DOI:
10.1039/C4TA01653J
(Paper)
J. Mater. Chem. A, 2014,
2, 12348-12354
Electron-deficient truxenone derivatives and their use in organic photovoltaics†
Received
4th April 2014
, Accepted 13th June 2014
First published on 16th June 2014
Abstract
A series of electron-deficient truxenone derivatives are investigated as fullerene alternatives in organic photovoltaic applications. These new electron-accepting molecules have easily tunable absorption profiles, more than ten-fold higher absorptivities than PCBM, slightly higher electron affinities than PCBM and clearly defined and highly reversible reductive characteristics. Fabrication of efficient bilayer solar cells with a subphthalocyanine (SubPc) donor illustrates the promise of this class of materials as electron acceptors in organic solar cells.
Introduction
In the field of organic photovoltaics, the development of novel oligomeric and polymeric donor materials has received a vast amount of attention,1–3 whereas design and synthesis of novel non-fullerene type acceptor materials have attracted much less attention.4–10 Although fullerene acceptors such as phenyl-C61-butyric acid methyl ester (PCBM) have several highly beneficial properties for organic photovoltaic (OPV) applications, non-fullerene acceptors are often superior to fullerenes in terms of optical absorptivity, ease of synthesis and ease of frontier molecular orbital energy level fine tuning. Additionally, in contrast to fullerenes, non-fullerene acceptors hold the potential to be processed from non-halogenated solvents, which is highly advantageous in relation to scale-up and commercialisation of organic photovoltaics.
We have recently shown that solution processable truxenone derivatives with highly electron-withdrawing functionalities such as the dicyanovinylene moiety can be used as efficient acceptors in bilayer OPV devices with evaporated phthalocyanine donor materials.11 Here, we investigate further truxenone acceptors for OPV applications and the role of the electron-withdrawing moiety as well as the effects of introducing one, two or three of these electron-withdrawing functionalities on the C3-symmetric truxenone core.
Synthesis and thermal properties
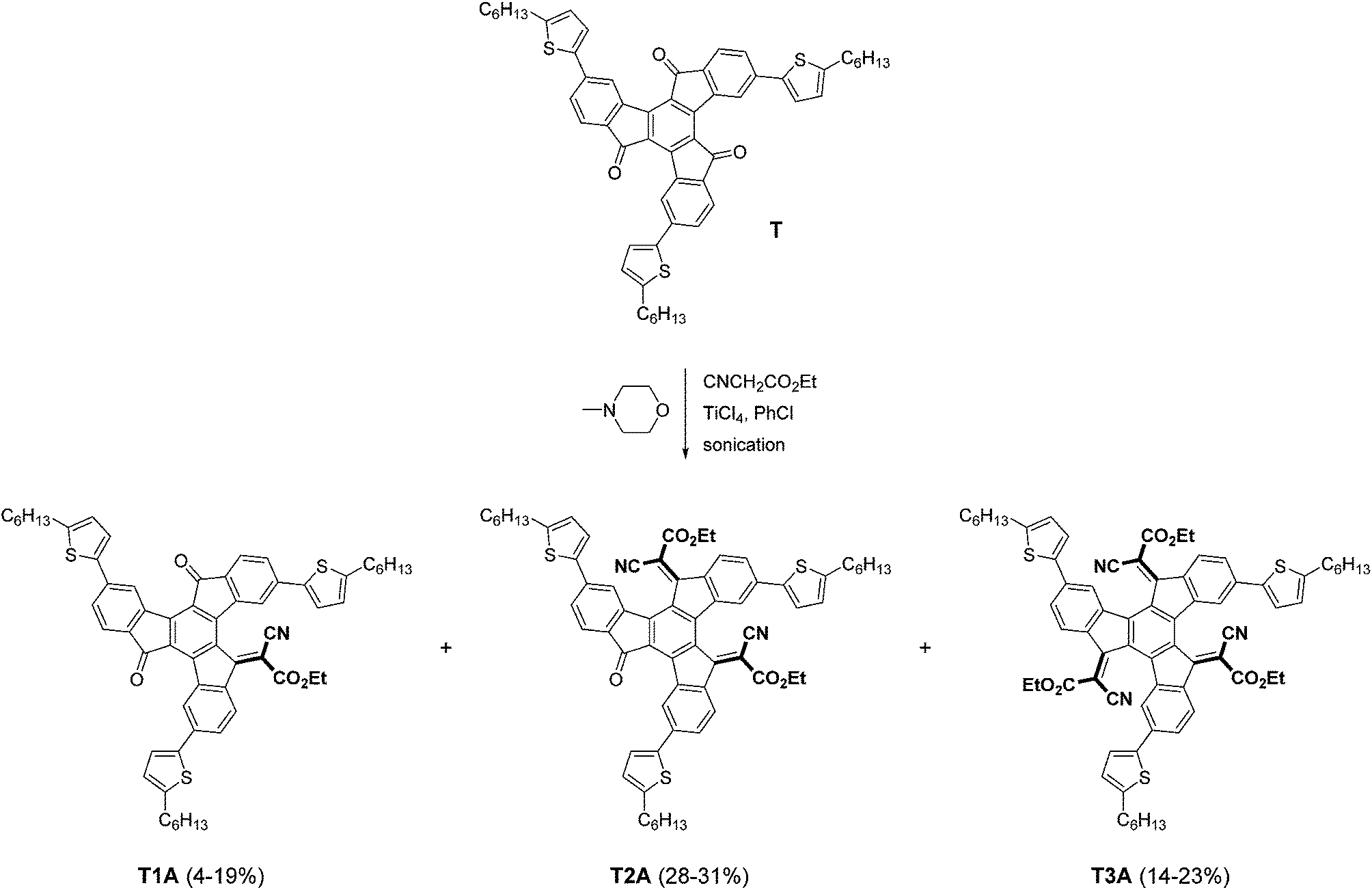
As illustrated in Scheme 1, mono-, bis- and tris-adducts T1A, T2A and T3A were obtained from the Knoevenagel reaction between the 5-hexyl-2-thienyl-substituted truxenone (T) and ethyl cyanoacetate. Depending on the exact reaction conditions, favouring either partial (mono and bis) or complete (tris) addition, T1A was obtained in yields of 4–19%, T2A in 28–31% and T3A in 14–23% with combined yields ranging from 58 to 61%. The three adducts were isolated by column chromatography as red solids and NMR spectroscopy was used to confirm the identity of the three adducts.
