
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Site specific supramolecular heterogeneous catalysis by optically patterned soft oxometalate–porous organic framework (SOM–POF) hybrid on a chip†
Preethi
Thomas
a,
Cuiying
Pei
b,
Basudev
Roy
c,
Subhrokoli
Ghosh
c,
Santu
Das
a,
Ayan
Banerjee
*c,
Teng
Ben
*b,
Shilun
Qiu
b and
Soumyajit
Roy
*a
aEFAML, Materials Science Centre, Department of Chemical Sciences, IISER-Kolkata, Mohanpur-741252, India. E-mail: s.roy@iiserkol.ac.in
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, Department of Chemistry, Jilin University, Changchun, People's Republic of China. E-mail: tben@jlu.edu.cn
cDepartment of Physical Sciences, Indian Institute of Science Education Research, Kolkata (IISER-Kolkata), Mohanpur-741252, India. E-mail: ayan@iiserkol.ac.in
First published on 23rd September 2014
Abstract
We have designed a supramolecularly bound multi-component catalytic material based on a soft oxometalate (SOM) and a porous organic framework (POF) material, which shows high catalytic conversion efficiency. We have also used this material for site directed supramolecular heterogeneous catalysis with a yield even higher than in the bulk, and with micron-level precision by controllably depositing the material on a glass substrate, making a reactor chip, using a thermo-optical tweezers. Various SOM–POF composites have been prepared in dispersion phase and patterned using thermo-optic tweezers and their catalytic activities have been compared with a benchmark molecular catalyst [PMo12]. This work can lead to further explorations for hybrid materials that are formed out of well-defined molecular level precursors which can be controllably micro-patterned to simultaneously catalyze targeted reactions.
Introduction
An immediate challenge in supramolecular catalysis is to design supramolecularly bound multi-component catalytic materials with high surface area in a controlled fashion. In this type of design, it is desired that distinct multifunctional components are held together by supramolecular interactions and they catalytically function in synergy to emulate enzymes.1 Moreover, for efficient catalysis, high surface area materials are required.Therefore, the need of the hour for supramolecular catalysis1 translates to the following questions. Is it possible to bring multiple functional components together? Can they be held together such that they can act in synergy? Can these components be patterned as ultrahigh surface area materials that show catalytic activities? In this paper we simultaneously address these challenges. We started with a soft oxometalate, (SOM) dispersion2 of molybdenum containing nanotubes to that we added porous organic framework (POF) material to get a stable soft oxometalate–porous organic framework, (SOM–POF) dispersion. We then exposed this dispersion to a thermo-optic laser tweezers to induce a phase transition from a dispersion to a crystalline state – thus simultaneously patterning3–11 the material in solid state by translating the laser beam.12 We further used this patterned material to carry out site-directed supramolecular catalysis.13,14 It is perhaps now apt to separately and briefly introduce the components of this material and the physical techniques that were used.
Soft oxometalates or SOMs are oxometalates endowed with soft matter properties2 of transition metal-oxide clusters known as polyoxometalates (POMs).15,16 POMs have been extensively studied because of their potential applications in various fields such as catalysis, electrical conductivity, sensing, medicinal chemistry and material science.16–31 SOMs are primarily governed by non-covalent interactions and can be moved with precision in an optical field,2a and they show topological transformation.2e Soft oxometalates have large surface areas, which can be easily manipulated and exploited in catalysis. In this work, we have used ammonium heptamolybdate tetrahydrate as the crystalline precursor to soft oxomolybdate.
The second component used in our work was POF or porous organic framework. POFs are porous materials32 with high surface area per unit mass as compared to metal–organic frameworks,33 which are extremely chemically as well as thermally stable. These materials provide accessible space within their pores for the reactant molecules to react.34–40 These porous frameworks are thus known for their applications in gas storage41–46 and separation.47 These porous frameworks are known to have affinity towards greenhouse gases such as carbon dioxide and methane, and exhibit high selectivity towards them in gas sorption studies.48a While SOMs provide catalytic potential; POFs provide high surface area and accessible space within the framework when these two components are combined together. JUC-Z4 used in this work was an electroactive porous organic framework synthesized from tris(4-chlorophenyl)phosphine, in which the radical redox centre of phosphorous was stabilized by an aromatic organic framework. The pore size, BET surface area and polarity of these organic frameworks can be manipulated and controlled by redox reactions.48a Furthermore, we also synthesized composites using other POF materials such as PAF1, JUC-Z2 and JUC-Z5 to form SOM–PAF1, SOM–JUC-Z2, SOM–JUC-Z5 composites, respectively. PAF1 is an extensively studied POF with exceptionally high surface area and thermal stability. It has a diamondoid structure with a tetrahedral carbon surrounded by phenyl rings. JUC-Z2 is similar to JUC-Z4 except for a hetero atom in this porous material, which is nitrogen instead of phosphorous. JUC-Z5, on the other hand, is the oxidised form of JUC-Z4 with phosphorous in the +5 oxidation state.48
It is the redox activity of JUC-Z4 coupled with the presence of phosphorous in it that prompted us to ask the following questions. Phosphomolybdates are known to be catalysts for several reactions.18,49 Can a supramolecular composite in dispersion state comprising of both molybdenum and phosphorous show comparable catalytic property as that of [PMo12] Keggin? Can the catalytic activity of this type of a SOM–POF composite (SOM–JUC-Z4) be compared with other SOM–POF composites? Can we further design high surface area patterned surfaces starting from such SOM–POF supramolecular composite dispersion? In addition to addressing these questions, we further compared and contrasted the catalytic activity of SOM–POF composites in dispersions. We demonstrated the catalytic activity of various thermo-optically patterned composite SOM–POF catalyst chips. We showed the stability of such catalytic chips before and after reaction and over days. We asked: can both aliphatic and aromatic aldehydes be oxidized by our method? Can other oxidants be used? How efficient is our SOM–POF chip compared to that of [PMo12] benchmark catalyst?
Materials and methods
Ammonium heptamolybdate tetrahydrate was used as obtained. JUC-Z4 was prepared following a previously published procedure.48 Benzaldehyde, hydrogen peroxide and DMSO were used as obtained. All the chemicals were obtained from Merck.Synthesis of SOMs
1.236 g (0.001 moles) of ammonium heptamolybdate tetrahydrate was dissolved in 10 mL of distilled water to prepare 0.1 M solution. Then, 0.2 M solution was prepared by dissolving 2.472 g (0.002 moles) of the heptamolybdate in 10 mL of distilled water. The two solutions of 0.1 M and 0.2 M were then warmed until dispersions were obtained that scatter laser light. These SOM dispersions were refrigerated for about 10 minutes for further experiments.Preparation of SOM–POF dispersions
Mole ratios of SOM and JUC-Z4 dispersions were varied from 1.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 to 51.8:1 (SOM:JUC-Z4), and 10 colloidal dispersions of SOM–POF hybrids were prepared. The same procedure was followed to prepare the dispersions of SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5.
:1 to 51.8:1 (SOM:JUC-Z4), and 10 colloidal dispersions of SOM–POF hybrids were prepared. The same procedure was followed to prepare the dispersions of SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5.
Description of optical tweezers set up
The optical patterning was done exactly similar to the procedure adopted in our previous work.12 The SOM–POF dispersion was patterned by exposing it to thermo-optical tweezers. The dispersion was placed between a 1 mm thick standard glass slide (top surface) and a 160 μm thick glass cover-slip (bottom surface). The spacing between the two surfaces was controlled by applying a double-sided sticky tape on the sides of the cover-slip. This set-up of slide and cover-slip stuck together with double-sided sticky tape constitute the sample holder. The dispersion in this holder was impinged with a CW laser beam which was focused to a diffraction limited spot by a high numerical aperture objective lens. The thermo-optical tweezers set up was built around an inverted microscope (Zeiss Axiovert. A1Observer), and it has a 100×, 1.4 N.A. oil immersion microscope objective (Zeiss planoapochromat, infinity corrected) which was used to focus light from a diode laser (Lasever LSR1064ML) of 1064 nm wavelength on the sample holder to attain a spot size of waist radius ∼500 nm. The power of the laser can be varied from 0 to 100 mW after the objective. Before every experimental run, the slide and coverslip constituting the sample holder were rinsed with methanol and dried to remove any adsorbed impurity. The sample holder containing about 75 μL of the dispersion was translated using the microscope scanning stage, which was stepper motor controlled with a Ludl MAC5000 XY stage controller that was operated by a joystick. The total travel range of the stage was 130 × 100 mm with a resolution of 100 nm. The patterning process was observed on a monitor with the help of a CCD camera (Axiovision) running at a fastest rate of 30 frames per second. A combination of 10×/40×/100× objectives and external optics were arranged outside the microscope to assist in imaging which was done at the back focal part of microscope.Zeta potential measurements
Malvern Zetasizer instrument coupled with Malvern Zetasizer software was used to measure the zeta potential of the prepared SOM–POF dispersions in a folded capillary cell.FTIR spectroscopy
Perkin Elmer Spectrum RX1 spectrophotometer was used to record the FTIR spectra in the range of 400–4000 cm−1.Low pressure N2 adsorption measurements
N2 sorption isotherm measurements were performed with a Micromeritics Tristar II 3020 surface area and pore size analyser. Before the measurement, the sample was subjected to a vacuum degassing system under ultra-high vacuum (90 mTorr) at 100 °C for 12 h. The sample was backfilled with nitrogen and transferred to the analysis system. A sample of 50 mg and ultrahigh purity grade nitrogen (99.999%) gas source were used in the nitrogen sorption measurements at 77 K, which was maintained with liquid nitrogen throughout the measurement. Helium was used for the free space determination after sorption analysis, both at ambient temperature and at 77 K. Apparent surface areas were calculated from nitrogen adsorption data by multi-point Brunauer–Emmett–Teller (BET) analysis. Apparent micropore distributions were calculated from carbon dioxide adsorption data by the density functional theory (DFT) method.Thermogravimetric analysis
The TGA was performed using a Shimadzu DTG-60 thermal analyser system at a heating rate of 10 °C min−1 to 800 °C in a dried air atmosphere and the air flow rate was 30 mL min−1. The sample was loaded in an alumina pan.Raman spectroscopy
A LABRAM HR800 Raman spectrometer was employed using the 633 nm line of a He–Ne ion laser as the excitation source to analyze the sample.Transmission electron microscopy
The characterization of the composite was primarily done by the use of a Tecnai 20 transmission electron microscope (FEI Company) operated at an accelerating voltage of 200 kV. The TEM micrographs were processed using SIS software (Soft Imaging System).Nuclear magnetic resonance
Time dependent 1H NMR measurements were performed using 400 MHz Bruker Ultrashield Plus System.Results and discussion
On the formation of SOM–POF composites and their characterization
We first analyzed the formation of the supramolecular composite between molybdenum containing soft oxometalate and the phosphorous containing porous organic framework. Therefore, we first checked whether a stable dispersion can be obtained using soft oxometalates and porous organic frameworks in synergy. We now systematically check what happens when SOM dispersion was mixed with a phosphorous containing POF (JUC-Z4). Upon mixing, a colloidal dispersion of SOM–POF composite is obtained. The colloidal nature of these stable dispersions was confirmed by passing laser light through these dispersions and checking for the scattering of light. We further saw that as we varied the relative loading of JUC-Z4 to heptamolybdate, the system changed from unstable to a stable colloidal regime [Fig. 1(b)]. We observed that the dispersions of 3.86 × 10−5, 7.72 × 10−5 and 1.15 × 10−4 moles of JUC-Z4 gave stable dispersions when mixed with 0.1 M and 0.2 M ammonium heptamolybdate based SOMs in 2:1 ratio of volume. The stable dispersion was negatively charge stabilized [Fig. 1(b)].
