 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Redox-responsive, reversibly fluorescent nanoparticles from sustainable cellulose derivatives†
Wei
Li
a,
Wei
Wang
a,
Yongbiao
Yang
b and
Kai
Zhang
*a
aErnst-Berl-Institute for Chemical Engineering and Macromolecular Science, Technische Universität Darmstadt, Alarich-Weiss-Straße 8, 64287 Darmstadt, Germany. E-mail: zhang@cellulose.tu-darmstadt.de; Fax: +49 6151 16 2479; Tel: +49 6151 16 75831
bEduard-Zintl-Institute of Inorganic and Physical Chemistry, Technische Universität Darmstadt, Alarich-Weiss-Straße 4, 64287 Darmstadt, Germany
First published on 18th June 2014
Abstract
In comparison to single-stimuli responsive cellulose derivatives, multi-stimuli and reversibly responsive compounds from cellulose are still scarce. In this report, the fabrication of redox-controllable nanoparticles (NPs) from novel cellulose derivatives containing thiol groups and rhodamine spiroamide showing reversible fluorescence is described. The thiol groups were introduced into cellulose chains after esterification by 3,3′-dithiodipropionic acid and further reductive cleavage of disulfide bonds. Then, rhodamine spiroamide was immobilized via thiol–ene reaction between cellulose thiopropionyl ester and rhodamine B methacrylamide. The obtained cellulose derivative containing rhodamine spiroamide (cellulose-RhBMA) could be transformed into NPs in aqueous medium and dissolved again via redox reactions on thiol groups. At the same time, cellulose-RhBMA exhibited reversible fluorescence that could be switched using pH (protons) or UV-illumination/heating as external stimuli. In total, we demonstrated the fabrication of redox-controllable NPs with reversible fluorescence, and a novel platform for the chemical modification of cellulose via thiol–ene reaction.
Introduction
Stimuli-responsive polymers have attracted much attention in recent years, and most used extra triggers include light, pH, temperature and magnetic field.1–6 Just as examples, poly(dimethyl acrylamide) (PDMAA) or polyglycerols containing nitrospiropyran groups change their physical properties in response to light,7–9 synthetic polypeptides respond to pH variation,10 poly(N-isopropylacrylamide) (PNIPAM) responds to temperature,11 and cross-linked poly(vinyl alcohol) containing magnetite (Fe3O4) particles respond to the magnetic field.12 However, most of the stimuli-responsive polymers are hitherto still synthetic polymers, which contain corresponding functional groups for the stimuli-responsiveness.5In comparison, stimuli-responsive polymers based on naturally occurring polysaccharides, in particular cellulose, are still limited to conventional stimuli, mostly pH and temperature.13,14 Cellulose, one of the most abundant native polysaccharides, consists of β-(1,4)-linked anhydroglucose units (AGUs) and contains three hydroxyl groups in each AGU. This chemical structure provides a platform for the derivatization with diverse functional groups.15 Methyl-, ethyl- and hydroxypropylcellulose are probably the most common temperature-responsive cellulose derivatives.13 Their hydrophobicity in the dissolved status increases with increasing temperature, and they show thermo-reversible gelation behavior. More studies focused on the grafting of stimuli-responsive polymer chains onto the cellulose backbone,16e.g. with PNIPAM or poly(N,N-dimethyl aminoethyl methacrylate) into hydroxypropyl- or ethylcellulose, leading to pH- and thermo-sensitive copolymers.17–19 By using this strategy, cellulose-based copolymers were adopted with the stimuli-responsiveness of synthetic polymers. Recently, fluorescent and light-responsive cellulose derivatives containing coumarin groups were reported and the coumarin moieties maintained their response to UV-illumination.20,21
In comparison to many other functional groups,22 the thiol moiety as a reactive group for facile thiol–ene groups has attracted more attention for the surface-modification of micro-/nanofibrillated cellulose,23,24 but much less attention for polymeric cellulose derivatives.25,26
In this study, we report the first synthesis of multi-responsive cellulose derivatives via thiol–ene reaction, which can form redox-controllable nanoparticles (NPs) showing reversible fluorescence. Cellulose derivatives containing thiol groups were synthesized after the reaction with 3,3′-dithiodipropionic acid in ionic liquids, 1-butyl-3-methylimidazolium acetate (BMIMAc), and N,N-dimethylacetamide (DMAc). Then, rhodamine B methacrylamide was introduced into polymer chains via thiol–ene reaction on thiol groups. The synthesized cellulose derivative, cellulose-rhodamine B methacrylamide (cellulose-RhBMA), formed NPs after the nanoprecipitation into water and these NPs can be dissolved or reconstructed again based on the redox properties of thiol groups on cellulose chains. The rhodamine spiroamide immobilized on thiol groups showed reversible fluorescence in response to pH (in the presence and absence of protons) or UV-illumination/heating.