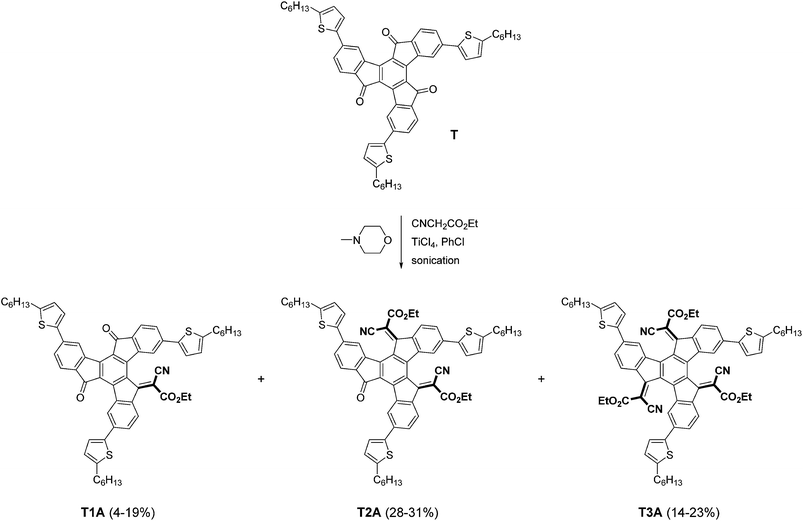 |
| | Scheme 1 Synthesis of truxenone adducts. | |
Thermogravimetric analysis revealed good thermal stability for all three adducts as depicted in Fig. 1 with 5% weight loss in all cases observed above 365 °C. When compared to the truxenone starting material (T), it is evident that each subsequent Knoevenagel adduct formation with ethyl cyanoacetate results in a thermogravimetric weight loss of approximately 4–5% around 400–410 °C. It should be noted that, although less thermally stable, the higher adducts, with no more than 2% weight loss observed at 350 °C, are still perfectly suitable for organic electronic applications.
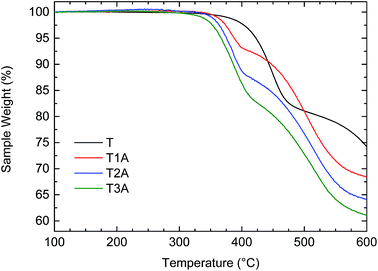 |
| | Fig. 1 TGA traces for the truxenone precursor (T) and the three truxenone adducts recorded in a nitrogen atmosphere at a heating rate of 10 °C min−1. | |
Differential scanning calorimetry (DSC, Fig. 2) showed that while the unsubstituted truxenone (T) had a very distinct thermal transition around 230 °C, all three truxenone adducts had DSC traces with no clear thermal transitions. This observation is most likely a result of the contortion of the planar truxenone core upon Knoevenagel adduct formation as will be discussed in the following section in greater detail.