 | ||
| Fig. 1 (a) Schematic representation of ammonium heptamolybdate tetrahydrate (red-blue clusters) soft oxometalate incorporated into JUC-Z4 (phosphorous represented in green colour) porous organic framework. (b) Ψ, zeta potential of various SOM–POF dispersions of SOM–JUC-Z4 composite versus SOM–POF (mole ratio). (c) FTIR spectra of SOM–POF composite catalyst showing shifts in the N–H (1405 cm−1) and C–H (845 cm−1) bending frequencies and C–H (806 cm−1) and C–P (1123 cm−1) stretching frequencies. | ||
Now, the stable dispersion was used for further characterization and catalytic studies. We first closely observe the FTIR spectra of the dried SOM–POF dispersion and compare it with the peaks of its individual components [Fig. 1(c)]. The fingerprint peaks of ammonium heptamolybdate were retained almost unchanged in the SOM–POF composite. The peak at 1400 cm−1 intensified in the composite as compared to that of ammonium heptamolybdate. This peak was attributed to the bending mode (δN–H) of N–H vibration. This intensification was due to the condensation of NH4+ counter ions around the composite. We also observed shifts in the peaks of JUC-Z4 in the composite. For instance, we observed a shift in the bending mode (δC–H) and stretching mode (νC–H) of C–H vibration at 845 cm−1 and 806 cm−1, respectively, in the composite. This indicates that the aromatic C–H bonds of the benzene rings of POF are getting stiffened due to the restriction brought about by the surrounding heptamolybdate clusters. We also observed a slight blue shift in the band around 1123 cm−1. This was attributed to the stretching mode (νC–P) of C–P vibration in the SOM–POF composite, which occurs due to electron donation from heptamolybdate to the positive centre of phosphorous of JUC-Z4, which in turn strengthens the C–P bond.
The FTIR spectra of other SOM–POF composites, namely SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5, also showed the characteristic peaks of SOM as well as their respective constituting POF materials with slight shifts in the peak values, indicating the successful formation of SOM–POF composites (see ESI† for details). The loading of PAF-1, JUC-Z2 and JUC-Z5 was accordingly varied and the variation of the zeta potential was studied for obtaining a correspondingly stable SOM–POF composite (see ESI† for details).
The Raman spectroscopic signature of the SOM–JUC-Z4 composite was further checked and compared with the dispersion of soft oxometalates. In the Raman spectrum of the composite, there was a shift of the Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O peak towards higher wavenumber (∼26 cm−1) [Fig. 2(a)]. This implies the successful formation of the SOM–JUC-Z4 composite. This shift was because of the polarizability of the MoO bond in the composite being considerably less because it was attached to the electron deficient JUC-Z4, as compared to SOM alone.
O peak towards higher wavenumber (∼26 cm−1) [Fig. 2(a)]. This implies the successful formation of the SOM–JUC-Z4 composite. This shift was because of the polarizability of the MoO bond in the composite being considerably less because it was attached to the electron deficient JUC-Z4, as compared to SOM alone.
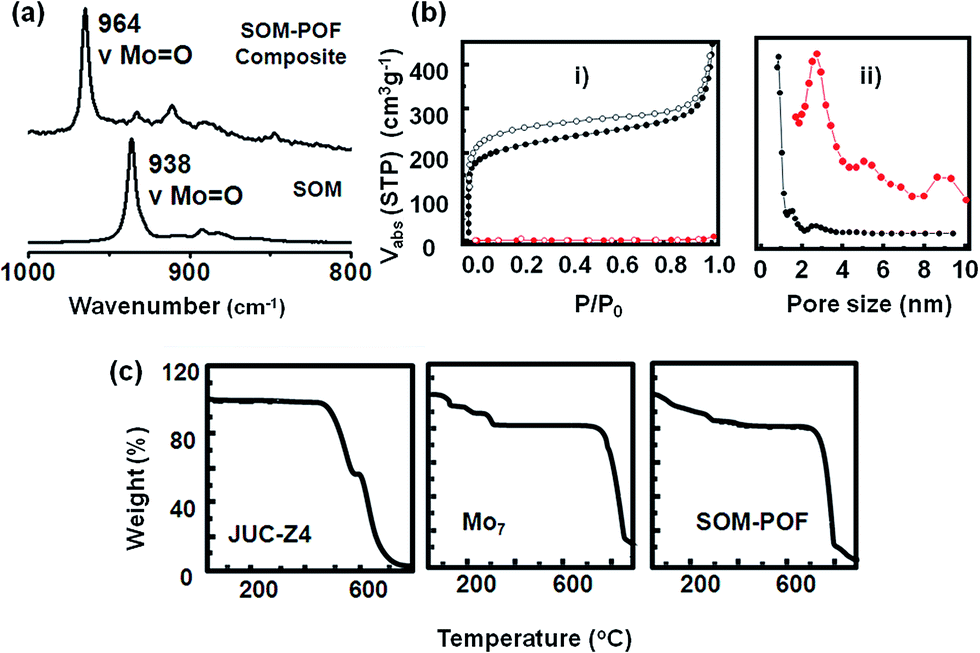 | ||
| Fig. 2 (a) Raman spectra of the composite SOM–POF and ammonium heptamolybdate. (b) (i) N2 sorption isotherms of activated JUC-Z4 (black) and SOM–POF (red) (solid symbols, adsorption; open symbols, desorption). (ii) Pore size distribution for JUC-Z4 (black) and SOM–POF (red) derived from N2 adsorption calculated by DFT method. (c) TGA plot of JUC-Z4, Mo7 (ammonium heptamolybdate tetrahydrate) and SOM–POF under atmospheric conditions. | ||
The TGA measurements showed the decomposition patterns of SOM–POF material as compared to that of the starting materials, viz., ammonium heptamolybdate and JUC-Z4 [Fig. 2(c)]. It was observed that the thermal stability of the SOM–POF hybrid was enhanced to 750 °C from that of 475 °C of individual JUC-Z4. The same trend was observed for SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5, where the decomposition temperature of each of the composites was increased by 200–300 °C than their POF counterparts (see ESI† for details). The capacity of SOM–POF composites for gas adsorption were further tested from the BET and Langmuir surface area values, and the pore size distribution profile were checked and the values were compared with those of the precursor POFs. To perform sorption measurements, it was made sure that there are no guest molecules such as carbon dioxide, water, and oxygen in the SOM–POF material. This type of a sample is called the activated sample. To ensure the purity of sample it was degassed at 100 °C and then the N2 sorption measurements were performed. From the sorption curves [Fig. 2(b)] the BET and Langmuir surface area of the SOM–JUC-Z4 composite were calculated to be 7.49 m2 g−1 and 10.10 m2 g−1, respectively. Moreover, the calculated BET and Langmuir surface area of JUC-Z4 was found to be 793 m2 g−1 and 979 m2 g−1, respectively. This implied that in the case of the composite, the surface area reduced almost 100 times, which was attributed to the incorporation of heptamolybdate clusters in the POF pores. The pore size distribution derived from N2 adsorption calculated by DFT method [Fig. 2(b)] showed the average pore size of the composite to be around 2.73 nm, which was inherited from its POF component-JUC-Z4. The 0.93 nm pore of JUC-Z4, however, was not observed in the composite. This indicates that the SOMs blocked the micropores of the POF, whereas the mesopores remained vacant. The existence of these mesopores of 2.73 nm provided the fine hierarchical pore structure, which was suitable for catalysis. N2 adsorption measurements for SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5 composites were also performed and their BET surface area were determined to be around 9.610 m2 g−1, 4.928 m2 g−1 and 14.693 m2 g−1, respectively. In all these cases it was observed that the surface area was reduced almost 100 fold, as in case of SOM–JUC-Z4, implying the impregnation of oxometalate units in the POF core pores (see ESI† for details).
