 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA transformative route to nanoporous manganese oxides of controlled oxidation states with identical textural properties†
Jae Hwa
Lee‡
a,
Young Jin
Sa‡
a,
Tae Kyung
Kim
a,
Hoi Ri
Moon
*a and
Sang Hoon
Joo
*b
aDepartment of Chemistry, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 689-798, Republic of Korea. E-mail: hoirimoon@unist.ac.kr
bSchool of Energy and Chemical Engineering and KIER-UNIST Advanced Center for Energy, Ulsan National Institute of Science and Technology (UNIST), 50 UNIST-gil, Ulsan 689-798, Republic of Korea. E-mail: shjoo@unist.ac.kr; Fax: +82-52-217-2509; Tel: +82-52-217-2522
First published on 29th April 2014
Abstract
Nanoporous nanocrystalline metal oxides with tunable oxidation states are crucial for controlling their catalytic, electronic, and optical properties. However, previous approaches to modulate oxidation states in nanoporous metal oxides commonly lead to the breakdown of the nanoporous structure as well as involve concomitant changes in their morphology, pore size, surface area, and nanocrystalline size. Herein, we present a transformative route to nanoporous metal oxides with various oxidation states using manganese oxides as model systems. Thermal conversion of Mn-based metal–organic frameworks (Mn-MOFs) at controlled temperature and atmosphere yielded a series of nanoporous manganese oxides with continuously tuned oxidation states: MnO, Mn3O4, Mn5O8, and Mn2O3. This transformation enabled the preparation of low-oxidation phase MnO and metastable intermediate phase Mn5O8 with nanoporous architectures, which were previously rarely accessible. Significantly, nanoporous MnO, Mn3O4, and Mn5O8 had a very similar morphology, surface area, and crystalline size. We investigated the electrocatalytic activity of nanoporous manganese oxides for oxygen reduction reaction (ORR) to identify the role of oxidation states, and observed oxidation state-dependent activity and kinetics for the ORR.
1. Introduction
Nanoporous metal oxide materials with crystalline frameworks have been of prime importance in various fields, including adsorption, catalysis, and energy conversion and storage.1–5 In these materials, the control of oxidation states is crucial for tailoring their catalytic, electronic, and optical properties. Over the last two decades, diverse soft and hard templating strategies have been developed to prepare nanoporous metal oxides with a rich variety of compositions and structures.6–14 However, these methods usually involve solution processes and subsequent high-temperature annealing steps, which have limited the range of stable metal oxide phases predominantly to those with high oxidation numbers. In addition, post-synthetic thermal treatments aimed at changing the oxidation states of nanoporous metal oxides often result in the growth of nanocrystalline domain size, which consequently leads to the breakdown of the nanoporous structure. Hence, the preparation of nanoporous metal oxides with broadly tunable oxidation states, particularly those with low oxidation or metastable intermediate phases, has been challenging.Recently, conversion reactions that transform pre-synthesized nanoscale or microscale inorganic solid precursors into new materials have emerged as new synthetic toolboxes in materials chemistry.15–19 Conversion reactions have been explored extensively with various types of precursors, and have enabled the synthesis of new materials with a high degree of compositional and structural complexity. The versatility of conversion reactions can be exemplified in the preparation of thermodynamically unfavorable hollow nanoparticles from solid analogues by exploiting the nanoscale Kirkendall effect20 or galvanic replacement reaction.21 In the context of conversion reactions, metal–organic frameworks (MOFs), in which inorganic and organic building blocks are alternatively arranged in a periodic manner, can serve as excellent precursors, and accordingly, conversion reactions utilizing MOFs are currently being actively pursued.22–31 In this line of effort, we recently reported the preparation of nanoporous non-siliceous metal oxides with nanocrystalline frameworks via thermal conversion of MOFs for the first time.30
Herein, we demonstrate that conversion reactions using MOFs as precursors can be successfully exploited to prepare nanoporous nanocrystalline metal oxides with tunable oxidation states. Manganese oxides were chosen as a model system because they can exist in various oxidation states32 and are the centers of great interest in energy conversion and storage as well as catalysis.33–36 We show that sequential conversion reactions are capable of transforming a Mn-based MOF (Mn-MOF) into nanoporous nanocrystalline manganese oxides with continuously tuned oxidation states, MnO, Mn3O4, Mn5O8, and Mn2O3, with conservation of the original morphology of the Mn-MOF crystals (Fig. 1). Importantly, these conversion reactions enabled the preparation of low-oxidation-state MnO and metastable intermediate Mn5O8 phases with nanoporous nanocrystalline architecture, which was previously difficult to access. Nanoporous MnO, Mn3O4, and Mn5O8 have almost identical textural properties, including their morphology, surface areas, pore volumes, and the size of nanocrystals composing nanoporous frameworks. The textural similarity among the series of manganese oxides allowed them to be used as a system for studying the oxidation-state effect in catalysis; previous studies related to such an effect were mostly performed with a series of catalysts possessing various other variables that can affect catalytic performances.37–40 We investigated the electrocatalytic activity of these nanoporous manganese oxides for the oxygen reduction reaction (ORR), and observed oxidation-state-dependent catalytic activity and kinetics in the ORR.