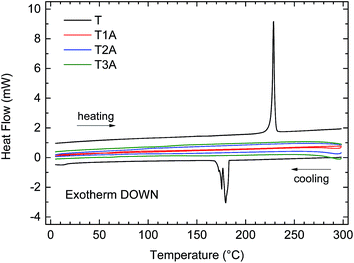 |
| | Fig. 2 DSC traces (exotherm down) for the truxenone precursor (T) and the three truxenone adducts recorded with a heating and cooling rate of 10 °C min−1. | |
Computational properties
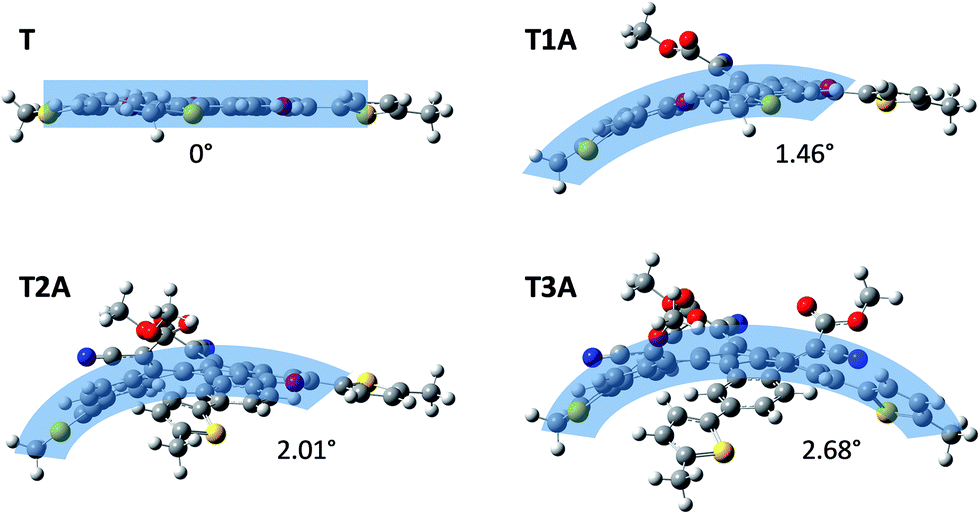
Semi-empirical calculations with Gaussian using the B3LYP/6-31G* model were carried out in order to investigate the effect of the adduct formation on the molecular conformation. Side-view images of the energy-minimised conformations (Fig. 3) clearly illustrates how the truxenone core of T as expected adopts a completely coplanar conformation (thiophene rings are twisted 13° relative to the core), whereas all three adducts adopt a curved conformation owing to the steric repulsions between the four substituents on the newly formed carbon–carbon double bonds. This is in full agreement with the crystal structure reported by Zhang and co-workers on a closely related truxenone tris-adduct without the 5-hexyl-2-thienyl substituents.12 For a closer inspection of the exact curvature of the three adducts, we have applied the π-orbital axis vector (POAV) analysis developed by Haddon and Scott.13–15 This method was used to derive pyramidalization angles for each carbon atom in the central ring of the truxenone core for all three adducts. For T1A, the largest pyramidalization angle, which relates directly to the curvature, was found to be 1.46°, while the corresponding angles were 2.01° for T2A and 2.68° for T3A (Fig. 3) illustrating the increased molecular curvature when going from T1A to T2A to T3A.
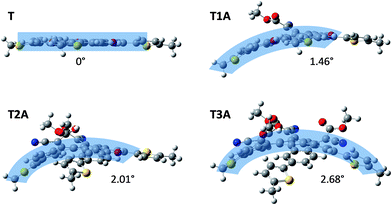 |
| | Fig. 3 Energy-minimised (Gaussian B3LYP/6-31G*) side-view conformations of the truxenone precursor and the three truxenone adducts with POAV pyramidalization angles indicating the curvature of each molecule. | |
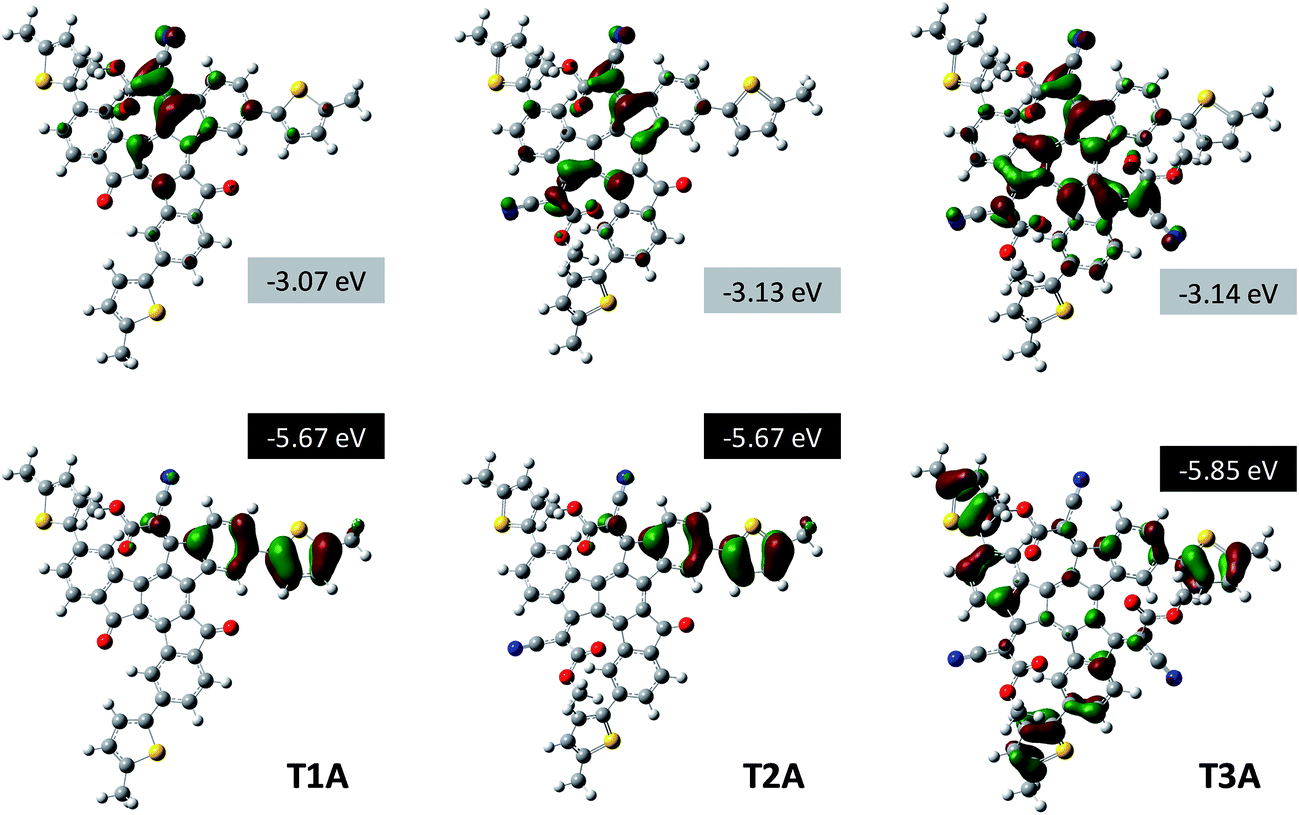
The highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) distributions are visualised for all three adducts in Fig. 4. T1A has its HOMO localised on one of the three electron-rich peripheral phenylene-thienylene “arms”, while the LUMO is localised around the electron-deficient cyanoacetate moiety on the truxenone core. In this context it is worth emphasising that it is the meta-linkage between the central benzene ring and the outer thiophene that disrupts the conjugation over the entire π-system. Compared to T1A, the bis-adduct T2A has a similar HOMO distribution reflected in the identical calculated HOMO energy values of −5.67 eV. The LUMO of T2A, on the other hand, is delocalised more than that of T1A due to the additional cyanoacetate moiety, which explains the stabilisation of the LUMO energy from −3.07 eV for T1A to −3.13 eV for T2A. By formation of the tris-adduct T3A, the C3-symmetry of the molecule is restored and as a consequence both the HOMO and the LUMO is found to be doubly degenerate. The HOMO is thus equally distributed on all three peripheral “arms” and therefore significantly stabilised (−5.85 eV) compared to T1A and T2A. The LUMO of T3A is likewise evenly distributed over the entire electron-deficient core causing a slight stabilisation (−3.14 eV) relative to T2A. Due to the stabilisation of the LUMO in T2A, the calculated HOMO–LUMO gap is smallest for the bis-adduct, while the stabilisation of the HOMO in T3A explains why the tris-adduct has the highest HOMO–LUMO gap of the three.
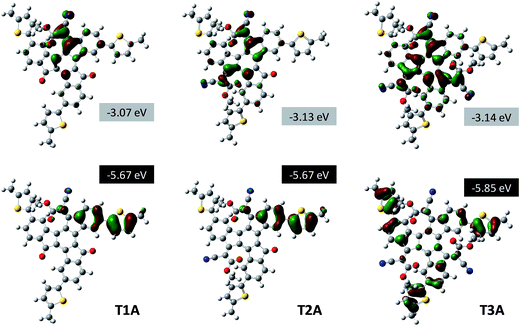 |
| | Fig. 4 Visualization of HOMO and LUMO distributions and the calculated frontal orbital energies (Gaussian B3LYP/6-31G*) for the three truxenone adducts. | |
Optical properties
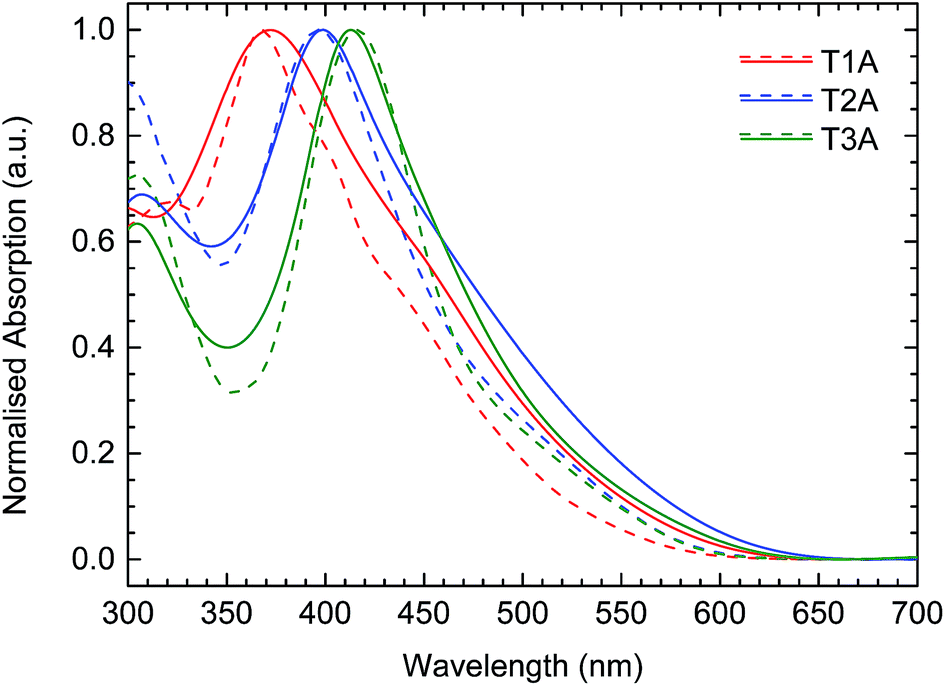
The UV-vis absorption spectra of the three truxenone adducts were recorded in dichloromethane (DCM) solution and as thin films spin-coated from chloroform solution; the obtained spectra are depicted in Fig. 5 and summarized in Table 1. A gradual red-shift of the absorption maximum of 20–30 nm was observed, both in solution and the solid state, when going from T1A to T2A to T3A (Table 1). For the transition from solution to solid state, we observe small red-shifts for the mono- and bis-adduct of 2–4 nm, while a small blue-shift of 3 nm is seen for the tris-adduct. In all three cases, a significant broadening of the absorption profile, especially on the low-energy side, is seen when going from solution to solid state. Optical bandgaps, as estimated from the onset of absorption in the solid state, are very similar for the three compounds, varying from 2.05 eV for T2A to 2.10 eV for T3A and 2.12 eV for T1A.
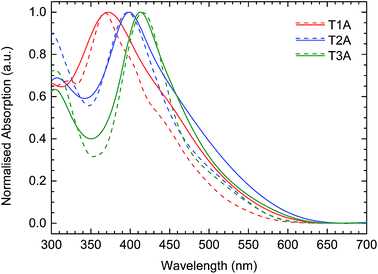 |
| | Fig. 5 UV-vis absorption spectra for the three truxenone adducts in dichloromethane solution (dashed lines) and as thin films spin-cast from chloroform (solid lines). | |
Table 1 Experimental and theoretical optical properties for truxenone adducts
|
|
λ
max sol'n exp. (nm) |
ε
max sol'n exp. (M−1 cm−1) |
λ
max sol'n calc. (nm) |
ε
max sol'n calc. (M−1 cm−1) |
λ
max film (nm) |
E
g film (eV) |
|
T1A
|
368 |
67![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
430 |
81900 |
372 |
2.12 |
|
T2A
|
397 |
74600 |
442 |
105400 |
399 |
2.05 |
|
T3A
|
416 |
89400 |
463 |
125300 |
413 |
2.10 |
Time-Dependent Density Function Theory (TDDFT, Gaussian B3LYP/6-31G* with DCM solvation) was used to compute the excitation energies and the oscillator strengths associated with the first twenty vertical excitations for each truxenone adduct (raw data in Tables S1–S3 in ESI†). With this data, the theoretical UV-vis spectra were calculated using the SWizard program as depicted in Fig. 6.16,17 The theoretical data nicely corroborates the experimental data (also obtained in DCM solution) with respect to the gradual red-shift when going from mono-adduct to bis- and subsequently tris-adduct. The calculations furthermore predict a gradual increase in molar absorptivity when going from T1A to T2A to T3A and this trend is also found experimentally with a maximum molar extinction coefficient of roughly 9 × 104 M−1 cm−1 measured for T3A (Table 1). In this context it should be noted that the C3-symmetry of T3A would imply that the S0–S1 transition is orbitally forbidden, but mixing with strong transitions nearby are likely responsible for the low-energy absorption.18
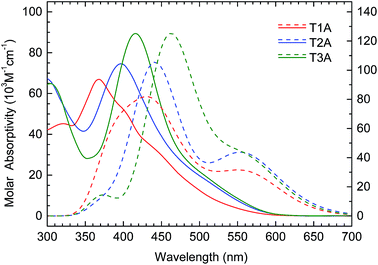 |
| | Fig. 6 Experimental (solid lines, left y-axis) and theoretical (dashed lines, right y-axis) UV-vis absorption spectra for the three truxenone adducts in dichloromethane solution. | |
We note that the absorption profile of these truxenone acceptors effectively can be blue- or red-shifted depending on the degree of functionalization; a highly advantageous feature when seeking complementarity of absorption profiles between the donor and the acceptor materials in OPV applications. Additionally, the molar extinction coefficients of 67000 to 89000 M−1 cm−1 exceed the value reported for PCBM by more than one order of magnitude.19
Electrochemical properties
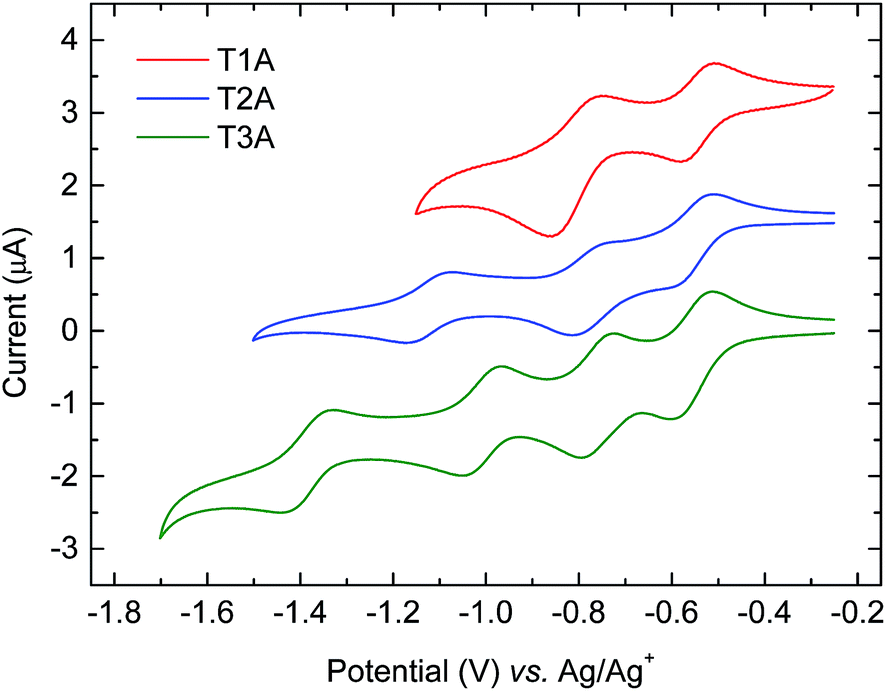
The electron-accepting behaviour of the truxenone adducts were investigated by reductive cyclic voltammetry (CV) in dichloromethane solution and the resulting voltammograms are depicted in Fig. 7 and results are summarised in Table 2. Congruent with the increasing number of cyanoestervinylene moieties, we observe two, three and four semi-reversible reduction waves for T1A, T2A and T3A, respectively. For all three truxenone adducts, we observe the first and second half-wave potentials around −0.55 V and −0.80 V, respectively. In addition, T2A shows a third reduction event with a half-wave potential at −1.13 V, while T3A has a third and a fourth reduction event at −1.01 V and −1.39 V, respectively. From the onset of reduction, we estimate the LUMO energy levels to be −3.87 to −3.88 eV for all three adducts, while the corresponding value for PC61BM measured under identical conditions was found at −3.75 eV. We thus find all three truxenone-adducts to be slightly stronger electron acceptors than PC61BM. It is also worth emphasising that the CV results clearly indicate that the LUMO level is not significantly altered when going from T1A to T2A and T3A. This is in good agreement with the semi-empirical calculations presented above (Fig. 4), where the three LUMO levels are predicted to differ no more than 0.07 eV. From a molecular point of view, the 1,3,5-positioning of the electron-withdrawing moieties around the central benzene ring and thus the limited electronic communication between them is likely responsible for the nearly identical LUMO levels. The distributions of the calculated LUMO+n orbitals (Fig. S5–S7 in ESI†) support the assumption that each cyanoestervinylene moiety will accept one electron upon reduction. Subsequently, another electron can be accepted by either the remaining carbonyl group (for T1A and T2A) or the aromatic core of the truxenone system (for T3A), which explains why two reduction events are observed for the mono-adduct, three reduction events for the bis-adduct and four reduction events for the tris-adduct. We also note that the area of the second reduction wave of T1A is roughly twice that of the area of the first reduction wave, in agreement with one cyanoestervinylene moiety being reduced in the first reduction process and two carbonyl groups being reduced in the second event.
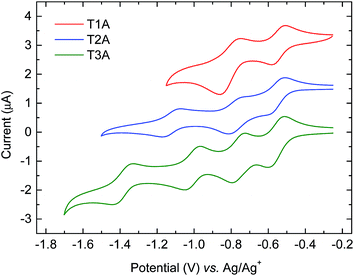 |
| | Fig. 7 Reductive cyclic voltammograms for the three truxenone adducts in dichloromethane solution with tetrabutylammonium hexafluorophosphate as the supporting electrolyte (curves are shifted vertically for improved clarity). | |
Table 2 Reductive electrochemical properties for truxenone adducts
|
|
E
1/2(1) (V) |
E
1/2(2) (V) |
E
1/2(3) (V) |
E
1/2(4) (V) |
E
LUMO (eV) |
|
T1A
|
−0.55 |
−0.81 |
— |
— |
−3.87 |
|
T2A
|
−0.55 |
−0.78 |
−1.13 |
— |
−3.87 |
|
T3A
|
−0.56 |
−0.77 |
−1.01 |
−1.39 |
−3.88 |
Photovoltaic properties
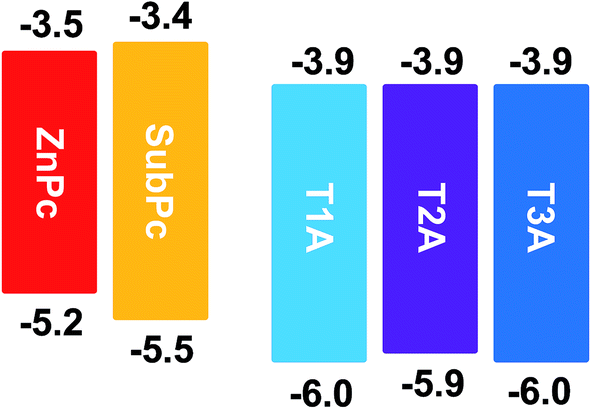
To investigate the fundamental photovoltaic properties of these electron-accepting truxenone adducts, a bilayer device configuration was chosen over a bulk heterojunction configuration in order to eliminate any potential interference from blend morphology. Thus, with an inverted bilayer device configuration, these new solution-processable truxenone derivatives were spin-coated onto TiO2-coated ITO substrates after which either subphthalocyanine (SubPc) or zinc phthalocyanine (ZnPc) was applied as the donor material by evaporation.20,21 Both phthalocyanine compounds have complementary absorption to the truxenone derivatives as SubPc absorbs between 450 and 600 nm, while ZnPc between 600 and 800 nm. Finally a thin MoO3 and Ag top contact was deposited. As illustrated in Fig. 8, both phthalocyanine donor materials used in conjunction with the truxenone-based acceptors herein have sufficiently high-lying LUMO levels (−3.5 eV for ZnPc and −3.4 eV for SubPc) to allow for energetically favorable charge transfer to the acceptor material.
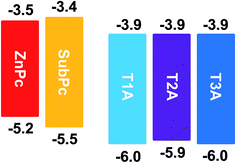 |
| | Fig. 8 Frontier energy levels (all values in eV) of electron donor and acceptor materials used herein. | |
As illustrated in Fig. 9 and summarised in Table 3, the solar cells with SubPc (30 nm) show much higher open circuit voltages (Voc) than the cells with the ZnPc donor (50 nm) due to the much lower HOMO value of SubPc relative to ZnPc (Fig. 8). We also note that there is basically no variation of the Voc with the three different truxenone acceptors, which is in good agreement with the nearly identical LUMO levels determined for the three compounds. Fill factors (FF) for these bilayer devices are in all cases rather low with values ranging from 19 to 25% with slightly higher values observed for T3A than for T1A and T2A. With both SubPc and ZnPc donors, a clear trend of increasing short circuit current (Jsc) when going from T1A to T2A to T3A is discernible. The highest Jsc-values are obtained with SubPc as the donor ranging from 0.53 mA cm−2 with T1A to 0.85 mA cm−2 with T3A (for JV and EQE curves, see ESI). Consequently, the best bilayer OPV devices are obtained with the subphthalocyanine donor and the tris-adduct giving a power conversion efficiency (PCE) of 0.28% while the best ZnPc-device with T3A has a PCE of 0.08%. In comparison, reference OPV devices with PCBM as the electron acceptor material gave power conversion efficiencies of 0.73% with SubPc and 1.30% with ZnPc in the investigated device configuration (Table 3).
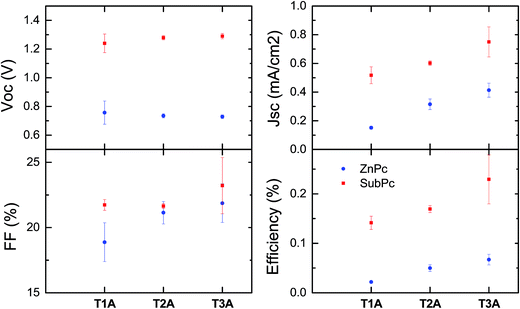 |
| | Fig. 9 Photovoltaic device parameters of bilayer OPV devices with the three truxenone adducts (T1A, T2A and T3A) tested with both ZnPc (blue circles) and SubPc (red squares) donor materials. Values are averages and standard deviations measured on 4–8 cells under 100 mW cm−2 and AM1.5G simulated solar illumination. | |
Table 3 Photovoltaic properties for truxenone adducts
| Acceptor |
Donor |
V
oc (V) |
J
sc (mA cm−2) |
FF |
PCE (%) |
|
T1A
|
SubPc |
1.30 |
0.53 |
0.21 |
0.15 |
| ZnPc |
0.79 |
0.16 |
0.19 |
0.02 |
|
T2A
|
SubPc |
1.30 |
0.62 |
0.22 |
0.18 |
| ZnPc |
0.72 |
0.35 |
0.22 |
0.06 |
|
T3A
|
SubPc |
1.29 |
0.85 |
0.25 |
0.28 |
| ZnPc |
0.72 |
0.48 |
0.23 |
0.08 |
| PCBM |
SubPc |
0.98 |
1.50 |
0.50 |
0.73 |
| ZnPc |
0.59 |
3.45 |
0.63 |
1.30 |
In light of the above results, we set out to investigate further the underlying reasons for the inferior OPV device performance of these truxenone derivatives despite favourable frontier energy levels and high optical extinction coefficients. Attempts to incorporate the truxenone adducts in bottom-gate bottom-contact organic field-effect transistors (OFETs) were in all cases unsuccessful; no transistor behaviour could be observed neither for n-type nor for p-type charge transport. This is in good agreement with the very low fill factors observed for the OPV devices and we thus conclude that very poor charge transport properties is most likely the limiting factor for this class of materials. Further to this observation, we turn our attention once again to Fig. 4 and the highly localised LUMO distributions around the truxenone core for all three truxenone adducts. While this strong spatial separation of HOMO (localised around periphery of molecule) and LUMO (localised around the core of the molecule) has proven beneficial for independently altering the HOMO and LUMO energy levels, we believe that it can also explain the very poor electron transport properties of these truxenone acceptors. Numerous reports in the recent literature have highlighted a strong spatial overlap of HOMO and LUMO distributions for highly efficient electron-transporting materials.22,23 Furthermore, while the contortion of the planar truxenone core is beneficial for reducing pi–pi stacking and hence crystallinity (for improved bulk-heterojunction blend morphology with donor materials), it is likely to strongly limit intermolecular charge transport as well.
Conclusions
In conclusion, we have introduced a truxenone moiety with peripheral thiophene groups for extended pi-conjugation and presented a synthetic route that allows us to access three different Knoevenagel adducts of this molecule in order to form highly electron-deficient materials with electron affinities slightly higher than PCBM. The three adducts are thermally stable and show a lack of crystallinity due to the contortion of the otherwise planar truxenone motif. In addition, these novel truxenone derivatives have promising optical properties with high molar extinction coefficients and easily tunable absorption maxima, while they also show good electrochemical stability during reduction with clearly defined electrochemical events. Satisfactorily, initial assessment in photovoltaic devices showed that all three adducts behaved as electron acceptor materials with phthalocyanine based donor materials. Modest device performances were obtained in an inverted bilayer device architecture with the tris-adduct T3A showing the best performance (PCE of 0.28%) with an evaporated subphthalocyanine donor.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Acknowledgements
This work was in part carried out with financial support from EPSRC (EP/G037515/1) and EC FP7 Project X10D with support from BASF.
References
- C. B. Nielsen, B. C. Schroeder, A. Hadipour, B. P. Rand, S. E. Watkins and I. McCulloch, J. Mater. Chem., 2011, 21, 17642 RSC.
- J. Kirkpatrick, C. B. Nielsen, W. Zhang, H. Bronstein, R. S. Ashraf, M. Heeney and I. McCulloch, Adv. Energy Mater., 2012, 2, 260 CrossRef CAS.
- L. Dou, J. You, Z. Hong, Z. Xu, G. Li, R. A. Street and Y. Yang, Adv. Mater., 2013, 25, 6642 CrossRef CAS PubMed.
- A. a. F. Eftaiha, J.-P. Sun, I. G. Hill and G. C. Welch, J. Mater. Chem. A, 2014, 2, 1201 CAS.
- X. Zhang, Z. Lu, L. Ye, C. Zhan, J. Hou, S. Zhang, B. Jiang, Y. Zhao, J. Huang, S. Zhang, Y. Liu, Q. Shi, Y. Liu and J. Yao, Adv. Mater., 2013, 25, 5791 CrossRef CAS PubMed.
- W. Jiang, L. Ye, X. Li, C. Xiao, F. Tan, W. Zhao, J. Hou and Z. Wang, Chem. Commun., 2014, 50, 1024 RSC.
- J. E. Anthony, A. Facchetti, M. Heeney, S. R. Marder and X. Zhan, Adv. Mater., 2010, 22, 3876 CrossRef CAS PubMed.
- J. E. Anthony, Chem. Mater., 2011, 23, 583 CrossRef CAS.
- G. M. Su, T. V. Pho, N. D. Eisenmenger, C. Wang, F. Wudl, E. J. Kramer and M. L. Chabinyc, J. Mater. Chem. A, 2014, 2, 1781 CAS.
- G. Ren, E. Ahmed and S. A. Jenekhe, Adv. Energy Mater., 2011, 1, 946 CrossRef CAS.
- C. B. Nielsen, E. Voroshazi, S. Holliday, K. Cnops, B. P. Rand and I. McCulloch, J. Mater. Chem. A, 2013, 1, 73 CAS.
- X.-R. Zhang, W. Chao, Y.-T. Chuai, Y. Ma, R. Hao, D.-C. Zou, Y.-G. Wei and Y. Wang, Org. Lett., 2006, 8, 2563 CrossRef CAS PubMed.
- R. C. Haddon, J. Am. Chem. Soc., 1986, 108, 2837 CrossRef CAS.
- L. T. Scott and R. C. Haddon, Pure Appl. Chem., 1986, 58, 137 Search PubMed.
- R. C. Haddon, J. Am. Chem. Soc., 1990, 112, 3385 CrossRef CAS.
- S. I. Gorelsky and A. B. P. Lever, J. Organomet. Chem., 2001, 635, 187 CrossRef CAS.
- S. I. Gorelsky, University of Ottawa, Ottawa, Canada, http://www.sg-chem.net/, 2013.
-
N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed.
- S. T. Kim, D.-H. Hwang, X. C. Li, J. Grüner, R. H. Friend, A. B. Holmes and H. K. Shim, Adv. Mater., 1996, 8, 979 CrossRef CAS.
- D. Cheyns, B. P. Rand and P. Heremans, Appl. Phys. Lett., 2010, 97, 033301 CrossRef PubMed.
- B. P. Rand, D. Cheyns, K. Vasseur, N. C. Giebink, S. Mothy, Y. Yi, V. Coropceanu, D. Beljonne, J. Cornil, J.-L. Brédas and J. Genoe, Adv. Funct. Mater., 2012, 22, 2987 CrossRef CAS.
- A. J. Kronemeijer, E. Gili, M. Shahid, J. Rivnay, A. Salleo, M. Heeney and H. Sirringhaus, Adv. Mater., 2012, 24, 1558 CrossRef CAS PubMed.
- C. B. Nielsen, M. Turbiez and I. McCulloch, Adv. Mater., 2013, 25, 1859 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, AFM micrographs, JV and EQE curves, molecular orbital distributions, calculated excitation energies and oscillator strengths. See DOI: 10.1039/c4ta01653j |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.  Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a