Experimental section
Materials
Microcrystalline cellulose with a granule size of 50 μm, 3,3′-dithiodipropionic acid, rhodamine B base (97%), 1-butyl-3-methylimidazolium acetate (BMIMAc), aqueous ammonium thioglycolate solution (AmTG, 60%), ethylenediamine and methacryloyl chloride were obtained from Sigma-Aldrich (Steinheim, Germany). 1,1′-Carbonyldiimidazol (CDI, >97%) and 1,3-propanedithiol (PDT) were purchased from Merck (Darmstadt, Germany) and triethylamine from Fischer Scientific (Loughborough, UK). Other chemicals were all of analytical grade and used without further treatment. The dialysis membrane with a molecular weight cut-off of 3500 was received from VWR International GmbH (Darmstadt, Germany).Synthesis of thiolated cellulose (thiolcellulose 4) via a dithiodipropionic ester of cellulose
Cellulose 1 (1 g, 6.17 mmol) in 50 mL BMIMAc was heated to 100 °C to dissolve it. Then, 80 mL DMAc was added to the solution. The mixture was cooled to 80 °C, and the solution of 3,3′-dithiodipropionic acid 2 (12.97 g, 61.73 mmol) and CDI (5 g, 30.87 mmol) in 40 mL DMAc was added to the mixture. The mixture was allowed to react for 22 h at 80 °C under stirring. The product was precipitated in 500 mL ethanol, centrifuged and the solid was washed with ethanol. Then, the product was dissolved in 50 mL water, filtered, dialyzed in deionized water and freeze-dried, leading to a dithiodipropionic ester of cellulose (yield: 1.2 g). The dithiodipropionic ester of cellulose (1 g dissolved in 60 mL water) was further reduced by 10 g ammonium thioglycolate aqueous solution (60%) at RT for 3 h. Then, the solution was precipitated in ethanol (400 mL) and the product was collected via centrifugation. After that, the precipitate was purified via dissolution in water, and precipitated in ethanol, followed by washing with acetone (2 × 50 mL). Finally, a white powder was obtained after the removal of ethanol at RT, leading to thiolcellulose 4 (yield: 0.68 g).Synthesis of cellulose-rhodamine B methacrylamide (cellulose-RhBMA, 5)
Rhodamine B methacrylamide (RhBMA) S3 was synthesized in two steps from rhodamine B as reported before (Scheme S1†).27,28 Then, RhBMA was introduced into cellulose chains via thiol–ene reaction on the thiol groups within thiolcellulose 4. The freeze-dried solid (0.5 g) in 30 mL DMSO was mixed with RhBMA (3, 0.48 g) and triethylamine (0.28 mL) at 50 °C for 48 h. Then, the reaction was stopped by adding 250 mL THF and the precipitate was collected via centrifugation. After repeated dissolution in DMSO and precipitation using THF, the DMSO-soluble product 5a was obtained after washing with THF (2 × 30 mL) and acetone (3 × 30 mL) before drying at 60 °C (yield: 0.4 g). Freshly synthesized 5a was further reduced using ammonium thioglycolate aqueous solution (60%) at RT and precipitated in acetone, leading to water-soluble 5b.Fabrication of NPs from cellulose-RhBMA (5a/5b)
5a was dissolved in DMSO at the concentration of 5, 10, 20 and 25 mg mL−1. Then, 1 mL of each solution was dropped into 10 mL water. After 30 min, the mixture was dialyzed in water. To show the revisable dissolution and formation of NPs, 0.5 mL ammonium thioglycolate aqueous solution (60%) was added into 3 mL NP suspension of 5a (2 mg NPs per mL water), and the NP suspension turned clear, indicating the formation of water-soluble 5b. After the dialysis in water, the solution was diluted to 6 mL (with the concentration of 1 mg mL−1). To 2 mL of this solution, 100 μL DMSO and 20 μL PDT were added. After 15 min ultrasonic treatment, the NP suspension was dialyzed in water.Characterization
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and 80 were accumulated for 13C and 1H NMR spectroscopy, respectively.
000 and 80 were accumulated for 13C and 1H NMR spectroscopy, respectively.