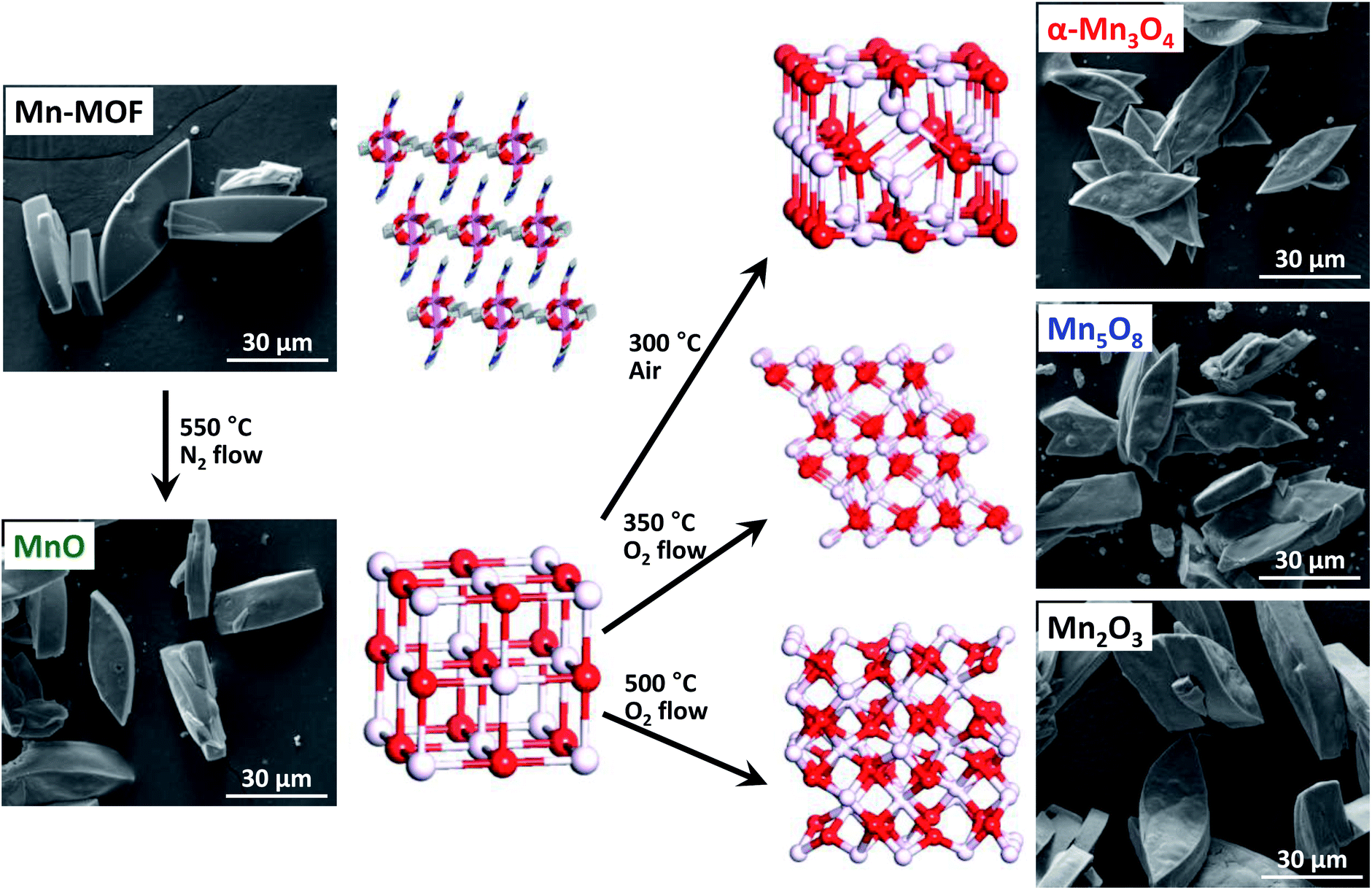 | ||
| Fig. 1 SEM images and atomic structures of Mn-MOF and converted manganese oxides during structural transformation of Mn-MOF into MnO, Mn3O4, Mn5O8 and Mn2O3. | ||
2. Experimental section
2.1. Materials
All chemicals and solvents were of reagent grade and used without further purification. meso-Butane-1,2,3,4-tetracarboxylic acid (H4BuTC, >98%) and N,N-dimethylacetamide (DMA, >99%) were supplied by TCI. Mn(NO3)2·4H2O (>97%) was purchased from Aldrich.2.2. Synthesis of Mn-MOF ([Mn2(BuTC)(DMA)2]n)
H4BuTC (0.037 g, 0.16 mmol) was dissolved in DMA (2.5 mL) and mixed with a DMA (2.5 mL) solution of Mn(NO3)2·4H2O (0.080 g, 0.32 mmol). The solution was sealed in a glass jar, heated to 100 °C for 24 h, and then cooled to room temperature. Yellowish leaf-shaped crystals formed, which were filtered and washed briefly with the mother liquor. Yield: 35%. FT-IR (Nujol mull): νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O(DMA), 1652 (s); νO–CO, 1615, 1418 (s) cm−1. Anal. calcd for Mn2C16H24O10N2: C, 37.37; H, 4.70; N, 5.45. Found: C, 37.53; H, 4.59; N, 5.44%.
O(DMA), 1652 (s); νO–CO, 1615, 1418 (s) cm−1. Anal. calcd for Mn2C16H24O10N2: C, 37.37; H, 4.70; N, 5.45. Found: C, 37.53; H, 4.59; N, 5.44%.