On heterogeneous catalysis by SOM–POF composites
We now reiterate the question: can the simultaneous abundance of phosphorous and molybdenum in a supramolecular structure lead to the emergence of catalytic behaviour comparable to that of the molecular [PMo12] Keggin? By considering this issue, we undertook a model reaction namely the oxidation of benzaldehyde to benzoic acid with hydrogen peroxide using the SOM–JUCZ4 dispersion as a catalyst [Fig. 3].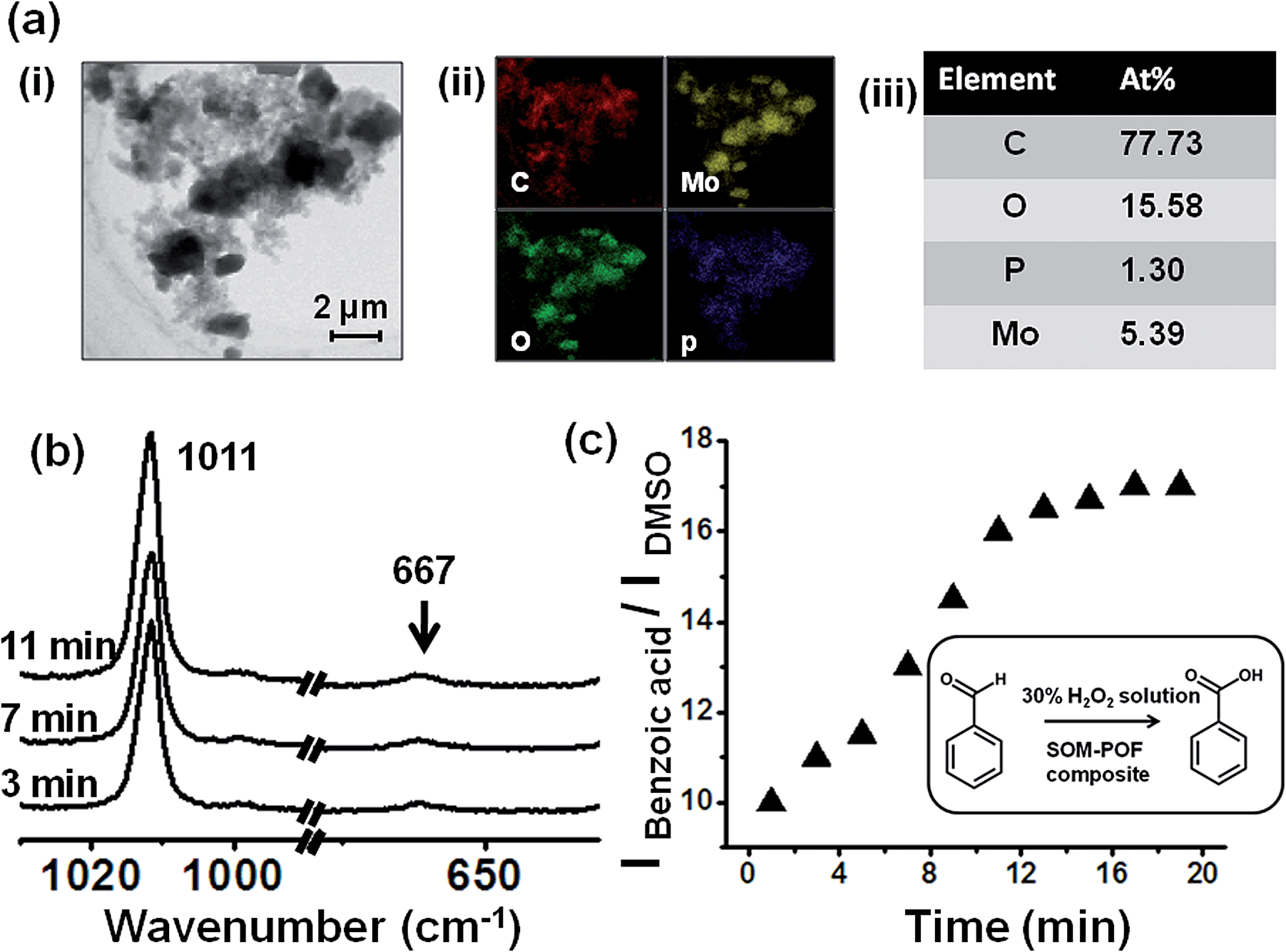 | ||
| Fig. 3 (a) (i) Close up on the SOM–POF composite particle in dispersion. (ii) Elemental mapping via EDX-TEM of the SOM–POF dispersion showing various elements present. (iii) Table depicting atomic abundance of each element present in the dispersion. (b) Time resolved Raman spectroscopic study to show the catalysis of benzaldehyde oxidation using the composite SOM–POF dispersion as the catalyst and DMSO as an external standard. (c) IBenzoic acid/IDMSOversus time plot indicating increase in the intensity of the ring breathing mode of benzoic acid (1011 cm−1 peak); thus, confirming the catalytic oxidation of benzaldehyde to benzoic acid. | ||
It was observed that the SOM–JUC-Z4 dispersion was effective as a catalyst for the oxidation of benzaldehyde in the presence of 30% aqueous hydrogen peroxide using dimethyl sulfoxide (DMSO) as an external standard. We monitored the reaction by time resolved Raman spectroscopy [Fig. 3(b)].
The rate of formation of the product benzoic acid increases with time in a sigmoid fashion [Fig. 3(c)] with respect to the added external standard DMSO, and it shows an initial lag phase of few minutes. Such a presence of lag phase and the sigmoidal nature of product formation indicate the operation of heterogeneous catalysis similar to the one shown very nicely in the literature.50–61 Indeed, in the SOM–JUC-Z4 dispersion we observed composite particles, which are a mixture of irregular shaped particles and nanotubes. They possess C, P, Mo and O on a nanoscopic scale in a homogeneous manner, as observed from EDX-TEM of the SOM–POF dispersion, which catalyses the reaction [Fig. 3(a)].
We speculate that these are the active particles that are responsible for the heterogeneous catalysis that we observe in the reaction. It is also worth noting the polydispersity in the system as well. In short, we speculate that in a heterogeneous catalytic mode, these particles of SOM–JUC-Z4 actively catalyze the system. Apart from SOM–JUC-Z4 composite, other SOM–POF composites such as SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5 were used to catalyse the oxidation of benzaldehyde in the presence of hydrogen peroxide as oxidant in dispersion phase. These materials were compared with molecular [PMo12] for their catalytic activity.
Catalytic activity of SOM–JUC-Z4 composite, SOM, JUC-Z4 and benchmark molecular [PMo12]
We now compare catalytic activity of SOM, JUC-Z4, SOM–JUC-Z4 composite and [PMo12], which are our benchmark catalysts. Therefore, the oxidation of benzaldehyde was studied. The catalytic studies were done by time dependent 1H NMR studies in dispersion. The reaction mixture comprising of benzaldehyde along with hydrogen peroxide, DMSO (used as an external standard) and SOM–JUC-Z4 composite catalyst were monitored by1H NMR at regular time intervals.The POF material i.e. JUC-Z4 alone did not show any activity, whereas the SOM did show some catalytic activity but it was considerably low as compared to the catalytic activity of the SOM–JUC-Z4 composite [Fig. 5]. This means that the SOM-component in the composite was facilitating the catalysis; however, its efficiency was amplified in the presence of a porous material with fine accessible mesopores of 2.7 nm, which provide the site for catalysis. In fact, the supramolecular composite of SOM–JUC-Z4 shows a sort of synergistic effect arising out of SOM and POF, and its catalytic activity even surpasses the activity of our benchmark [PMo12] polyoxometalate.
