 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA multifunctional phosphite-containing electrolyte for 5 V-class LiNi0.5Mn1.5O4 cathodes with superior electrochemical performance†
Young-Min
Song
,
Jung-Gu
Han
,
Soojin
Park
,
Kyu Tae
Lee
and
Nam-Soon
Choi
*
School of Green Energy, Ulsan National Institute of Science and Technology (UNIST), Ulsan 689-798, Republic of Korea. E-mail: nschoi@unist.ac.kr
First published on 14th March 2014
Abstract
We report a highly promising organophosphorus compound with an organic substituent, tris(trimethylsilyl)phosphite (TMSP), to improve the electrochemical performance of 5 V-class LiNi0.5Mn1.5O4 cathode materials. Our investigation reveals that TMSP alleviates the decomposition of LiPF6 by hydrolysis, effectively eliminates HF promoting Mn/Ni dissolution from the cathode, and forms a protective layer on the cathode surface against severe electrolyte decomposition at high voltages. Remarkable improvements in the cycling stability and rate capability of high voltage cathodes were achieved in the TMSP-containing electrolyte. After 100 cycles at 60 °C, the discharge capacity retention was 73% in the baseline electrolyte, whereas the TMSP-added electrolyte maintained 90% of its initial discharge capacity. In addition, the LiNi0.5Mn1.5O4 cathode with TMSP delivers a superior discharge capacity of 105 mA h g−1 at a high rate of 3 C and an excellent capacity retention of 81% with a high coulombic efficiency of over 99.6% is exhibited for a graphite/LiNi0.5Mn1.5O4 full cell after 100 cycles at 30 °C.
Introduction
Lithium-ion batteries (LIBs) are one of the most promising energy storage devices because of their high power and energy densities, current technical maturity, and economic considerations.1 Although LIBs have been successfully commercialized, a noticeable improvement in the energy densities of Li-ion cells will be necessary to satisfy society's needs for high power and/or capacity, for applications such as power tools and electric vehicles, or the efficient use of renewable energies. High energy density in a battery can be attained by increasing the reversible capacity of the electrodes, by decreasing the working potential of the anode, or by increasing the working potential of the cathode. Among various high-voltage cathode materials, LiNi0.5Mn1.5O4, which operates in the vicinity of 4.7 V vs. Li/Li+, has been investigated as a promising material for LIBs with high energy densities. However, the practical application of this material in LIBs is still quite challenging because LiNi0.5Mn1.5O4 suffers from the severe oxidative decomposition of electrolyte solutions beyond the upper voltage limit of LiPF6-based conventional electrolytes, around 4.3 V vs. Li/Li+. This leads to the formation of a resistive and unstable surface film consisting of inorganic lithium salts and organic carbonates on the cathode.2,3 As a result, large irreversible capacity and low coulombic efficiency are observed for the LiNi0.5Mn1.5O4 cathode with conventional electrolytes.4,5To date, several approaches toward the development of suitable electrolytes for high voltage cathodes in LIBs have been explored. To alleviate the oxidative decomposition of electrolytes, sulfone-based solvents, ionic liquids, and dinitrile solvents with high anodic stabilities have been explored.6–10 Unfortunately, these solvents suffer from high intrinsic viscosities and severe reductive decomposition on carbonaceous anode materials. One approach to resolve these problems by the Zhang group employed fluorinated carbonates as solvents to improve the electrochemical performance of LiNi0.5Mn1.5O4 at elevated temperature. The high anodic stability of the fluorinated carbonate solvent-based electrolytes was supported by the electrochemical evaluation results obtained using LiNi0.5Mn1.5O4/Li and LiNi0.5Mn1.5O4/Li4Ti5O12 electrochemical couples.11 According to recent reports, the formation of surface films through the use of reducible and oxidative additives in the electrolytes is thought to be one of the most effective strategies to stabilize the electrode–electrolyte interface. To preserve carbonate-based electrolytes on 5 V-class cathode surfaces at room temperature, a highly fluorinated phosphate ester additive was investigated.12 Lithium bis(oxalato)borate (LiBOB) has been recognized as one of the effective additives and salts that form a stable solid electrolyte interphase (SEI), not only on Si electrodes, but also on those based on graphite.13,14 Using LiBOB as an additive led to the formation of an SEI film which protected the graphite anode material against exfoliation by the propylenecarbonate-rich electrolyte.15 Very recently, our group and the Kim group reported the beneficial influence of the LiBOB additive on the cycling performance of high-voltage LiNi0.5Mn1.5O4 cathodes at elevated temperatures.16,17 However, the slightly higher polarization induced by the LiBOB-generated SEI at the end of the charge and discharge process may pose unavoidable problems (such as inferior rate capability), and should be remedied by other surface modifications. Since protection by the SEI against cathode-electrolyte reactions affects the electrochemical properties of LiNi0.5Mn1.5O4 cathodes, the nature of the SEI on the cathode is critically important. Although remarkable improvements in the electrochemical performance of Li/LiNi0.5Mn1.5O4 half-cells have been achieved, considerable capacity fading of full-cells with a graphite anode still occurs because of the metal (Mn and Ni) ions dissolved from the LiNi0.5Mn1.5O4 cathode in LiPF6-based electrolytes. The electron consumption via reduction of metal ions on the graphite anode results in the reduction of the reversible capacity and the deposited metal provokes unwanted side reactions at the anode.18,19 This is the main factor limiting the commercialization of high voltage LiNi0.5Mn1.5O4 cathodes. It is obvious that building innovative electrolytes is an immediate technological solution for high-performance LIBs with a high-voltage cathode and graphite anode.
Herein, we present, for the first time, the highly promising multifunctional additive, tris(trimethylsilyl) phosphite (TMSP), to scavenge HF promoting the Mn and Ni dissolution from a 5 V-class high voltage LiNi0.5Mn1.5O4 cathode, and to alleviate severe electrolyte decomposition at a high charge potential of 5.0 V vs. Li/Li+. Furthermore, from an investigation of the surface chemistry of the SEI on high voltage cathodes via ex situ X-ray photoelectron spectroscopy (XPS), we propose possible mechanisms of action of the TMSP additive, based on an organic–inorganic molecular architecture, drastically improving the cycling stability of LiNi0.5Mn1.5O4 cathodes in half- and full-cells.