2.3. Conversion reactions of Mn-MOFs into manganese oxides
2.4. Single-crystal X-ray crystallography
A single-crystal of Mn-MOF was coated with paratone-N oil, and the diffraction data were obtained at 100 K with synchrotron radiation (λ = 0.69999 Å) on an ADSC Quantum-210 detector at 2D SMC with a silicon (111) double crystal monochromator (DCM) at the Pohang Accelerator Laboratory, Republic of Korea. The ADSC Q210 ADX program41 was used for data collection, and HKL3000sm (Ver. 703r)42 was used for cell refinement, reduction, and absorption correction. The crystal structures were solved by direct methods and refined by full-matrix least-squares calculations with the SHELX-97 computer program.43 The positions of all non-hydrogen atoms were refined with anisotropic displacement factors. The hydrogen atoms were positioned geometrically using a riding model.2.5. Characterization methods
All characterization processes were carried out at the Ulsan National Institute of Science and Technology (UNIST) Central Research Facilities (UCRF) Center. Infrared spectra were recorded with a Thermo Fisher Scientific iS10 FT-IR spectrometer. Elemental analyses (for C, H, N, S, and O) were performed by using a Thermo Scientific Flash 2000 series CHNS/O analyzer. Thermogravimetric analyses (TGA) were performed under nitrogen gas at a scan rate of 5 °C min−1, using a TGA Q50 from TA Instruments. X-ray photoelectron spectroscopy (XPS) was performed on a K-alpha from Thermo Fisher. X-ray powder diffraction (XRPD) data were recorded on a Bruker D8 advance diffractometer at 40 kV and 40 mA and a Bruker D2 phaser diffractometer at 30 kV and 10 mA using Cu Kα (λ = 1.541 Å) radiation, with a step size of 0.02° in 2θ. Scanning electron microscope (SEM) images were taken using a Quanta 200 microscope (FEI) operating at 18 kV. Transmission electron microscope (TEM) images and energy dispersive X-ray spectra were obtained with a JEOL JEM-2100F microscope. The nitrogen adsorption–desorption isotherms were measured at −196 °C using liquid nitrogen on a BELSORP-MAX. Prior to the adsorption measurement, the samples were evacuated at 200 °C under vacuum (p < 10−5 mbar) overnight. The specific surface area was determined in the relative pressure range from 0.05 to 0.3 of the Brunauer–Emmett–Teller (BET) plot, and the total pore volume was calculated from the amount adsorbed at a relative pressure of about 0.98–0.99. Pore size distribution curves were obtained from the corresponding isotherms using the Barrett–Joyner–Halenda (BJH) method.2.6. Electrochemical measurements
Electrochemical characterization of the samples was carried out using a high-power potentiostat (IviumStat, Ivium Technologies, Netherlands). The electrochemical experiments were performed at room temperature (25 °C) and under atmospheric pressure using a three-compartment electrochemical cell. A graphite rod counter electrode and Hg/HgO reference electrode (XR400, Radiometer Analytical) with 1 M KOH (99.999%, Aldrich) filling solution were used. The Hg/HgO reference electrode was calibrated with respect to the reversible hydrogen electrode (RHE) before use. For this purpose, Pt wires were used as both the working electrode and counter electrode, and the potential cycle between −0.93 V and −0.87 V (vs. Hg/HgO) was applied with a scan rate of 1 mV s−1 in H2-saturated 0.1 M KOH solution. The resulting potential–current (V–I) curve after averaging the anodic and cathodic currents gave a result of E(RHE) = E(Hg/HgO) + 0.887 V. All potentials in this report were reported in the RHE scale.For ORR performance measurement, a rotating ring disk electrode (RRDE) with a glassy carbon disk (GC, 4 mm in diameter) and a Pt ring (5 mm in inner diameter and 7 mm in outer diameter) was used as a working electrode (ALS, Cat. no. 012613). The RRDE was polished with 1.0 and 0.3 μm alumina suspensions to generate a mirror finish. For preparation of a catalyst ink, Nafion (5 wt% in isopropyl alcohol (IPA), Aldrich) was neutralized by 0.1 M NaOH (99.99%, Aldrich) at a volume ratio of Nafion![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.1 M NaOH = 2:1 in order to prevent oxide passivation.44 Commercial carbon black (Vulcan XC-72R) was added to the catalyst ink to overcome the low electrical conductivity of oxides. The catalyst ink was prepared by mixing 5 mg of catalyst, 12 mg of Vulcan, 116 μL of neutralized Nafion, 100 μL of deionized water, and 0.98 mL of IPA (>99.9%, Aldrich) with ultra-sonication for at least 30 min. 3 μL of the catalyst ink was deposited onto the GC electrode with a micro-syringe. The resulting catalyst loading was 0.1 mgoxide cm−2. Prior to the electrochemical measurements, a potential cycle from 0.7 to 1.0 V was applied in a N2-saturated 0.1 M KOH solution for 50 cycles at a scan rate of 100 mV s−1 for electrochemical cleaning. Subsequently, the cyclic voltammogram was obtained under the same conditions used for electrochemical cleaning, except for the change in the scan rate of 20 mV s−1 for 3 cycles. Linear sweep voltammetry (LSV) curves for the ORR were obtained by cycling the potential from 1.1 to 0.2 V in an O2-saturated 0.1 M KOH solution with O2 purging at different rotating speeds. The anodic and cathodic sweeps for the ORR were averaged in order to correct the capacitive-current contribution.44 The ORR measurement was independently repeated three times, and the averaged currents were used.
:0.1 M NaOH = 2:1 in order to prevent oxide passivation.44 Commercial carbon black (Vulcan XC-72R) was added to the catalyst ink to overcome the low electrical conductivity of oxides. The catalyst ink was prepared by mixing 5 mg of catalyst, 12 mg of Vulcan, 116 μL of neutralized Nafion, 100 μL of deionized water, and 0.98 mL of IPA (>99.9%, Aldrich) with ultra-sonication for at least 30 min. 3 μL of the catalyst ink was deposited onto the GC electrode with a micro-syringe. The resulting catalyst loading was 0.1 mgoxide cm−2. Prior to the electrochemical measurements, a potential cycle from 0.7 to 1.0 V was applied in a N2-saturated 0.1 M KOH solution for 50 cycles at a scan rate of 100 mV s−1 for electrochemical cleaning. Subsequently, the cyclic voltammogram was obtained under the same conditions used for electrochemical cleaning, except for the change in the scan rate of 20 mV s−1 for 3 cycles. Linear sweep voltammetry (LSV) curves for the ORR were obtained by cycling the potential from 1.1 to 0.2 V in an O2-saturated 0.1 M KOH solution with O2 purging at different rotating speeds. The anodic and cathodic sweeps for the ORR were averaged in order to correct the capacitive-current contribution.44 The ORR measurement was independently repeated three times, and the averaged currents were used.
For the evaluation of four-electron selectivity, the potential of the Pt ring was fixed at 1.3 V (vs. RHE) during LSV scans for the ORR. The number of electrons transferred was calculated by the following equation:

The four-electron selectivity was also assessed by the Koutecký–Levich equation:

Before electrochemical impedance spectroscopy (EIS) measurement, the potential was cycled from 0.7 to 1.0 V for 50 cycles in N2-saturated 0.1 M KOH solution. Then, impedance spectra were obtained in O2-saturated 0.1 M KOH at a fixed potential of 0.7 V (vs. RHE) in the frequency range varying from 10 kHz to 0.05 Hz.