Results and discussion
One major reason for scarce multi-stimuli responsive cellulose derivatives is the complicated multi-step synthesis for the introduction of stimuli-responsive functional groups into cellulose chains. In order to introduce a general functional group into the cellulose backbone that could be further used for the coupling of versatile groups, the thiol moiety was chosen due to its highly specific reaction with a carbon–carbon double bond and mild reaction conditions, e.g. UV-illumination at room temperature.29,30 For this purpose, 3,3′-dithiodipropionic acid (DTDPA) was used to esterify OH-groups in AGUs for the introduction of protected thiol groups. The esterification of OH-groups by DTDPA was carried out in 1-butyl-3-methylimidazolium acetate (BMIMAc)/DMAc solution and catalyzed by 1,1′-carbonyldiimidazol (CDI) (Scheme 1).31,32 The introduction of DTDPA into cellulose chains was confirmed by FTIR spectroscopy (Fig. S1†) and the disulfide groups within DTDPA can be cleaved using active reducing agents, such as ammonium thioglycolate. Then, thiolcellulose with thiol groups along cellulose chains was obtained (Scheme 1) and the degree of substitution ascribed to thiol groups was determined to be 0.62 based on elemental analysis. | ||
| Scheme 1 Schematic illustration for the synthesis of cellulose-RhBMA, 5a/5b. (i) (1) 1-Butyl-3-methylimidazolium acetate (BMIMAc), DMAc, CDI, 80 °C, 22 h; (2) aqueous ammonium thioglycolate (60%), 3 h; (ii) rhodamine B methacrylamide (RhBMA, S3), DMSO, 50 °C, 48 h; (iii) reduction in DMSO with aqueous ammonium thioglycolate (60%), 3 h. (iv) Oxidation by dissolution in DMSO. | ||
The presence of thiol groups provides the feasibility to further react with versatile functional groups containing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C-bonds via thiol–ene reactions, in particular in our study, rhodamine B methacrylamide (Scheme 1(ii)). Rhodamine is a photostable fluorescent dye with wide potential applications in biosensing and imaging.33,34 In order to immobilize rhodamine spiroamide on thiol groups in thiolcellulose 4, rhodamine spiroamide in ring-closed form was decorated with methacrylamide groups (Scheme S1†).27,28 After the thiol–ene reaction, obtained cellulose-RhBMA 5a shows clearly the presence of both rhodamine spiroamide and the cellulose backbone according to 1H, 13C NMR and FTIR spectra (Fig. 1 and S3†). Moreover, the presence of signals at 173, 99.5 and 63 ppm ascribed to C in carboxyl groups, C1′ as well as C6′ indicates the derivatization of the hydroxyl groups at C6 and C2 positions by thiopropionic acid.32,35,36 The degree of substitution ascribed to rhodamine groups was calculated to be 0.12 based on elemental analysis, indicating that about 20% of total thiol groups were involved in the thiol–ene reaction.
C-bonds via thiol–ene reactions, in particular in our study, rhodamine B methacrylamide (Scheme 1(ii)). Rhodamine is a photostable fluorescent dye with wide potential applications in biosensing and imaging.33,34 In order to immobilize rhodamine spiroamide on thiol groups in thiolcellulose 4, rhodamine spiroamide in ring-closed form was decorated with methacrylamide groups (Scheme S1†).27,28 After the thiol–ene reaction, obtained cellulose-RhBMA 5a shows clearly the presence of both rhodamine spiroamide and the cellulose backbone according to 1H, 13C NMR and FTIR spectra (Fig. 1 and S3†). Moreover, the presence of signals at 173, 99.5 and 63 ppm ascribed to C in carboxyl groups, C1′ as well as C6′ indicates the derivatization of the hydroxyl groups at C6 and C2 positions by thiopropionic acid.32,35,36 The degree of substitution ascribed to rhodamine groups was calculated to be 0.12 based on elemental analysis, indicating that about 20% of total thiol groups were involved in the thiol–ene reaction.
 | ||
| Fig. 1 13C NMR spectrum of 5a in d6-DMSO (the 1H NMR spectrum in Fig. S3†). | ||
5a is readily soluble in DMSO, but not in water. By the addition of an aqueous solution of ammonium thioglycolate (60%) into the DMSO solution of 5a, water-soluble, reduced 5b was obtained. Thus, the non-solubility of 5a in water is due to the presence of disulfide linkages.25 A competing reaction such as the disulfide formation took place during the thiol–ene reaction, due to the oxidizing feasibility of thiol groups by DMSO/O2.25,37
The solubility of 5a in DMSO but not in water allows the formation of NPs from 5a in water via nanoprecipitation.38–40 The addition of the DMSO solution of 5a into excess water led to a stable, milky NP suspension (Movie S1†). The average diameters of these NPs were measured to be between 180 and 360 nm, depending positively on the concentrations of the 5a solutions (Fig. 2). Due to the redox feasibility of the disulfide groups, these NPs could be dissolved again via the addition of a reducing agent, such as ammonium thioglycolate or dithiolthreitol (Movie S2†). The reduction and thus the cleavage of the disulfide bonds of 5a led to water-soluble 5b (Scheme 1). 5b is water-soluble due to the presence of thiol groups along cellulose chains. These thiol groups can be further oxidized using oxidizing agents for thiol groups including O2 gas and aqueous NaClO2,25 resulting in 5a containing disulfide linkages. However, these oxidations did not lead to the formation of NPs, but only white flocculants. In contrast, if 1,3-propanedithiol (PDT) in DMSO was added into the aqueous solution of 5b, NPs could be reconstructed again after the removal of DMSO. Nevertheless, these NPs from a 5b solution of 1 mg mL−1 exhibited a large average diameter of 384 ± 14 nm, compared to the NPs from 5a solutions of higher concentrations after the nanoprecipitation (Fig. 2b). Thus, the NPs from cellulose-RhBMA can be reversibly dissolved and reconstructed again by using the corresponding redox-reaction (Fig. 3).