Results and discussion
The oxidation tendency of organic molecules can be predicted by calculating the highest occupied molecular orbital (HOMO) energy level. Fig. 1 shows the HOMO energy as well as the lowest unoccupied molecular orbital (LUMO) energy of ethylene carbonate (EC) as a conventional carbonate solvent and reducible additives such as vinylene carbonate (VC) and fluoroethylene carbonate (FEC), which have been identified as the most efficient anode SEI formers.1 Appropriate additives making a protective layer on the high voltage LiNi0.5Mn1.5O4 cathode without the reductive decomposition should have analogous LUMO energy and higher HOMO energy relative to carbonate solvents. In this point of view, tris(trimethylsilyl) phosphite (TMSP) including an inorganic phosphorous (III) core surrounded by three –O–Si–(CH3)3 moieties is predicted to oxidize on the cathode without appreciable reduction on the anode (Fig. 1). The HOMO of TMSP is situated at the P–O groups displaying the donor character. It is expected that these donor groups will undergo the electrochemical oxidation at high potential and contribute to the formation of the SEI on the cathode. Moreover, hydrofluoric acid (HF), which is inevitably generated from the decomposition of PF5 in the presence of water traces, may readily attack the Si–C groups with a low bond energy of 318 kJ mol−1 and thereby HF promoting Mn/Ni dissolution from LiNi0.5Mn1.5O4 cathodes may be eliminated from the electrolyte.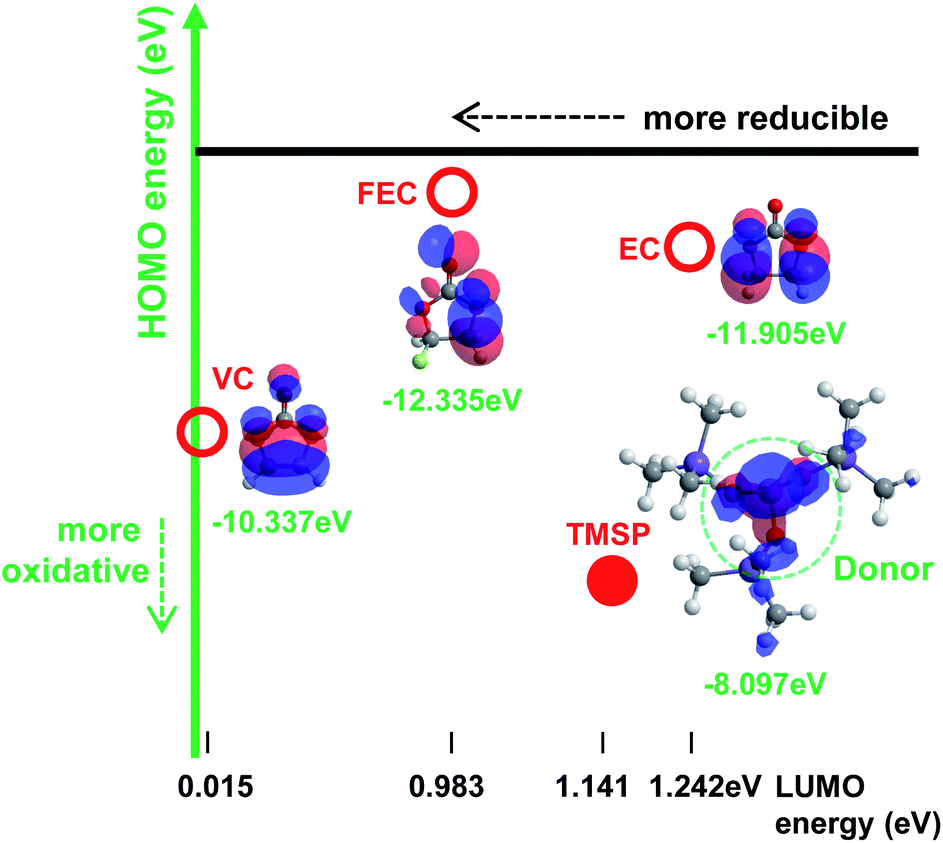 | ||
| Fig. 1 The calculated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels of TMSP, VC, FEC additives and an EC solvent. The HOMO is situated at the electron-rich moieties showing the donor character. | ||
A comparison of the cycling stabilities of LiNi0.5Mn1.5O4 cathodes with and without the TMSP additive at a current density of 60 mA g−1 is presented in Fig. 2. Surprisingly, an excellent cycling stability of the LiNi0.5Mn1.5O4 cathode at 60 °C and a current density of 60 mA g−1 was achieved in the TMSP-added electrolyte, delivering a capacity of 110 mA h g−1 without a noticeable capacity loss over 100 cycles. The discharge capacity retention was drastically improved from 66.5% (baseline electrolyte) to 82.1% (TMSP-containing electrolyte) after 160 cycles at 60 °C. The cell cycled in the TMSP-added electrolyte displayed a high capacity retention of ca. 90% in the 100th cycle. This lower capacity fade with cycling can be explained by the formation of a TMSP-derived SEI layer on the LiNi0.5Mn1.5O4 cathode.
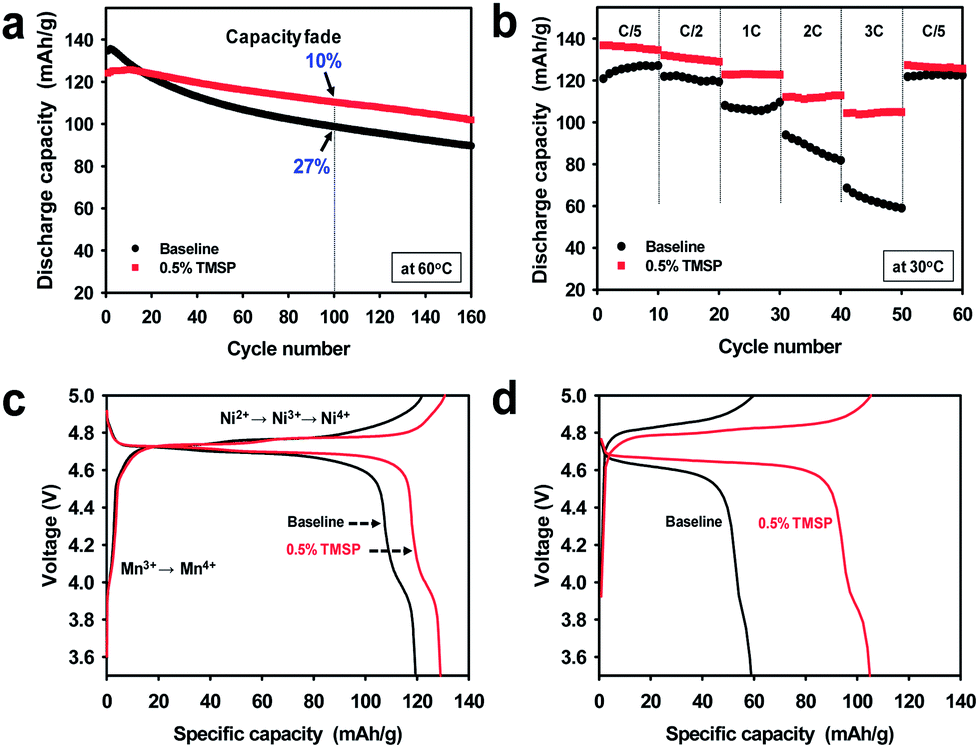 | ||
| Fig. 2 Electrochemical performance of LiNi0.5Mn1.5O4 cathodes: (a) cycling stability when cycled between 3.5 and 5.0 V at a rate of C/2, (b) rate capability at different C rates, (c) charge and discharge curves at a rate of C/2, and (d) charge and discharge curves at a rate of 3 C. | ||
This SEI layer inhibits direct contact of the electrolyte components with the high-voltage cathode and, thus, effectively diminishes the continuous oxidative decomposition of the electrolyte solution upon prolonged cycling at 60 °C. The discharge capacity fading of cells with the conventional (baseline) electrolyte was much stronger than that of cells using the TMSP-added electrolyte in subsequent cycles (Fig. 2a). This is consistent with the result that a fully charged LixNi0.5Mn1.5O4 cathode stored in the TMSP-added electrolyte at 60 °C shows better storage performance (Fig. S1†). The LiNi0.5Mn1.5O4 cathodes cycled in FEC- and VC-containing electrolytes exhibited considerably reduced discharge capacities and inferior cycling stabilities compared to the cells with TMSP, which showed a noticeable improvement in cycling stability (Fig. S2†). The cathodes with VC- and FEC-added electrolytes delivered very low discharge capacities of 72 and 42 mA h g−1 in the 160th cycle, respectively. This implies that the FEC and VC additives could not preserve the electrochemical properties of the high voltage cathodes because of the vulnerabilities of the FEC- and VC-derived SEI layers under high voltage conditions.
To further confirm the positive impact of the TMSP additive on the electrochemical properties of the cathode, galvanostatic cycling of Li/LiNi0.5Mn1.5O4 cells was performed at a current density of 60 mA g−1 and 30 °C (Fig. S3†). Clearly, the discharge capacity of the cell with the TMSP-added electrolyte during cycling was comparable with that of the cathode with the baseline electrolyte. A major challenge for the implementation of LiNi0.5Mn1.5O4 cathodes is undesirable electrolyte decomposition at high voltage, as evidenced by the previously reported low coulombic efficiency of ca. 97% for Li/LiNi0.5Mn1.5O4 cells at room temperature.20 Interestingly, the Li/LiNi0.5Mn1.5O4 half cells cycled in the TMSP-added electrolyte displayed a significant coulombic efficiency improvement of ca. 99% during cycling at 30 °C (Fig. S3†). This result suggested that the TMSP-derived SEI was sufficiently robust to suppress continuous electrolyte decomposition at 30 °C and effectively reduced irreversible capacity during cycling.