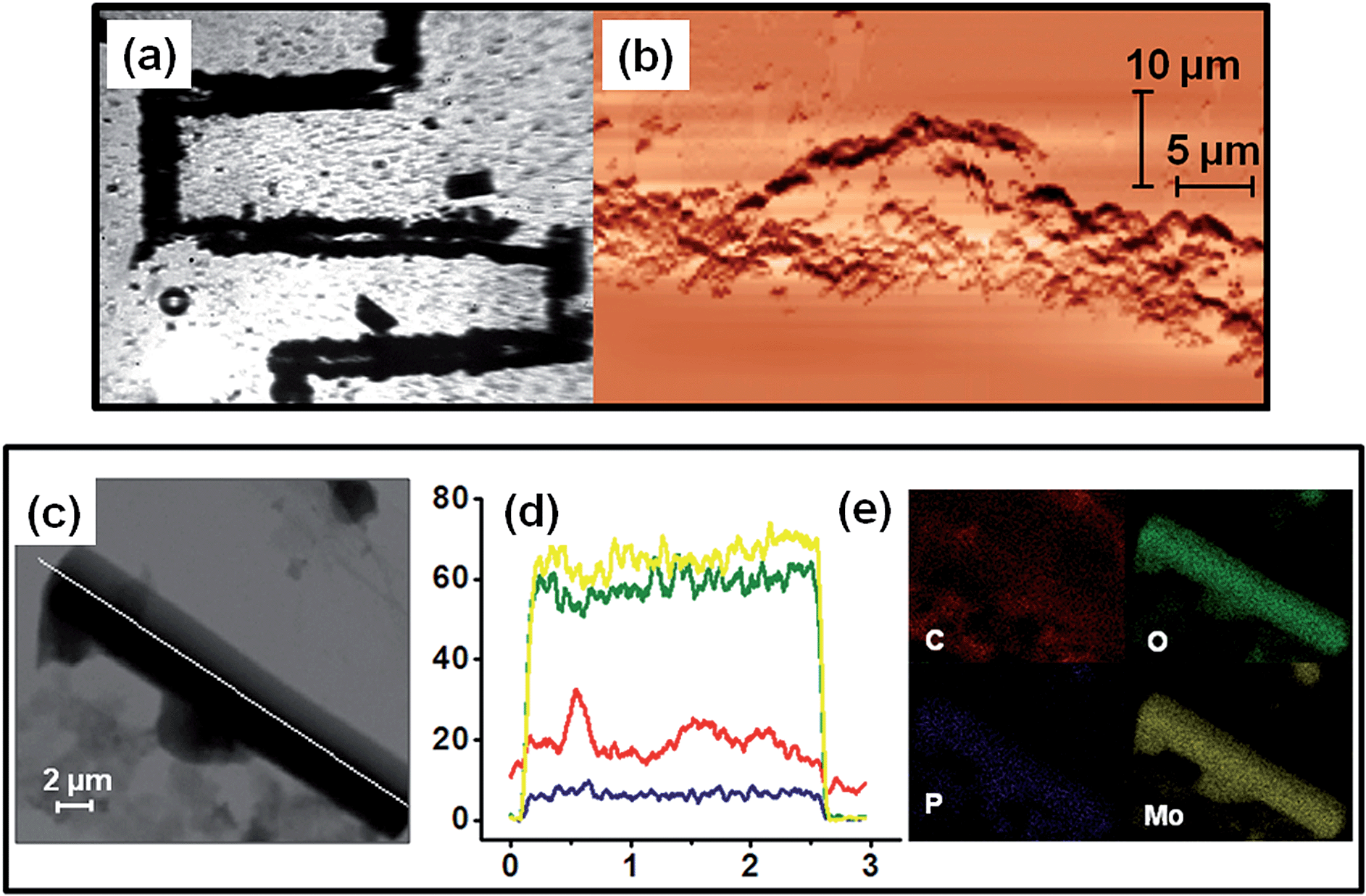 | ||
| Fig. 4 (a) Close up on the catalytic reactor chip: trail formation by the composite SOM–POF under the laser of the optical tweezers of 1064 nm wavelength. (b) AFM image showing the topography of the trail. (c) Elemental mapping via line scan of a rod-like structure of SOM–POF composite. (d) Count versus distance plot indicating the abundance of each element, namely carbon (red), oxygen (green), phosphorous (blue) and molybdenum (yellow), in the composite catalyst. (e) Elemental mapping of each of the elements shown in (d). | ||
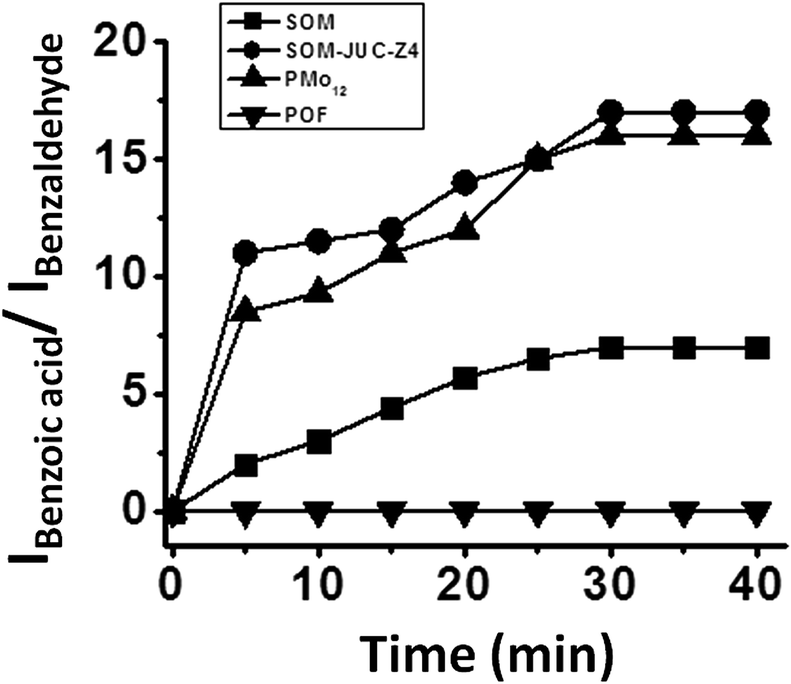 | ||
| Fig. 5 Plot of the ratio of the intensity of the acidic proton of benzoic acid and the aldehydic proton of benzaldehyde versus time using SOM, JUC-Z4, SOM–JUC-Z4 composite and PMo12 obtained by time dependent 1H NMR studies. | ||
Comparative catalytic activity of SOM–POF composites in dispersions and with benchmark [PMo12]
To understand the effect of catalyst loading on the yield of benzoic acid, NMR studies were done on dispersions containing the oxidation product i.e. benzoic acid with different loading of different composite catalysts. From the plot between the ratio of the intensity of the acidic proton of benzoic acid peak and the intensity of aldehydic proton of benzaldehyde versus the amount of catalyst loading, it was observed that SOM–JUC-Z4 showed maximum catalytic activity, which was even greater than molecular [PMo12] (benchmark catalyst) followed by SOM–JUC-Z5, and almost negligible activity was shown by SOM–PAF1 and SOM–JUC-Z2 [Fig. 6]. Clearly, the higher activity of SOM–JUC-Z4 implies the following: (1) the supramolecular composite SOM–JUC-Z4 is an effective catalyst. (2) Because it has mesopores, which are not available in [PMo12], SOM–JUC-Z4 is more efficient than [PMo12], the benchmark catalyst. | ||
| Fig. 6 Plot between the ratio of the intensity of the acidic proton of benzoic acid peak and the intensity of the aldehydic proton of benzaldehyde peak versus catalyst loading from 1H NMR studies. The reactions are in dispersions. | ||
Comparative kinetic study of aliphatic and aromatic aldehydes oxidation using different SOM–POF composites in dispersion
We selected the benzaldehyde oxidation with hydrogen peroxide as an oxidant and DMSO as an external standard as our model reaction system. This reaction system was used for oxidation using different SOM–POF composites, viz., SOM–PAF1, SOM–JUC-Z2, SOM–JUCZ-4, SOM–JUC-Z5, and molecular [PMo12], where [PMo12] was the benchmark catalyst because it is a standard catalyst for oxidation reactions. The kinetics of different reactions was monitored by time dependent 1H NMR spectroscopy [Fig. 7]. This comparative study showed that SOM–JUC-Z4 has maximum catalytic activity followed by considerable catalytic activity by molecular [PMo12] and SOM–JUC-Z5, and almost no activity by SOM–JUC-Z2 and SOM–PAF1. This high activity by SOM–JUC-Z4 implies that indeed the supramolecular combination of phosphonium and molybdate is similarly effective in catalysis as elemental P and Mo in [PMo12]. However, it is worth noting that SOM–JUC-Z4 composite showed greater catalytic activity than the standard catalyst molecular [PMo12], which is a known catalyst as well as a catalyst precursor for oxidation reactions [Fig. 7]. Such high activity of SOM–JUC-Z4 compared to [PMo12] can be attributed to the higher accessible mesopores of the composite compared to that of [PMo12], as mentioned before.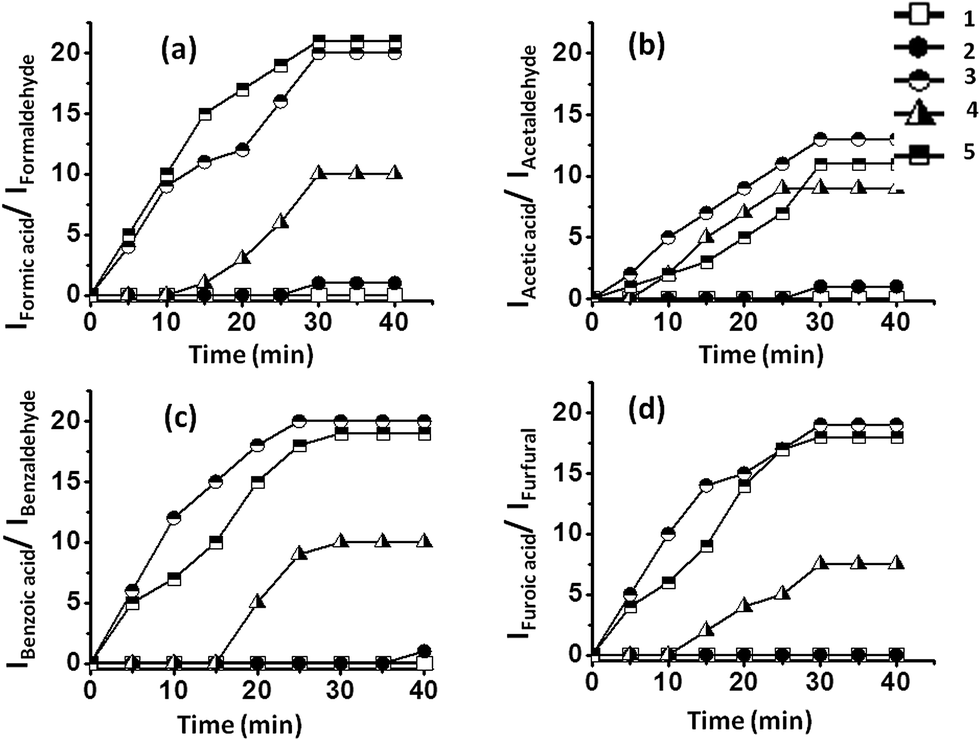 | ||
| Fig. 7 Plot between the ratio of intensity of the acidic proton peak of the product (i.e. carboxylic acid) and the intensity of the aldehydic proton peak of the reactant (i.e. aldehyde) and time. The results are obtained by time dependent 1H NMR studies of the oxidation of: (a) formaldehyde (b) acetaldehyde (c) benzaldehyde (d) furfural by SOM–POF composites. SOM–POF composites shown in the figure are SOM–PAF1 (1), SOM–JUC-Z2 (2), SOM–JUC-Z4 (3), SOM–JUC-Z5 (4) composites and molecular PMo12 (5). The reactions are in dispersions. | ||
Optically patterned SOM–POF composite on a chip and its electron microscopic characterization
Now, having shown catalysis by SOM–POF dispersion, we wanted to take the next step. We ask: can we induce a phase transition and pattern the SOM–POF dispersion and see the effect it has on catalysis in a designed catalytic chip? Can we achieve a site-specific heterogeneous catalysis on such a designed catalytic chip? Now we address these questions.Recently, we have shown that it is possible to write patterns using thermo-optical tweezers starting from SOM dispersion by inducing soft to crystalline phase transition.12 A similar patterning by inducing phase transition in the freshly synthesized SOM–POF dispersion should also be possible. Thus, we placed an aqueous SOM–POF dispersion in our sample holder that was described earlier. We placed this sample holder on a movable scanning stage and then focused 1064 nm laser light on the top surface of the sample holder. The SOM–JUC-Z4 composite has an absorptivity of around 105 mol−1 cm−1 at 1064 nm. Therefore, the laser light was absorbed by the composite such that heating occurs at the focused spot in a localized manner. This, in turn, led to the formation of a water vapour bubble from the water present in the medium. Because no such heating had occurred in the vicinity of the bubble, a temperature gradient was generated, which caused a convection current. These currents circulated the SOM–POF material and eventually got deposited at the base of the bubble. Because the laser beam was translated such that it focuses at a nearby new spot, there can be two possibilities: the first is that a new bubble is generated and the second possibility is that the previously formed bubble migrates to the new spot. Because the latter was thermodynamically more favourable, the initial bubble was also translated to the new location of the laser focus and the chain of events that led to the deposition of SOM–POF composite continued. Thus, we were able to design a catalytic reactor chip comprising a patterned trail [Fig. 4(a)] having a width of few micrometres – a fact that was confirmed by the AFM studies [Fig. 4(b)]. The TEM measurement of the patterned material was also carried out by scraping off the active catalytic material from the slide.