3. Results and discussion
3.1. Synthesis of Mn-MOF precursor
We began with the synthesis of the Mn-MOF precursor. A solvothermal reaction of Mn(NO3)2·4H2O and aliphatic ligand, meso-butane-1,2,3,4-tetracarboxylic acid (H4BuTC), in N,N-dimethylacetamide (DMA) solvent at 100 °C for 24 h afforded yellowish leaf-shaped crystals of [Mn2(BuTC)(DMA)2]n (Fig. 1 and 2). Single-crystal X-ray crystallographic data48 (Fig. 2 and S1 and Tables S1 and S2†) revealed that Mn-MOF is constructed with Mn2(COO)4 paddle-wheel clusters as secondary building units interconnected by the BuTC ligand to form a 2-dimensional layer. In the Mn-MOF, each MnII ion is coordinated with four oxygen atoms of four different BuTC ligands in the equatorial positions and an oxygen atom of a DMA molecule at the axial position to show a square-pyramidal coordination geometry. Four carboxylate groups of four different BuTC ligands bridge between the two MnII centers in a syn–syn bidentate mode, forming a paddle-wheel shaped SBU. The Mn–Mn distance in a SBU is 3.051(1) Å. Each BuTC4− acts as a tetra-bidentate bridging ligand by coordinating to eight different MnII centers. The shortest interlayer distance is 3.8 Å, based on the van der Waals radii. The XRPD pattern of [Mn2(BuTC)(DMA)2]n showed good agreement with the simulated pattern (Fig. S2†). In the TGA trace of the as-synthesized Mn-MOF in a N2 atmosphere (Fig. S3†), the two coordinating DMA molecules per formula ([Mn2(BuTC)(DMA)2]n) were removed over the range 200–300 °C, and decomposition of Mn-MOF ensued above ∼400 °C. | ||
| Fig. 2 Synthesis and single-crystal X-ray structure of Mn-MOF. (a) Synthetic scheme of Mn-MOF. (b) Side view of one layer of Mn-MOF. Hydrogen atoms and guest molecules are omitted for clarity (color scheme: C, white; O, purple; N, light blue; Mn, pink). (c) Entire structure of Mn-MOF (color scheme: 2-D layer, white; coordinating DMA, blue). | ||
3.2. Conversion reactions of Mn-MOF into manganese oxides and their characterization
We explored the conversion reactions of Mn-MOF into manganese oxides. Based on the above TGA result, we first heated as-synthesized Mn-MOF under N2 flow to 550 °C and maintained this temperature for 12 h to achieve complete thermal decomposition of the MOF structure (see the ESI†). Scanning electron microscope (SEM) images of pristine Mn-MOF and converted black powder revealed a pseudomorphic conversion process (Fig. 1). The converted material was nanoporous, as its cross-sectional SEM and TEM images (Fig. 3a and d) clearly showed the macropores and mesopores, respectively. The TEM image revealed that the frameworks of the converted material were composed of small nanoparticles of ∼5 nm. A high-resolution (HR) TEM image of this material (Fig. 3d, inset) exhibited distinct lattice fringes indicating the highly crystalline nature of these nanoparticles. The XRPD pattern of the converted material identifies its structure as single-phase cubic manganosite (MnO), which is coincident with JCPDS no. 71-4748 (Fig. 4, bottom). The cubic MnO phase could be confirmed by its electron diffraction pattern (Fig. 3d, inset) as well. The crystalline size of MnO nanoparticles, calculated by applying the Scherrer equation to the (200) reflection, was 5.0 nm in diameter. The elemental analysis revealed that the nanoporous MnO contained residual carbon (18 wt%) and nitrogen (0.6 wt%). | ||
| Fig. 3 Characterization of converted manganese oxides. (a–c) SEM images, (d–f) TEM images, and (d–f, insets) HR-TEM images and electron diffraction patterns of nanoporous MnO (a and d), Mn3O4 (b and e), and Mn5O8 (c and f). | ||
 | ||
| Fig. 4 XRPD patterns of converted manganese oxides. | ||
Using the nanoporous MnO that has the lowest oxidation state among the manganese oxide structures as a precursor, we further carried out sequential conversion reactions in an oxidizing environment to generate manganese oxides with higher oxidation states. First, nanoporous MnO was heated in ambient air. The XRPD patterns monitored at incremental heating temperatures (Fig. 5a) indicated that the transformation of cubic MnO into tetragonal spinel-phase hausmannite-Mn3O4 (JCPDS no. 24-0734, hereafter Mn3O4) started at 100 °C, and the latter phase was maintained up to 300 °C. At 350 °C, several new peaks started to appear in the XRPD pattern, which correspond to bixbyite Mn2O3 (JCPDS no. 73-1826). Further heating to ≥450 °C yielded the pure phase of Mn2O3. Meanwhile, conversion reactions in a more oxidative environment allowed for the stabilization of metastable intermediate Mn5O8; we found that the nanoporous MnO can be transformed into a pure, layered Mn5O8 phase by way of the Mn3O4 phase, when it was heated to 350 °C under oxygen flow at a very slow ramping rate of 1 °C min−1 (Fig. 5b and c). The heating of MnO to the same temperature but with higher ramping rates (3 or 5 °C min−1) afforded a mixture of Mn3O4 and Mn5O8 phases instead of the sequential formation of pure Mn3O4 and Mn5O8 phases (Fig. 5c). Hence, the degree of oxidation environment and the change of heating rate can adjust oxidation states and purities of the resultant manganese oxides.
