on the observed photocatalytic activities of the prepared photocatalysts, the QE for CO2 generation was calculated using the following equation: QE = Rrp/Rap= 6RCO2/Rap, where Rrp is the reaction rate of photons involved in CO2 generation, RCO2is the CO2 generation rate, and Rap is the absorption rate of incident photons. The details of this calculation are described in the literature5b and ESI (Fig. S11†), and the data used in the calculations are summarized in Table 1. Under the same light irradiation conditions, the NbxTi1−xO2 sample exhibited a high absorption rate of incident photons, but a low CO2 generation rate of only 0.015 μmol h−1, indicating that this photocatalyst has a low charge separation efficiency. Although the Cu(II)–TiO2 nanocomposites displayed a high QE (27.7%), indicating that efficient IFCT and a multi-electron reduction reaction proceeded on the surface,5–7 the visible-light absorption rate of this material was relatively low and can be attributed to the limited light absorption by the IFCT process (Table 1). Interestingly, the grafting of Cu(II) nanoclusters onto the surface of NbxTi1−xO2 resulted in strong visible-light absorption by the synthesized Cu(II)–NbxTi1−xO2 nanocomposite, which also exhibited a high QE of 25.3%. The high reaction rate of Cu(II)–NbxTi1−xO2 is due to the efficient light absorption by doped Nb ions and electron transfer between the doped Nb and surface-grafted Cu(II) nanoclusters, as well as the efficient multi-electron reduction of oxygen on the surface Cu(II) nanoclusters.5–7 The QE of Cu(II)–NbxTi1−xO2 is markedly higher than that of Cu(II)-grafted, Ti3+ self-doped TiO2,5d indicating that efficient charge transfer proceeds between the dopants and surface Cu(II) nanoclusters because of similar energy levels. Due to these excellent properties, the Cu(II)–NbxTi1−xO2 nanocomposites exhibited a CO2 generation rate of 0.20 μmol h−1, which is much higher than those of NbxTi1−xO2 and Cu(II)–TiO2 under the same visible-light irradiation conditions.
| Sample | TiO2−xNx | NbxTi1−xO2 | Cu(II)–TiO2 | Cu(II)–NbxTi1−xO2 |
|---|---|---|---|---|
| a R ip, rate of incident photons; Rap, absorbed photon number; RCO2, CO2 generation rate; QE, quantum efficiency. | ||||
| R ip (quanta per s) | 1.30 × 1016 | 1.30 × 1016 | 1.30 × 1016 | 1.30 × 1016 |
| R ap (quanta per s) | 4.10 × 1015 | 6.53 × 1014 | 4.69 × 1014 | 7.91 × 1014 |
| R CO2 (μmol h−1) | 0.16 | 0.015 | 0.13 | 0.20 |
| QE (%) | 3.9 | 2.3 | 27.7 | 25.3 |
We also investigated the photocatalytic activities of TiO2 modified with Cu(II) surface nanoclusters and various metal ions as dopants (Fig. 6c). In addition to Nb dopants, we have also checked the photocatalytic performances of Fe and W doped TiO2 with various doping densities (Fig. S12†). The results showed that 0.1 wt% was the optimal amount for Fe and W doped TiO2. The synthesized nanocomposites were all single phases of the rutile TiO2 crystal structure. Among the examined metal dopants, Cu(II)–NbxTi1−xO2 nanocomposites exhibited the best performance, indicating that well energy level matching occurred between doped Nb ions and grafted Cu(II) nanoclusters. Further, the visible-light activity of Cu(II)–NbxTi1−xO2 was superior to that of TiO2−xNx, which is widely recognized as one of the most efficient visible-light photocatalysts reported to date (Fig. 6d and Table 1).3a TiO2−xNx exhibited a rather low QE of 3.9% and required a much longer incubation period, over 300 h, to completely decompose the gaseous IPA. The low activity of this system originates from the lower oxidation power of the photogenerated holes in the nitrogen levels than those in the VB.4
Finally, we attempted to optimize the experimental conditions for enhancing the activity and found that 0.1 wt% was the optimal amount for both doped Nb and surface-grafted Cu(II) nanoclusters (Fig. S13 and S14†). A good junction between the surface-grafted Cu(II) nanoclusters and bulk-doped Nb ions was also critical for the efficient charge transfer. Notably, if a thin layer was introduced between the surface Cu(II) nanoclusters and doped Nb ions (Fig. S15†), the visible-light activity was markedly reduced (Fig. S16†). The Cu(II)–NbxTi1−xO2 sample was also very active under UV-light irradiation. Taken together, these findings suggest that Cu(II)–NbxTi1−xO2 nanocomposites are promising visible-light-sensitive photocatalysts for practical applications.