We now check the elemental composition of the patterned trail and the nature of the active catalytic material. Therefore, we re-dispersed a part of the catalytic trail in water and drop-casted it on a TEM grid. On subjecting it to elemental mapping we indeed observed a molecular level composite of phosphorous and molybdenum to exist in the structures found in the dispersions that were obtained from the patterned material of SOM–POF. Both elemental mapping and line scan analysis showed the presence of phosphorous and molybdenum at atomic level in the structures of the patterned material; thus, proving that we indeed have a composite on a chip, whose composition on nanoscopic length scale shows the presence of both phosphorous and molybdenum [Fig. 4(c)–(e)].
Catalysis by optically patterned SOM–JUC-Z4 catalyst chip: on heterogeneous catalysis
Now we test the catalysis of benzaldehyde oxidation by optically patterned SOM–JUC-Z4 composite on the chip. We used the patterned material as a catalyst for benzaldehyde oxidation. We observed by time resolved Raman spectroscopy that the peak intensities of the product, benzoic acid, with respect to external standard DMSO increased with time [Fig. 8(a)]. However, in addition to general bulk catalysis we checked the possibility of site-specific catalysis on the patterned material surface using dimethyl sulfoxide (DMSO) as an external standard. Here, we observed that indeed the reaction was catalysed much more efficiently on the patterned material surface in a site specific fashion. There was also a sigmoidal increase in the formation of the product with respect to external standard DMSO as shown in the (IBenzoic acid/IDMSO) versus time plot on the trail [Fig. 8(b)]. However, this reaction on the trail was more efficient than that of the dispersion, although the nature of catalysis was heterogeneous like that by the dispersion [Fig. 3(c) and 8(b)]. It was also worth noting that the catalysis by the reactor chip of the patterned surface clearly showed a lag phase of few minutes with regards to the catalysis [Fig. 8(b)]. This lag phase coupled with the sigmoidal pattern of the kinetics of product formation (benzoic acid) [Fig. 8(b)] clearly points to the operation of a heterogeneous catalysis on the trail surface in this system.50–61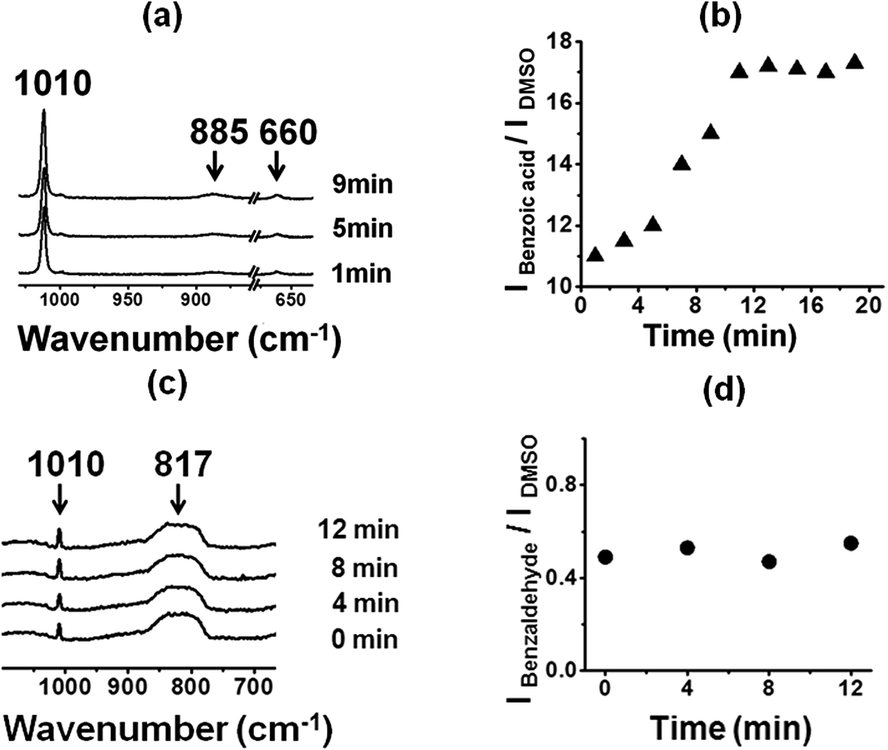 | ||
| Fig. 8 (a) Time dependent Raman spectroscopy showing catalysis at trail site. (b) Intensity versus time plot clearly depicting the increase in the intensity of the peak corresponding to the oxidation product, benzoic acid. (c) Time resolved Raman spectroscopy showing no catalysis away from SOM–POF trail site. (d) Intensity versus time plot clearly depicting the constant intensity of the peak corresponding to the reactant benzaldehyde. Note: DMSO acts as an external standard. | ||
We now compare and contrast the catalysis by the trail with the catalysis by the background using DMSO as an external standard. We observed the excess of unreacted benzaldehyde from the non-patterned background using Raman spectroscopy [Fig. 8(c) and (d)]. This was because after the nucleation in the system and formation of the trail, there was a lack of active catalyzing particles in the non-patterned background, which loses the ability to catalyze the reaction as obvious from the AFM image of the background, where no active catalytic particles were present [Fig. 4(b)].
These observations have the following aspects: (1) the trail was more efficient in catalysis and performs site specific catalysis. (2) The active particles of the catalysis were the SOM–POF hybrid particles, which are nanotubes and irregular particles in the dispersion and only nanotubes on the crystallized trail. It should also be noted that we observed crystallization after laser irradiation from SAED-TEM, as we have previously shown it elsewhere.12 (3) Catalysis by both dispersion and the trail were heterogeneous in nature. (4) We can further infer that the active species in this catalysis were the rod-shaped SOM–POF nanotubes and irregular particles, as seen and characterized in detail by TEM-EDX elemental mapping from the active catalyst trail of the reactor catalyst chip [Fig. 4(c)–(e)]. Because these particles were not present in the background of the catalytic chip, the background cannot catalyze the reaction [Fig. 7(d)]. Both the sigmoidal kinetics of the reaction and the presence of active particles showed that the catalysis in dispersion and on the chip were heterogeneous in nature.