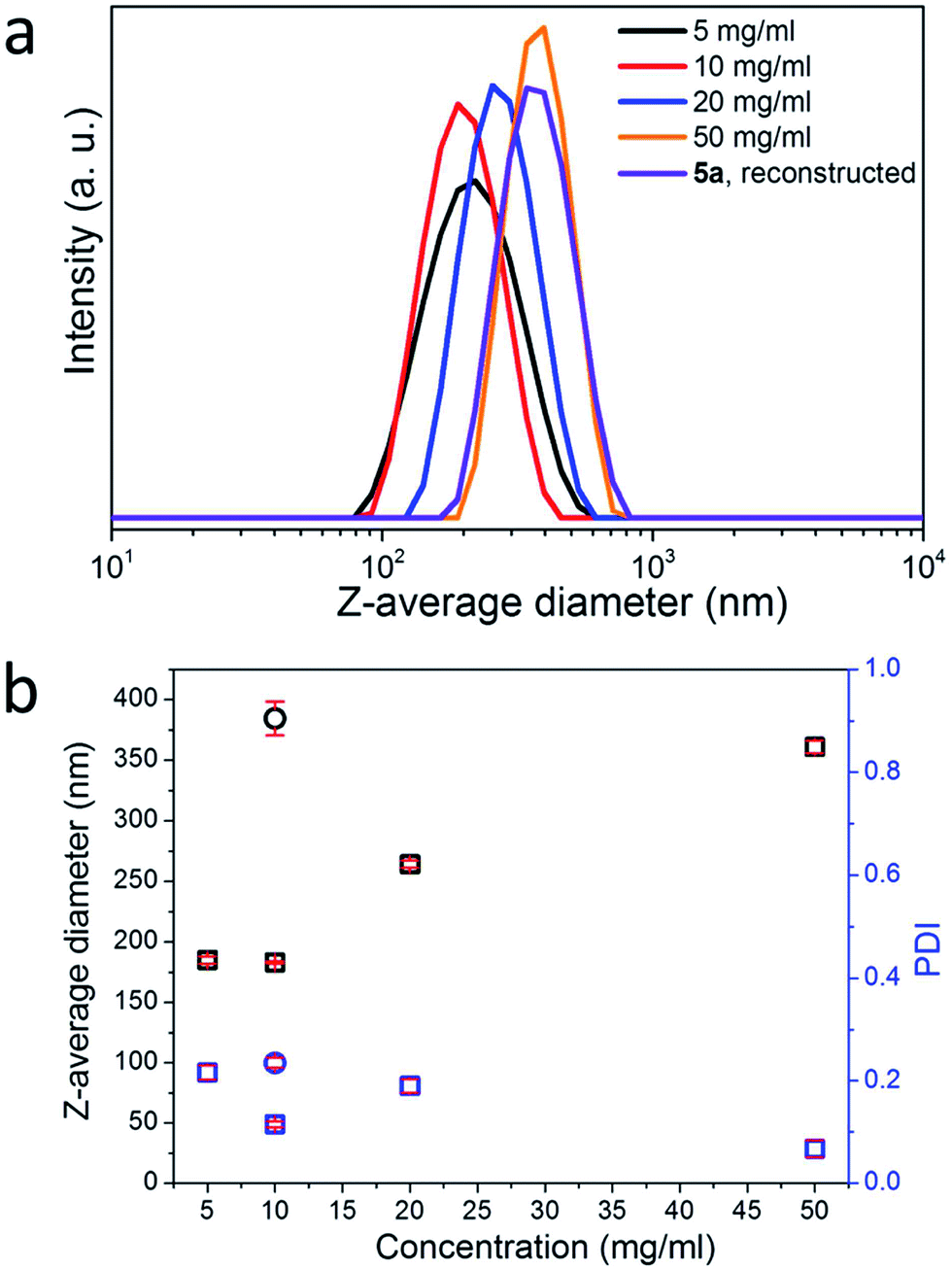 | ||
| Fig. 2 (a) DLS curves of aqueous NP suspensions of cellulose-RhBMA 5a from its solutions in DMSO at diverse concentrations as well as reconstructed NPs. (b) Z-Average diameters and PDI of NPs. Square: from solution of 5a in DMSO and circle: reconstructed NPs from dissolved 5b in water using 1,3-propanedithiol/DMSO. The red lines are error bars. | ||
 | ||
| Fig. 3 Schematic representation of the formation and dissolution of NPs via nanoprecipitation/dissolution of 5a or oxidation/reduction of 5b. The insets show representative images. | ||
The fluorescence of 5a dissolved in DMSO can be altered by changing the pH of its solution via addition of a low amount of aqueous HCl or NaOH.28 At acidic conditions, strong fluorescence is visible due to the presence of the intensive emission signal at 590 nm ascribed to rhodamine, while very weak fluorescence is observed under alkaline conditions according to the fluorescence spectroscopic analysis (Fig. 4a). After drying, 5a with rhodamine spiroamide in ring-closed form is a light-yellow compound showing weak fluorescence, while UV-illumination of 5a led to the emergence of pink color and thus stronger fluorescence (Fig. 4b). A further heating treatment led to the decrease of the color intensity and accompanying fluorescence. The reason is that the switching ability of rhodamine spiroamide between ring-closed form (weakly fluorescent) and ring-open form (strongly fluorescent) depends on the external stimuli, e.g. UV-illumination, heating or pH.28,41 In comparison, the switching of the fluorescence of 5a in DMSO could not be conducted using UV and heating as external stimuli (Fig. S4†). Reduced 5b dissolved in water also did not show alteration of the fluorescence after the UV-illumination (Fig. S5†). This effect is due to the presence of a preferential open ring of rhodamine spiroamide while dissolved in polar solvents such as DMSO or H2O.
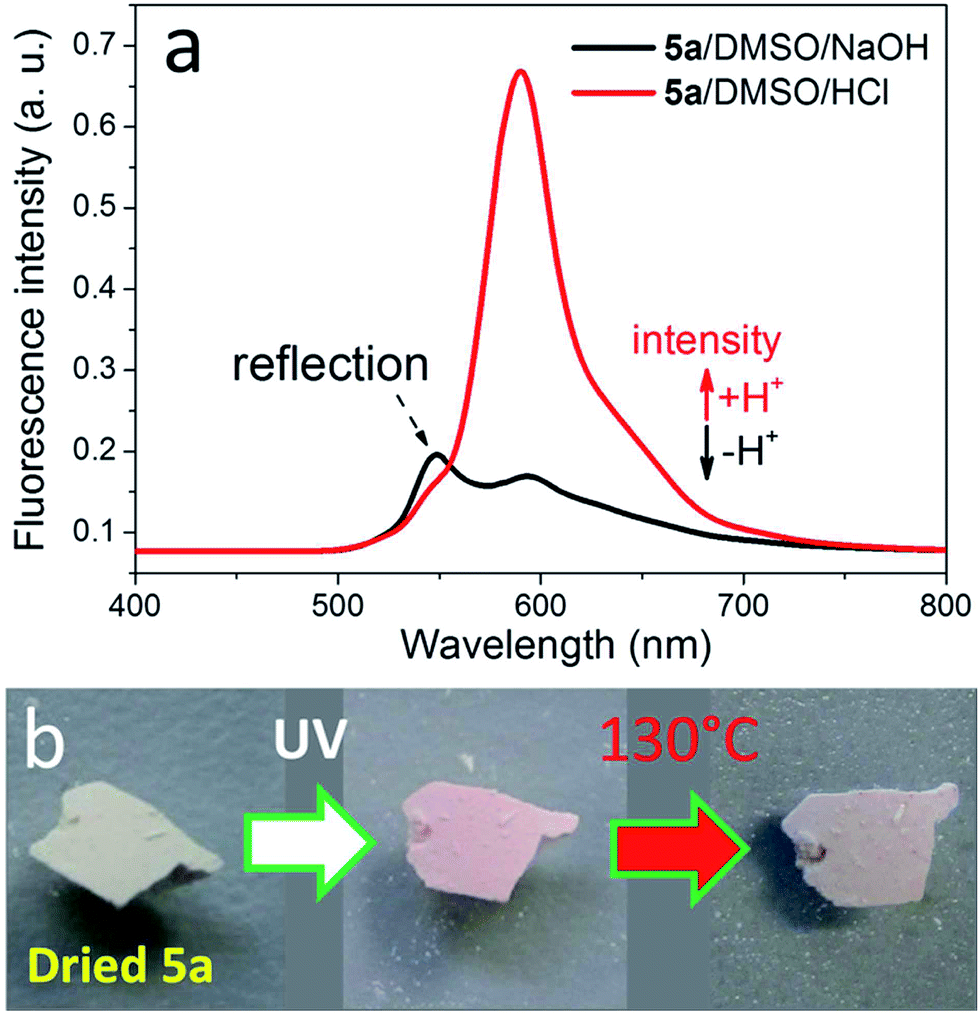 | ||
| Fig. 4 Reversible fluorescence of 5a. (a) Fluorescence spectra of 5a in DMSO with the addition of 50 μL aqueous HCl or NaOH (5 wt%). (b) Photo images of freshly dried 5a, UV-illuminated 5a (at 365 nm for 30 min) and subsequently heated 5a (at 130 °C for 30 min). | ||
As well, the NPs of 5a were also endowed with the reversible fluorescence, which is displayed using fluorescence microscopy. By nanoprecipitating DMSO solution of 5a into water followed by drying, the obtained NPs with non-activated rhodamine exhibits very low fluorescence (Fig. 5a). As further illustrated with the fluorescence microscopic analysis, the fluorescent intensity of dried 5a NPs could be switched via UV-illumination and heating at 130 °C (Fig. 5a–d). After UV-illumination at 365 nm for 30 min, the fluorescence becomes stronger, while subsequent 30 min treatment at 130 °C resulted in weaker fluorescence. The switching of the fluorescence intensity can also be realized after shorter time, such as 10 min UV-illumination or heating (Fig. S6†). Thus, by applying these external stimuli, the fluorescence intensities of the NPs can be reversibly adjusted (Fig. 5e). It should be noted that dried 5a already exhibited slight pink color and associated fluorescence. Further switching using UV-illumination and heating led to steady lowering of switching feasibility. Furthermore, the UV-illumination and heating are effective stimuli for the samples at dried status, while they could not significantly change the fluorescence of dissolved samples. The fluorophores with switchable fluorescence, such as rhodamine B and spiropyrane, prefer a particular chemical structure if dissolved in a certain solvent, e.g. they demonstrate strong fluorescence if they are dissolved in polar solvents.42,43 Nevertheless, the pH value or precisely the presence or absence of protons is another stimulus for switching the fluorescence of the dissolved fluorophore.
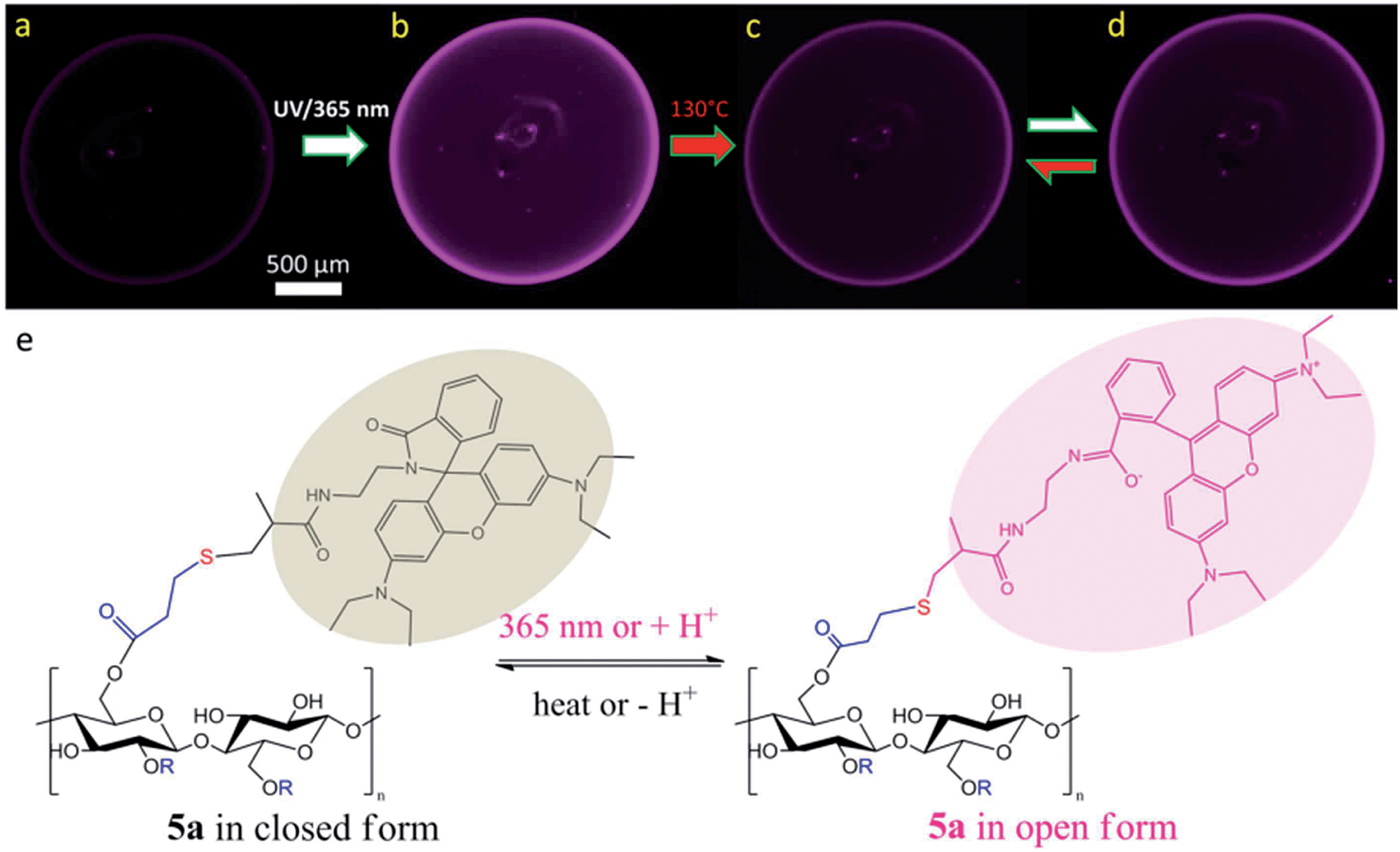 | ||
| Fig. 5 Fluorescence microscopy images of dried NPs from 5a: (a) freshly prepared and dried NPs, (b) after UV-treatment at 365 nm for 30 min, (c) the same NPs after heating at 130 °C for 30 min, and (d) the same NPs after the second UV-treatment at 365 nm for 30 min as well as schematic illustration of the reversibility between weakly and strongly fluorescent NPs (short-time treatments in Fig. S6†). Scale bar: 500 μm. (e) Schematic representation of the structural change of 5a in response to corresponding treatments. | ||
Moreover, these multi-responsive NPs from biocompatible and sustainable cellulose are promising candidates for biomedical sensing and imaging.44,45 In particular, they can potentially be used for site-specific detection and delivery of drugs.6,46–48
Conclusion
In summary, we reported the synthesis of novel, multi-stimuli responsive and reversibly fluorescent cellulose derivatives containing thiol groups and rhodamine spiroamide, which can form NPs or be dissolved again under redox-controllable conditions. The thiol groups were introduced into cellulose chains after esterification with 3,3′-dithiodipropionic acid, which provide a platform for further modifications via thiol–ene reaction, and in the present study, rhodamine B methacrylamide. Cellulose-RhBMA 5a containing disulfide groups were transformed into NPs via nanoprecipitation of its DMSO solution into water. The NPs could be dissolved or reconstructed through the reduction or oxidation, respectively. The rhodamine spiroamide endows the cellulose-RhBMA 5a and the NPs reversible fluorescence, in response to UV-illumination/heating or pH (presence or absence of protons). These multi-responsive NPs from biocompatible and sustainable cellulose with reversible fluorescence are of particular interest for biomedical sensing and imaging. Moreover, the facile thiol–ene reaction can be applied for the construction of novel cellulose derivatives.Acknowledgements
The authors thank the Hessian excellence initiative LOEWE – research cluster SOFT CONTROL for the financial support. W. L. thanks the China Scholarship Council (CSC) for the financial support. K. Z. thanks Prof. M. Biesalski for the kind support. Mrs S. Starke and Dr J. Appelt (TU Bergakademie Freiberg, Germany) are gratefully acknowledged for the elemental analysis.References
- S.-K. Ahn, R. M. Kasi, S.-C. Kim, N. Sharma and Y. Zhou, Soft Matter, 2008, 4, 1151–1157 RSC.
- A. P. Esser-Kahn, S. A. Odom, N. R. Sottos, S. R. White and J. S. Moore, Macromolecules, 2011, 44, 5539–5553 CrossRef CAS.
- H. Meng and G. Li, J. Mater. Chem. A, 2013, 1, 7838–7865 CAS.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS PubMed.
- P. Schattling, F. D. Jochum and P. Theato, Polym. Chem., 2014, 5, 25–36 RSC.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- H. Schenderlein, A. Voss, R. W. Stark and M. Biesalski, Langmuir, 2013, 29, 4525–4534 CrossRef CAS PubMed.
- S. Son, E. Shin and B. S. Kim, Biomacromolecules, 2014, 15, 628–634 CrossRef CAS PubMed.
- Y. Zhao, Macromolecules, 2012, 45, 3647–3657 CrossRef CAS.
- C. He, X. Zhuang, Z. Tang, H. Tian and X. Chen, Adv. Healthcare Mater., 2012, 1, 48–78 CrossRef CAS PubMed.
- H. G. Schild, Prog. Polym. Sci., 1992, 17, 163–249 CrossRef CAS.
- D. Szabo, G. Szeghy and M. Zrinyi, Macromolecules, 1998, 31, 6541–6548 CrossRef CAS.
- Y. Jing and P. Wu, Cellulose, 2012, 20, 67–81 CrossRef.
- M. Prabaharan and J. F. Mano, Macromol. Biosci., 2006, 6, 991–1008 CrossRef CAS PubMed.
- D. Klemm, B. Heublein, H. P. Fink and A. Bohn, Angew. Chem., Int. Ed., 2005, 44, 3358–3393 CrossRef CAS PubMed.
- D. Roy, M. Semsarilar, J. T. Guthrie and S. Perrier, Chem. Soc. Rev., 2009, 38, 2046–2064 RSC.
- L. Ma, R. Liu, J. Tan, D. Wang, X. Jin, H. Kang, M. Wu and Y. Huang, Langmuir, 2010, 26, 8697–8703 CrossRef CAS PubMed.
- F. J. Xu, Y. Zhu, F. S. Liu, J. Nie, J. Ma and W. T. Yang, Bioconjugate Chem., 2010, 21, 456–464 CrossRef CAS PubMed.
- Q. Yan, J. Yuan, F. Zhang, X. Sui, X. Xie, Y. Yin, S. Wang and Y. Wei, Biomacromolecules, 2009, 10, 2033–2042 CrossRef CAS PubMed.