4. Conclusions
Efficient visible-light-sensitive TiO2 photocatalysts were developed based on the concept of energy level matching between surface-grafted Cu(II) nanoclusters and bulk-doped Nb ions. Bulk-doped Nb ions produce energy levels below the CB of TiO2, which matches well with the redox potential of Cu2+/Cu+ in surface-grafted Cu(II) nanoclusters. Both grafted Cu(II) nanoclusters and doped Nb ions induce similar increases in light absorption in the wavelength region from 420 to 550 nm. In this photocatalytic system, Ti ions were substituted for doped Nb ions that existed in the Nb4+ oxidation state, which avoided the generation of Ti3+ species. The doping of Nb ions enhanced the visible-light absorption of TiO2, whereas the grafting of Cu(II) nanoclusters retained the high QE of this system. The present Cu(II)–NbxTi1−xO2 nanocomposites exhibited strong visible-light absorption and maintained a high QE, leading to high visible-light photocatalytic performance for the decomposition of gaseous organic compounds. Thus, our findings demonstrate that Cu(II)–NbxTi1−xO2 is a suitable visible-light-sensitive photocatalyst for practical applications, and that the concept of energy level matching is an effective approach for the construction of advanced visible-light photocatalysts.Acknowledgements
This work was performed under the management of the Project to Create Photocatalysts Industry for Recycling-Oriented Society supported by the New Energy and Industrial Technology Development Organization (NEDO) in Japan. This research was also supported by the ACT-C program of the Japan Science and Technology (JST) Agency.References
- (a) A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS; (b) T. L. Thompson and J. T. Yates, Chem. Rev., 2006, 106, 4428–4453 CrossRef CAS PubMed; (c) B. O'Regan and M. Gratzel, Nature, 1991, 353, 737–740 CrossRef; (d) R. Liu, P. Wang, X. F. Wang, H. G. Yu and J. G. Yu, J. Phys. Chem. C, 2012, 116, 17721–17728 CrossRef CAS; (e) P. Wang, J. Wang, X. F. Wang, H. G. Yu and J. G. Yu, Appl. Catal., B, 2013, 132–133, 452–459 CrossRef CAS PubMed.
- (a) M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS; (b) X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed; (c) X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed; (d) M. Zhong, Y. Sato, M. Kurniawan, A. Apostoluk, B. Masenelli, E. Maeda, Y. Ikuhara and J. J. Delaunay, Nanotechnology, 2012, 23, 495602 CrossRef PubMed; (e) M. Liu, W. M. Lu, L. Zhao, C. L. Zhou, H. L. Li and W. J. Wang, Trans. Nonferrous Met. Soc. China, 2010, 20, 2299–2302 CrossRef CAS.
- (a) R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed; (b) D. F. Wang, J. H. Ye, T. Kako and T. Kimura, J. Phys. Chem. B, 2006, 110, 15824–15830 CrossRef CAS PubMed; (c) A. Kudo, R. Niishiro, A. Iwase and H. Kato, Chem. Phys., 2007, 339, 104–110 CrossRef CAS PubMed; (d) H. Kato and A. Kudo, J. Phys. Chem. B, 2002, 106, 5029–5034 CrossRef CAS; (e) H. Irie, Y. Maruyama and K. Hashimoto, J. Phys. Chem. C, 2007, 111, 1847–1852 CrossRef CAS; (f) T. H. Xie, X. Y. Sun and J. Lin, J. Phys. Chem. C, 2008, 112, 9753–9759 CrossRef CAS; (g) M. Miyauchi, M. Takashio and H. Tobimatsu, Langmuir, 2004, 20, 232–236 CrossRef CAS; (h) X. Y. Sun and J. Lin, J. Phys. Chem. C, 2009, 113, 4970–4975 CrossRef CAS; (i) M. Mrowetz, W. Balcerski, A. J. Colussi and M. R. Hoffmann, J. Phys. Chem. B, 2004, 108, 17269–17273 CrossRef CAS; (j) M. Liu, L. Piao, S. Ju, W. Lu, L. Zhao, C. Zhou and W. Wang, Mater. Lett., 2010, 64, 1204–1207 CrossRef CAS PubMed.