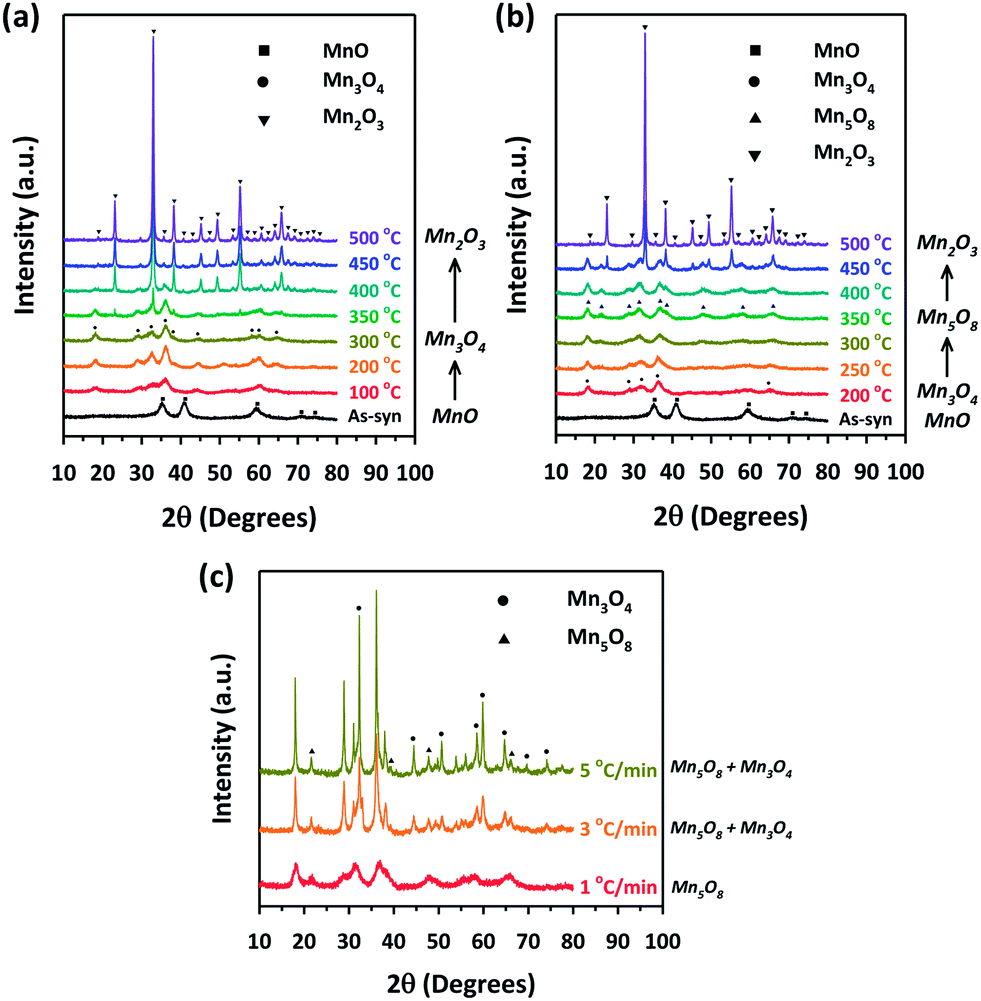 | ||
| Fig. 5 Monitoring structural transformations from MnO to manganese oxides with higher oxidation numbers. XRPD patterns for MnO before and after heat treatment at various temperatures (a) in a box furnace with a ramping rate of 5 °C min−1 and (b) in a tube furnace under an oxygen flow of 100 mL min−1 with a ramping rate of 1 °C min−1. (c) XRPD patterns of MnO after heat treatment at 350 °C in a tube furnace under an oxygen flow of 100 mL min−1 and with different ramping rates of 1 °C min−1, 3 °C min−1, and 5 °C min−1. | ||
Under the optimized experimental conditions based on the above experiments, we could obtain Mn3O4 and Mn5O8 in pure phases via pseudomorphic transformation of nanoporous MnO (Fig. 1). Further examination of these two manganese oxides with cross-sectional SEM and TEM images (Fig. 3b, c, e and f) revealed that the nanoporous structure of MnO was retained despite the structural transformation processes. The crystalline sizes of Mn3O4 and Mn5O8, calculated by applying the Scherrer equation to the (101) and (−201) reflections of their XRPD patterns (Fig. 4), respectively, were 5.6 nm and 5.2 nm, similar to those of MnO. The crystallinity and pure phases of nanoporous Mn3O4 and Mn5O8 were also confirmed by their HR-TEM images and electron diffraction patterns (Fig. 3e and f, insets), respectively. However, Mn2O3 showed a marked increase in the nanocrystal size, from 5.0 to ∼20 nm, regardless of the reaction conditions such as the gas environment or heating rate (Fig. 5a and b and S5†). Hereafter, we will focus on the three phases—MnO, Mn3O4, and Mn5O8—that have almost identical textural properties, for further discussion of the converted manganese oxides.
Fig. 6 shows the nitrogen adsorption–desorption isotherms of the three manganese oxides, which feature type IV isotherms with H3-type hysteresis loops over all three samples, indicating well-developed mesoporosity. The highly porous nature of the three nanoporous manganese oxides provide similarly large Brunauer–Emmett–Teller (BET) surface areas (∼145 m2 g−1) and pore volumes (∼0.3 cm3 g−1). The pore size distribution curves obtained by the BJH method exhibited similar pores sizes (5 to 20 nm) of manganese oxides (Fig. S4†). The detailed textural data are summarized in Table S3.† While the N2 adsorption data reveal similar porous properties of the three manganese oxides, the X-ray photoelectron spectroscopy (XPS) data clearly establish a marked difference in their oxidation states (Fig. 7 and Table S4†). The Mn 3s XPS spectra (Fig. 7) revealed that the position of the lower binding energy peak gradually shifted to higher energy with the increase in the oxidation number of the sample (MnO → Mn3O4 → Mn5O8), which is consistent with previous results.49–51 This systematic peak shift results in a corresponding decrease in the 3s multiplet splitting (ΔE3s) of these manganese oxides (Table S4†).
 | ||
| Fig. 6 Nitrogen adsorption–desorption isotherms of converted manganese oxides. | ||
 | ||
| Fig. 7 Mn 3s XPS spectra of converted manganese oxides. The blue asterisk indicates the presence of low-valent (e.g. Mn2+) manganese species in Mn5O8. | ||
Nanoporous manganese oxides thus far reported have been predominantly limited to those with high oxidation states such as MnO2 and Mn2O3.9,52–58 Toward the low-oxidation-state manganese oxide, Bruce et al. prepared nanoporous Mn3O4 by reducing pre-synthesized nanoporous Mn2O3 with hydrogen gas.58 Likewise, other nanoporous metal oxides with low oxidation states, for instance, CoO and Fe3O4, could be prepared by the reduction of the corresponding metal oxides with higher oxidation numbers.59,60 In contrast, in our approach, the oxidative conversion reactions of Mn-MOF could sequentially generate nanoporous MnO, Mn3O4, Mn5O8, and Mn2O3, by progressively increasing the degree of oxidation. Significantly, our MOF-driven route enabled the isolation of the low-oxidation-state MnO and metastable intermediate Mn5O8 as pure phases with nanoporous nanocrystalline structures, which were previously rarely attainable.61,62 In particular, to our knowledge, nanoporous Mn5O8 with such a large surface area and well-developed porosity is prepared for the first time.