Comparison of catalytic activity of various optically patterned SOM–POF composite chips
The catalytic activity of SOM–JUC-Z4 needs to be compared with other composites to perform a comparative study of the catalytic activity of SOM–POF materials. To accomplish this, the patterned trails of SOM–PAF1, SOM–JUC-Z2 and SOM–JUC-Z5 were designed, and these trails were used as sites for benzaldehyde oxidation in the presence of hydrogen peroxide as oxidant. Their catalytic activity was compared in terms of the ratio of the intensity of benzoic acid peak and the intensity of DMSO peak (external standard) obtained after the reaction by Raman spectroscopy. We observed that SOM–JUC-Z4 patterned chip was the most effective in catalysis, even more than [PMo12] benchmark [Fig. 9]. This clearly indicates that the supramolecular composite of patterned phosphonium with molybdate in SOM–JUC-Z4 is effective in catalysis. Because it has higher mesoscopic accessibility as compared to [PMo12], the SOM–JUC-Z4 patterned chip is more efficient in catalysis.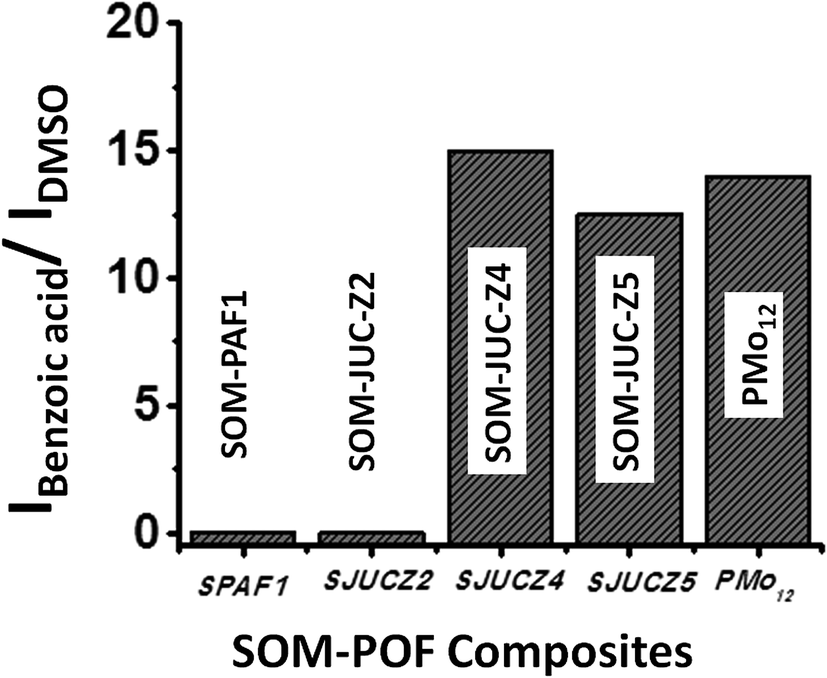 | ||
| Fig. 9 Catalysis by SOM–POF composite chips: graph showing the ratio of the intensity of benzoic acid peak and the intensity of DMSO peak from Raman spectroscopy obtained for SOM–PAF1 (SPAF1), SOM–JUC-Z2 (SJUCZ2), SOM–JUC-Z4 (SJUCZ4), SOM–JUC-Z5 (SJUCZ5) composites and molecular [PMo12]. | ||
We now place the reaction mixture comprising benzaldehyde, hydrogen peroxide as an oxidant and DMSO as the external standard on the patterned SOM–POF chip and monitor the kinetics of the reaction by Raman spectroscopy [Fig. 10]. The kinetics of all the patterned chips showed sigmoidal behaviour. Catalysis was observed only on the trail and not elsewhere; thus, it was site-specific. Moreover, the patterned trails of SOM–JUC-Z4 showed the maximum efficiency as a catalyst for benzaldehyde oxidation followed by the catalytic activity of the trails of molecular [PMo12] and SOM–JUC-Z5. There was almost no catalytic activity by the trails of SOM–JUC-Z2 and SOM–PAF1 [Fig. 10].
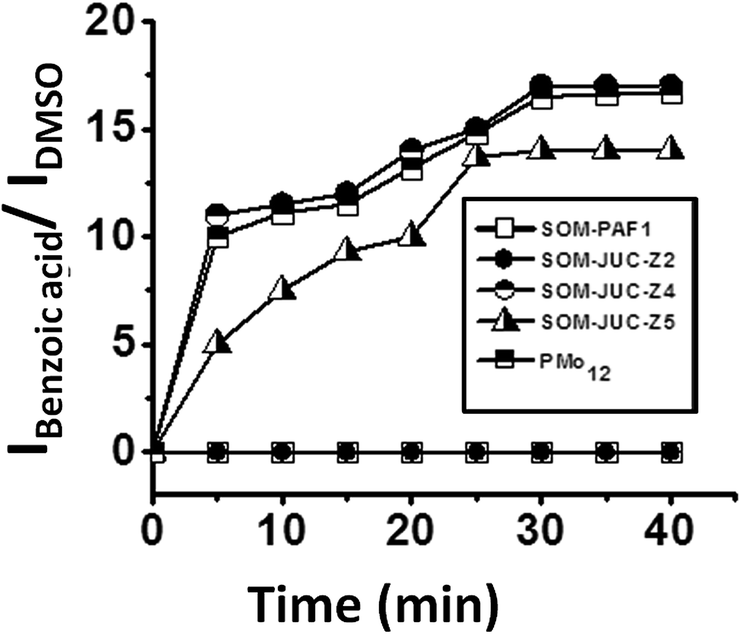 | ||
| Fig. 10 Kinetics of SOM–POF composite catalyst chip: plot of the ratio of the intensity of benzoic acid peak and the ratio of the intensity of external standard DMSO peak versus time obtained by time dependent Raman studies using different SOM–POF composites and hydrogen peroxide as oxidant. | ||
Stability of the optically patterned catalyst chip
After investigating the catalytic potential of the SOM–JUC-Z4 patterned chip, the subsequent step was to assess the stability of this catalyst chip. For this purpose the trail was checked for its intactness by Raman spectroscopy post-reaction. It was found that the trail formed by SOM–JUC-Z4 gave the same Raman characteristics as it showed before the catalytic reaction; thus, indicating that the chip was stable and reusable after the reaction [Fig. 11]. The chip was also stable for weeks after its fabrication.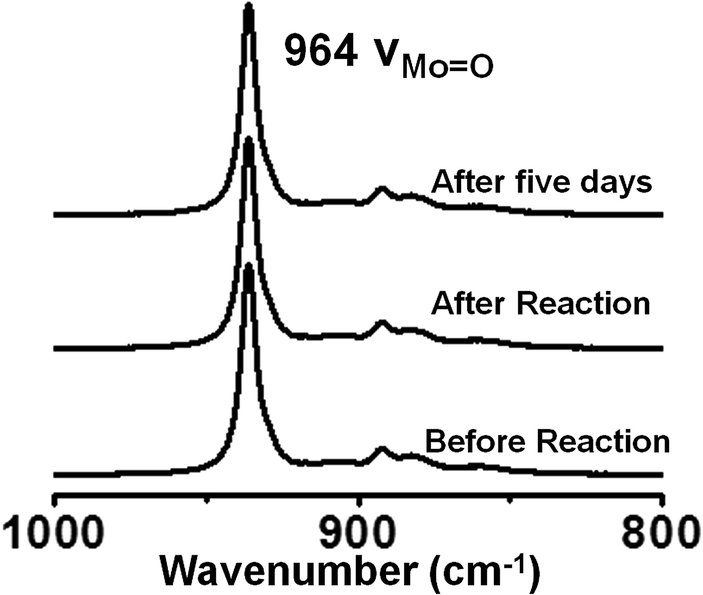 | ||
| Fig. 11 Raman spectra of SOM–JUC-Z4 composite chip recorded before the reaction, immediately after the reaction and 5 days after the reaction. | ||
Comparative kinetic study of aromatic and aliphatic aldehydes oxidation using the dispersion and patterned chips of SOM–POF composites as catalysts
The oxidation of different aldehydes, viz., formaldehyde, acetaldehyde, furfural and benzaldehyde, using all the five catalysts namely SOM–PAF1, SOM–JUC-Z2, SOM–JUCZ-4, SOM–JUC-Z5 and molecular [PMo12] were studied in dispersion phase in the presence of hydrogen peroxide as oxidant. With the assistance of time dependent 1H NMR studies, it was found that SOM–JUC-Z4 dispersion showed the maximum catalytic activity for the oxidation of all the four aldehydes [Fig. 7]. Among all the aldehydes, the SOM–POF dispersion showed higher selectivity towards the oxidation of benzaldehyde followed by formaldehyde, furfural and acetaldehyde, respectively [Fig. 12]. For all the composites, we obtained sigmoidal curves in conversion kinetics, which suggest heterogeneous catalysis. SOM–JUC-Z4 patterned chip was then used to catalyse the oxidation of aliphatic aldehydes such as formaldehyde, acetaldehyde and aromatic aldehydes, namely furfural and benzaldehyde, and it was monitored by time dependent Raman spectroscopy [Fig. 13]. The results obtained from the patterned catalyst chip were also considerably similar to that of the dispersion phase catalysis. The rate of the reaction of formaldehyde oxidation was faster than that of furfural, followed by acetaldehyde oxidation but the fastest oxidation was that of the benzaldehyde reaction. This implies that the chip formed by SOM–JUC-Z4 composite catalyzes benzaldehyde oxidation at the fastest rate [Fig. 12].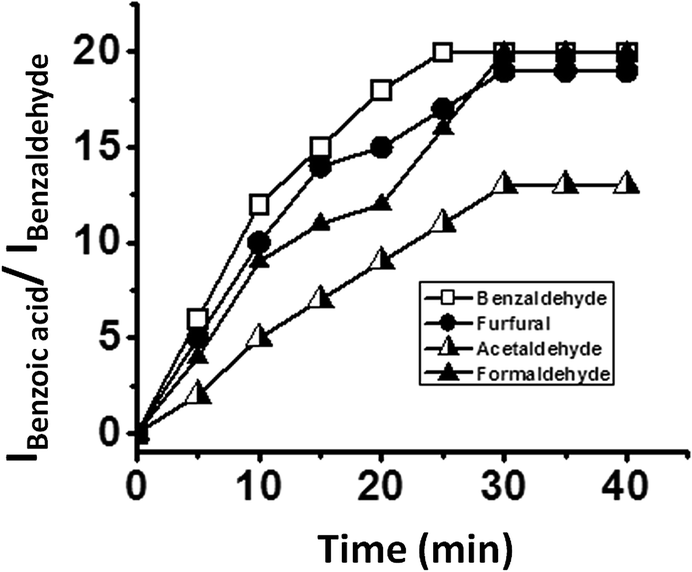 | ||
| Fig. 12 The ratio of the intensity of benzoic acid peak and the intensity of benzaldehyde peak versus time plot clearly depicting the catalytic activity of SOM–JUC-Z4 composites in dispersion for the oxidation of different aldehydes obtained by time dependent 1H NMR studies. The reactions are in dispersion. | ||
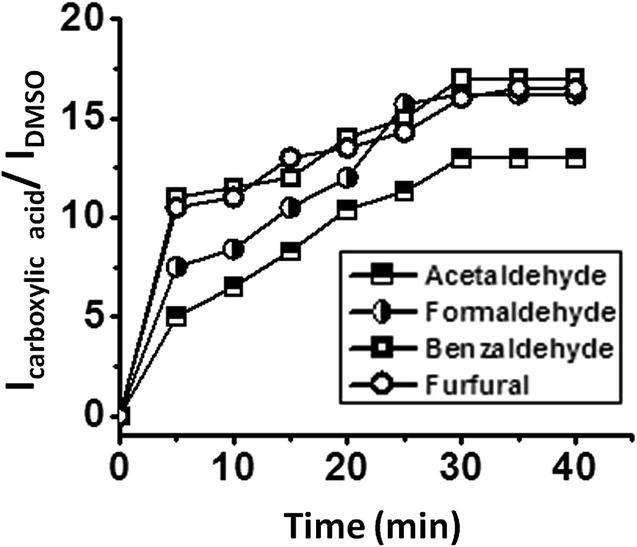 | ||
| Fig. 13 Catalysis of the oxidation of different aldehydes by the SOM–JUC-Z4 patterned catalyst chip: the ratio of the intensity of the peak of carboxylic acid and the intensity of DMSO peak versus time plot clearly depicting the catalytic activity of different SOM–JUC-Z4 composite catalyst chip in the oxidation of different aldehydes monitored by time dependent Raman studies. | ||
Comparative study of benzaldehyde oxidation using different oxidants
The effect of different oxidising agents on the rate of reaction was studied by using different oxidants such as hydrogen peroxide, dilute sulphuric acid, dilute nitric acid and silver(I) oxide for the oxidation of benzaldehyde in the presence of all five catalysts i.e. SOM–PAF1, SOM–JUC-Z2, SOM–JUCZ-4, SOM–JUC-Z5 and molecular [PMo12]. The kinetics of the reactions were monitored by 1H NMR and the ratio of the intensity of the acidic proton of benzoic acid peak and the intensity of the aldehydic proton of benzaldehyde peak was plotted against time. These plots were sigmoidal suggesting the heterogeneous nature of catalysis, and it showed that the highest rate of reaction for benzaldehyde oxidation was with dilute sulphuric acid. Hydrogen peroxide acted as the subsequent efficient oxidant, followed by dilute nitric acid and, finally, silver(I) oxide (see ESI† for more details). With the maximum product formation obtained in the case of SOM–JUC-Z4 catalyst for benzaldehyde oxidation in the dispersion phase, the patterned chip of SOM–JUC-Z4 was chosen to obtain a comparative study of aldehyde oxidation with the same set of four different oxidising agents, viz., silver(I) oxide, dilute nitric acid, dilute sulphuric acid and hydrogen peroxide. The reactions were monitored by time dependent Raman spectroscopic experiments. It was observed that dilute sulphuric acid acted as the strongest oxidising agent. Hydrogen peroxide and dilute nitric acid showed comparable oxidising ability, whereas silver(I) oxide oxidised benzaldehyde to a lesser extent [Fig. 14].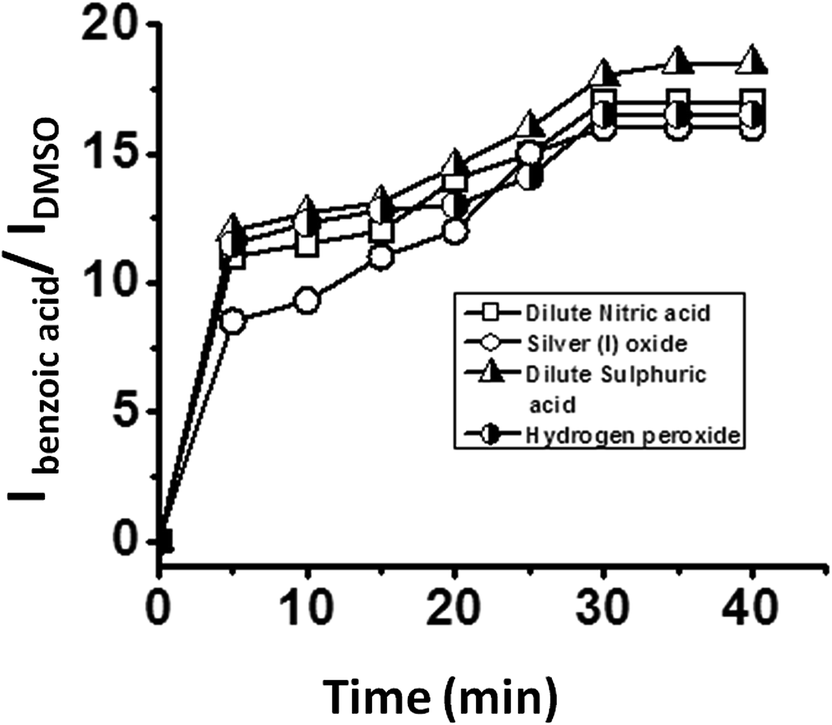 | ||
| Fig. 14 Kinetics of oxidation by various oxidants on a catalyst chip: plot of the ratio of the intensity of the formed benzoic acid peak and the ratio of the intensity of external standard DMSO peak versus time obtained by time dependent Raman studies using SOM–JUC-Z4 composite as catalyst chip and different oxidants. | ||
Conclusions
To summarize, we have shown that starting from a well-defined molecular precursor of a heptamolybdate polyoxometalate, we first synthesized in a controlled way a soft oxomolybdate (SOM) nanotube. This, in turn, formed another stable dispersion with a well-defined porous aromatic framework, namely JUC-Z4.The composite material was characterized by detailed spectroscopic, gas sorption and microscopy experiments and indeed showed that in the structure constituting the solid phase of the dispersion, there exist a simultaneous abundance of phosphorous and molybdenum at the nano scale.
Therefore, we tested our hypothesis: can this composite catalytically act in a comparable way to that of [PMo12] Keggin? Thus, we used this composite for catalyzing the oxidation of benzaldehyde to benzoic acid using hydrogen peroxide. We observed that not only the dispersion but also the patterned material comprising of SOM–POF composite efficiently catalyzes the reaction. Moreover, the catalysis on the surface of SOM–POF patterned material was more pronounced than that of the dispersion. We further showed the operation of heterogeneous mode in the catalysis, both in the dispersion and on the trail of the chip, by the observation of the sigmoidal kinetics of the product formation and by identifying the active catalytic particles. We also showed that when these particles are not present, as in the background of the catalytic chip, there was no catalysis. A comparative study of different composite materials as dispersions and as chips for the oxidation of different aldehydes using different oxidants were done, and based on the kinetic studies we can conclude that SOM–JUC-Z4 composite catalyzes the oxidation of benzaldehyde in the presence of hydrogen peroxide to the maximum extent, even greater than molecular [PMo12]. Such a high activity of SOM–JUC-Z4 indicates two points: (1) supramolecular composite comprising of phosphonium and molybdate is effective as catalyst. (2). The highly accessible mesopores of such composite compared to that of molecular [PMo12] is the reason for its high catalytic activity. This work, in turn, implies that it is possible to design a patterned composite material to deliberately catalyze targeted reactions starting from well-defined molecular level precursors in a completely controlled fashion.
Author contribution
PT synthesized and characterized the SOMs as well as the SOM–POF composites in SR's laboratory. With the involvement of SD, PT tested their catalytic activity in dispersion as well as on the chip, which was also performed in SR's laboratory. CP was instrumental in POF designs, SOM–POF microscopy and porosity measurements at TB's laboratory in SQ's facility. BR and SG were involved in designing the chips along with PT in AB's laboratory. SR initiated the project with TB and AB and coordinated the project, as well as wrote the paper with inputs from PT, AB and TB.Acknowledgements
Financial support of DST Fast track, BRNS DAE, India and IISER-Kolkata, India, NSFC grant of the P. R. China is gratefully acknowledged. This paper is dedicated to Prof. J. M. Lehn.References
- W. N. M. Piet van Leeuwen, Supramolecular Catalysis, Wiley-VCH, Weinheim, Germany 2008 Search PubMed.
- (a) S. Roy, Comments Inorg. Chem., 2011, 32, 113 CrossRef CAS; (b) S. Roy, CrystEngComm, 2014, 16, 4667–4676 RSC; (c) S. Roy (Ed.), Special Issue on Soft-Oxometalates – Soft-Oxometalates (SOMs): Towards New Oxometalate-Based Materials, J. Mol. Eng. Mater., 2014, 2(1) Search PubMed; (d) B. Roy, N. Ghosh, S. D. Gupta, S. Roy and A. Banerjee, New J. Phys., 2014, 16, 083037 CrossRef; (e) S. Das, P. Thomas and S. Roy, Eur. J. Inorg. Chem., 2014, 27, 4551–4557 CrossRef.
- A. Ashkin, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 4853–4860 CrossRef CAS.
- T. Leong, Z. Gu, T. Koh and D. H. Gracias, J. Am. Chem. Soc., 2006, 128, 11336–11337 CrossRef CAS PubMed.
- J. C. Love, A. R. Urbach, M. G. Prentiss and G. M. Whitesides, J. Am. Chem. Soc., 2003, 125, 12696–12697 CrossRef CAS PubMed.
- H. F. Hamann, S. I. Woods and S. Sun, Nano Lett., 2003, 3, 1643–1645 CrossRef CAS.
- K. G. Yager and C. J. Barrett, Macromolecules, 2006, 39, 9320–9326 CrossRef CAS.
- R. S. Singh, V. Nalla, W. Chen, A. T. S. Wee and W. Ji, ACS Nano, 2011, 5, 5969–5975 CrossRef CAS PubMed.
- A. Dzwilewski, T. Wågberg and L. Edman, J. Am. Chem. Soc., 2009, 131, 4006–4011 CrossRef CAS PubMed.
- J. D. Gerding, D. M. Willard and A. V. Orden, J. Am. Chem. Soc., 2005, 127, 1106–1107 CrossRef CAS PubMed.
- W. R. Childs and R. G. Nuzzo, J. Am. Chem. Soc., 2002, 124, 13583–13596 CrossRef CAS PubMed.
- B. Roy, M. Arya, P. Thomas, J. K. Jürgschat, V. Rao, A. Banerjee, C. M. Reddy and S. Roy, Langmuir, 2013, 29, 14733–14742 CrossRef CAS PubMed.
- L. D. Zarzar, B. S. Swartzentruber, J. C. Harper, D. R. Dunphy, C. J. Brinker, J. Aizenberg and B. Kaehr, J. Am. Chem. Soc., 2012, 134, 4007–4010 CrossRef CAS PubMed.
- N. M. Andoy, X. Zhou, E. Choudhary, H. Shen, G. Liu and P. Chen, J. Am. Chem. Soc., 2013, 135, 1845–1852 CrossRef CAS PubMed.
- M. T. Pope, Heteropoly and IsopolyOxometalates, Springer, Berlin, Germany, 1983 Search PubMed.
- A. Müller and S. Roy, in The Chemistry of Nanomaterials: Synthesis, Properties and Applications, ed. C. N. R. Rao, A. Müller and A. K. Cheetham, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2005, ch. 14 Search PubMed.
- (a) A. Müller and S. Roy, Russ. Chem. Rev., 2002, 71, 981–991 CrossRef PubMed; (b) A. A. Verhoeff, M. L. Kistler, A. Bhatt, J. Pigga, J. Groenewold, M. Klokkenburg, S. Veen, S. Roy, T. Liu and W. K. Kegel, Phys. Rev. Lett., 2007, 99, 066104 CrossRef.
- C. L. Hill, Chem. Rev., 1998, 98, 1–2 CrossRef CAS PubMed.
- M. T. Pope and A. Müller, Angew. Chem., Int. Ed. Engl., 1991, 30, 34 CrossRef.
- Inorganic Synthesis, ed. A. P. Ginsberg, Wiley, New York, USA, 1990, vol. 27 Search PubMed.
- C. L. Hill and D. A. Bouchard, J. Am. Chem. Soc., 1985, 107, 5148–5157 CrossRef CAS.
- Comprehensive Coordination Chemistry, ed. L. Cronin, J. A. McCleverty and T. J. Meyer, Elsevier, Amsterdam, Netherlands, 2004 Search PubMed.
- Polyoxometalate Chemistry for Nanocomposite Design, ed. T. Yamase and M. T. Pope, Kluwer Academic Publishers, New York, USA, 2002 Search PubMed.
- Introduction to Polyoxometalate Chemistry: From Topology via Self-Assembly to Applications, ed. M. T. Pope and A. Müller, Kluwer Academic Publishers, NewYork, USA, 2001 Search PubMed.
- Polyoxometalate Molecular Science NATO Science series, ed. J. J. Borrás-Alamenar, E. Coronado, A. Müller and M. T. Pope, Kluwer Academic Publishers, Dordrecht, Netherlands, 2003 Search PubMed.
- S. Roy, K. L. Planken, R. Kim, D. v. d. Mandele and W. K. Kegel, Inorg. Chem., 2007, 46, 8469–8471 CrossRef CAS PubMed.
- S. Roy, M. T. Rijneveld-Ockers, J. Groenewold, B. W. M. Kuipers, H. Meeldijk and W. K. Kegel, Langmuir, 2007, 23, 5292–5295 CrossRef CAS PubMed.
- S. Roy, H. J. D. Meeldijk, A. V. Petukhov, M. Versluijs and F. Soulimani, Dalton Trans., 2008, 2861–2865 RSC.
- S. Roy, M. C. D. Mourad and M. T. Rijneveld-Ockers, Langmuir, 2007, 23, 399–401 CrossRef CAS PubMed.
- S. Roy, L. C. A. M. Bossers, H. J. D. Meeldijk, B. W. M. Kuipers and W. K. Kegel, Langmuir, 2008, 24, 666–669 CrossRef CAS PubMed.
- (a) A. Proust, R. Thouvenot and P. Gouzerh, Chem. Commun., 2008, 1837–1852 RSC; (b) D. Chen, A. Sahasrabudhe, P. Wang, A. Dasgupta, R. Yuan and S. Roy, Dalton Trans., 2013, 10587–10596 RSC.
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- D. Farrusseng, Metal-Organic Frameworks: Applications from Catalysis to Gas Storage, Wiley-VCH, Weinheim, Germany, 2011 Search PubMed.
- T. Ben and S. Qiu, CrystEngComm, 2013, 15, 17–26 RSC.
- S. J. Garibay, CrystEngComm, 2013, 15, 1515–1519 RSC.
- W. Wang, H. Ren, F. Sun, K. Cai, H. Ma, J. Du, H. Zhao and G. Zhu, Dalton Trans., 2012, 3933–3936 RSC.
- K. Konstas, J. W. Taylor, A. W. Thornton, C. M. Doherty, W. X. Lim, T. J. Bastow, D. F. Kennedy, C. D. Wood, B. J. Cox, J. M. Hill, A. J. Hill and M. R. Hill, Angew. Chem., Int. Ed., 2012, 51, 6639–6642 CrossRef CAS PubMed.
- T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., Int. Ed., 2009, 48, 9457–9460 CrossRef CAS PubMed.
- R. Babarao, S. Dai and D. E. Jiang, Langmuir, 2011, 27, 3451–3460 CrossRef CAS PubMed.
- B. Lukose, J. Phys. Chem. C, 2012, 116, 22878–22884 CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- H. Ren, T. Ben, E. Wang, X. Jing, M. Xue, B. Liu, Y. Cui, S. Qiu and G. Zhu, Chem. Commun., 2010, 46, 291–293 RSC.
- T. Ben, C. Pei, D. Zhang, J. Xu, F. Deng, X. Jing and S. Qiu, Energy Environ. Sci., 2011, 4, 3991–3999 CAS.
- J. Lan, D. Cao, W. Wang, T. Ben and G. Zhu, J. Phys. Chem. Lett., 2010, 1, 978–981 CrossRef CAS.
- H. Ren, T. Ben, F. Sun, M. Guo, X. Jing, H. Ma, K. Cai, S. Qiu and G. Zhu, J. Mater. Chem., 2011, 21, 10348–10353 RSC.
- T. Ben, Y. Li, L. Zhu, D. Zhang, D. Cao, Z. Xiang, X. Yao and S. Qiu, Energy Environ. Sci., 2012, 5, 8370–8376 CAS.
- K. A. Kraus, A. E. Marcinkowsky, J. S. Johnson and A. J. Shor, Science, 1966, 151, 194–195 CrossRef CAS PubMed.
- (a) C. Pei, T. Ben, H. Guo, J. Xu, F. Deng, Z. Xiang, D. Cao and S. Qiu, Philos. Trans. R. Soc., A, 2013, 371, 20120312 CrossRef PubMed; (b) T. Ben, H. Ren, S. Ma, D. Cao, J. Lan, X. Jing, W. Wang, J. Xu, F. Deng, J. M. Simmons, S. Qiu and G. Zhu, Angew. Chem., 2009, 121, 9621–9624 CrossRef; (c) C. Pei, T. Ben, Y. Cui and S. Qiu, Adsorption, 2012, 18, 375–380 CrossRef CAS PubMed.
- L. C. W. Baker and J. S. Figgis, J. Am. Chem. Soc., 1970, 92, 3794–3797 CrossRef CAS.
- J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317–341 CrossRef CAS.
- J. P. Collman, L. S. Hegedus, J. R. Norton and R. G. Finke, Principles and Applications of Organotransition Metal Chemistry, University Science Books, Mill Valley, 1987, ch. 10 Search PubMed.
- G. M. Whitesides, M. Hackett, R. L. Brainard, J. P. P. M. Lavalleye, A. F. Sowinski, A. N. Izumi, S. S. Moore, D. W. Brown and E. M. Staudt, Organometallics, 1985, 4, 1819–1830 CrossRef CAS.
- Y. Lin and R. G. Finke, Inorg. Chem., 1994, 33, 4891–4910 CrossRef CAS.
- K. S. Weddle, J. D. Aiken III and R. G. Finke, J. Am. Chem. Soc., 1998, 120, 5653–5666 CrossRef CAS.
- J. A. Widegren, M. A. Bennett and R. G. Finke, J. Am. Chem. Soc., 2003, 125, 10301–10310 CrossRef CAS PubMed.
- M. A. Watzky and R. G. Finke, J. Am. Chem. Soc., 1997, 119, 10382–10400 CrossRef CAS.
- J. A. Widegren, J. D. Aiken III, S. Özkar and R. G. Finke, Chem. Mater., 2001, 13, 312–324 CrossRef CAS.
- Y. Lin and R. G. Finke, J. Am. Chem. Soc., 1994, 116, 8335–8353 CrossRef CAS.
- J. D. Aiken III and R. G. Finke, Chem. Mater., 1999, 11, 1035–1047 CrossRef.
- S. Özkar and R. G. Finke, J. Am. Chem. Soc., 2002, 124, 5796–5810 CrossRef PubMed.
- V. Artero and M. Fontecave, Chem. Soc. Rev., 2013, 42, 2338–2356 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta01304b |
| This journal is © The Royal Society of Chemistry 2015 |