- (a) H. Irie, Y. Watanabe and K. Hashimoto, J. Phys. Chem. B, 2003, 107, 5483–5486 CrossRef CAS; (b) M. Miyauchi, A. Ikezawa, H. Tobimatsu, H. Irie and K. Hashimoto, Phys. Chem. Chem. Phys., 2004, 6, 865–870 RSC.
- (a) H. Irie, S. Miura, K. Kamiya and K. Hashimoto, Chem. Phys. Lett., 2008, 457, 202–205 CrossRef CAS PubMed; (b) H. Irie, K. Kamiya, T. Shibanuma, S. Miura, D. A. Tryk, T. Yokoyama and K. Hashimoto, J. Phys. Chem. C, 2009, 113, 10761–10766 CrossRef CAS; (c) H. Yu, H. Irie, Y. Shimodaira, Y. Hosogi, Y. Kuroda, M. Miyauchi and K. Hashimoto, J. Phys. Chem. C, 2010, 114, 16481–16487 CrossRef CAS; (d) M. Liu, X. Q. Qiu, M. Miyauchi and K. Hashimoto, Chem. Mater., 2011, 23, 5282–5286 CrossRef CAS; (e) M. Liu, X. Q. Qiu, M. Miyauchi and K. Hashimoto, J. Am. Chem. Soc., 2013, 135, 10064–10072 CrossRef CAS PubMed; (f) X. Q. Qiu, M. Miyauchi, H. Yu, H. Irie and K. Hashimoto, J. Am. Chem. Soc., 2010, 132, 15259–15267 CrossRef CAS PubMed.
- (a) C. Creutz, B. S. Brunschwig and N. Sutin, J. Phys. Chem. B, 2005, 109, 10251–10260 CrossRef CAS PubMed; (b) C. Creutz, B. S. Brunschwig and N. Sutin, J. Phys. Chem. B, 2006, 110, 25181–25190 CrossRef CAS PubMed.
- (a) F. Himo, L. A. Eriksson, F. Maseras and P. E. M. Siegbahn, J. Am. Chem. Soc., 2000, 122, 8031–8036 CrossRef CAS; (b) S. Goldstein, G. Czapski, R. Eldik, H. Cohen and D. Meyerstein, J. Phys. Chem., 1991, 95, 1282–1285 CrossRef CAS; (c) N. Kitajima and Y. Morooka, Chem. Rev., 1994, 94, 737–757 CrossRef CAS; (d) A. P. Cole, D. E. Root, P. Mukherjee, E. I. Solomon and T. D. P. Stack, Science, 1996, 273, 1848–1850 CrossRef; (e) Y. Nosaka, S. Takahashi, H. Sakamoto and A. Nosaka, J. Phys. Chem. C, 2011, 115, 21283–21290 CrossRef CAS; (f) M. Nishikawa, Y. Mitani and Y. Nosaka, J. Phys. Chem. C, 2012, 116, 14900–14907 CrossRef CAS.
- (a) S. Anandan and M. Miyauchi, Phys. Chem. Chem. Phys., 2011, 13, 14937–14945 RSC; (b) Y. Nosaka, S. Takahashi, Y. Mitani, X. Qiu and M. Miyauchi, Appl. Catal., B, 2012, 111, 636–640 CrossRef PubMed; (c) S. Anandan and M. Miyauchi, Electrochemistry, 2011, 79, 842–844 CrossRef CAS PubMed; (d) S. Anandan, N. Ohashi and M. Miyauchi, Appl. Catal., B, 2010, 100, 502–509 CrossRef CAS PubMed.
- (a) K. Sunada, T. Watanabe and K. Hashimoto, Environ. Sci. Technol., 2003, 37, 4785–4789 CrossRef CAS; (b) K. Sunada, M. Minoshima and K. Hashimoto, J. Hazard. Mater., 2012, 235–236, 265–270 CrossRef CAS PubMed; (c) X. Q. Qiu, M. Miyauchi, K. Sunada, M. Minoshima, M. Liu, Y. Lu, D. Li, Y. Shimodaira, Y. Hosogi, Y. Kuroda and K. Hashimoto, ACS Nano, 2012, 6, 1609–1618 CrossRef CAS PubMed.
- (a) H. Y. Lee and J. Robertson, J. Appl. Phys., 2013, 113, 213706 CrossRef PubMed; (b) K. K. Ghuman and C. V. Singh, J. Phys. Condens. Matter., 2013, 25, 475501 CrossRef PubMed; (c) K. C. OK, Y. Park, K. B. Chung and J. S. Park, J. Phys. D: Appl. Phys., 2013, 46, 295102 CrossRef; (d) K. N. Song, X. P. Han and G. S. Shao, J. Alloys Compd., 2013, 551, 118–124 CrossRef CAS PubMed.
- (a) J. Baumard and E. Tani, J. Chem. Phys., 1977, 67, 857–860 CrossRef CAS PubMed; (b) M. C. Carotta, M. Ferroni, D. Gnani, V. Guidi, M. Merli, G. Martinelli, M. C. Casale and M. Notaro, Sens. Actuators, B, 1999, 58, 310–317 CrossRef CAS; (c) Y. Furubayashi, T. Hitosugi, Y. Yamamoto, K. Inaba, G. Kinoda, Y. Hirose, T. Shimada and T. Hasegawa, Appl. Phys. Lett., 2005, 86, 252101 CrossRef PubMed; (d) X. Lu, W. Yang, Z. Quan, T. Lin, L. Bai, L. Wang, F. Huang and Y. Zhao, J. Am. Chem. Soc., 2014, 136, 419–426 CrossRef CAS PubMed; (e) T. Nikolay, L. larina, O. Shevaleevskiy and B. T. Ahn, Energy Environ. Sci., 2011, 4, 1480–1486 RSC; (f) X. Lu, X. Mou, J. Wu, D. Zhang, L. Zhang, F. Huang, F. Xu and S. Huang, Adv. Funct. Mater., 2010, 20, 509–515 CrossRef.
- (a) K. Sakata, J. Phys. Soc. Jpn., 1969, 26, 1067 CrossRef CAS; (b) A. M. Ruiz, G. Dezanneau, J. Arbiol, A. Cornet and J. R. Morante, Chem. Mater., 2004, 16, 862–871 CrossRef CAS.
- (a) H. H. Wang, C. S. Xie, W. Zhang, S. Z. Cai, Z. H. Yang and Y. H. Gui, J. Hazard. Mater., 2007, 14, 645–652 CrossRef PubMed; (b) D. Li and H. Haneda, Chemosphere, 2003, 51, 129–137 CrossRef CAS.
- M. Oku, K. Wagatsuma and S. Kohiki, Phys. Chem. Chem. Phys., 1999, 1, 5327–5331 RSC.
- (a) T. Hitosugi, H. Kamisaka, K. Yamashita, H. Nogawa, Y. Furubayashi, S. Nakao, N. Yamada, A. Chikamatsu, H. Kumigashira, M. Oshima, Y. Hirose, T. Shimada and T. Hasegawa, Appl. Phys. Express, 2008, 1, 111203 CrossRef; (b) D. A. khoviv, S. V. Zaytsev and V. M. Levlev, Thin Solid Films, 2012, 520, 4796–4799 CrossRef PubMed.
- (a) P. F. Chester, J. Appl. Phys., 1961, 32, 866–868 CrossRef CAS PubMed; (b) P. H. Zimmerman, Phys. Rev. B: Solid State, 1973, 8, 3917–3927 CrossRef.
- L. R. Sheppard, J. Phys. Chem. C, 2013, 117, 3407–3413 CAS.
- (a) P. Deak, B. Aradi and T. Frauenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155207 CrossRef; (b) T. Yamamoto and T. Ohno, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 033104 CrossRef; (c) J. K. Yang, X. T. Zhang, C. H. Wang, P. P. Sun, L. L. Wang, B. Xia and Y. C. Liu, Solid State Sci., 2012, 14, 139–144 CrossRef CAS PubMed.
- H. Kamisaka, T. Hitosugi and K. Yamashita, J. Surf. Sci. Soc. Jpn., 2010, 31, 343 CrossRef CAS.
- D. C. Valentin, G. Pacchioni and A. Selloni, J. Phys. Chem. C, 2009, 113, 20543 Search PubMed.
- (a) S. A. Chambers, Y. Gao, Y. J. Kim, M. A. Henderson, S. Thevuthasan, S. Wen and K. L. Merkle, Surf. Sci., 1996, 365, 625–637 CrossRef CAS; (b) S. A. Chambers, Y. Gao, S. Thevuthasan, Y. Liang, N. R. Shivaparan and R. J. Smith, J. Vac. Sci. Technol., A, 1996, 14, 1387–1394 CrossRef CAS; (c) Y. Gao and S. A. Chambers, J. Mater. Res., 1996, 11, 1025–1029 CrossRef CAS; (d) Y. Gao, Y. Liang and S. A. Chambers, Surf. Sci., 1996, 348, 17–27 CrossRef CAS; (e) Y. Gao, S. Thevuthasan and D. E. McCready, J. Gryst. Growth., 2000, 212, 178–190 CrossRef CAS; (f) A. Sasahara and M. Tomitori, J. Phys. Chem. C, 2013, 117, 17680–17686 CrossRef CAS; (g) M. Chiesa, M. C. Paganini, S. livraghi and E. Giamello, Phys. Chem. Chem. Phys., 2013, 15, 9435 RSC; (h) Z. H. Zhang, S. Y. Wu, P. Xu, L. L. Li and S. X. Zhang, Eur. Phys. J.: Appl. Phys., 2011, 53, 20902 CrossRef.
- (a) J. B. Goodenough, Les oxydes des metaux de transition, Gauthier-Villars, Paris, 1973 Search PubMed; (b) J. Claverie, J. Verniolle, G. Campet, J. P. Doumerc and P. Hagenmuller, Mater. Res. Bull., 1981, 16, 1019–1025 CrossRef CAS; (c) T. Okamura and H. Okushi, Jpn. J. Appl. Phys., 1993, 32, L454–L457 CrossRef CAS; (d) K. A. Michalow, D. Flak, A. Heel, M. P. Wojtan, M. Rekas and T. Graule, Environ. Sci. Pollut. Res., 2012, 19, 3696–3708 CrossRef CAS PubMed; (e) H. O. Finklea, Semiconductor electrodes, Elsevier, Amsterdam, 1988, pp. 58–61 Search PubMed.
- (a) H. Y. Lee and J. Robertson, J. Appl. Phys., 2013, 113, 213706 CrossRef PubMed; (b) T. Yamamoto and T. Ohno, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 033104 CrossRef; (c) P. Deak, B. Aradi and T. Frauenheim, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 155207 CrossRef.
- D. Morris, Y. Dou, J. Rebane, C. E. J. Mitchell, R. G. Egdell, D. S. L. Law, A. Vittadini and M. Casarin, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 61, 13445–13457 CrossRef CAS.
- Y. Ohko, K. Hashimoto and A. Fujishima, J. Phys. Chem. A, 1997, 101, 8057–8062 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Table S1, the amount of Nb in the samples derived from ICP-AES. Table S2, BET surface areas of the samples. Fig. S1, XRD patterns of NbxTi1−xO2. Fig. S2, SEM images of NbxTi1−xO2. Fig. S3, TEM images and the EDS pattern of Cu(II)–NbxTi1−xO2. Fig. S4, full-scale XPS spectra of the prepared samples. Fig. S5, Ti 2p core-level spectra of NbxTi1−xO2. Fig. S6, ESR spectra of NbxTi1−xO2. Fig. S7, Nb 3d core-level spectra of NbxTi1−xO2, Nb2O5 and the mixture of Nb2O5 and TiO2. Fig. S8, valence band XPS spectra of the samples. Fig. S9, band gap of the samples. Fig. S10, light source for the visible-light irradiation. Fig. S11, details of the quantum efficiency calculations. Fig. S12, CO2 generation curve for Cu(II)–FexTi1−xO2 and Cu(II)–WxTi1−xO2 with different doping densities. Fig. S13, comparative evaluation of CO2 generation by Cu(II)–NbxTi1−xO2 with different doping densities. Fig. S14, comparative evaluation of CO2 generation by Cu(II)–NbxTi1−xO2 with different amounts of Cu(II) nanoclusters. Fig. S15, preparation of Cu(II)–TiO2@NbxTi1−xO2. Fig. S16, CO2 generation curve for the Cu(II)–NbxTi1−xO2 and Cu(II)–TiO2@NbxTi1−xO2 samples. See DOI: 10.1039/c4ta02211d |
| This journal is © The Royal Society of Chemistry 2014 |