We note that the transformations between different structures, from cubic (MnO) to spinel (Mn3O4), and to layered (Mn5O8), occurred while preserving a nanoporous structure and textural properties. As demonstrated in our recent work,30 the conversion reactions exploiting aliphatic ligand-based MOFs allow for the formation of thermally stable nanoporous metal oxide frameworks that are composed of small nanoparticles. In this approach, labile aliphatic ligands can act as self-templates during the thermal conversion process, and finally evaporate to generate nanopores. We also found that the infinite coordination between inorganic and organic moieties in the MOFs is critical for suppressing the aggregation of metallic entities into large particles; instead, the thermal conversion of a single coordination compound generated very large agglomerates.31 In this way, the prepared nanoporous manganese oxide frameworks are constructed with small (∼5 nm) manganese oxide nanoparticles, which have a high density of defects or grain boundaries. Compared to bulk metal oxides, nanometer scale manganese oxide nanoparticles could relieve or minimize the structural stress during the transformations between different structures, which could lead to a minimal change in their textural properties.
3.3. Electrocatalytic properties of nanoporous manganese oxides for oxygen reduction reaction
Among the transformed nanoporous manganese oxides, Mn3O4 and Mn5O8 have nearly identical textural properties (such as morphology, surface area, and crystallite size of framework nanoparticles) and are free of residual carbon, but differ only in their oxidation states. As such, these two manganese oxides can serve as a system for the study of the oxidation state effect in catalysis. We investigated the electrocatalytic properties of the two nanoporous manganese oxides for the ORR (Fig. 8, S6, and S7†), because the latter is a key reaction in energy conversion devices such as fuel cells and metal–air batteries. The LSV curves for the two nanoporous manganese oxides (Fig. 8a) measured in the 0.1 M KOH electrolyte at a rotating speed of 1600 rpm revealed that the nanoporous Mn3O4 had higher on-set potential (0.75 V vs. RHE) and current density at 0.75 V (−0.26 mA cm−2) than Mn5O8 (0.73 V and −0.15 mA cm−2, respectively). In addition, nanoporous Mn3O4 showed a higher limiting current density than Mn5O8. We note that nanoporous MnO showed higher current density than the other oxides in the kinetic-diffusion controlled region of the LSV curve (Fig. S8†). This result does not correspond to the previous report where MnO exhibited inferior ORR activity to Mn3O4.63 We attribute this higher activity of the nanoporous MnO to the presence of residual carbon and nitrogen. The kinetic parameters of the two catalysts for the ORR were deduced by measurement of the ring currents during the ORR (Fig. 8b and c) and the Koutecký–Levich analyses (Fig. 8d and S6†). Results from both the methods consistently indicated that the electron numbers transferred by the nanoporous Mn3O4 were higher (3.8) than those of Mn5O8 (3.1–3.5). The excellent kinetics observed for nanoporous Mn3O4 could also be confirmed with its smaller charge transfer resistance at 0.7 V in EIS data (Fig. S7†). | ||
| Fig. 8 Electrocatalytic activity of a series of nanoporous manganese oxides for the ORR. (a) LSV curves of nanoporous manganese oxides on glassy carbon electrodes. (b) The numbers of electrons transferred at 0.4 V (vs. RHE) during the ORR obtained from the RRDE measurements. (c) Plot of the number of transferred electrons against potential during the ORR obtained by RRDE measurement. (d) Koutecký–Levich plots at 0.4 V (vs. RHE). The dotted lines show the theoretical slopes when two and four electrons are transferred during ORR. The numbers in the parentheses indicate the number of transferred electrons. | ||
Collectively, nanoporous Mn3O4 had higher diffusion-limited current, electron transfer number, and lower charge transfer resistance than Mn5O8. For the ORR in an alkaline electrolyte, the most efficient way is the direct four electron transfer mechanism, which is, however, kinetically impeded due to its high activation barrier. Instead, a two-step mechanism is favored, wherein oxygen is first reduced via a two electron pathway producing peroxide species (HO2−) and the catalytic decomposition of HO2− produces half of oxygen molecule that is further reduced. Importantly, rapidly repeating the latter cycles lead to the quasi-four-electron transfer mechanism. Comparing the oxidation state of the two manganese oxides, the spinel Mn3O4 structure consists of Mn cations with +2 and +3 oxidation numbers, whereas layered Mn5O8 has +2 and +4 oxidation states. The decomposition of HO2− can be facilitated by the facile redox mechanism between +2 and +3 Mn cations on the surface of the Mn3O4 structure.64 Such enhanced HO2− decomposition results in quasi-four-electron transfer with nanoporous Mn3O4. It is suggested that the disparity in the framework structure between Mn3O4 and Mn5O8 could also contribute to the difference in their ORR activity. Layered Mn5O8 is constructed with dense frameworks, which can impede facile adsorption of oxygen molecules. In contrast, spinel Mn3O4 with a relatively opened framework is favorable for oxygen adsorption, thus expediting catalytic cycles. Likewise, a recent study revealed that among a series of calcium manganese oxides, Ca2Mn3O8 that has an analogous crystal structure to Mn5O8 exhibited lowest ORR activity.65
4. Conclusions
In summary, conversion reactions using the Mn-MOF precursor yielded a series of nanoporous manganese oxide structures via sequential transformations. Significantly, this transformative route allowed for the preparation of low-oxidation-state (MnO) and metastable intermediate (Mn5O8) phases while having nanoporous architecture, which were otherwise difficult to synthesize. Additionally, this method enabled facile structural transformations between different manganese oxide structures while preserving their structural properties. In comparative investigation of the electrocatalytic activity of nanoporous manganese oxides, Mn3O4 showed better ORR activity and kinetics than nanoporous Mn5O8, demonstrating an important role of the oxidation state for enhanced performance of the ORR.Together with our recent work,30 conversion reactions exploiting aliphatic ligand-based MOFs have been demonstrated as a versatile route to nanoporous metal oxides with nanocrystalline frameworks. This work, in particular, has shown that this transformative route can enable the preparation of nanoporous architectures with unexplored compositions. We envisage that further investigations into the MOF-driven conversion reactions can enrich the composition, structure, and function of nanoporous metal oxide materials.