To investigate the suitability of the TMSP-derived SEI for facilitating charge transfer at the cathode surface, the cycling stability of the LiNi0.5Mn1.5O4 cathode was investigated at high current densities (Fig. 2b). The TMSP-added electrolyte clearly exhibited a much superior rate capability than the baseline electrolyte. The LiNi0.5Mn1.5O4 cathode with the TMSP additive delivered a superior discharge capacity (105 mA h g−1) at a very high current density (360 mA g−1, corresponding to 3 C) at 30 °C. However, the cell cycled in the baseline electrolyte showed rapid capacity fading as a function of the applied current density, and delivered a very low discharge capacity of only 60 mA h g−1 at 3 C.
The LiNi0.5Mn1.5O4 cathode cycled in the TMSP-added electrolyte exhibited a lower charge plateau and a higher discharge plateau compared to the cathode with the baseline electrolyte, indicating faster kinetics (Fig. 2c and d). A plateau at 4.0 V attributed to the Mn3+/Mn4+ redox couple was clearly observed in the TMSP-added electrolyte, even at a high C rate of 3 C. This finding suggested that the TMSP-derived SEI allowed fast charge transfer at high C rates, while the SEI formed by the baseline electrolyte impeded the diffusion of Li ions. Further evidence is given via comparison of the resistance of the cathode after precycling (Fig. S4†). The interfacial resistance including Rf and Rct components was smaller for the LiNi0.5Mn1.5O4 cathode precycled in the TMSP-added electrolyte. The discharge capacities of 122 mA h g−1 in the baseline electrolyte and 126 mA h g−1 in the TMSP-added electrolyte were recovered at a rate of C/5 (Fig. 2b). Considering the high potential of 5.0 V at which Li ions are extracted from the LiNi0.5Mn1.5O4 cathode, electrolyte components will be electrochemically oxidized and the SEI may be generated on the cathode surface.
The effects of TMSP on the surface chemistry of the LiNi0.5Mn1.5O4 cathode were confirmed by a comparison of the XPS spectra obtained from cathodes cycled in the baseline and TMSP-added electrolytes after precycling at 30 °C and 5 cycles at 60 °C. Fig. 3a shows the F 1s and P 2p spectra of the SEIs formed on LiNi0.5Mn1.5O4 cathodes cycled in the baseline and the TMSP-added electrolytes at 30 and 60 °C. The F 1s core level peaks assigned to the P–F moiety and LiF were clearly shown on the cathode surface, and the LiF peak intensity increased remarkably after 5 cycles at 60 °C in the baseline electrolyte, compared to that after the precycle at 30 °C. The labile P–F bonds of the LiPF6 salt are highly susceptible to hydrolysis even if trace amounts of moisture are present in the electrolyte solution: i.e., LiPF6 (sol.) + H2O → POF3 (sol.) + LiF (s) + 2HF (sol.), and PF5 (sol.) + H2O → POF3 (sol.) + 2HF (sol.).21–23 The resulting HF severely consumes Li ions to form LiF as a cathode SEI component in the baseline electrolyte during cycling and causes the loss of active Li+ ions. Moreover, the dissolution of Mn2+ ions from LiNi0.5Mn1.5O4 can take place in the presence of HF and additional LiF may be formed on the cathode, as displayed in Fig. 3. However, the LiF peak in the SEI on the cathode cycled in the TMSP-added electrolyte had a much weaker intensity than that of the SEI on the cathode cycled in the baseline electrolyte.
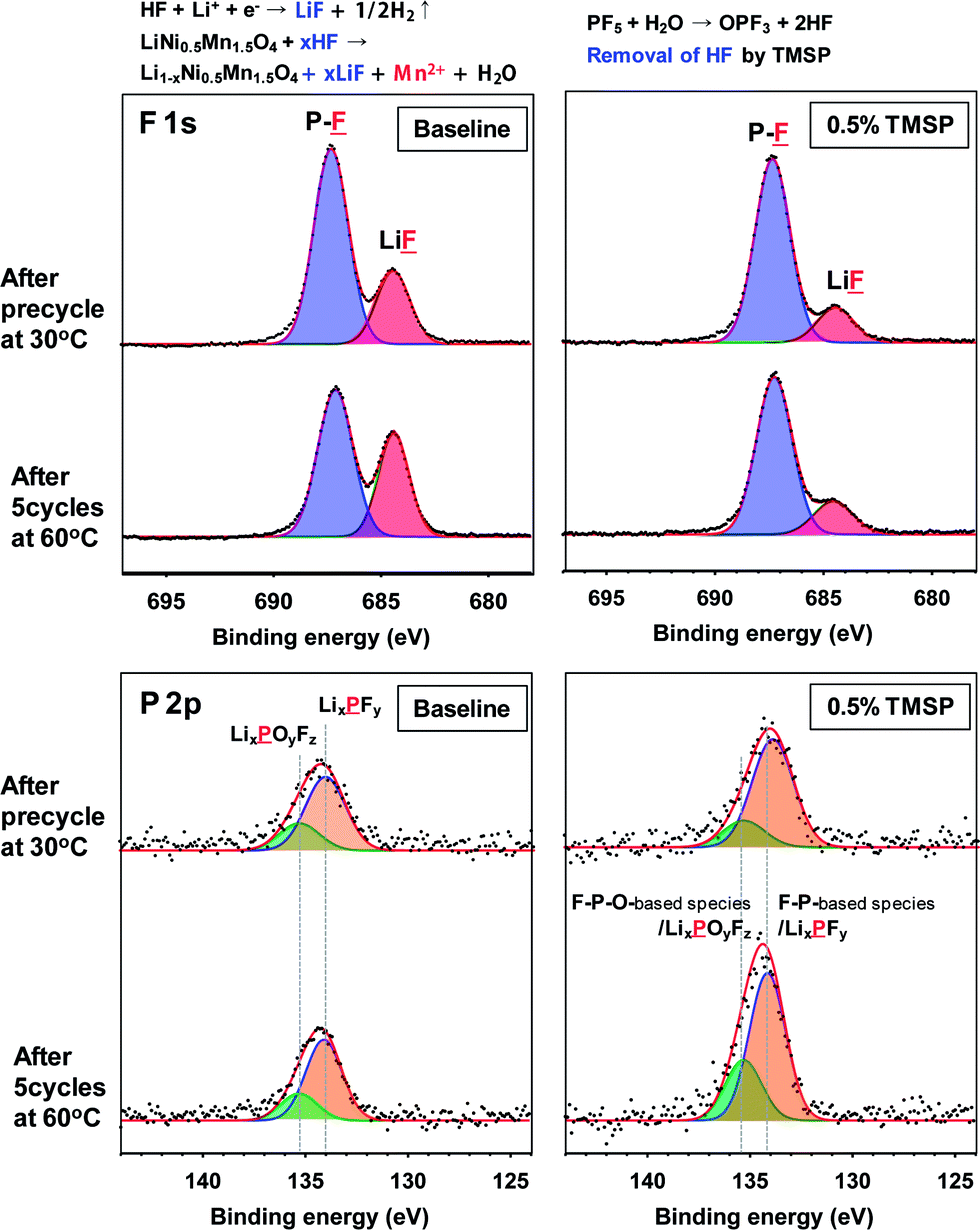 | ||
| Fig. 3 F 1s and P 2p XPS spectra of the LiNi0.5Mn1.5O4 cathodes after precycling at 30 °C and 5 cycles at 60 °C. | ||
HF produced from the LiPF6-based electrolyte solution may be eliminated by electrochemically oxidized TMSP and by directly reacting with TMSP, as depicted in Fig. 4. This HF removal is thought to suppress LiF formation on the cathode. This mechanism of action for HF removal is expected to suppress the dissolution of metal ions (Mn and Ni) from the cathode. The possible mechanisms for the HF removal by the TMSP additive are displayed in Fig. 4.