- H. Wondraczek and T. Heinze, Macromol. Biosci., 2008, 8, 606–614 CrossRef CAS PubMed.
- H. Wondraczek, A. Pfeifer and T. Heinze, Cellulose, 2012, 19, 1327–1335 CrossRef CAS.
- D. Klemm, B. Philipp, T. Heinze, U. Heinze and W. Wagenknecht, Comprehensive Cellulose Chemistry, Functionalization of Cellulose, Wiley-VCH Verlag GmbH, Weinheim, 1998, vol. 2 Search PubMed.
- P. Tingaut, R. Hauert and T. Zimmermann, J. Mater. Chem., 2011, 21, 16066–16076 RSC.
- A. R. Lokanathan, A. Nykanen, J. Seitsonen, L. S. Johansson, J. Campbell, O. J. Rojas, O. Ikkala and J. Laine, Biomacromolecules, 2013, 14, 2807–2813 CrossRef CAS PubMed.
- D. Aoki, Y. Teramoto and Y. Nishio, Biomacromolecules, 2007, 8, 3749–3757 CrossRef CAS PubMed.
- D. F. S. Petri, S. Choi, H. Beyer, T. Schimmel, M. Bruns and G. Wenz, Polymer, 1999, 40, 1593–1601 CrossRef CAS.
- C. G. Schäfer, M. Gallei, J. T. Zahn, J. Engelhardt, G. P. Hellmann and M. Rehahn, Chem. Mater., 2013, 25, 2309–2318 CrossRef.
- Y. Shiraishi, R. Miyamoto, X. Zhang and T. Hirai, Org. Lett., 2007, 9, 3921–3924 CrossRef CAS PubMed.
- C. E. Hoyle, T. Y. Lee and T. Roper, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5301–5338 CrossRef CAS.
- C. E. Hoyle and C. N. Bowman, Angew. Chem., Int. Ed., 2010, 49, 1540–1573 CrossRef CAS PubMed.
- H. Thomas, L. Tim and K. Andreas, Esterification of Polysaccharides, Springer-Verlag Berlin Heidelberg, Heidelberg, Germany, 2006 Search PubMed.
- T. F. Liebert and T. Heinze, Biomacromolecules, 2005, 6, 333–340 CrossRef CAS PubMed.
- V. N. Belov, M. L. Bossi, J. Folling, V. P. Boyarskiy and S. W. Hell, Chemistry, 2009, 15, 10762–10776 CrossRef CAS PubMed.
- V. N. Belov, C. A. Wurm, V. P. Boyarskiy, S. Jakobs and S. W. Hell, Angew. Chem., Int. Ed., 2010, 49, 3520–3523 CrossRef CAS PubMed.
- N. Kar, H. Liu and K. J. Edgar, Biomacromolecules, 2011, 12, 1106–1115 CrossRef CAS PubMed.
- K. Zhang, S. Fischer, A. Geissler, E. Brendler and K. Gebauer, Cellulose, 2013, 20, 2069–2080 CrossRef CAS.
- K. F. Rasmussen, A. A. Smith, P. Ruiz-Sanchis, K. Edlund and A. N. Zelikin, Macromol. Biosci., 2014, 14, 33–44 CrossRef CAS PubMed.
- A. Geissler, M. Biesalski, T. Heinze and K. Zhang, J. Mater. Chem. A, 2014, 2, 1107–1116 CAS.
- S. Hornig and T. Heinze, Biomacromolecules, 2008, 9, 1487–1492 CrossRef CAS PubMed.
- S. Schubert, J. J. T. Delaney and U. S. Schubert, Soft Matter, 2011, 7, 1581–1588 RSC.
- M. Beija, C. A. Afonso and J. M. Martinho, Chem. Soc. Rev., 2009, 38, 2410–2433 RSC.
- H. Görner, Phys. Chem. Chem. Phys., 2001, 3, 416–423 RSC.
- D. A. Hinckley, P. G. Seybold and D. P. Borris, Spectrochim. Acta, Part A, 1986, 42, 747–754 CrossRef.
- H. Sun, B. Guo, X. Li, R. Cheng, F. Meng, H. Liu and Z. Zhong, Biomacromolecules, 2010, 11, 848–854 CrossRef CAS PubMed.
- J. Pauli, K. Licha, J. Berkemeyer, M. Grabolle, M. Spieles, N. Wegner, P. Welker and U. Resch-Genger, Bioconjugate Chem., 2013, 24, 1174–1185 CrossRef CAS PubMed.
- M. M. Russew and S. Hecht, Adv. Mater., 2010, 22, 3348–3360 CrossRef CAS PubMed.
- E. Vlashi, L. E. Kelderhouse, J. E. Sturgis and P. S. Low, ACS Nano, 2013, 7, 8573–8582 CrossRef CAS PubMed.
- R. Cheng, F. Meng, C. Deng, H. A. Klok and Z. Zhong, Biomaterials, 2013, 34, 3647–3657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta02126f |
| This journal is © The Royal Society of Chemistry 2014 |