Acknowledgements
This work was supported by the Korea CCS R&D Center (KCRC) grant funded by the Korean Ministry of Science, ICT & Future Planning (NRF-2013M1A8A1039968) and by the Basic Science Research Program through the National Research Foundation of Korea (2013R1A1A2012960). J.H.L. and Y.J.S. acknowledge the Global PhD Fellowship (NRF-2013H1A2A1033501 and NRF-2013H1A2A1032644, respectively). The authors acknowledge PAL for beamline use (Grant 2013-2nd-2D-003).Notes and references
- F. Schüth, Chem. Mater., 2001, 13, 3184–3195 CrossRef.
- G. J. A. A. Soler-Illia, C. Sanchez, B. Lebeau and J. Patarin, Chem. Rev., 2002, 102, 4093–4138 CrossRef PubMed.
- H. Yang and D. Zhao, J. Mater. Chem., 2005, 15, 1217–1231 CAS.
- Y. Ren, Z. Ma and P. G. Bruce, Chem. Soc. Rev., 2012, 41, 4909–4927 RSC.
- D. Gu and F. Schüth, Chem. Soc. Rev., 2013, 43, 313–344 RSC.
- Q. Huo, D. I. Margolese, U. Ciesla, P. Feng, T. E. Gier, P. Sieger, R. Leon, P. M. Petroff, F. Schüth and G. D. Stucky, Nature, 1994, 368, 317–321 CrossRef CAS.
- D. M. Antonelli and J. Y. Ying, Angew. Chem., Int. Ed., 1995, 34, 2014–2017 CrossRef CAS.
- P. Yang, D. Zhao, D. I. Margolese, B. F. Chmelka and G. D. Stucky, Nature, 1998, 396, 152–155 CrossRef CAS.
- D. Grosso, C. Boissière, B. Smarsly, T. Brezesinski, N. Pinna, P. A. Albouy, H. Amenitsch, M. Antonietti and C. Sanchez, Nat. Mater., 2004, 3, 787–792 CrossRef CAS PubMed.
- B. Tian, X. Liu, H. Yang, S. Xie, C. Yu, B. Tu and D. Zhao, Adv. Mater., 2003, 15, 1370–1374 CrossRef CAS.
- S. C. Laha and R. Ryoo, Chem. Commun., 2003, 2138–2139 RSC.
- J. Lee, M. C. Orilall, S. C. Warren, M. Kamperman, F. J. DiSalvo and U. Wiesner, Nat. Mater., 2008, 7, 222–228 CrossRef CAS PubMed.
- A. Corma, P. Atienzar, H. Garcia and J.-Y. Chane-Ching, Nat. Mater., 2004, 3, 394–397 CrossRef CAS PubMed.
- R. Buonsanti, T. E. Pick, N. Krins, T. J. Richardson, B. A. Helms and D. J. Milliron, Nano Lett., 2012, 12, 3872–3877 CrossRef CAS PubMed.
- Y. Vasquez, A. E. Henkes, J. C. Bauer and R. E. Schaak, J. Solid State Chem., 2008, 181, 1509–1523 CrossRef CAS.
- M. Kim, V. N. Phan and K. Lee, CrystEngComm, 2012, 14, 7535–7548 RSC.
- B. J. Beberwyck, Y. Surendranath and A. P. Alivisatos, J. Phys. Chem. C, 2013, 117, 19759–19770 CAS.
- W. Wang, M. Dahl and Y. Yin, Chem. Mater., 2013, 25, 1179–1189 CrossRef CAS.
- J. H. Han, S. Lee and J. Cheon, Chem. Soc. Rev., 2013, 42, 2581–2591 RSC.
- Y. Yin, R. M. Rioux, C. K. Erdonmez, S. Hughes, G. A. Somorjai and A. P. Alivisatos, Science, 2004, 304, 711–714 CrossRef CAS PubMed.
- Y. Sun and Y. Xia, Science, 2002, 298, 2176–2179 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- S. J. Yang and C. R. Park, Adv. Mater., 2012, 24, 4010–4013 CrossRef CAS PubMed.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. M. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 2864–2867 CrossRef CAS PubMed.
- W. Cho, S. Park and M. Oh, Chem. Commun., 2011, 47, 4138–4140 RSC.
- X. Xu, R. Cao, S. Jeong and J. Cho, Nano Lett., 2012, 12, 4988–4991 CrossRef CAS PubMed.
- L. Zhang, H. B. Wu, S. Madhavi, H. H. Hng and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 17388–17391 CrossRef CAS PubMed.
- M. Hu, A. A. Belik, M. Imura and Y. Yamauchi, J. Am. Chem. Soc., 2013, 135, 384–391 CrossRef CAS PubMed.
- A. S. Hall, A. Kondo, K. Maeda and T. E. Mallouk, J. Am. Chem. Soc., 2013, 135, 16276–16279 CrossRef CAS PubMed.
- T. K. Kim, K. J. Lee, J. Y. Cheon, J. H. Lee, S. H. Joo and H. R. Moon, J. Am. Chem. Soc., 2013, 135, 8940–8946 CrossRef CAS PubMed.
- L. Guo, H. Arafune and N. Teramae, Langmuir, 2013, 29, 4404–4412 CrossRef CAS PubMed.
- A. H. Reidies, Manganese Compounds, in Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 6 edn, 2002, vol. 20, pp. 543–564 Search PubMed.
- H. Wang, L.-F. Cui, Y. Yang, H. S. Casalongue, J. T. Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- J.-S. Lee, G. S. Park, H. I. Lee, S. T. Kim, R. Cao, M. Liu and J. Cho, Nano Lett., 2011, 11, 5362–5366 CrossRef CAS PubMed.
- W. Wei, X. Cui, W. Chen and D. G. Ivey, Chem. Soc. Rev., 2011, 40, 1697–1721 RSC.
- F. Jiao and H. Frei, Chem. Commun., 2010, 46, 2920–2922 RSC.
- L. Mao, T. Sotomura, K. Nakatsu, N. Koshiba, D. Zhang and T. Ohsaka, J. Electrochem. Soc., 2002, 149, A504–A507 CrossRef CAS.
- L. Mao, D. Zhang, T. Sotomura, K. Nakatsu, N. Koshiba and T. Ohsaka, Electrochim. Acta, 2003, 48, 1015–1021 CrossRef CAS.