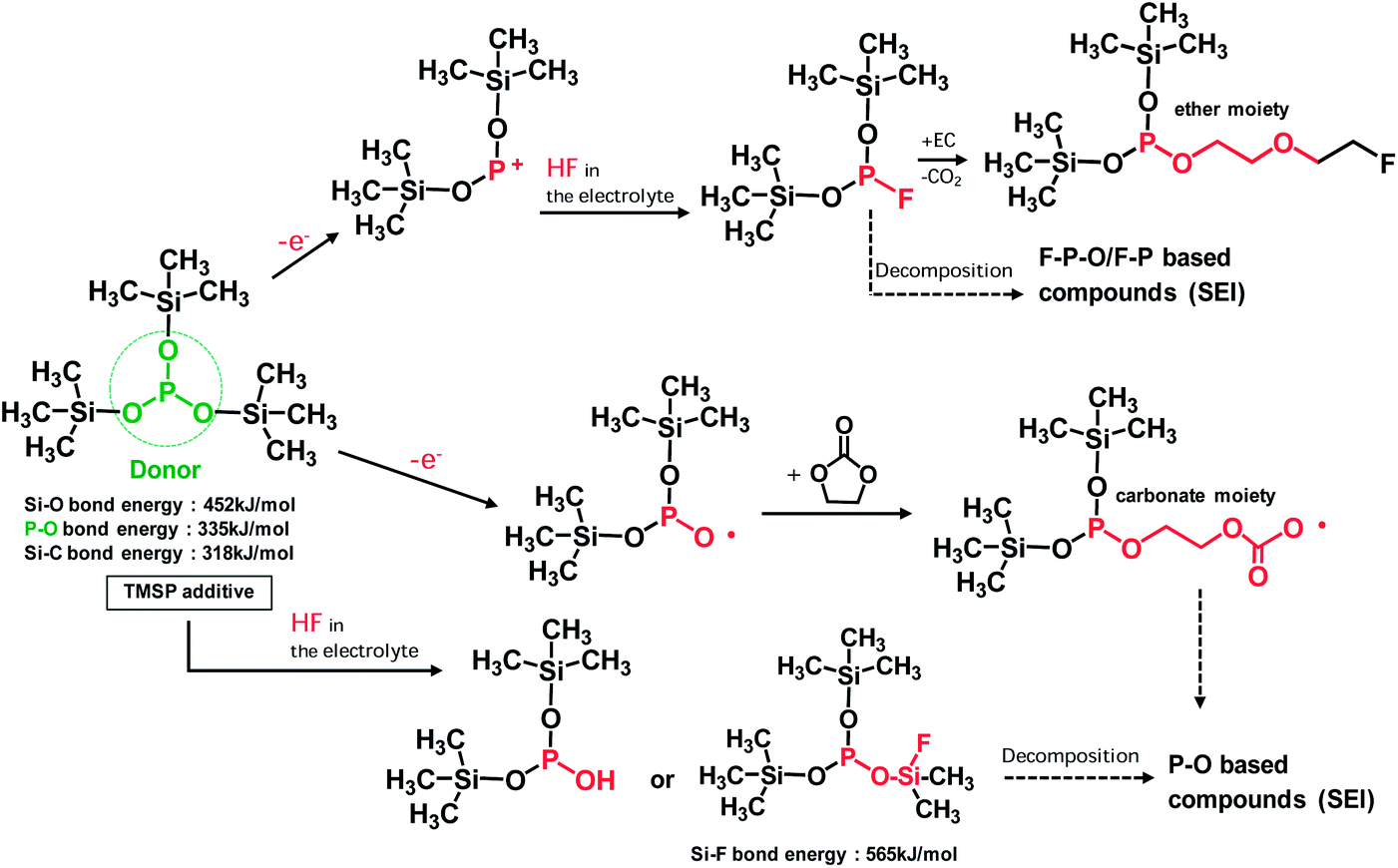 | ||
| Fig. 4 Schematic representation of possible mechanisms for electrochemical oxidative decomposition of TMSP and unique function of TMSP scavenging HF from the electrolyte. The products produced by TMSP decomposition will contribute to the formation of the SEI on the LiNi0.5Mn1.5O4 cathode. | ||
If TMSP is not entirely consumed during SEI formation on the cathode, it can react with HF in the electrolyte solution and lead to the formation of products with –P–OH or –P–O–Si(CH3)3−xFx groups (Fig. 4). Additionally, TMSP can produce F–Si(CH3)3 or –Si(CH3)3−xFx-containing compounds via the direct reaction with HF. The products derived from F–Si(CH3)3 or Si(CH3)3−xFx were not clearly detectable in the Si 2p XPS spectra of the SEI on the cathode (Fig. S5†).
To clarify the significant role of TMSP on HF removal from the electrolyte, 5 vol% water was added to the baseline and TMSP-added electrolyte solutions and the resulting solutions were stored for 22 h at room temperature. The 19F NMR spectrum of the baseline electrolyte with 5 vol% water showed three different doublets consistent with a single phosphorous coupled to each fluorine (Fig. S6†). The pronounced doublet peaks at −72 and −72.7 ppm corresponded to the PF6− anion. The peaks near −75 and −83 ppm could be assigned to PO3F2− and PO2F2−, respectively; these were produced by the hydrolysis of LiPF6 in the presence of water.24–26 Noticeable features of the TMSP-added electrolyte with 5 vol% water were the disappearance of the PO3F2− resonance near −75 ppm and prevention of LiF formation on the cathode surface by TMSP through HF produced by the hydrolysis of LiPF6. Indeed, the characteristic resonance of HF at −153 ppm apparently disappeared in the electrolyte with TMSP (see the 19F NMR spectrum of Fig. S6†). This result was persuasive evidence of TMSP effectively eliminating HF from the electrolyte solution, as displayed in Fig. 3b. It should be noted that a considerable degree of Mn dissolution from the LiMn2O4 cathode into the electrolyte takes place in the presence of HF that is formed by the hydrolysis of LiPF6 salts, and thereby, fast capacity fading of the cathode cannot be restrained.27–31 Similarly, Mn and Ni dissolution from the LiNi0.5Mn1.5O4 cathode can be driven by HF attack in the electrolyte.32 Surprisingly, the amount of Mn and Ni dissolution from the non-cycled LiNi0.5Mn1.5O4 cathode stored in the TMSP-containing electrolyte was negligible compared to the cathode in contact with the baseline electrolyte at 60 °C (Fig. 6d). This revealed that the removal of HF by the TMSP additive was very effectual in alleviating Mn and Ni dissolution from the LiMn1.5Ni0.5O4 cathode. The P 2p spectra in Fig. 3 clearly showed peaks for LixPFy (F–P) (at 134 eV) and LixPFyOz (F–P–O) (at 135.5 eV) in the SEI on the cathodes cycled in the baseline and TMSP-added electrolytes. The intensities of these peaks discernibly increased in the TMSP-added electrolyte. This was likely because F–P–O and F–P intermediates were generated by the decomposition of TMSP upon cycling at 60 °C (see possible reactions in Fig. 4). Comparison of the O 1s XPS spectra for the SEI on similarly cycled cathodes revealed that the relative fractions of carboxylate/carbonates (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) and ethers (C–O–C) in the TMSP-added electrolyte increased compared to the baseline electrolyte (Fig. S7†). Ethers can be formed by the reaction between –P–F species and organic solvents such as EC, and carbonate/carboxylates can be generated by the reaction between the oxy radical (–P–O˙) and EC (Fig. 4). From the XPS results, we confirmed that TMSP modified the surface chemistry of the SEI; the resulting SEI prevented further electrolyte decomposition during cycling and preserved the electrochemical performance of the LiNi0.5Mn1.5O4 cathode.
O) and ethers (C–O–C) in the TMSP-added electrolyte increased compared to the baseline electrolyte (Fig. S7†). Ethers can be formed by the reaction between –P–F species and organic solvents such as EC, and carbonate/carboxylates can be generated by the reaction between the oxy radical (–P–O˙) and EC (Fig. 4). From the XPS results, we confirmed that TMSP modified the surface chemistry of the SEI; the resulting SEI prevented further electrolyte decomposition during cycling and preserved the electrochemical performance of the LiNi0.5Mn1.5O4 cathode.
The discharge capacity retention of full cells constructed with the graphite anode, the LiNi0.5Mn1.5O4 cathode, and the electrolyte with or without 0.5% TMSP at 30 °C is shown in Fig. 5a. The capacity retention at the 100th cycle is much higher for the cell containing TMSP (81%) than for the cell without TMSP (73%). Interestingly, the graphite/LiNi0.5Mn1.5O4 full cell cycled in the TMSP-added electrolyte exhibited a discernible improvement in coulombic efficiency (over 99.6%) upon cycling at 30 °C (Fig. 5b). This result was in good agreement with Fig. S3.† Galvanostatic charge–discharge curves of a full cell with TMSP clearly shows much higher discharge capacity for 1st, 30th, 50th and 80th cycles compared to the baseline electrolyte (Fig. 5c and d). Initial discharge and charge curves of the graphite anodes with or without 0.5% TMSP are shown in Fig. S8.† The ICE of the graphite anode precycled in a 0.5% TMSP-added electrolyte slightly decreased from 95.3 to 95.1%. This is probably because HF removal by TMSP modified the SEI on the graphite anode. In addition, the discharge capacity retention of graphite/LiNi0.5Mn1.5O4 full cells with the TMSP-added electrolyte was significantly improved from 48.2 to 81.0% after 100 cycles at 45 °C (Fig. S9†). It is thus strongly believed that TMSP predominantly modifies the cathode surface and the resulting TMSP-derived SEI on the cathode is sufficiently robust to suppress severe electrolyte decomposition under high voltage conditions.
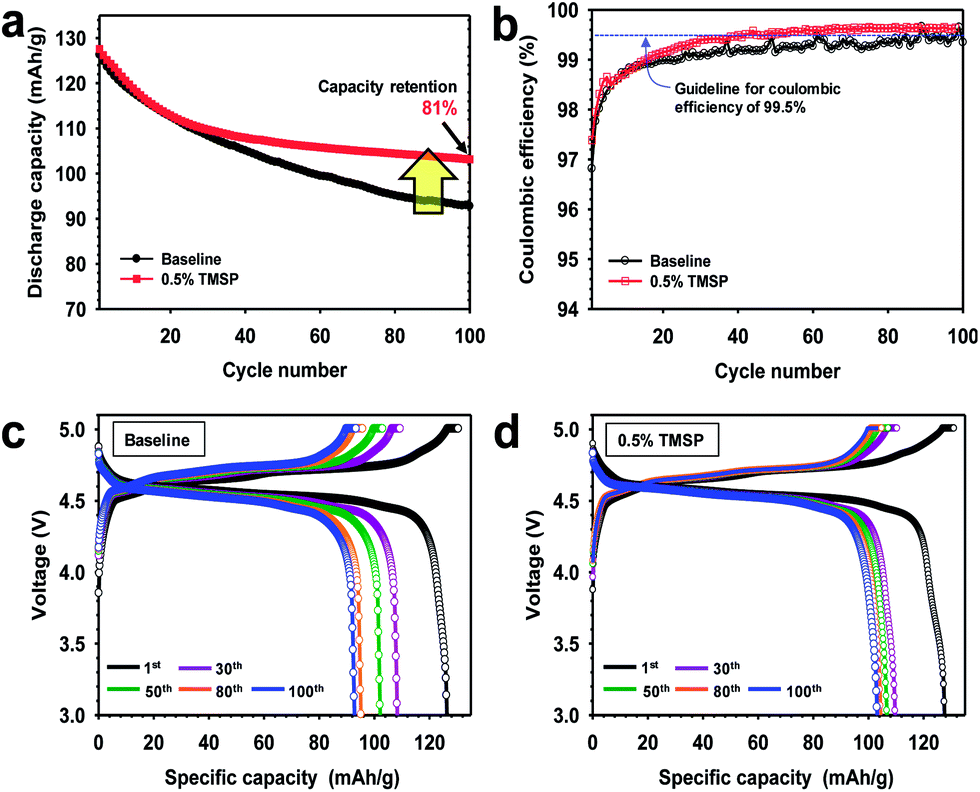 | ||
| Fig. 5 Electrochemical performance of graphite/LiNi0.5Mn1.5O4 full cells at C/2 and 30 °C when cycled between 3.0 and 5.0 V: (a) cycling stability and (b) coulombic efficiency, charge and discharge curves of the full cell for 1st, 30th, 50th, 80th and 100th cycles in (c) baseline electrolyte and (d) TMSP-added electrolyte. A very high coulombic efficiency (red square) of over 99.5%, which is vital for practical applications, was obtained. | ||
The significant capacity fading of the graphite/LiNi0.5Mn1.5O4 full cell due to Mn and Ni dissolution from the cathode was similarly expected to be suppressed in the presence of TMSP eliminating HF from the electrolyte. It should be noted that a small amount of Mn and Ni ions in the electrolyte causes a significant fading of the capacity of a graphite anode because the Mn and Ni deposits are formed by consuming the electrons for intercalation of Li+ ions into the graphite and the metallic Mn and Ni can lead to the formation of thick SEI layers.
To verify this, the graphite anode from a graphite/LiNi0.5Mn1.5O4 full cell after 100 cycles was analyzed by EDS measurements. Mn and Ni signals were clearly observed on the graphite anode in a full cell that was cycled during 100 cycles in the baseline electrolyte, whereas the presence of TMSP excluded the peaks assigned to Mn and Ni elements (see Fig. 6b and c). This is probably because TMSP eliminated HF from the electrolyte and thereby Mn and Ni dissolution from the cathode was effectively restrained (see Fig. 6e). Further evidence, which proves that the Mn and Ni dissolution by HF attack is drastically alleviated in the presence of TMSP, is shown in Fig. 6d. It is also possible that the TMSP-derived SEI on the cathode imparts resistance to the metal dissolution under high voltage conditions. Unlike the pristine graphite showing a very clean surface, the graphite anode from a full cell cycled in the baseline electrolyte during 100 cycles revealed a bumpy surface due to non-uniform SEI layers (Fig. 6b and e).
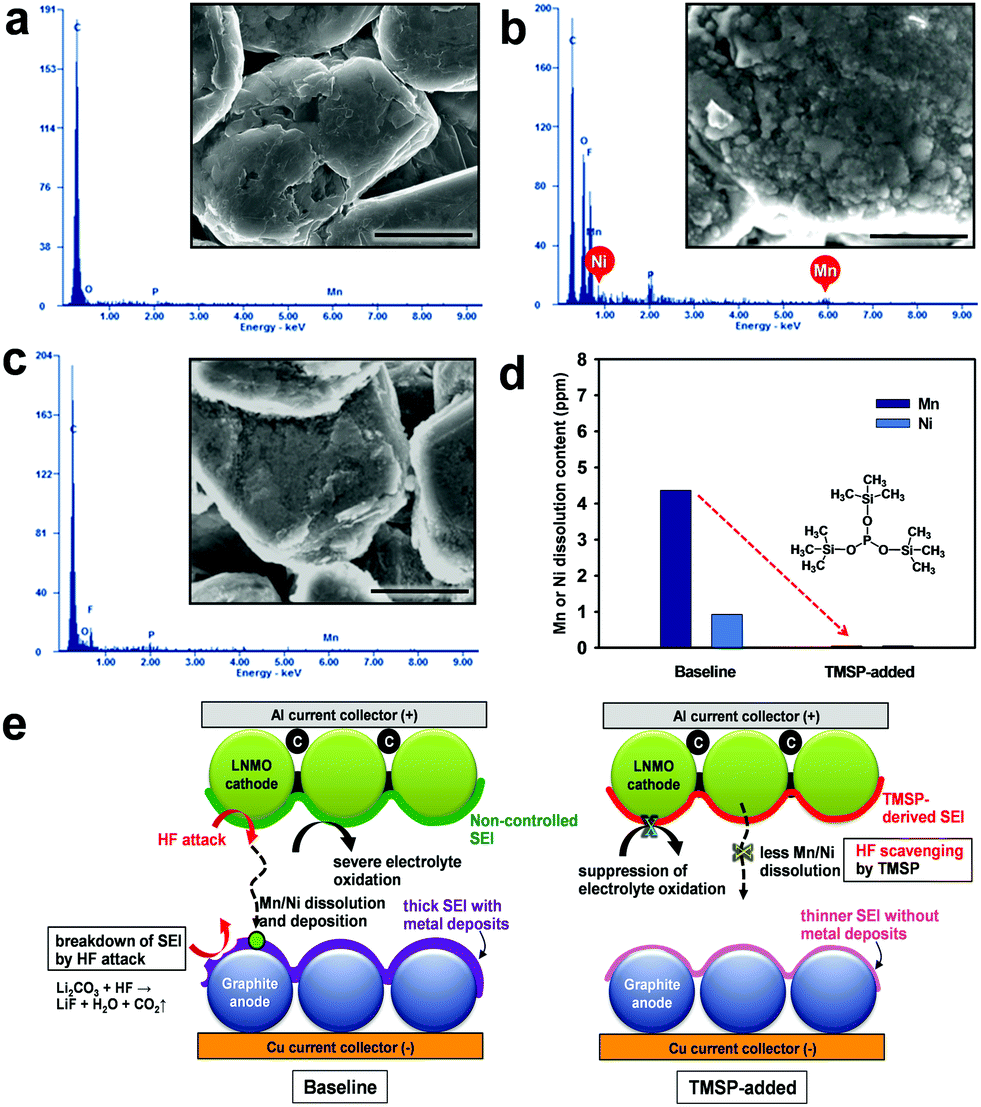 | ||
| Fig. 6 EDS patterns and SEM images of (a) pristine graphite and graphite anodes retrieved from graphite/LiNi0.5Mn1.5O4 full cells after 100 cycles at 30 °C in (b) baseline electrolyte and (c) TMSP-added electrolyte. Scale bar represents 5 μm. (d) ICP result showing the amount of Mn and Ni dissolution from non-cycled LiNi0.5Mn1.5O4 cathodes in the electrolytes with and without TMSP at 60 °C for 12 h. The use of TMSP inhibited the Mn and Ni dissolution from the pristine cathode. (e) Schematic illustration of unique functions of TMSP in a full cell. | ||
It should be noted that metallic Mn and Ni deposited on the surface of the graphite anode lead to a significant decomposition of the electrolyte and result in the formation of thick SEI layers (Fig. 6e). On the other hand, the graphite cycled in a TMSP-containing electrolyte did not display the rough surface. Therefore, it can be thought that the TMSP-containing electrolyte effectively suppresses the migration of Mn and Ni ions toward the graphite anode.
Comparison of the initial charge and discharge profiles of the Li/LiNi0.5Mn1.5O4 half cells in electrolytes with and without additives at 30 °C is presented in Fig. 7. Significant overcharging of the LiNi0.5Mn1.5O4 cathode took place in a 1% VC-added electrolyte when charged up to 5.0 V; thereby, the initial coulombic efficiency (ICE) of the cathode drastically decreased to 71.2% compared with cells precycled in baseline, and FEC-added and TMSP-added electrolytes. The voltage plateau around 4.85 V and the low ICE of the Li/LiNi0.5Mn1.5O4 cell with 1% VC could be explained by severe VC decomposition on the high voltage cathode surface at 30 °C.
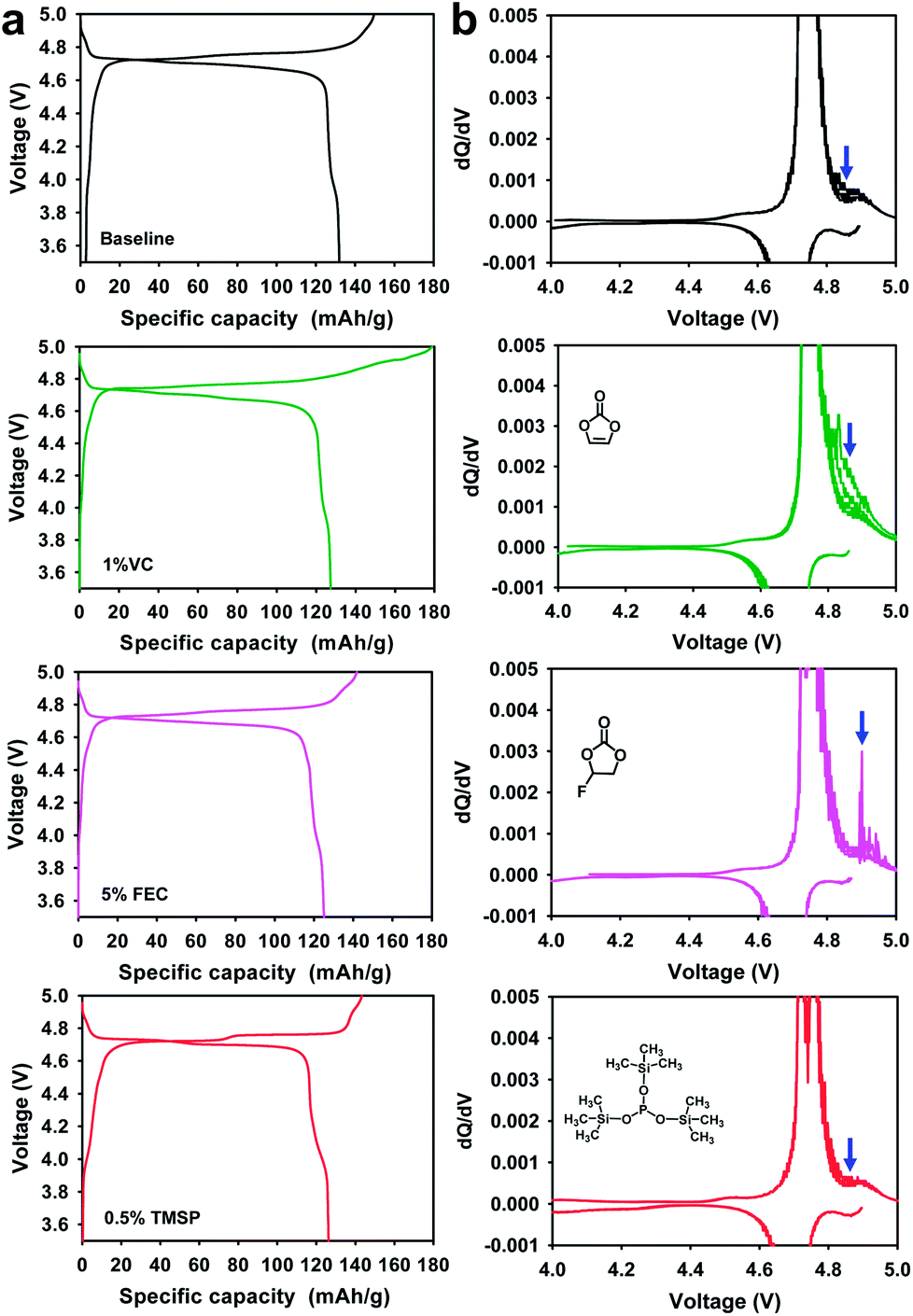 | ||
| Fig. 7 (a) Initial charge and discharge profiles at 30 °C and (b) dQ/dV graphs of 5 V-class LiNi0.5Mn1.5O4 cathodes in various electrolytes during 5 cycles at 60 °C after initial cycling at 30 °C. | ||
Moreover, it was clear that the VC additive continuously underwent oxidative decomposition which resulted in a significant oxidative differential capacity at over 4.8 V during 5 cycles at 60 °C (see dQ/dV in Fig. 7b). This is because the VC additive is prone to oxidize at a high charge potential of 5.0 V due to its relatively high HOMO energy level, compared to conventional carbonate solvents such as EC (Fig. 1). By field-emission scanning electron microscopy (FE-SEM), the cathode cycled in the baseline electrolyte seemed to be partly covered with small particles, whereas the surface film originating from VC decomposition appeared to be discontinuous and relatively thick on the cathode surface after cycling at 60 °C (Fig. S10b and c†). Although preferential reduction of VC as the most effective additive prior to other electrolyte solvents resulted in a stable SEI on the anode,33,34 VC showed the worst oxidation stability toward high voltage cathodes. Moreover, the VC-derived SEI was not maintained during cycling and was appreciably peeled away from the cathode surface, as shown in Fig. S10c.† To investigate the cathode SEI formed by VC, the surface of the cathode after 5 cycles at 60 °C was examined by ex situ XPS (Fig. S11†). The C 1s and O 1s XPS spectra clearly showed that VC produced ether (C–O–C) and carboxylate groups as the dominant species in the LiNi0.5Mn1.5O4 cathode surface layer. Interestingly, peaks that could be assigned to poly(VC), which would generally be observed on the anode, were not detected on the LiNi0.5Mn1.5O4 cathode cycled in the VC-added electrolyte (see O 1s and C 1s XPS of Fig. S11†). This implied that VC did not decompose into poly(VC) on the cathode charged up to 5.0 V. Comparing the initial charge and discharge profiles of the LiNi0.5Mn1.5O4 cathodes in electrolytes with and without a 5% FEC-added electrolyte at 30 °C, there were no significant differences in charge and discharge capacities (Fig. 7a). However, the dQ/dV graph obtained during 5 cycles at 60 °C clearly showed that appreciable differential capacity was generated by the severe oxidative decomposition of the FEC-added electrolyte (Fig. 7b). It is thought that FEC undergoes considerable oxidative decomposition on the high voltage cathode surface at 60 °C compared to the baseline electrolyte.
Although an FEC-derived surface film was formed on the cathode (see Fig. S10d†), it did not preserve the electrochemical performance of the LiNi0.5Mn1.5O4 cathode during cycling at 60 °C (Fig. S2†). The dQ/dV graphs confirmed that the VC and FEC additives were detrimental to the interfacial stability of LiNi0.5Mn1.5O4 cathodes charged up to 5 V.
The TMSP-added electrolyte exhibited a slightly reduced ICE of 88.0% compared to the baseline electrolyte (88.2%). This was probably because the TMSP decomposition reaction on the cathode resulted in a capacity loss during the first charge and discharge process. Since the HOMO energy (−8.097 eV) of TMSP is higher than that of EC (−11.905 eV) (Fig. 1), TMSP is more prone to lose electrons relative to the EC solvent when charged up to 5.0 V. Thus, TMSP oxidation on the LiNi0.5Mn1.5O4 cathode is thought to take place prior to that of other electrolyte components (solvents and salt), forming the cathodic SEI. The characteristics of the SEI formed on the cathode represent a key parameter that influences the kinetics of delithiation–lithiation and the interfacial stability during long-term cycling. Importantly, the LiNi0.5Mn1.5O4 cathode cycled in the TMSP-added electrolyte showed no differential capacity attributed to the oxidative decomposition of the electrolyte when charged up to 5.0 V at 60 °C (see the TMSP-added sample in Fig. 7b). Moreover, the cathode cycled in the TMSP-added electrolyte at 60 °C had a very clean and smooth surface (Fig. S10e†). This implied that the TMSP-derived SEI formed during precycling was maintained and effectively alleviated further electrolyte decomposition during cycling at 60 °C. The distinction between the TMSP-added and other electrolytes can be ascribed to the differences in the electrochemical natures of the SEIs formed on the high voltage cathodes.
To verify the superior anodic stability of the TMSP-added electrolyte, the leakage current of Li/LiNi0.5Mn1.5O4 cells was monitored at a constant charging voltage of 5.0 V for 5 h. The baseline electrolyte showed a much larger leakage current, which indicated significant oxidative decomposition of the electrolyte, whereas the presence of TMSP greatly reduced the leakage current (Fig. S12a†). Linear sweep voltammetry (LSV) results confirmed that the anodic stability of the electrolyte could be improved by TMSP (Fig. S12b†). These results suggested that the TMSP-derived SEI formed on the cathode surface helped the electrolyte tolerate a high voltage of 5.0 V.
Conclusions
We have demonstrated for the first time that the TMSP-containing electrolyte, which eliminated HF from the electrolyte and modified the surface chemistry of high voltage cathodes, significantly improved the cycling stability and rate capability of LiNi0.5Mn1.5O4 cathodes in half-cells and full-cells with the graphite anode. Cycling tests of LiNi0.5Mn1.5O4 cathodes at elevated temperature confirmed that robust SEIs were essential to inhibit unwanted electrolyte decomposition, to alleviate Mn/Ni dissolution, and to preserve the electrochemical properties of high voltage cathodes. Furthermore, the effects of TMSP on the chemical structure of the SEI layer, composed of inorganic phosphorous-based species with organic ether and carboxylate/carbonate moieties and reduced LiF, were clearly revealed by ex situ XPS analyses after cycling. The unique function of the TMSP additive-eliminating HF from the electrolyte solution was elucidated by 19F and 31P NMR studies. We believe that the results of this study and the associated analysis will contribute to the design of suitable electrolyte systems for 5 V-class LiNi0.5Mn1.5O4 cathodes, with the promise of further improvements in electrochemical performance.Experimental method
The electrolyte with and without 5 wt% FEC (Soulbrain Co. Ltd.), 1 wt% VC (Soulbrain Co. Ltd.), or TMSP (Aldrich) was composed of commercially available 1.0 M lithium hexafluorophosphate (LiPF6) dissolved in a solvent mixture of ethylene carbonate (EC), ethyl methyl carbonate (EMC), and dimethyl carbonate (DMC) in a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:3 volume ratio. FEC, VC, and TMSP were used as received and introduced as additives for LiNi0.5Mn1.5O4 cathodes. Molecular orbital calculations were performed by MOPAC, a semi-empirical molecular orbital program. A slurry was prepared by mixing 90 wt% LiNi0.5Mn1.5O4 particles (GS Energy and Materials Co. Ltd.), 5 wt% carbon black as a conducting material, and a 5 wt% polyvinylidene fluoride (PVDF) binder dissolved in anhydrous N-methyl-2-pyrrolidinone (NMP). The resulting slurry was cast on aluminum foil. The composite cathode was then dried in a convection oven at 110 °C for 30 min. The electrode was next pressed; its thickness was around 34 μm. The specific capacity of the cathode and the active material loading were 0.45 mA h cm−2 and 3.97 mg cm−2.
:4:3 volume ratio. FEC, VC, and TMSP were used as received and introduced as additives for LiNi0.5Mn1.5O4 cathodes. Molecular orbital calculations were performed by MOPAC, a semi-empirical molecular orbital program. A slurry was prepared by mixing 90 wt% LiNi0.5Mn1.5O4 particles (GS Energy and Materials Co. Ltd.), 5 wt% carbon black as a conducting material, and a 5 wt% polyvinylidene fluoride (PVDF) binder dissolved in anhydrous N-methyl-2-pyrrolidinone (NMP). The resulting slurry was cast on aluminum foil. The composite cathode was then dried in a convection oven at 110 °C for 30 min. The electrode was next pressed; its thickness was around 34 μm. The specific capacity of the cathode and the active material loading were 0.45 mA h cm−2 and 3.97 mg cm−2.
The specific capacity of the thick cathode for a full cell coupled with the graphite anode was 2.24 mA h cm−2. The anode was composed of 97 wt% natural graphite and a 2.5 wt% binder (1.5 wt% styrene-butadiene rubber +1 wt% sodium carboxymethyl cellulose).
A coin-type half cell (2032) with a LiNi0.5Mn1.5O4 cathode and a Li metal electrode was assembled in an argon-filled glove box with less than 1 ppm of either oxygen or moisture. The thickness and porosity of the microporous polyethylene film (SK Innovation Co., Ltd.) used as a separator were 20 μm and 38%, respectively. The coin-type half cells were galvanostatically precycled at a current density of 24 mA g−1 (corresponding to 0.2 C) between 3.5 and 5.0 V at 30 °C using a computer-controlled battery measurement system (WonATech WBCS 3000). Thereafter, charge and discharge cycling for the cells was performed for a current density of 60 mA g−1 (corresponding to 0.5 C) at 30 and 60 °C. To demonstrate the impact of TMSP on the rate capability of the LiNi0.5Mn1.5O4 cathodes, cells were charged and discharged at 30 °C using various C rates: 0.2, 0.5, 1, 2, and 3 C. dQ/dV graphs were obtained by computing the differential capacity versus the potential of the cells during 5 cycles at 60 °C. The coin-type full cells were galvanostatically precycled at a rate of 0.1 C between 3.0 and 5.0 V at 30 °C. Thereafter, the graphite/LiNi0.5Mn1.5O4 full cells were cycled at a rate of 0.5 C at 30 °C.
After cycling, the cells were carefully opened in a glove box to retrieve their electrodes. The electrodes were rinsed with dimethyl carbonate (DMC) to remove the residual LiPF6-based electrolyte, and the resulting materials were dried at room temperature for analysis using ex situ XPS (Thermo Scientific K-Alpha System). The XPS measurements were performed with Al Kα (hν = 1486.6 eV) radiation under ultrahigh vacuum, using a 0.10 eV step and 50 eV pass energy. All XPS spectra were energy calibrated by the hydrocarbon peak at a binding energy of 284.8 eV. The surface morphologies of the electrodes were observed by FE-SEM (FEI Nanonova 230). Samples were prepared in a glove box and sealed with an aluminum pouch film under vacuum before use. Then, samples were rapidly transferred into the chamber of the XPS or FE-SEM instrument to minimize any possible contamination. 19F and 31P NMR spectra were recorded on an Agilent VNMRS-600 spectrometer with THF-d8 as a solvent. 19F and 31P NMR resonances were referenced to LiPF6 at −72.4 ppm and −146 ppm, respectively. To understand the dissolution behaviors of manganese and nickel ions from LiNi0.5Mn1.5O4 cathodes at 60 °C, a typical experiment was performed as follows. A pristine LiNi0.5Mn1.5O4 cathode was soaked in EC/DMC/EMC (3/4/3, volume ratio) with 1.0 M LiPF6 in a polyethylene bottle under argon. After sealing with an aluminum pouch film, the polyethylene bottle was stored in a convection oven at 60 °C for 12 h. The amounts of manganese (Mn) and nickel (Ni) ions in the electrolytes were then measured via inductively coupled plasma (ICP).
Acknowledgements
This research was supported by the MSIP (Ministry of Science, ICT & Future Planning), Korea, under the C-ITRC (Convergence Information Technology Research Center) support program (NIPA-2013-H0301-13-1009) supervised by the NIPA(National IT Industry Promotion Agency) and by the IT R&D program of MKE/KEIT (KI001810039182, development of a 5 V cathode material whose capacity is 125 mA g−1 and high voltage electrolyte whose decomposition is over 5 V for a lithium secondary battery). This study was partly supported by a National Research Foundation of Korea Grant funded by the Korean Government (MEST) (NRF-2013-C1AAA001-0030538).Notes and references
- N.-S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef CAS PubMed.
- K. Xu, Chem. Rev., 2004, 104, 4303 CrossRef CAS.
- L. Yang, B. Ravdel and B. Lucht, Electrochem. Solid-State Lett., 2010, 13, A95 CrossRef CAS PubMed.
- S. Brutti, V. Gentili, P. Reale, L. Carbone and S. Panero, J. Power Sources, 2011, 196, 9792 CrossRef CAS PubMed.
- L. Baggetto, R. R. Unocic, N. J. Dudney and G. M. Veith, J. Power Sources, 2012, 211, 108 CrossRef CAS PubMed.
- K. Xu and C. A. Angell, J. Electrochem. Soc., 2002, 149, A920 CrossRef CAS PubMed.
- N. Shao, X.-G. Sun, S. Dai and D. Jiang, J. Phys. Chem. B, 2012, 116, 3235 CrossRef CAS PubMed.
- S. K. Martha, E. Markevich, V. Burgel, G. Salitra, E. Zinigrad, B. Markovsky, H. Sclar, Z. Pramovich, O. Heik, D. Aurbach, I. Exnar, H. Buqa, T. Drezen, G. Semrau, M. Schmidt, D. Kovacheva and N. Saliyski, J. Power Sources, 2009, 189, 288 CrossRef CAS PubMed.
- V. Borgel, E. Markevich, D. Aurbach, G. Semrau and M. Schmidt, J. Power Sources, 2009, 189, 331 CrossRef CAS PubMed.
- Y. A. Lebdeh and I. Davidson, J. Electrochem. Soc., 2009, 156, A60 CrossRef PubMed.
- Z. Zhang, L. Hu, H. Wu, W. Weng, M. Koh, P. C. Redfern, L. A. Curtiss and K. Amine, Energy Environ. Sci., 2013, 6, 1806 CAS.
- A. V. Cresce and K. Xu, J. Electrochem. Soc., 2011, 158, A337 CrossRef PubMed.
- K. Xu, S. Zhang, T. R. Jow, W. Xu and C. A. Angell, Electrochem. Solid-State Lett., 2005, 8, A365 CrossRef CAS PubMed.
- N.-S. Choi, K. H. Yew, H. Kim, S.-S. Kim and W.-U. Choi, J. Power Sources, 2007, 172, 404 CrossRef CAS PubMed.
- K. Xu, S. S. Zhang, U. Lee, J. L. Allen and T. R. Jow, J. Power Sources, 2005, 146, 79 CrossRef CAS PubMed.
- S.-Y. Ha, J.-G. Han, Y.-M. Song, M.-J. Chun, S.-I. Han, W.-C. Shin and N.-S. Choi, Electrochim. Acta, 2013, 104, 170 CrossRef CAS PubMed.
- N. P. W. Pieczonka, L. Yang, M. P. Balogh, B. R. Powell, K. Chemelewski, A. Manthiram, S. A. Krachkovskiy, G. R. Goward, M. Liu and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 22603 CAS.
- A. Manthiram, K. Chemelewski and E. lee, Energy Environ. Sci., 2014, 7, 1339 CAS.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947 CAS.
- H. Duncan, Y. Abu-Lebdeh and I. J. Davidson, J. Electrochem. Soc., 2010, 157, A528 CrossRef CAS PubMed.
- K. Tasaki, A. Goldberg, J.-J. Lian, M. Walker, A. Timmons and S. J. Harris, J. Electrochem. Soc., 2009, 156, A1019 CrossRef CAS PubMed.
- N.-S. Choi, Y. Yao, Y. Cui and J. Cho, J. Mater. Chem., 2011, 21, 9825 RSC.
- M.-J. Chun, H. Park, S. Park and N.-S. Choi, RSC Adv., 2013, 3, 21320 RSC.
- H.-B. Han, S.-S. Zhou, D.-J. Zhang, S.-W. Feng, L.-F. Li, K. Liu, W.-F. Feng, J. Nie, H. Li, X.-J. Huang, M. Armand and Z.-B. Zhou, J. Power Sources, 2011, 196, 3623 CrossRef CAS PubMed.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152, A2327 CrossRef CAS PubMed.
- S. Dalavi, M. Xu, B. Ravdel, L. Zhou and B. L. Lucht, J. Electrochem. Soc., 2010, 157, A1113 CrossRef CAS PubMed.
- I. H. Cho, S.-S. Kim, S. C. Shin and N.-S. Choi, Electrochem. Solid-State Lett., 2010, 13, A168 CrossRef CAS PubMed.
- R. J. Gummow and A. de Kock, Solid State Ionics, 1994, 69, 59 CrossRef CAS.
- E. Wang, D. Ofer, W. Bowden, N. Iltchev, R. Moses and K. Brandt, J. Electrochem. Soc., 2000, 147, 4023 CrossRef CAS PubMed.
- H. Tsunekawa, S. Tanimoto, R. Marubayashi, M. Fujita, K. Kifune and M. Sano, J. Electrochem. Soc., 2002, 149, A1326 CrossRef CAS PubMed.
- N.-S. Choi, J.-T. Yeon, Y.-W. Lee, J.-G. Han, K. T. Lee and S.-S. Kim, Solid State Ionics, 2012, 219, 41 CrossRef CAS PubMed.
- N. P. W. Pieczonka, Z. Liu, P. Lu, K. L. Olson, J. Moote, B. R. Powell and J.-H. Kim, J. Phys. Chem. C, 2013, 117, 15947 CAS.
- S.-K. Jeong, M. Inaba, R. Mogi, Y. Iriyama, T. Abe and Z. Ogumi, Langmuir, 2001, 17, 8281 CrossRef CAS.
- M. Itagaki, S. Yotsuda, N. Kobari, K. Watanabe, S. Kinoshita and M. Ue, Electrochim. Acta, 2006, 51, 1629 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ta01129e |
| This journal is © The Royal Society of Chemistry 2014 |