- F. H. B. Lima, M. L. Calegaro and E. A. Ticianelli, J. Electroanal. Chem., 2006, 590, 152–160 CrossRef CAS.
- F. H. B. Lima, M. L. Calegaro and E. A. Ticianelli, Electrochim. Acta, 2007, 52, 3732–3738 CrossRef CAS.
- A. J. Arvai and C. Nielsen, ADSC Quantum-210 ADX Program, Area Detector System Corporation, Poway, CA, USA, 1983 Search PubMed.
- Z. Otwinowski and W. Minor, Methods in Enzymology, Part A, in Macromolecular Crystallography, ed. C. W. Carter Jr. and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- G. M. Sheldrick, SHELXTL-PLUS, Crystal Structure Analysis Package, Bruker Analytical X-Ray, Madison, WI, USA, 1997 Search PubMed.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi and Y. Shao-Horn, J. Electrochem. Soc., 2010, 157, B1263–B1268 CrossRef CAS.
- Z.-W. Liu, F. Peng, H.-J. Wang, H. Yu, W.-X. Zheng and J. Yang, Angew. Chem., Int. Ed., 2011, 50, 3257–3261 CrossRef PubMed.
- W. Yang, T.-P. Fellinger and M. Antonietti, J. Am. Chem. Soc., 2011, 133, 206–209 CrossRef CAS PubMed.
- S. Wang, D. Yu and L. Dai, J. Am. Chem. Soc., 2011, 133, 5182–5185 CrossRef CAS PubMed.
- Crystal data for Mn2C16N2O10H24, Mr = 514.25, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 6.473(13), b = 7.970(16), c = 10.577(2) Å, α = 101.69(3)°, β = 102.84(3)°, γ = 105.70(3)°, V = 491.7(17) Å3, Z = 1, dcalcd = 1.737 g cm−3, T = 100(2) K, crystal size 0.2 × 0.2 × 0.1 mm3, λ = 0.69999 Å, 2θ = 67.92, 136 parameters, R1 = 0.0379 (I > 2σ(I), 2865 reflections), wR2 = 0.1082 (all data, 5533 reflections), GOF = 1.093. Crystallographic data are summarized in the ESI.†.
(no. 2), a = 6.473(13), b = 7.970(16), c = 10.577(2) Å, α = 101.69(3)°, β = 102.84(3)°, γ = 105.70(3)°, V = 491.7(17) Å3, Z = 1, dcalcd = 1.737 g cm−3, T = 100(2) K, crystal size 0.2 × 0.2 × 0.1 mm3, λ = 0.69999 Å, 2θ = 67.92, 136 parameters, R1 = 0.0379 (I > 2σ(I), 2865 reflections), wR2 = 0.1082 (all data, 5533 reflections), GOF = 1.093. Crystallographic data are summarized in the ESI.†. - M. Oku, K. Hirokawa and S. Ikeda, J. Electron Spectrosc. Relat. Phenom., 1975, 7, 465–473 CrossRef CAS.
- V. Di Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117–123 CrossRef CAS.
- T. Gao, P. Norby, F. Krumeich, H. Okamoto, R. Nesper and H. Fjellvag, J. Phys. Chem. C, 2010, 114, 922–928 CAS.
- Z.-R. Tian, W. Tong, J.-Y. Wang, N.-G. Duan, V. V. Krishnan and S. L. Suib, Science, 1997, 276, 926–930 CrossRef CAS.
- A.-S. Malik, M. J. Duncan and P. G. Bruce, J. Mater. Chem., 2003, 13, 2123–2126 RSC.
- A. K. Sinha, K. Suzuki, M. Takahara, H. Azuma, T. Nonaka and K. Fukumoto, Angew. Chem., Int. Ed., 2007, 46, 2891–2894 CrossRef CAS PubMed.
- M. Imperor-Clerc, D. Bazin, M. D. Appay, P. Beaunier and A. Davidson, Chem. Mater., 2004, 16, 1813–1821 CrossRef CAS.
- J.-Y. Luo, J.-J. Zhang and Y.-Y. Xia, Chem. Mater., 2006, 18, 5618–5623 CrossRef CAS.
- F. Jiao and P. G. Bruce, Adv. Mater., 2007, 19, 657–660 CrossRef CAS.
- F. Jiao, A. Harrison, A. H. Hill and P. G. Bruce, Adv. Mater., 2007, 19, 4063–4066 CrossRef CAS.
- F. Jiao, J.-C. Jumas, M. Womes, A. V. Chadwick, A. Harrison and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 12905–12909 CrossRef CAS PubMed.
- H. Tüysüz, Y. Liu, C. Weidenthaler and F. Schüth, J. Am. Chem. Soc., 2008, 130, 14108–14110 CrossRef PubMed.
- G.-L. Xu, Y.-F. Xu, J.-C. Fang, F. Fu, H. Sun, L. Huang, S. Yang and S.-G. Sun, ACS Appl. Mater. Interfaces, 2013, 5, 6316–6323 CAS.
- S. S. T. Bastos, J. J. M. Orfao, M. M. A. Freitas, M. F. R. Pereira and J. L. Figueiredo, Appl. Catal., B, 2009, 93, 30–37 CrossRef CAS.
- Y. Gorlin, C.-J. Chung, D. Nordlund, B. M. Clemens and T. F. Jaramillo, ACS Catal., 2012, 2, 2687–2694 CrossRef CAS.
- T. Rhadfi, J.-Y. Piquemal, L. Sicard, F. Herbst, E. Briot, M. Benedetti and A. Atlamsani, Appl. Catal., A, 2010, 386, 132–139 CrossRef CAS.
- X. Han, T. Zhang, J. Du, F. Cheng and J. Chen, Chem. Sci., 2013, 4, 368–376 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: X-ray crystallographic data, XRPD pattern and TGA data for Mn-MOF, the pore size distribution of the nanoporous MnO, Mn3O4, and Mn5O8, characterization data for Mn2O3, LSV polarization curves of the nanoporous manganese oxides at different rotating speeds, Nyquist plots of the nanoporous manganese oxides, and LSV polarization curves of the nanoporous manganese oxides including MnO. CCDC 978436. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ta01272k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |